Simvastatin Induces Unfolded Protein Response and Enhances Temozolomide-Induced Cell Death in Glioblastoma Cells

 , ,
, ,  , and
, and
Abstract
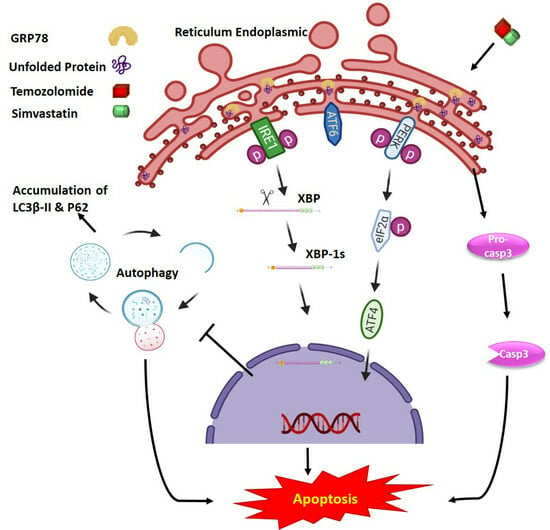
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Co-Treatment with Simva-TMZ Increases Mevalonate Caspase Independet Cytotoxicity and Autophagy Flux Inhibtion in GBM Cells
2.2. Co-Treatment with Simva–TMZ Induces UPR in GBM Cells
2.3. IRE-1α RNase Inhibition Does Not Impact the Effects of Simva, TMZ, or Simva–TMZ on Cell Viability of GBM Cells
2.4. Inhibition of IRE-1α RNase Activity Regulates the Effect of Co-Treatment with Simva and TMZ on Autophagic Flux Inhibition without Affecting the Overall Cytotoxic Effect
2.5. PERK Inhibition Regulates the Effect of Co-Treatment with Simva–TMZ on Autophagy Flux in U87 and U251 Cells and Increased Simva–TMZ-Induced Cell Death in U87 Cells
2.6. Phospho-eIF2α Phosphatase Inhibitor Did Not Change Simva-TMZ-Induced Cell Death in U87 and U251 Cells
3. Discussion
4. Materials and Methods
4.1. Reagents and Drugs
4.2. Cell Lines, Culture, and Treatment
4.3. MTT Assay
4.4. Evaluation of Cell Death by Flow Cytometry
4.5. Immunoblotting
4.6. Inhibition of Specific UPR Signaling Arms (PERK & IRE1)
4.7. Quantitative Real-Time PCR
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AMPK | AMP-activated protein kinase; |
| ATF4 | activating transcription factor 4; |
| ATF-6 | activated transcription factor 6; |
| ATG | autophagy-related protein; |
| CHOP | C/EBP homologous protein; |
| eIF2α | eukaryote initiation factor 2α; |
| ER | endoplasmic reticulum; |
| ERAD | ER-associated protein degradation; |
| GBM | glioblastoma; |
| GGPP | geranylgeranyl pyrophosphate; |
| GRP78 | glucose-related peptide 78 kD; |
| hATF | human atrial fibroblasts; |
| HMG–CoA | hydroxy-3-methyl-glutaryl–CoA; |
| IRE1 | inositol-requiring enzyme 1α; |
| JNK | c-Jun N-terminal kinase; |
| LC3 | microtubule-associated proteins light chain 3; |
| MEV | mevalonate; |
| MPG | methylpurine DNA glycosylase; |
| OEP | our experimental protocol; |
| PERK | protein kinase RNA-like ER kinase; |
| PERKi | PERK kinase activity inhibitor; |
| Simva | simvastatin; |
| TMZ | temozolomide; |
| UPR | unfolded protein response; |
| XBP-1s | x-box binding protein-1 spliced; |
References
- Rulseh, A.M.; Vymazal, J. Whole brain apparent diffusion coefficient measurements correlate with survival in glioblastoma patients. J. Neuroncol. 2020, 146, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Gittleman, H.; Liao, P.; Vecchione-Koval, T.; Wolinsky, Y.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS statistical report: Primary brain and other central nervous system tumors diagnosed in the United States in 2010–2014. Neuro Oncol. 2017, 19, v1–v88. [Google Scholar] [CrossRef] [PubMed]
- Samiei, E.; Seyfoori, A.; Toyota, B.; Ghavami, S.; Akbari, M. Investigating Programmed Cell Death and Tumor Invasion in a Three-Dimensional (3D) Microfluidic Model of Glioblastoma. Int. J. Mol. Sci. 2020, 21, 3162. [Google Scholar] [CrossRef] [PubMed]
- Sharifzad, F.; Yasavoli-Sharahi, H.; Mardpour, S.; Fakharian, E.; Nikuinejad, H.; Heydari, Y.; Mardpour, S.; Taghikhani, A.; Khellat, R.; Vafaei, S.; et al. Neuropathological and genomic characterization of glioblastoma-induced rat model: How similar is it to humans for targeted therapy? J. Cell. Physiol. 2019, 234, 22493–22504. [Google Scholar] [CrossRef]
- Sharifzad, F.; Mardpour, S.; Mardpour, S.; Fakharian, E.; Taghikhani, A.; Sharifzad, A.; Kiani, S.; Heydarian, Y.; Los, M.J.; Azizi, Z.; et al. HSP70/IL-2 Treated NK Cells Effectively Cross the Blood Brain Barrier and Target Tumor Cells in a Rat Model of Induced Glioblastoma Multiforme (GBM). Int. J. Mol. Sci. 2020, 21, 2263. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; Bauchet, L.; Davis, F.G.; Deltour, I.; Fisher, J.L.; Langer, C.E.; Pekmezci, M.; Schwartzbaum, J.A.; Turner, M.C.; Walsh, K.M. The epidemiology of glioma in adults: A “state of the science” review. Neuro Oncol. 2014, 16, 896–913. [Google Scholar] [CrossRef]
- Sayegh, E.T.; Kaur, G.; Bloch, O.; Parsa, A.T. Systematic review of protein biomarkers of invasive behavior in glioblastoma. Mol. Neurobiol. 2014, 49, 1212–1244. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Moghadam, A.R.; da Silva Rosa, S.C.; Samiei, E.; Alizadeh, J.; Field, J.; Kawalec, P.; Thliveris, J.; Akbari, M.; Ghavami, S.; Gordon, J.W. Autophagy modulates temozolomide-induced cell death in alveolar Rhabdomyosarcoma cells. Cell Death Discov. 2018, 4, 52. [Google Scholar] [CrossRef]
- Likus, W.; Siemianowicz, K.; Bienk, K.; Pakula, M.; Pathak, H.; Dutta, C.; Wang, Q.; Shojaei, S.; Assaraf, Y.G.; Ghavami, S.; et al. Could drugs inhibiting the mevalonate pathway also target cancer stem cells? Drug Resist. Updat 2016, 25, 13–25. [Google Scholar] [CrossRef]
- Ahmadi, M.; Amiri, S.; Pecic, S.; Machaj, F.; Rosik, J.; Los, M.J.; Alizadeh, J.; Mahdian, R.; da Silva Rosa, S.C.; Schaafsma, D.; et al. Pleiotropic effects of statins: A focus on cancer. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165968. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-C.; Wu, M.-L.; Huang, K.-C.; Lin, W.-W. HMG-CoA reductase inhibitors activate the unfolded protein response and induce cytoprotective GRP78 expression. Cardiovasc. Res. 2008, 80, 138–150. [Google Scholar] [CrossRef]
- Gaist, D.; Hallas, J.; Friis, S.; Hansen, S.; Sørensen, H. Statin use and survival following glioblastoma multiforme. Cancer Epidemiol. 2014, 38, 722–727. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, S.F.; Nordestgaard, B.G.; Bojesen, S.E. Statin use and reduced cancer-related mortality. N. Engl. J. Med. 2012, 367, 1792–1802. [Google Scholar] [CrossRef] [PubMed]
- Boudreau, D.M.; Yu, O.; Johnson, J. Statin use and cancer risk: A comprehensive review. Expert Opin. Drug Saf. 2010, 9, 603–621. [Google Scholar] [CrossRef] [PubMed]
- Altwairgi, A.K. Statins are potential anticancerous agents (review). Oncol. Rep. 2015, 33, 1019–1039. [Google Scholar] [CrossRef]
- Shojaei, S.; Alizadeh, J.; Thliveris, J.; Koleini, N.; Kardami, E.; Hatch, G.; Xu, F.; Hombach-Klonisch, S.; Klonisch, T.; Ghavami, S. Statins: A new approach to combat temozolomide chemoresistance in glioblastoma. J. Investig. Med. 2018, 66, 1083–1087. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Papa, F.R. The unfolded protein response and cell fate control. Mol. Cell. 2018, 69, 169–181. [Google Scholar] [CrossRef]
- Green, D.R.; Levine, B. To be or not to be? How selective autophagy and cell death govern cell fate. Cell 2014, 157, 65–75. [Google Scholar] [CrossRef]
- Senft, D.; Ze’ev, A.R. UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem. Sci. 2015, 40, 141–148. [Google Scholar] [CrossRef]
- Peñaranda-Fajardo, N.M.; Meijer, C.; Liang, Y.; Dijkstra, B.M.; Aguirre-Gamboa, R.; den Dunnen, W.F.; Kruyt, F.A. ER stress and UPR activation in glioblastoma: Identification of a noncanonical PERK mechanism regulating GBM stem cells through SOX2 modulation. Cell Death Dis. 2019, 10, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Hombach-Klonisch, S.; Mehrpour, M.; Shojaei, S.; Harlos, C.; Pitz, M.; Hamai, A.; Hoshyar, R. Glioblastoma and chemoresistance to alkylating agents: Involvement of apoptosis, autophagy, and unfolded protein response. Pharm. Ther. 2018, 184, 13–41. [Google Scholar] [CrossRef]
- Urra, H.; Dufey, E.; Avril, T.; Chevet, E.; Hetz, C. Endoplasmic reticulum stress and the hallmarks of cancer. Trends Cancer 2016, 2, 252–262. [Google Scholar] [CrossRef]
- Flynn, A.L.; Schiemann, W.P. Autophagy in breast cancer metastatic dormancy: Tumor suppressing or tumor promoting functions? J. Cancer Metastasis Treat. 2019, 5, 43. [Google Scholar] [PubMed]
- Kimmelman, A.C. The dynamic nature of autophagy in cancer. Genes Dev. 2011, 25, 1999–2010. [Google Scholar] [CrossRef] [PubMed]
- Ghavami, S.; Mutawe, M.M.; Sharma, P.; Yeganeh, B.; McNeill, K.D.; Klonisch, T.; Unruh, H.; Kashani, H.H.; Schaafsma, D.; Los, M.; et al. Mevalonate cascade regulation of airway mesenchymal cell autophagy and apoptosis: A dual role for p53. PLoS ONE 2011, 6, e16523. [Google Scholar] [CrossRef] [PubMed]
- Shojaei, S.; Suresh, M.; Klionsky, D.J.; Labouta, H.I.; Ghavami, S. Autophagy and SARS-CoV-2 infection: Apossible smart targeting of the autophagy pathway. Virulence 2020, 11, 805–810. [Google Scholar] [CrossRef] [PubMed]
- Alizadeh, J.; Shojaei, S.; Sepanjnia, A.; Hashemi, M.; Eftekharpour, E.; Ghavami, S. Simultaneous Detection of Autophagy and Epithelial to Mesenchymal Transition in the Non-small Cell Lung Cancer Cells. Methods Mol. Biol. 2019, 1854, 87–103. [Google Scholar] [CrossRef]
- Hsu, S.-K.; Chiu, C.-C.; Dahms, H.-U.; Chou, C.-K.; Cheng, C.-M.; Chang, W.-T.; Cheng, K.-C.; Wang, H.-M.D.; Lin, I. Unfolded protein response (UPR) in survival, dormancy, immunosuppression, metastasis, and treatments of cancer cells. Int. J. Mol. Sci. 2019, 20, 2518. [Google Scholar] [CrossRef]
- Sureda, A.; Alizadeh, J.; Nabavi, S.F.; Berindan-Neagoe, I.; Cismaru, C.A.; Jeandet, P.; Los, M.J.; Clementi, E.; Nabavi, S.M.; Ghavami, S. Endoplasmic reticulum as a potential therapeutic target for covid-19 infection management? Eur. J. Pharm. 2020, 882, 173288. [Google Scholar] [CrossRef]
- Mehrbod, P.; Ande, S.R.; Alizadeh, J.; Rahimizadeh, S.; Shariati, A.; Malek, H.; Hashemi, M.; Glover, K.K.M.; Sher, A.A.; Coombs, K.M.; et al. The roles of apoptosis, autophagy and unfolded protein response in arbovirus, influenza virus, and HIV infections. Virulence 2019, 10, 376–413. [Google Scholar] [CrossRef]
- Suh, D.H.; Kim, M.-K.; Kim, H.S.; Chung, H.H.; Song, Y.S. Unfolded protein response to autophagy as a promising druggable target for anticancer therapy. Ann. N. Y. Acad. Sci. 2012, 1271, 20. [Google Scholar] [CrossRef] [PubMed]
- Logue, S.E.; McGrath, E.P.; Cleary, P.; Greene, S.; Mnich, K.; Almanza, A.; Chevet, E.; Dwyer, R.M.; Oommen, A.; Legembre, P.; et al. Inhibition of IRE1 RNase activity modulates the tumor cell secretome and enhances response to chemotherapy. Nat. Commun. 2018, 9, 3267. [Google Scholar] [CrossRef]
- Ghavami, S.; Yeganeh, B.; Zeki, A.A.; Shojaei, S.; Kenyon, N.J.; Ott, S.; Samali, A.; Patterson, J.; Alizadeh, J.; Moghadam, A.R.; et al. Autophagy and the unfolded protein response promote profibrotic effects of TGF-beta1 in human lung fibroblasts. Am. J. Physiol Lung Cell. Mol. Physiol. 2018, 314, L493–L504. [Google Scholar] [CrossRef] [PubMed]
- Yeganeh, B.; Rezaei Moghadam, A.; Alizadeh, J.; Wiechec, E.; Alavian, S.M.; Hashemi, M.; Geramizadeh, B.; Samali, A.; Bagheri Lankarani, K.; Post, M.; et al. Hepatitis B and C virus-induced hepatitis: Apoptosis, autophagy, and unfolded protein response. World J. Gastroenterol. 2015, 21, 13225–13239. [Google Scholar] [CrossRef] [PubMed]
- Ghavami, S.; Yeganeh, B.; Stelmack, G.L.; Kashani, H.H.; Sharma, P.; Cunnington, R.; Rattan, S.; Bathe, K.; Klonisch, T.; Dixon, I.M.; et al. Apoptosis, autophagy and ER stress in mevalonate cascade inhibition-induced cell death of human atrial fibroblasts. Cell Death Dis. 2012, 3, e330. [Google Scholar] [CrossRef] [PubMed]
- Ghavami, S.; Sharma, P.; Yeganeh, B.; Ojo, O.O.; Jha, A.; Mutawe, M.M.; Kashani, H.H.; Los, M.J.; Klonisch, T.; Unruh, H.; et al. Airway mesenchymal cell death by mevalonate cascade inhibition: Integration of autophagy, unfolded protein response and apoptosis focusing on Bcl2 family proteins. Biochim. Biophys. Acta 2014, 1843, 1259–1271. [Google Scholar] [CrossRef] [PubMed]
- Moon, S.Y.; Kim, H.S.; Nho, K.W.; Jang, Y.J.; Lee, S.K. Endoplasmic reticulum stress induces epithelial-mesenchymal transition through autophagy via activation of c-Src kinase. Nephron Exp. Nephrol. 2014, 126, 127–140. [Google Scholar] [CrossRef] [PubMed]
- Corazzari, M.; Rapino, F.; Ciccosanti, F.; Giglio, P.; Antonioli, M.; Conti, B.; Fimia, G.M.; Lovat, P.E.; Piacentini, M. Oncogenic BRAF induces chronic ER stress condition resulting in increased basal autophagy and apoptotic resistance of cutaneous melanoma. Cell Death Differ. 2015, 22, 946–958. [Google Scholar] [CrossRef]
- Shojaei, S.; Alizadeh, J.; Thliveris, J.; Koleini, N.; Kardami, E.; Hatch, G.; Xu, F.; Hombach-Klonisch, S.; Klonisch, T.; Ghavami, S. 08 Inhibition of autophagy by mevalonate pathway inhibitors, a new therapeutic approach to sensitize glioblastoma cells to temozolomide induced apoptosis. Can. J. Neurol. Sci. 2018, 45, S1–S2. [Google Scholar] [CrossRef]
- Shojaei, S.; Koleini, N.; Samiei, E.; Aghaei, M.; Cole, L.K.; Alizadeh, J.; Islam, M.I.; Vosoughi, A.R.; Albokashy, M.; Butterfield, Y.; et al. Simvastatin increases temozolomide-induced cell death by targeting the fusion of autophagosomes and lysosomes. FEBS J. 2020, 287, 1005–1034. [Google Scholar] [CrossRef] [PubMed]
- Dastghaib, S.; Kumar, P.S.; Aftabi, S.; Damera, G.; Dalvand, A.; Sepanjnia, A.; Kiumarsi, M.; Aghanoori, M.R.; Sohal, S.S.; Ande, S.R.; et al. Mechanisms Targeting the Unfolded Protein Response in Asthma. Am. J. Respir. Cell Mol. Biol. 2020, in press. [Google Scholar] [CrossRef]
- Sheikholeslami, K.; Ali Sher, A.; Lockman, S.; Kroft, D.; Ganjibakhsh, M.; Nejati-Koshki, K.; Shojaei, S.; Ghavami, S.; Rastegar, M. Simvastatin induces apoptosis in medulloblastoma brain tumor cells via mevalonate cascade prenylation substrates. Cancers 2019, 11, 994. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.J.; Xia, Y.; Liu, L.; Liu, M.; Gu, N.; Guang, H.; Zhang, F. Comparison of MTT assay, flow cytometry, and RT-PCR in the evaluation of cytotoxicity of five prosthodontic materials. J. Biomed. Mater. Res. B Appl. Biomater. 2010, 95, 357–364. [Google Scholar] [CrossRef]
- Van Tonder, A.; Joubert, A.M.; Cromarty, A.D. Limitations of the 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyl-2H-tetrazolium bromide (MTT) assay when compared to three commonly used cell enumeration assays. BMC Res. Notes 2015, 8, 47. [Google Scholar] [CrossRef]
- Emami, A.; Shojaei, S.; da Silva Rosa, S.C.; Aghaei, M.; Samiei, E.; Vosoughi, A.R.; Kalantari, F.; Kawalec, P.; Thliveris, J.; Sharma, P.; et al. Mechanisms of simvastatin myotoxicity: The role of autophagy flux inhibition. Eur. J. Pharm. 2019, 862, 172616. [Google Scholar] [CrossRef]
- Madden, E.; Logue, S.E.; Healy, S.J.; Manie, S.; Samali, A. The role of the unfolded protein response in cancer progression: From oncogenesis to chemoresistance. Biol. Cell 2019, 111, 1–17. [Google Scholar] [CrossRef]
- Sovolyova, N.; Healy, S.; Samali, A.; Logue, S.E. Stressed to death—Mechanisms of ER stress-induced cell death. Biol. Chem. 2014, 395, 1–13. [Google Scholar] [CrossRef]
- Gupta, S.; Giricz, Z.; Natoni, A.; Donnelly, N.; Deegan, S.; Szegezdi, E.; Samali, A. NOXA contributes to the sensitivity of PERK-deficient cells to ER stress. FEBS Lett. 2012, 586, 4023–4030. [Google Scholar] [CrossRef]
- Brun, P.; Tarricone, E.; Di Stefano, A.; Mattiuzzo, E.; Mehrbod, P.; Ghavami, S.; Leonardi, A. The regulatory activity of autophagy in conjunctival fibroblasts and its possible role in vernal keratoconjunctivitis. J. Allergy Clin. Immunol. 2020, 17, 1390–1402. [Google Scholar] [CrossRef]
- McAlinden, K.D.; Kota, A.; Haghi, M.; Ghavami, S.; Sharma, P. Pharmacologic Inhibition of Vacuolar H(+)ATPase Attenuates Features of Severe Asthma in Mice. Am. J. Respir. Cell Mol. Biol. 2020, 62, 117–120. [Google Scholar] [CrossRef] [PubMed]
- McAlinden, K.D.; Deshpande, D.A.; Ghavami, S.; Xenaki, D.; Sohal, S.S.; Oliver, B.G.; Haghi, M.; Sharma, P. Autophagy Activation in Asthma Airways Remodeling. Am. J. Respir. Cell Mol. Biol. 2019, 60, 541–553. [Google Scholar] [CrossRef] [PubMed]
- Antonioli, M.; Di Rienzo, M.; Piacentini, M.; Fimia, G.M. Emerging mechanisms in initiating and terminating autophagy. Trends Biochem. Sci. 2017, 42, 28–41. [Google Scholar] [CrossRef] [PubMed]
- Golbabapour, S.; Bagheri-Lankarani, K.; Ghavami, S.; Geramizadeh, B. Autoimmune Hepatitis and Stellate Cells; an Insight into the Role of Autophagy. Curr. Med. Chem. 2019. [Google Scholar] [CrossRef]
- Lee, A.S.; Hendershot, L.M. ER stress and cancer. Cancer Biol. Ther. 2006, 5, 721–722. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, K.; Li, Z. Unfolded protein response in cancer: The physician’s perspective. J. Hematol. Oncol. 2011, 4, 8. [Google Scholar] [CrossRef]
- Yeganeh, B.; Jager, R.; Gorman, A.; Samali, A.; Ghavami, S. Induction of autophagy: Role of endoplasmic reticulum stress and unfolded protein response. In Autophagy: Cancer, Other Pathologies, Inflammation, Immunity, Infection, and Aging; Hayat, M., Ed.; Elsevier: Amsterdam, The Netherlands, 2015; Volume 7, pp. 91–101. [Google Scholar]
- Healy, S.J.; Gorman, A.M.; Mousavi-Shafaei, P.; Gupta, S.; Samali, A. Targeting the endoplasmic reticulum-stress response as an anticancer strategy. Eur. J. Pharm. 2009, 625, 234–246. [Google Scholar] [CrossRef]
- Zhao, H.; Liao, Y.; Minamino, T.; Asano, Y.; Asakura, M.; Kim, J.; Asanuma, H.; Takashima, S.; Hori, M.; Kitakaze, M. Inhibition of cardiac remodeling by pravastatin is associated with amelioration of endoplasmic reticulum stress. Hypertens. Res. 2008, 31, 1977–1987. [Google Scholar] [CrossRef]
- Schweitzer, M.; Mitmaker, B.; Obrand, D.; Sheiner, N.; Abraham, C.; Dostanic, S.; Chalifour, L.E. Atorvastatin mediates increases in intralesional BAX and BAK expression in human end-stage abdominal aortic aneurysms. Can. J. Physiol. Pharmacol. 2009, 87, 915–922. [Google Scholar] [CrossRef]
- Zhu, Z.; Zhang, P.; Li, N.; Kiang, K.M.Y.; Cheng, S.Y.; Wong, V.K.-W.; Leung, G.K.-K. Lovastatin enhances cytotoxicity of temozolomide via impairing autophagic flux in glioblastoma cells. BioMed Res. Int. 2019, 2019, 2710693. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, J.; Li, K.; Deng, L.; Wang, H. Combination of an Autophagy Inducer and an Autophagy Inhibitor: A Smarter Strategy Emerging in Cancer Therapy. Front. Pharm. 2020, 11, 408. [Google Scholar] [CrossRef]
- Luo, X.N.; Yao, H.L.; Song, J.; Song, Q.Q.; Shi, B.T.; Xia, D.; Han, J. Coxsackievirus B3 Infection Triggers Autophagy through 3 Pathways of Endoplasmic Reticulum Stress. Biomed. Environ. Sci. 2018, 31, 867–875. [Google Scholar] [CrossRef]
- Kim, T.W.; Lee, S.Y.; Kim, M.; Cheon, C.; Ko, S.G. Kaempferol induces autophagic cell death via IRE1-JNK-CHOP pathway and inhibition of G9a in gastric cancer cells. Cell Death Dis. 2018, 9, 875. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chen, Y.; Huang, H.; Shi, M.; Yang, W.; Kuang, J.; Yan, J. Autophagy mediated by endoplasmic reticulum stress enhances the caffeine-induced apoptosis of hepatic stellate cells. Int. J. Mol. Med. 2017, 40, 1405–1414. [Google Scholar] [CrossRef] [PubMed]
- Deegan, S.; Koryga, I.; Glynn, S.A.; Gupta, S.; Gorman, A.M.; Samali, A. A close connection between the PERK and IRE arms of the UPR and the transcriptional regulation of autophagy. Biochem. Biophys. Res. Commun. 2015, 456, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Liu, H.; Jiang, C.C.; Fang, L.; Chen, C.; Zhang, X.D.; Jiang, Z.W. Connecting endoplasmic reticulum stress to autophagy through IRE1/JNK/beclin-1 in breast cancer cells. Int. J. Mol. Med. 2014, 34, 772–781. [Google Scholar] [CrossRef]
- Lee, H.; Noh, J.Y.; Oh, Y.; Kim, Y.; Chang, J.W.; Chung, C.W.; Lee, S.T.; Kim, M.; Ryu, H.; Jung, Y.K. IRE1 plays an essential role in ER stress-mediated aggregation of mutant huntingtin via the inhibition of autophagy flux. Hum. Mol. Genet. 2012, 21, 101–114. [Google Scholar] [CrossRef]
- Hu, Q.; Mao, Y.; Liu, M.; Luo, R.; Jiang, R.; Guo, F. The active nuclear form of SREBP1 amplifies ER stress and autophagy via regulation of PERK. FEBS J. 2020, 287, 2348–2366. [Google Scholar] [CrossRef]
- Hou, L.; Dong, J.; Zhu, S.; Yuan, F.; Wei, L.; Wang, J.; Quan, R.; Chu, J.; Wang, D.; Jiang, H.; et al. Seneca valley virus activates autophagy through the PERK and ATF6 UPR pathways. Virology 2019, 537, 254–263. [Google Scholar] [CrossRef]
- Zheng, W.; Xie, W.; Yin, D.; Luo, R.; Liu, M.; Guo, F. ATG5 and ATG7 induced autophagy interplays with UPR via PERK signaling. Cell Commun. Signal. 2019, 17, 42. [Google Scholar] [CrossRef]
- Chen, L.; Zhang, J.; Han, L.; Zhang, A.; Zhang, C.; Zheng, Y.; Jiang, T.; Pu, P.; Jiang, C.; Kang, C. Downregulation of miR-221/222 sensitizes glioma cells to temozolomide by regulating apoptosis independently of p53 status. Oncol. Rep. 2012, 27, 854–860. [Google Scholar] [PubMed]
- Kawtharani, Z. The Antitumor Effect of the Atypical Retinoid ST1926 in Human Glioblastoma. Master’s Thesis, American University of Beirut, Beirut, Lebanon, 2020. [Google Scholar]
- Roos, W.; Batista, L.; Naumann, S.; Wick, W.; Weller, M.; Menck, C.; Kaina, B. Apoptosis in malignant glioma cells triggered by the temozolomide-induced DNA lesion O 6-methylguanine. Oncogene 2007, 26, 186–197. [Google Scholar] [CrossRef] [PubMed]
- Blough, M.D.; Beauchamp, D.C.; Westgate, M.R.; Kelly, J.J.; Cairncross, J.G. Effect of aberrant p53 function on temozolomide sensitivity of glioma cell lines and brain tumor initiating cells from glioblastoma. J. Neuro Oncol. 2011, 102, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y. Temozolomide resistance in glioblastoma multiforme. Genes Dis. 2016, 3, 198–210. [Google Scholar] [CrossRef]
- Ghavami, S.; Mutawe, M.M.; Schaafsma, D.; Yeganeh, B.; Unruh, H.; Klonisch, T.; Halayko, A.J. Geranylgeranyl transferase 1 modulates autophagy and apoptosis in human airway smooth muscle. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 302, L420–L428. [Google Scholar] [CrossRef]
- Tasdemir, E.; Chiara Maiuri, M.; Morselli, E.; Criollo, A.; D’Amelio, M.; Djavaheri-Mergny, M.; Cecconi, F.; Tavernarakis, N.; Kroemer, G. A dual role of p53 in the control of autophagy. Autophagy 2008, 4, 810–814. [Google Scholar] [CrossRef]
- Namba, T.; Chu, K.; Kodama, R.; Byun, S.; Yoon, K.W.; Hiraki, M.; Mandinova, A.; Lee, S.W. Loss of p53 enhances the function of the endoplasmic reticulum through activation of the IRE1α/XBP1 pathway. Oncotarget 2015, 6, 19990. [Google Scholar] [CrossRef]
- Fusee, L.T.S.; Marin, M.; Fahraeus, R.; Lopez, I. Alternative Mechanisms of p53 Action During the Unfolded Protein Response. Cancers 2020, 12, 401. [Google Scholar] [CrossRef]
- Alizadeh, J.; Glogowska, A.; Thliveris, J.; Kalantari, F.; Shojaei, S.; Hombach-Klonisch, S.; Klonisch, T.; Ghavami, S. Autophagy modulates transforming growth factor beta 1 induced epithelial to mesenchymal transition in non-small cell lung cancer cells. Biochim. Biophys. Acta 2018, 1865, 749–768. [Google Scholar] [CrossRef]
- Hashemi, M.; Ghavami, S.; Eshraghi, M.; Booy, E.P.; Los, M. Cytotoxic effects of intra and extracellular zinc chelation on human breast cancer cells. Eur. J. Pharm. 2007, 557, 9–19. [Google Scholar] [CrossRef]
- Ghavami, S.; Asoodeh, A.; Klonisch, T.; Halayko, A.J.; Kadkhoda, K.; Kroczak, T.J.; Gibson, S.B.; Booy, E.P.; Naderi-Manesh, H.; Los, M. Brevinin-2R(1) semi-selectively kills cancer cells by a distinct mechanism, which involves the lysosomal-mitochondrial death pathway. J. Cell. Mol. Med. 2008, 12, 1005–1022. [Google Scholar] [CrossRef] [PubMed]
- Ghavami, S.; Mutawe, M.M.; Hauff, K.; Stelmack, G.L.; Schaafsma, D.; Sharma, P.; McNeill, K.D.; Hynes, T.S.; Kung, S.K.; Unruh, H.; et al. Statin-triggered cell death in primary human lung mesenchymal cells involves p53-PUMA and release of Smac and Omi but not cytochrome c. Biochim. Biophys. Acta 2010, 1803, 452–467. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.S.; Zeglinski, M.R.; Rattan, S.G.; Landry, N.M.; Ghavami, S.; Wigle, J.T.; Klonisch, T.; Halayko, A.J.; Dixon, I.M. Inhibition of autophagy inhibits the conversion of cardiac fibroblasts to cardiac myofibroblasts. Oncotarget 2016, 7, 78516–78531. [Google Scholar] [CrossRef] [PubMed]
- Ghavami, S.; Eshraghi, M.; Kadkhoda, K.; Mutawe, M.M.; Maddika, S.; Bay, G.H.; Wesselborg, S.; Halayko, A.J.; Klonisch, T.; Los, M. Role of BNIP3 in TNF-induced cell death--TNF upregulates BNIP3 expression. Biochim. Biophys. Acta 2009, 1793, 546–560. [Google Scholar] [CrossRef]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dastghaib, S.; Shojaei, S.; Mostafavi-Pour, Z.; Sharma, P.; Patterson, J.B.; Samali, A.; Mokarram, P.; Ghavami, S. Simvastatin Induces Unfolded Protein Response and Enhances Temozolomide-Induced Cell Death in Glioblastoma Cells. Cells 2020, 9, 2339. https://doi.org/10.3390/cells9112339
Dastghaib S, Shojaei S, Mostafavi-Pour Z, Sharma P, Patterson JB, Samali A, Mokarram P, Ghavami S. Simvastatin Induces Unfolded Protein Response and Enhances Temozolomide-Induced Cell Death in Glioblastoma Cells. Cells. 2020; 9(11):2339. https://doi.org/10.3390/cells9112339
Chicago/Turabian StyleDastghaib, Sanaz, Shahla Shojaei, Zohreh Mostafavi-Pour, Pawan Sharma, John B. Patterson, Afshin Samali, Pooneh Mokarram, and Saeid Ghavami. 2020. "Simvastatin Induces Unfolded Protein Response and Enhances Temozolomide-Induced Cell Death in Glioblastoma Cells" Cells 9, no. 11: 2339. https://doi.org/10.3390/cells9112339