A Single Amino Acid Residue Regulates PTEN-Binding and Stability of the Spinal Muscular Atrophy Protein SMN

 , , , ,
, , , ,  , and
, and
Abstract
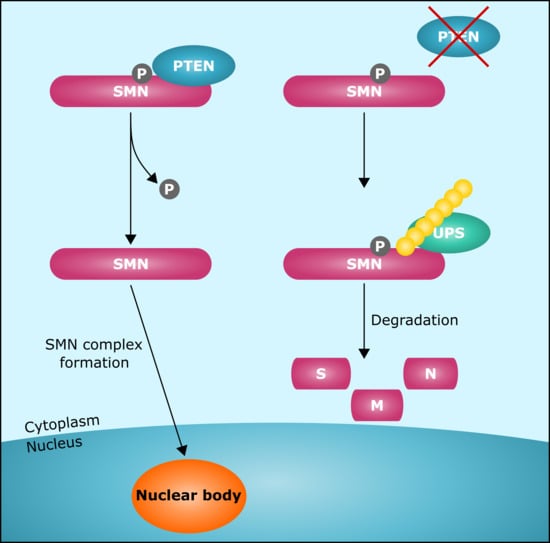
:
{kind=link}
{kind=link}
{kind=link}
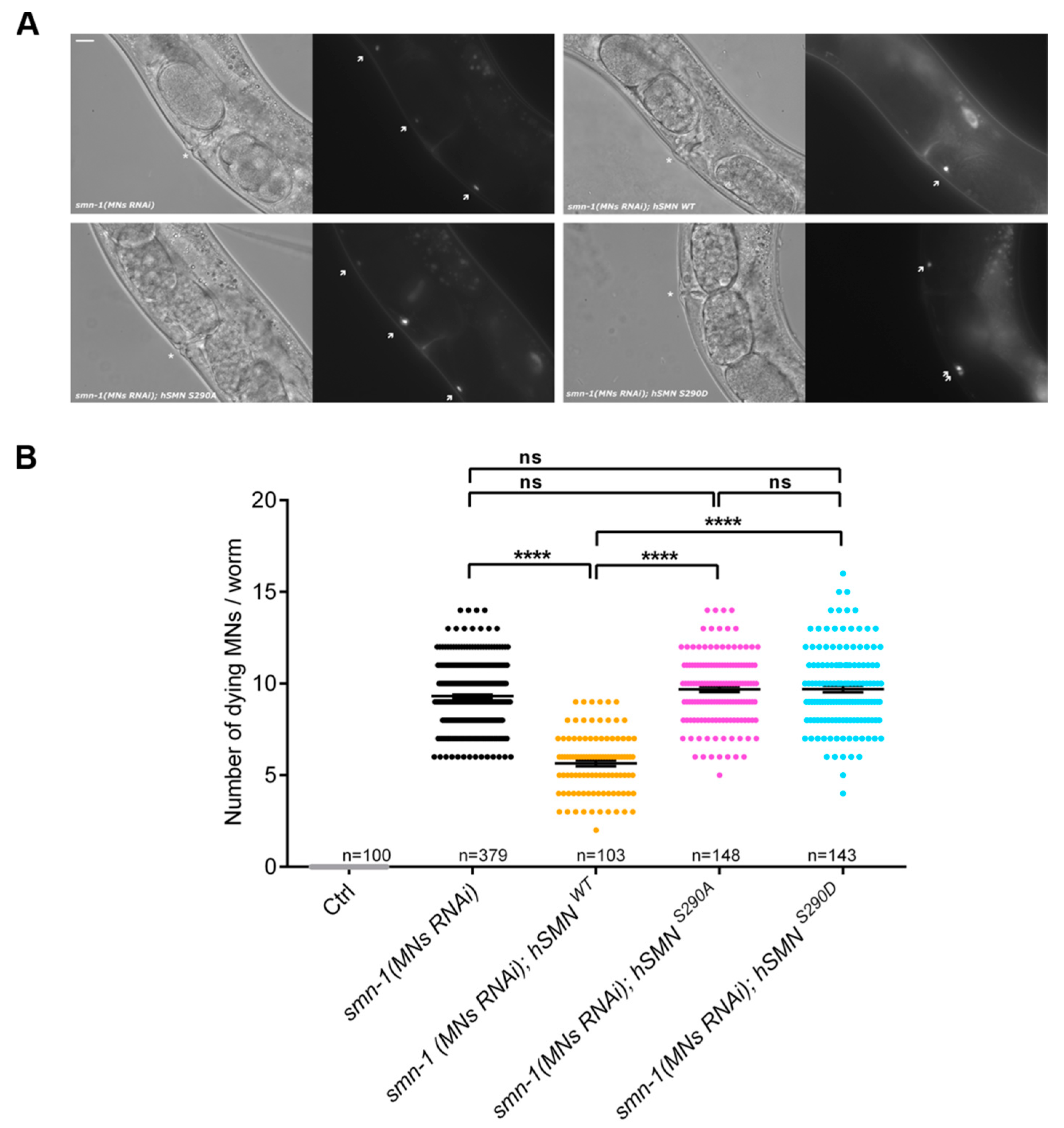{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Transfection
2.2. Plasmids, siRNAs and Inhibitors
2.3. Immunoprecipitation
2.4. Mass Spectrometry
2.5. Isoelectric Focusing
2.6. Western Blot Analysis
2.7. Pulse-Chase Assay
2.8. C. elegans Experiments
2.9. Quantitative Reverse-Transcription PCR
2.10. Immunocytochemistry
2.11. Microscopy and Evaluation of Images
2.12. Quantification of Nuclear Bodies
2.13. Statistics and Curve Fitting
3. Results
3.1. Identification of Novel Phosphorylation Sites in Human SMN
3.2. Characterization of hSMN S290 Phospho-Mutant and Phospho-Mimetic
3.3. PTEN is a Novel SMN-Interacting Partner
3.4. PTEN Dephosphorylates SMN In Vitro
3.5. Pten Knockdown Negatively Influences SMN Stability and Reduces the Number of SMN-Positive Nuclear Bodies
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lefebvre, S.; Burglen, L.; Reboullet, S.; Clermont, O.; Burlet, P.; Viollet, L.; Benichou, B.; Cruaud, C.; Millasseau, P.; Zeviani, M.; et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 1995, 80, 155–165. [Google Scholar] [CrossRef] [Green Version]
- Lorson, C.L.; Hahnen, E.; Androphy, E.J.; Wirth, B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc. Natl. Acad. Sci. USA 1999, 96, 6307–6311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vitte, J.; Fassier, C.; Tiziano, F.D.; Dalard, C.; Soave, S.; Roblot, N.; Brahe, C.; Saugier-Veber, P.; Bonnefont, J.P.; Melki, J. Refined characterization of the expression and stability of the SMN gene products. American J. Pathol. 2007, 171, 1269–1280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Amico, A.; Mercuri, E.; Tiziano, F.D.; Bertini, E. Spinal muscular atrophy. Orphanet J. Rare Dis. 2011, 6, 71. [Google Scholar] [CrossRef] [Green Version]
- Campbell, L.; Potter, A.; Ignatius, J.; Dubowitz, V.; Davies, K. Genomic variation and gene conversion in spinal muscular atrophy: Implications for disease process and clinical phenotype. Am. J. Hum. Genet. 1997, 61, 40–50. [Google Scholar] [CrossRef] [Green Version]
- Finkel, R.S.; Chiriboga, C.A.; Vajsar, J.; Day, J.W.; Montes, J.; De Vivo, D.C.; Yamashita, M.; Rigo, F.; Hung, G.; Schneider, E.; et al. Treatment of infantile-onset spinal muscular atrophy with nusinersen: A phase 2, open-label, dose-escalation study. Lancet 2016, 388, 3017–3026. [Google Scholar] [CrossRef]
- Finkel, R.S.; Mercuri, E.; Darras, B.T.; Connolly, A.M.; Kuntz, N.L.; Kirschner, J.; Chiriboga, C.A.; Saito, K.; Servais, L.; Tizzano, E.; et al. Nusinersen versus Sham Control in Infantile-Onset Spinal Muscular Atrophy. N. Engl. J. Med. 2017, 377, 1723–1732. [Google Scholar] [CrossRef] [Green Version]
- Mendell, J.R.; Al-Zaidy, S.; Shell, R.; Arnold, W.D.; Rodino-Klapac, L.R.; Prior, T.W.; Lowes, L.; Alfano, L.; Berry, K.; Church, K.; et al. Single-Dose Gene-Replacement Therapy for Spinal Muscular Atrophy. N. Engl. J. Med. 2017, 377, 1713–1722. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.N.; Howell, M.D.; Ottesen, E.W.; Singh, N.N. Diverse role of survival motor neuron protein. Biochim. Biophys. Acta Gene Regul. Mech. 2017, 1860, 299–315. [Google Scholar] [CrossRef] [Green Version]
- Meister, G.; Eggert, C.; Fischer, U. SMN-mediated assembly of RNPs: A complex story. Trends Cell Biol. 2002, 12, 472–478. [Google Scholar] [CrossRef]
- Paushkin, S.; Gubitz, A.K.; Massenet, S.; Dreyfuss, G. The SMN complex, an assemblyosome of ribonucleoproteins. Curr. Opin. Cell Biol. 2002, 14, 305–312. [Google Scholar] [CrossRef]
- Gruss, O.J.; Meduri, R.; Schilling, M.; Fischer, U. UsnRNP biogenesis: Mechanisms and regulation. Chromosoma 2017, 126, 577–593. [Google Scholar] [CrossRef]
- Otter, S.; Grimmler, M.; Neuenkirchen, N.; Chari, A.; Sickmann, A.; Fischer, U. A comprehensive interaction map of the human survival of motor neuron (SMN) complex. J. Biol. Chem. 2007, 282, 5825–5833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanek, D.; Neugebauer, K.M. The Cajal body: A meeting place for spliceosomal snRNPs in the nuclear maze. Chromosoma 2006, 115, 343–354. [Google Scholar] [CrossRef]
- Gangwani, L.; Mikrut, M.; Theroux, S.; Sharma, M.; Davis, R.J. Spinal muscular atrophy disrupts the interaction of ZPR1 with the SMN protein. Nat. Cell Biol. 2001, 3, 376–383. [Google Scholar] [CrossRef]
- Ahmad, S.; Bhatia, K.; Kannan, A.; Gangwani, L. Molecular Mechanisms of Neurodegeneration in Spinal Muscular Atrophy. J. Exp. Neurosci. 2016, 10, 39–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hensel, N.; Baskal, S.; Walter, L.M.; Brinkmann, H.; Gernert, M.; Claus, P. ERK and ROCK functionally interact in a signaling network that is compensationally upregulated in Spinal Muscular Atrophy. Neurobiol. Dis. 2017, 108, 352–361. [Google Scholar] [CrossRef]
- Hensel, N.; Claus, P. The Actin Cytoskeleton in SMA and ALS: How Does It Contribute to Motoneuron Degeneration? Neurosci. Rev. J. Bringing Neurobiol. Neurol. Psychiatry 2018, 24, 54–72. [Google Scholar] [CrossRef]
- Kannan, A.; Bhatia, K.; Branzei, D.; Gangwani, L. Combined deficiency of Senataxin and DNA-PKcs causes DNA damage accumulation and neurodegeneration in spinal muscular atrophy. Nucleic Acids Res. 2018, 46, 8326–8346. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.Y.; Curtis, A.; Choi, Y.S.; Maeda, M.; Xu, M.J.; Berg, A.; Joneja, U.; Mason, R.W.; Lee, K.H.; Wang, W. Identification of the phosphorylation sites in the survival motor neuron protein by protein kinase A. Biochim. Biophys. Acta Proteins Proteom. 2011, 1814, 1134–1139. [Google Scholar] [CrossRef] [Green Version]
- Petri, S.; Grimmler, M.; Over, S.; Fischer, U.; Gruss, O.J. Dephosphorylation of survival motor neurons (SMN) by PPM1G/PP2Cgamma governs Cajal body localization and stability of the SMN complex. J. Cell Biol. 2007, 179, 451–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimmler, M.; Bauer, L.; Nousiainen, M.; Korner, R.; Meister, G.; Fischer, U. Phosphorylation regulates the activity of the SMN complex during assembly of spliceosomal U snRNPs. EMBO Rep. 2005, 6, 70–76. [Google Scholar] [CrossRef] [Green Version]
- Renvoise, B.; Querol, G.; Verrier, E.R.; Burlet, P.; Lefebvre, S. A role for protein phosphatase PP1gamma in SMN complex formation and subnuclear localization to Cajal bodies. J. Cell Sci. 2012, 125, 2862–2874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Husedzinovic, A.; Oppermann, F.; Draeger-Meurer, S.; Chari, A.; Fischer, U.; Daub, H.; Gruss, O.J. Phosphoregulation of the human SMN complex. Eur. J. Cell Biol. 2014, 93, 106–117. [Google Scholar] [CrossRef]
- Husedzinovic, A.; Neumann, B.; Reymann, J.; Draeger-Meurer, S.; Chari, A.; Erfle, H.; Fischer, U.; Gruss, O.J. The catalytically inactive tyrosine phosphatase HD-PTP/PTPN23 is a novel regulator of SMN complex localization. Mol. Biol. Cell 2015, 26, 161–171. [Google Scholar] [CrossRef]
- Burnett, B.G.; Munoz, E.; Tandon, A.; Kwon, D.Y.; Sumner, C.J.; Fischbeck, K.H. Regulation of SMN protein stability. Mol. Cell Biol. 2009, 29, 1107–1115. [Google Scholar] [CrossRef] [Green Version]
- Harahap, N.I.F.; Nurputra, D.K.; Ar Rochmah, M.; Shima, A.; Morisada, N.; Takarada, T.; Takeuchi, A.; Tohyama, Y.; Yanagisawa, S.; Nishio, H. Salbutamol inhibits ubiquitin-mediated survival motor neuron protein degradation in spinal muscular atrophy cells. Biochem. Biophys. Rep. 2015, 4, 351–356. [Google Scholar] [CrossRef] [Green Version]
- Cashman, N.R.; Durham, H.D.; Blusztajn, J.K.; Oda, K.; Tabira, T.; Shaw, I.T.; Dahrouge, S.; Antel, J.P. Neuroblastoma x spinal cord (NSC) hybrid cell lines resemble developing motor neurons. Dev. Dyn. 1992, 194, 209–221. [Google Scholar] [CrossRef]
- Hensel, N.; Ratzka, A.; Brinkmann, H.; Klimaschewski, L.; Grothe, C.; Claus, P. Analysis of the fibroblast growth factor system reveals alterations in a mouse model of spinal muscular atrophy. PLoS ONE 2012, 7, e31202. [Google Scholar] [CrossRef] [Green Version]
- Van Bergeijk, J.; Rydel-Konecke, K.; Grothe, C.; Claus, P. The spinal muscular atrophy gene product regulates neurite outgrowth: Importance of the C terminus. FASEB J. 2007, 21, 1492–1502. [Google Scholar] [CrossRef]
- Nölle, A.; Zeug, A.; van Bergeijk, J.; Tonges, L.; Gerhard, R.; Brinkmann, H.; Al Rayes, S.; Hensel, N.; Schill, Y.; Apkhazava, D.; et al. The spinal muscular atrophy disease protein SMN is linked to the Rho-kinase pathway via profilin. Hum. Mol. Genet. 2011, 20, 4865–4878. [Google Scholar] [CrossRef] [Green Version]
- Lehmann, A.; Niewienda, A.; Jechow, K.; Janek, K.; Enenkel, C. Ecm29 fulfils quality control functions in proteasome assembly. Mol. Cell 2010, 38, 879–888. [Google Scholar] [CrossRef] [PubMed]
- Claus, P.; Doring, F.; Gringel, S.; Muller-Ostermeyer, F.; Fuhlrott, J.; Kraft, T.; Grothe, C. Differential intranuclear localization of fibroblast growth factor-2 isoforms and specific interaction with the survival of motoneuron protein. J. Biol. Chem. 2003, 278, 479–485. [Google Scholar] [CrossRef] [Green Version]
- Brenner, S. The genetics of Caenorhabditis elegans. Genetics 1974, 77, 71–94. [Google Scholar]
- Gallotta, I.; Mazzarella, N.; Donato, A.; Esposito, A.; Chaplin, J.C.; Castro, S.; Zampi, G.; Battaglia, G.S.; Hilliard, M.A.; Bazzicalupo, P.; et al. Neuron-specific knock-down of SMN1 causes neuron degeneration and death through an apoptotic mechanism. Hum. Mol. Genet. 2016, 25, 2564–2577. [Google Scholar] [CrossRef] [Green Version]
- Maduro, M.; Pilgrim, D. Identification and cloning of unc-119, a gene expressed in the Caenorhabditis elegans nervous system. Genetics 1995, 141, 977–988. [Google Scholar]
- Mello, C.C.; Kramer, J.M.; Stinchcomb, D.; Ambros, V. Efficient gene transfer in C. elegans: Extrachromosomal maintenance and integration of transforming sequences. EMBO J. 1991, 10, 3959–3970. [Google Scholar] [CrossRef]
- McGhee, J.D.; Fukushige, T.; Krause, M.W.; Minnema, S.E.; Goszczynski, B.; Gaudet, J.; Kohara, Y.; Bossinger, O.; Zhao, Y.; Khattra, J.; et al. ELT-2 is the predominant transcription factor controlling differentiation and function of the C. elegans intestine, from embryo to adult. Dev. Biol. 2009, 327, 551–565. [Google Scholar] [CrossRef] [Green Version]
- Schiff, M.; Weinhold, B.; Grothe, C.; Hildebrandt, H. NCAM and polysialyltransferase profiles match dopaminergic marker gene expression but polysialic acid is dispensable for development of the midbrain dopamine system. J. Neurochem. 2009, 110, 1661–1673. [Google Scholar] [CrossRef]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef]
- Blom, N.; Sicheritz-Ponten, T.; Gupta, R.; Gammeltoft, S.; Brunak, S. Prediction of post-translational glycosylation and phosphorylation of proteins from the amino acid sequence. Proteomics 2004, 4, 1633–1649. [Google Scholar] [CrossRef]
- Li, D.K.; Tisdale, S.; Lotti, F.; Pellizzoni, L. SMN control of RNP assembly: From post-transcriptional gene regulation to motor neuron disease. In Seminars in Cell & Developmental Biology; Academic Press: Cambridge, MA, USA, 2014; Volume 32, pp. 22–29. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Dreyfuss, G. A novel nuclear structure containing the survival of motor neurons protein. EMBO J. 1996, 15, 3555–3565. [Google Scholar] [CrossRef]
- Gabanella, F.; Butchbach, M.E.; Saieva, L.; Carissimi, C.; Burghes, A.H.; Pellizzoni, L. Ribonucleoprotein assembly defects correlate with spinal muscular atrophy severity and preferentially affect a subset of spliceosomal snRNPs. PLoS ONE 2007, 2, e921. [Google Scholar] [CrossRef] [Green Version]
- Lorson, C.L.; Strasswimmer, J.; Yao, J.M.; Baleja, J.D.; Hahnen, E.; Wirth, B.; Le, T.; Burghes, A.H.; Androphy, E.J. SMN oligomerization defect correlates with spinal muscular atrophy severity. Nat. Genet. 1998, 19, 63–66. [Google Scholar] [CrossRef] [PubMed]
- Pellizzoni, L.; Charroux, B.; Dreyfuss, G. SMN mutants of spinal muscular atrophy patients are defective in binding to snRNP proteins. Proc. Natl. Acad. Sci. USA 1999, 96, 11167–11172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, R.; Gupta, K.; Ninan, N.S.; Perry, K.; Van Duyne, G.D. The survival motor neuron protein forms soluble glycine zipper oligomers. Structure 2012, 20, 1929–1939. [Google Scholar] [CrossRef] [Green Version]
- Gangwani, L.; Flavell, R.A.; Davis, R.J. ZPR1 is essential for survival and is required for localization of the survival motor neurons (SMN) protein to Cajal bodies. Mol. Cell Biol. 2005, 25, 2744–2756. [Google Scholar] [CrossRef] [Green Version]
- Briese, M.; Esmaeili, B.; Fraboulet, S.; Burt, E.C.; Christodoulou, S.; Towers, P.R.; Davies, K.E.; Sattelle, D.B. Deletion of smn-1, the Caenorhabditis elegans ortholog of the spinal muscular atrophy gene, results in locomotor dysfunction and reduced lifespan. Hum. Mol. Genet. 2009, 18, 97–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miguel-Aliaga, I.; Culetto, E.; Walker, D.S.; Baylis, H.A.; Sattelle, D.B.; Davies, K.E. The Caenorhabditis elegans orthologue of the human gene responsible for spinal muscular atrophy is a maternal product critical for germline maturation and embryonic viability. Hum. Mol. Genet. 1999, 8, 2133–2143. [Google Scholar] [CrossRef]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [Green Version]
- Papa, A.; Pandolfi, P.P. The PTEN(-)PI3K Axis in Cancer. Biomolecules 2019, 9, 153. [Google Scholar] [CrossRef] [Green Version]
- Van Diepen, M.T.; Eickholt, B.J. Function of PTEN during the formation and maintenance of neuronal circuits in the brain. Dev. Neurosci. 2008, 30, 59–64. [Google Scholar] [CrossRef]
- Rademacher, S.; Eickholt, B.J. PTEN in autism and neurodevelopmental disorders. Cold Spring Harb. Perspect. Med. 2019, 9, a036780. [Google Scholar] [CrossRef] [Green Version]
- Eng, C. PTEN: One gene, many syndromes. Hum. Mutat. 2003, 22, 183–198. [Google Scholar] [CrossRef]
- Shen, S.M.; Ji, Y.; Zhang, C.; Dong, S.S.; Yang, S.; Xiong, Z.; Ge, M.K.; Yu, Y.; Xia, L.; Guo, M.; et al. Nuclear PTEN safeguards pre-mRNA splicing to link Golgi apparatus for its tumor suppressive role. Nat. Commun. 2018, 9, 2392. [Google Scholar] [CrossRef] [Green Version]
- Jochner, M.C.E.; An, J.; Lattig-Tunnemann, G.; Kirchner, M.; Dagane, A.; Dittmar, G.; Dirnagl, U.; Eickholt, B.J.; Harms, C. Unique properties of PTEN-L contribute to neuroprotection in response to ischemic-like stress. Sci. Rep. 2019, 9, 3183. [Google Scholar] [CrossRef] [PubMed]
- Rahdar, M.; Inoue, T.; Meyer, T.; Zhang, J.; Vazquez, F.; Devreotes, P.N. A phosphorylation-dependent intramolecular interaction regulates the membrane association and activity of the tumor suppressor PTEN. Proc. Natl. Acad. Sci. USA 2009, 106, 480–485. [Google Scholar] [CrossRef] [Green Version]
- Forthmann, B.; Brinkmann, H.; Ratzka, A.; Stachowiak, M.K.; Grothe, C.; Claus, P. Immobile survival of motoneuron (SMN) protein stored in Cajal bodies can be mobilized by protein interactions. Cell Mol. Life Sci. 2013, 70, 2555–2568. [Google Scholar] [CrossRef] [PubMed]
- Burghes, A.H.; Beattie, C.E. Spinal muscular atrophy: Why do low levels of survival motor neuron protein make motor neurons sick? Nat. Rev. Neurosci. 2009, 10, 597–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmid, A.C.; Byrne, R.D.; Vilar, R.; Woscholski, R. Bisperoxovanadium compounds are potent PTEN inhibitors. FEBS Lett. 2004, 566, 35–38. [Google Scholar] [CrossRef]
- Locatelli, D.; Terao, M.; Kurosaki, M.; Zanellati, M.C.; Pletto, D.R.; Finardi, A.; Colciaghi, F.; Garattini, E.; Battaglia, G.S. Different Stability and Proteasome-Mediated Degradation Rate of SMN Protein Isoforms. PLoS ONE 2015, 10, e0134163. [Google Scholar] [CrossRef]
- Han, K.J.; Foster, D.G.; Zhang, N.Y.; Kanisha, K.; Dzieciatkowska, M.; Sclafani, R.A.; Hansen, K.C.; Peng, J.; Liu, C.W. Ubiquitin-specific protease 9x deubiquitinates and stabilizes the spinal muscular atrophy protein-survival motor neuron. J. Biol. Chem. 2012, 287, 43741–43752. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Lanzas, R.; Castano, J.G. Lysine-Less Variants of Spinal Muscular Atrophy SMN and SMNDelta7 Proteins Are Degraded by the Proteasome Pathway. Int. J. Mol. Sci. 2017, 18, 2667. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.C.; Hung, W.C.; Chuang, Y.J.; Jong, Y.J. Degradation of survival motor neuron (SMN) protein is mediated via the ubiquitin/proteasome pathway. Neurochem. Int. 2004, 45, 1107–1112. [Google Scholar] [CrossRef]
- Ning, K.; Drepper, C.; Valori, C.F.; Ahsan, M.; Wyles, M.; Higginbottom, A.; Herrmann, T.; Shaw, P.; Azzouz, M.; Sendtner, M. PTEN depletion rescues axonal growth defect and improves survival in SMN-deficient motor neurons. Hum. Mol. Genet. 2010, 19, 3159–3168. [Google Scholar] [CrossRef] [Green Version]
- Little, D.; Valori, C.F.; Mutsaers, C.A.; Bennett, E.J.; Wyles, M.; Sharrack, B.; Shaw, P.J.; Gillingwater, T.H.; Azzouz, M.; Ning, K. PTEN depletion decreases disease severity and modestly prolongs survival in a mouse model of spinal muscular atrophy. Mol. Ther. 2015, 23, 270–277. [Google Scholar] [CrossRef] [Green Version]
- Godena, V.K.; Ning, K. Phosphatase and tensin homologue: A therapeutic target for SMA. Signal Transduct. Target. Ther. 2017, 2, 17038. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Chin-Sang, I.D.C. elegans as a model to study PTEN’s regulation and function. Methods 2015, 77–78, 180–190. [Google Scholar] [CrossRef]
- Byrne, A.B.; Walradt, T.; Gardner, K.E.; Hubbert, A.; Reinke, V.; Hammarlund, M. Insulin/IGF1 signaling inhibits age-dependent axon regeneration. Neuron 2014, 81, 561–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charroux, B.; Pellizzoni, L.; Perkinson, R.A.; Shevchenko, A.; Mann, M.; Dreyfuss, G. Gemin3: A novel DEAD box protein that interacts with SMN, the spinal muscular atrophy gene product, and is a component of gems. J. Cell Biol. 1999, 147, 1181–1194. [Google Scholar] [CrossRef]
- Chiriboga, C.A.; Swoboda, K.J.; Darras, B.T.; Iannaccone, S.T.; Montes, J.; De Vivo, D.C.; Norris, D.A.; Bennett, C.F.; Bishop, K.M. Results from a phase 1 study of nusinersen (ISIS-SMN(Rx)) in children with spinal muscular atrophy. Neurology 2016, 86, 890–897. [Google Scholar] [CrossRef] [Green Version]
- Bowerman, M.; Becker, C.G.; Yanez-Munoz, R.J.; Ning, K.; Wood, M.J.A.; Gillingwater, T.H.; Talbot, K.; Consortium, U.S.R. Therapeutic strategies for spinal muscular atrophy: SMN and beyond. Dis. Models Mech. 2017, 10, 943–954. [Google Scholar] [CrossRef] [Green Version]
- De Oliveira, P.S.; Ferraz, F.A.; Pena, D.A.; Pramio, D.T.; Morais, F.A.; Schechtman, D. Revisiting protein kinase-substrate interactions: Toward therapeutic development. Sci. Signal. 2016, 9. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rademacher, S.; Detering, N.T.; Schüning, T.; Lindner, R.; Santonicola, P.; Wefel, I.-M.; Dehus, J.; Walter, L.M.; Brinkmann, H.; Niewienda, A.; et al. A Single Amino Acid Residue Regulates PTEN-Binding and Stability of the Spinal Muscular Atrophy Protein SMN. Cells 2020, 9, 2405. https://doi.org/10.3390/cells9112405
Rademacher S, Detering NT, Schüning T, Lindner R, Santonicola P, Wefel I-M, Dehus J, Walter LM, Brinkmann H, Niewienda A, et al. A Single Amino Acid Residue Regulates PTEN-Binding and Stability of the Spinal Muscular Atrophy Protein SMN. Cells. 2020; 9(11):2405. https://doi.org/10.3390/cells9112405
Chicago/Turabian StyleRademacher, Sebastian, Nora T. Detering, Tobias Schüning, Robert Lindner, Pamela Santonicola, Inga-Maria Wefel, Janina Dehus, Lisa M. Walter, Hella Brinkmann, Agathe Niewienda, and et al. 2020. "A Single Amino Acid Residue Regulates PTEN-Binding and Stability of the Spinal Muscular Atrophy Protein SMN" Cells 9, no. 11: 2405. https://doi.org/10.3390/cells9112405