The Multimodal Serotonergic Agent Vilazodone Inhibits L-DOPA-Induced Gene Regulation in Striatal Projection Neurons and Associated Dyskinesia in an Animal Model of Parkinson’s Disease



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. 6-OHDA Lesions
2.3. Forelimb Stepping Test
2.4. Drug Treatment
2.5. Behavioral Analysis
2.6. Tissue Preparation and In Situ Hybridization Histochemistry
2.7. Analysis of Autoradiograms
2.8. Tyrosine Hydroxylase Immunohistochemistry
2.9. Statistics
3. Results
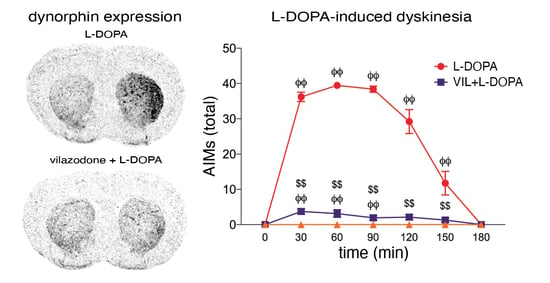
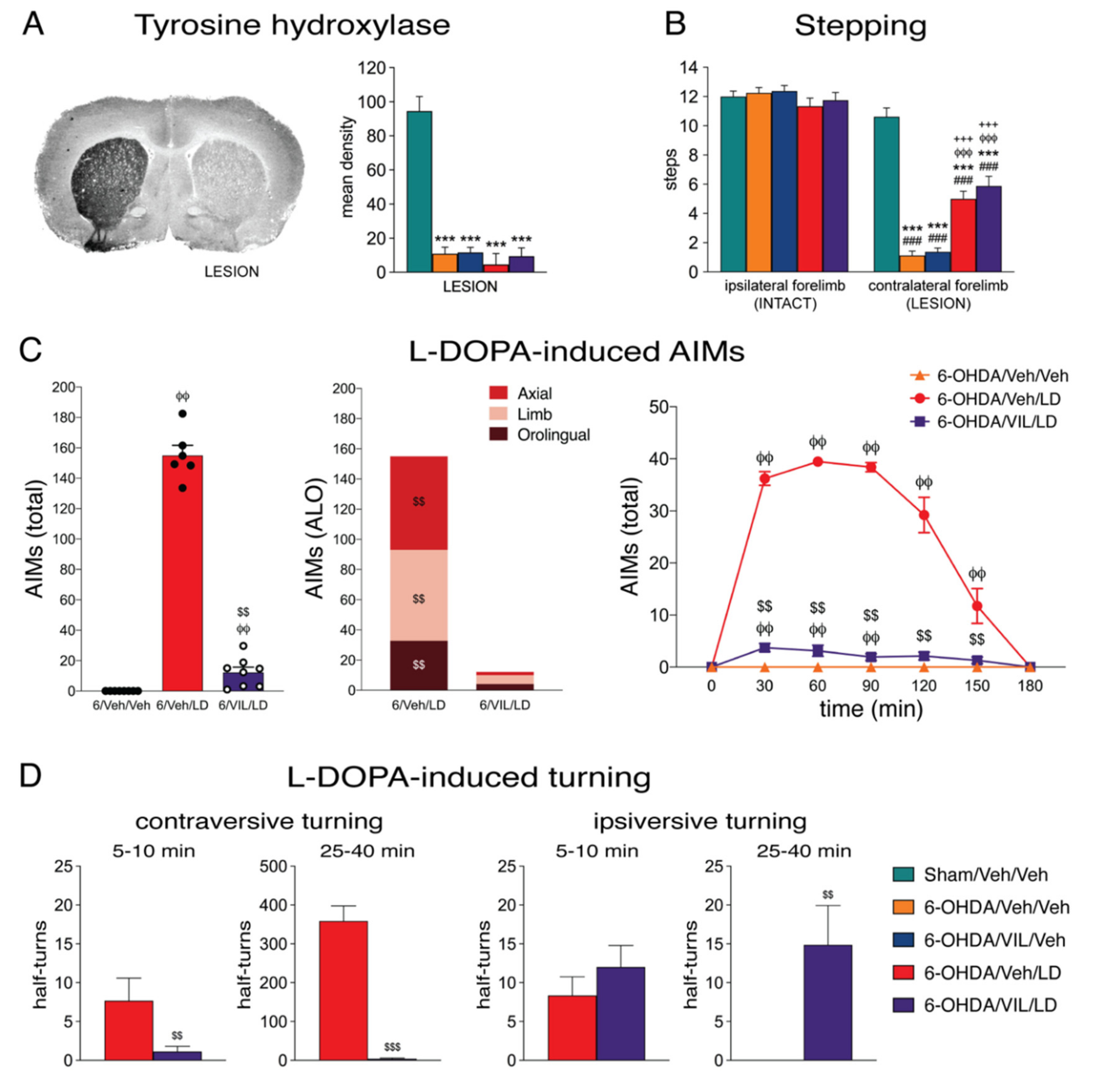
3.1. Behavioral Effects of Vilazodone
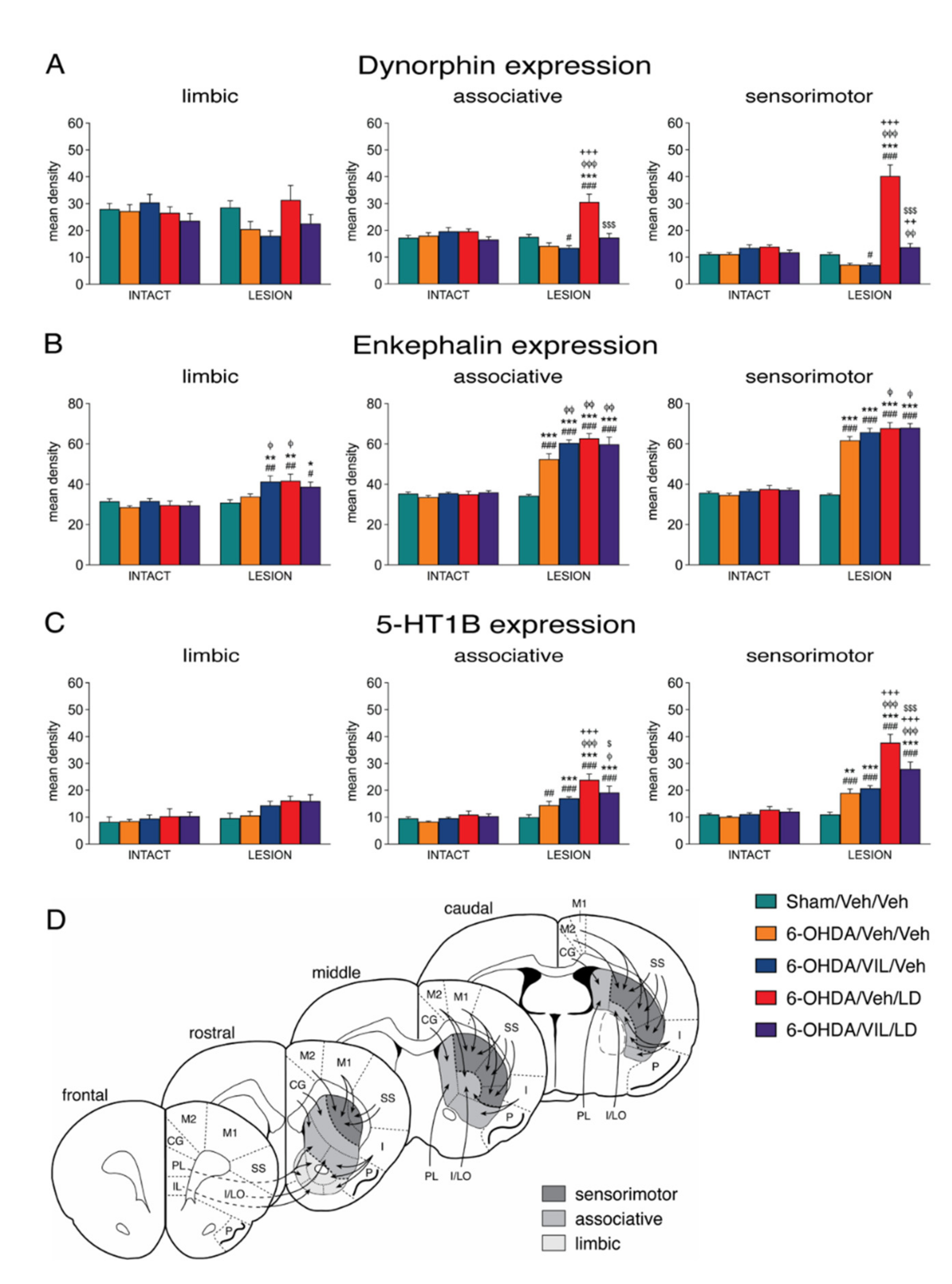
3.2. Effects of Dopamine Depletion and L-DOPA Treatment on Dynorphin and Enkephalin Expression
3.3. Effects of Vilazodone on Dynorphin and Enkephalin Expression after Dopamine Depletion and L-DOPA Treatment
3.4. Effects of Vilazodone on 5-HT1B Receptor Expression after Dopamine Depletion and L-DOPA Treatment
3.5. Effects of Vilazodone on Zif268 Expression after Dopamine Depletion and L-DOPA Treatment
4. Discussion
4.1. Vilazodone Attenuates L-DOPA-Induced Dyskinesia Without Inhibiting Its Prokinetic Effects
4.2. Vilazodone Inhibits L-DOPA-Induced Increase in Dynorphin Expression (dMSNs)
4.3. Vilazodone Does Not Inhibit Enkephalin Expression (iMSNs)
4.4. Vilazodone Inhibits L-DOPA-Induced Increases in 5-HT1B and Zif268 Expression
4.5. Possible Mechanisms Underlying the Effects of Vilazodone
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Birkmayer, W.; Hornykiewicz, O. The L-3,4-dioxyphenylalanine (DOPA)-effect in Parkinson-akinesia. Wiener Klinische Wochenschrift 1961, 73, 787–788. [Google Scholar] [PubMed]
- Cotzias, G.C.; Papavasiliou, P.S.; Gellene, R. L-dopa in parkinson’s syndrome. N. Engl. J. Med. 1969, 281, 272. [Google Scholar]
- Mones, R.J.; Elizan, T.S.; Siegel, G. L-dopa induced dyskinesias in 152 patients with Parkinson’s disease. Trans. Am. Neurol. Assoc. 1970, 95, 286–287. [Google Scholar]
- Chase, T.N. The significance of continuous dopaminergic stimulation in the treatment of Parkinson’s disease. Drugs 1998, 55 (Suppl. 1), 1–9. [Google Scholar] [CrossRef]
- Manson, A.; Stirpe, P.; Schrag, A. Levodopa-induced-dyskinesias clinical features, incidence, risk factors, management and impact on quality of life. J. Parkinsons Dis. 2012, 2, 189–198. [Google Scholar] [CrossRef] [Green Version]
- Marconi, R.; Lefebvre-Caparros, D.; Bonnet, A.M.; Vidailhet, M.; Dubois, B.; Agid, Y. Levodopa-induced dyskinesias in Parkinson’s disease phenomenology and pathophysiology. Mov. Disord. 1994, 9, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Obeso, J.A.; Olanow, W.; Nutt, J.G. Levodopa motor complications in Parkinson’s disease. Trends Neurosci. 2000, 23 (Suppl. 10), S2–S7. [Google Scholar] [CrossRef]
- Cenci, M.A. Transcription factors involved in the pathogenesis of L-DOPA-induced dyskinesia in a rat model of Parkinson’s disease. Amino Acids 2002, 23, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Cenci, M.A.; Konradi, C. Maladaptive striatal plasticity in L-DOPA-induced dyskinesia. Prog. Brain Res. 2010, 183, 209–233. [Google Scholar]
- Murer, M.G.; Moratalla, R. Striatal signaling in L-DOPA-induced dyskinesia: Common mechanisms with drug abuse and long term memory involving D1 dopamine receptor stimulation. Front Neuroanat. 2011, 5, 51. [Google Scholar] [CrossRef] [Green Version]
- Abercrombie, E.D.; Bonatz, A.E.; Zigmond, M.J. Effects of L-dopa on extracellular dopamine in striatum of normal and 6-hydroxydopamine-treated rats. Brain Res. 1990, 525, 36–44. [Google Scholar] [CrossRef]
- De la Fuente-Fernández, R.; Sossi, V.; Huang, Z.; Furtado, S.; Lu, J.Q.; Calne, D.B.; Ruth, T.J.; Stoessl, A.J. Levodopa-induced changes in synaptic dopamine levels increase with progression of Parkinson’s disease: Implications for dyskinesias. Brain 2004, 127, 2747–2754. [Google Scholar] [CrossRef] [Green Version]
- Lindgren, H.S.; Andersson, D.R.; Lagerkvist, S.; Nissbrandt, H.; Cenci, M.A. L-DOPA-induced dopamine efflux in the striatum and the substantia nigra in a rat model of Parkinson’s disease: Temporal and quantitative relationship to the expression of dyskinesia. J. Neurochem. 2010, 112, 1465–1476. [Google Scholar] [CrossRef]
- Politis, M.; Wu, K.; Loane, C.; Brooks, D.J.; Kiferle, L.; Turkheimer, F.E.; Bain, P.; Molloy, S.; Piccini, P. Serotonergic mechanisms responsible for levodopa-induced dyskinesias in Parkinson’s disease patients. J. Clin. Invest. 2014, 124, 1340–1349. [Google Scholar] [CrossRef]
- Arai, R.; Karasawa, N.; Geffard, M.; Nagatsu, I. L-DOPA is converted to dopamine in serotonergic fibers of the striatum of the rat: A double-labeling immunofluorescence study. Neurosci. Lett. 1995, 195, 195–198. [Google Scholar] [CrossRef]
- Tanaka, H.; Kannari, K.; Maeda, T.; Tomiyama, M.; Suda, T.; Matsunaga, M. Role of serotonergic neurons in L-DOPA-derived extracellular dopamine in the striatum of 6-OHDA-lesioned rats. Neuroreport 1999, 10, 631–634. [Google Scholar] [CrossRef] [PubMed]
- Cenci, M.A.; Lundblad, M. Post- versus presynaptic plasticity in L-DOPA-induced dyskinesia. J. Neurochem. 2006, 99, 381–392. [Google Scholar] [CrossRef] [PubMed]
- Carta, M.; Carlsson, T.; Kirik, D.; Björklund, A. Dopamine released from 5-HT terminals is the cause of L-DOPA-induced dyskinesia in parkinsonian rats. Brain 2007, 130, 1819–1833. [Google Scholar] [CrossRef] [Green Version]
- Navailles, S.; Bioulac, B.; Gross, C.; De Deurwaerdère, P. Serotonergic neurons mediate ectopic release of dopamine induced by L-DOPA in a rat model of Parkinson’s disease. Neurobiol. Dis. 2010, 38, 136–143. [Google Scholar] [CrossRef]
- Cenci, M.A. Molecular mechanisms of L-DOPA-induced dyskinesia. In Handbook of Basal Ganglia Structure and Function; Steiner, H., Tseng, K.Y., Eds.; Elsevier: London, UK, 2016; pp. 857–871. [Google Scholar]
- Moratalla, R.; Solis, O.; Suarez, L.M. Morphological plasticity in the striatum associated with dopamine dysfunction. In Handbook of Basal Ganglia Structure and Function; Steiner, H., Tseng, K.Y., Eds.; Elsevier: London, UK, 2016; pp. 755–770. [Google Scholar]
- Klawans, H.L.; Goetz, C.; Nausieda, P.A.; Weiner, W.J. Levodopa-induced dopamine receptor hypersensitivity. Trans. Am. Neurol. Assoc. 1977, 102, 80–83. [Google Scholar] [CrossRef]
- Gerfen, C.R. D1 dopamine receptor supersensitivity in the dopamine-depleted striatum animal model of Parkinson’s disease. Neuroscientist 2003, 9, 455–462. [Google Scholar] [CrossRef]
- Gerfen, C.R. D1 dopamine receptor supersensitivity in the dopamine-depleted striatum: Aberrant ERK1/2 signaling. In Handbook of Basal Ganglia Structure and Function; Steiner, H., Tseng, K.Y., Eds.; Academic Press/Elsevier: London, UK, 2010; pp. 491–500. [Google Scholar]
- Spigolon, G.; Fisone, G. Signal transduction in L-DOPA-induced dyskinesia: From receptor sensitization to abnormal gene expression. J. Neural. Transm. 2018, 125, 1171–1186. [Google Scholar] [CrossRef] [Green Version]
- Suarez, L.M.; Solis, O.; Aguado, C.; Lujan, R.; Moratalla, R. L-DOPA oppositely regulates synaptic strength and spine morphology in D1 and D2 striatal projection neurons in dyskinesia. Cereb. Cortex 2016, 26, 4253–4264. [Google Scholar] [CrossRef] [Green Version]
- Mela, F.; Marti, M.; Bido, S.; Cenci, M.A.; Morari, M. In vivo evidence for a differential contribution of striatal and nigral D1 and D2 receptors to L-DOPA induced dyskinesia and the accompanying surge of nigral amino acid levels. Neurobiol. Dis. 2012, 45, 573–582. [Google Scholar] [CrossRef]
- Konradi, C.; Westin, J.E.; Carta, M.; Eaton, M.E.; Kuter, K.; Dekundy, A.; Lundblad, M.; Cenci, M.A. Transcriptome analysis in a rat model of L-DOPA-induced dyskinesia. Neurobiol. Dis. 2004, 17, 219–236. [Google Scholar] [CrossRef] [Green Version]
- Heiman, M.; Heilbut, A.; Francardo, V.; Kulicke, R.; Fenster, R.J.; Kolaczyk, E.D.; Mesirov, J.P.; Surmeier, D.J.; Cenci, M.A.; Greengard, P. Molecular adaptations of striatal spiny projection neurons during levodopa-induced dyskinesia. Proc. Natl. Acad. Sci. USA 2014, 111, 4578–4583. [Google Scholar] [CrossRef] [Green Version]
- Södersten, E.; Feyder, M.; Lerdrup, M.; Gomes, A.L.; Kryh, H.; Spigolon, G.; Caboche, J.; Fisone, G.; Hansen, K. Dopamine signaling leads to loss of Polycomb repression and aberrant gene activation in experimental parkinsonism. PLoS Genet 2014, 10, e1004574. [Google Scholar] [CrossRef] [Green Version]
- Cenci, M.A.; Lee, C.S.; Bjorklund, A. L-DOPA-induced dyskinesia in the rat is associated with striatal overexpression of prodynorphin- and glutamic acid decarboxylase mRNA. Eur. J. Neurosci. 1998, 10, 2694–2706. [Google Scholar] [CrossRef]
- Andersson, M.; Hilbertson, A.; Cenci, M.A. Striatal fosB expression is causally linked with l-DOPA-induced abnormal involuntary movements and the associated upregulation of striatal prodynorphin mRNA in a rat model of Parkinson’s disease. Neurobiol. Dis. 1999, 6, 461–474. [Google Scholar] [CrossRef] [Green Version]
- Padovan-Neto, F.E.; Patterson, S.; Voelkner, N.M.; Altwal, F.; Beverley, J.A.; West, A.R.; Steiner, H. Selective regulation of 5-HT1B serotonin receptor expression in the striatum by dopamine depletion and repeated L-DOPA treatment: Relationship to L-DOPA-induced dyskinesias. Mol. Neurobiol. 2020, 57, 736–751. [Google Scholar] [CrossRef]
- Steece-Collier, K.; Collier, T.J.; Lipton, J.W.; Stancati, J.A.; Winn, M.E.; Cole-Strauss, A.; Sellnow, R.; Conti, M.M.; Mercado, N.M.; Nillni, E.A.; et al. Striatal Nurr1, but not FosB expression links a levodopa-induced dyskinesia phenotype to genotype in Fisher 344 vs. Lewis hemiparkinsonian rats. Exp. Neurol. 2020, 330, 113327. [Google Scholar] [CrossRef]
- Lanza, K.; Bishop, C. Serotonergic targets for the treatment of L-DOPA-induced dyskinesia. J. Neural. Transm. 2018, 125, 1203–1216. [Google Scholar] [CrossRef]
- Yamato, H.; Kannari, K.; Shen, H.; Suda, T.; Matsunaga, M. Fluoxetine reduces L-DOPA-derived extracellular DA in the 6-OHDA-lesioned rat striatum. Neuroreport 2001, 12, 1123–1126. [Google Scholar] [CrossRef]
- Tomiyama, M.; Kimura, T.; Maeda, T.; Kannari, K.; Matsunaga, M.; Baba, M. A serotonin 5-HT1A receptor agonist prevents behavioral sensitization to L-DOPA in a rodent model of Parkinson’s disease. Neurosci. Res. 2005, 52, 185–194. [Google Scholar] [CrossRef]
- Eskow, K.L.; Gupta, V.; Alam, S.; Park, J.Y.; Bishop, C. The partial 5-HT1A agonist buspirone reduces the expression and development of l-DOPA-induced dyskinesia in rats and improves L-DOPA efficacy. Pharmacol. Biochem. Behav. 2007, 87, 306–314. [Google Scholar] [CrossRef]
- Muñoz, A.; Li, Q.; Gardoni, F.; Marcello, E.; Qin, C.; Carlsson, T.; Kirik, D.; Di Luca, M.; Björklund, A.; Bezard, E.; et al. Combined 5-HT1A and 5-HT1B receptor agonists for the treatment of L-DOPA-induced dyskinesia. Brain 2008, 131, 3380–3394. [Google Scholar] [CrossRef] [Green Version]
- Bishop, C.; George, J.A.; Buchta, W.; Goldenberg, A.A.; Mohamed, M.; Dickinson, S.O.; Eissa, S.; Eskow Jaunarajs, K.L. Serotonin transporter inhibition attenuates l-DOPA-induced dyskinesia without compromising l-DOPA efficacy in hemi-parkinsonian rats. Eur. J. Neurosci. 2012, 36, 2839–2848. [Google Scholar] [CrossRef] [Green Version]
- Conti, M.M.; Ostock, C.Y.; Lindenbach, D.; Goldenberg, A.A.; Kampton, E.; Dell’isola, R.; Katzman, A.C.; Bishop, C. Effects of prolonged selective serotonin reuptake inhibition on the development and expression of l-DOPA-induced dyskinesia in hemi-parkinsonian rats. Neuropharmacology 2014, 77, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Fidalgo, C.; Ko, W.K.; Tronci, E.; Li, Q.; Stancampiano, R.; Chuan, Q.; Bezard, E.; Carta, M. Effect of serotonin transporter blockade on L-DOPA-induced dyskinesia in animal models of Parkinson’s disease. Neuroscience 2015, 298, 389–396. [Google Scholar] [CrossRef]
- Huot, P.; Johnston, T.H.; Fox, S.H.; Newman-Tancredi, A.; Brotchie, J.M. The highly-selective 5-HT1A agonist F15599 reduces L-DOPA-induced dyskinesia without compromising anti-parkinsonian benefits in the MPTP-lesioned macaque. Neuropharmacology 2015, 97, 306–311. [Google Scholar] [CrossRef]
- Bishop, C.; Krolewski, D.M.; Eskow, K.L.; Barnum, C.J.; Dupre, K.B.; Deak, T.; Walker, P.D. Contribution of the striatum to the effects of 5-HT1A receptor stimulation in l-DOPA-treated hemiparkinsonian rats. J. Neurosci. Res. 2009, 87, 1645–1658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kannari, K.; Kurahashi, K.; Tomiyama, M.; Maeda, T.; Arai, A.; Baba, M.; Suda, T.; Matsunaga, M. Tandospirone citrate, a selective 5-HT1A agonist, alleviates L-DOPA-induced dyskinesia in patients with Parkinson’s disease. No To Shinkei 2002, 54, 133–137. [Google Scholar] [PubMed]
- Goetz, C.G.; Damier, P.; Hicking, C.; Laska, E.; Müller, T.; Olanow, C.W.; Rascol, O.; Russ, H. Sarizotan as a treatment for dyskinesias in Parkinson’s disease: A double-blind placebo-controlled trial. Mov. Disord. 2007, 22, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Lindenbach, D.; Palumbo, N.; Ostock, C.Y.; Vilceus, N.; Conti, M.M.; Bishop, C. Side effect profile of 5-HT treatments for Parkinson’s disease and L-DOPA-induced dyskinesia in rats. Br. J. Pharmacol. 2015, 172, 119–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carta, M.; Björklund, A. The serotonergic system in L-DOPA-induced dyskinesia: Pre-clinical evidence and clinical perspective. J. Neural Transm. 2018, 125, 1195–1202. [Google Scholar] [CrossRef] [PubMed]
- Hughes, Z.A.; Starr, K.R.; Langmead, C.J.; Hill, M.; Bartoszyk, G.D.; Hagan, J.J.; Middlemiss, D.N.; Dawson, L.A. Neurochemical evaluation of the novel 5-HT1A receptor partial agonist/serotonin reuptake inhibitor, vilazodone. Eur. J. Pharmacol. 2005, 510, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Owen, R.T. Vilazodone: A new treatment option for major depressive disorder. Drugs Today (Barc) 2011, 47, 531–537. [Google Scholar] [CrossRef]
- Cruz, M.P. Vilazodone HCl (Viibryd): A serotonin partial agonist and reuptake inhibitor for the treatment of major depressive disorder. Pharm. Ther. 2012, 37, 28–31. [Google Scholar]
- Meadows, S.M.; Conti, M.M.; Gross, L.; Chambers, N.E.; Avnor, Y.; Ostock, C.Y.; Lanza, K.; Bishop, C. Diverse serotonin actions of Vilazodone reduce L-3,4-dihydroxyphenylalanine-induced dyskinesia in hemi-parkinsonian rats. Mov. Disord. 2018, 33, 1740–1749. [Google Scholar] [CrossRef]
- Steiner, H.; Gerfen, C.R. Role of dynorphin and enkephalin in the regulation of striatal output pathways and behavior. Exp. Brain Res. 1998, 123, 60–76. [Google Scholar] [CrossRef]
- Paxinos, G.; Watson, C. The Rat Brain in Stereotaxic Coordinates; Academic Press: New York, NY, USA, 1998. [Google Scholar]
- Olsson, M.; Nikkhah, G.; Bentlage, C.; Björklund, A. Forelimb akinesia in the rat Parkinson model: Differential effects of dopamine agonists and nigral transplants as assessed by a new stepping test. J. Neurosci. 1995, 15, 3863–3875. [Google Scholar] [CrossRef]
- Tseng, K.Y.; Caballero, A.; Dec, A.; Cass, D.K.; Simak, N.; Sunu, E.; Park, M.J.; Blume, S.R.; Sammut, S.; Park, D.J.; et al. Inhibition of striatal soluble guanylyl cyclase-cGMP signaling reverses basal ganglia dysfunction and akinesia in experimental parkinsonism. PLoS ONE 2011, 6, e27187. [Google Scholar] [CrossRef] [PubMed]
- Jayasinghe, V.R.; Flores-Barrera, E.; West, A.R.; Tseng, K.Y. Frequency-dependent corticostriatal disinhibition resulting from chronic dopamine depletion: Role of local striatal cGMP and GABA-AR signaling. Cereb. Cortex 2017, 27, 625–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winkler, C.; Kirik, D.; Björklund, A.; Cenci, M.A. L-DOPA-induced dyskinesia in the intrastriatal 6-hydroxydopamine model of parkinson’s disease: Relation to motor and cellular parameters of nigrostriatal function. Neurobiol. Dis. 2002, 10, 165–186. [Google Scholar] [CrossRef] [PubMed]
- Padovan-Neto, F.E.; Cavalcanti-Kiwiatkoviski, R.; Carolino, R.O.; Anselmo-Franci, J.; Del Bel, E. Effects of prolonged neuronal nitric oxide synthase inhibition on the development and expression of L-DOPA-induced dyskinesia in 6-OHDA-lesioned rats. Neuropharmacology 2015, 89, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Steiner, H.; Kitai, S.T. Unilateral striatal dopamine depletion: Time-dependent effects on cortical function and behavioural correlates. Eur. J. Neurosci. 2001, 14, 1390–1404. [Google Scholar] [CrossRef]
- Willuhn, I.; Sun, W.; Steiner, H. Topography of cocaine-induced gene regulation in the rat striatum: Relationship to cortical inputs and role of behavioural context. Eur. J. Neurosci. 2003, 17, 1053–1066. [Google Scholar] [CrossRef] [PubMed]
- Yano, M.; Steiner, H. Methylphenidate (Ritalin) induces Homer 1a and zif 268 expression in specific corticostriatal circuits. Neuroscience 2005, 132, 855–865. [Google Scholar] [CrossRef]
- Van Waes, V.; Ehrlich, S.; Beverley, J.A.; Steiner, H. Fluoxetine potentiation of methylphenidate-induced gene regulation in striatal output pathways: Potential role for 5-HT1B receptor. Neuropharmacology 2015, 89, 77–86. [Google Scholar] [CrossRef] [Green Version]
- Steiner, H.; Blum, M.; Kitai, S.T.; Fedi, P. Differential expression of ErbB3 and ErbB4 neuregulin receptors in dopamine neurons and forebrain areas of the adult rat. Exp. Neurol. 1999, 159, 494–503. [Google Scholar] [CrossRef]
- Eskow, K.L.; Dupre, K.B.; Barnum, C.J.; Dickinson, S.O.; Park, J.Y.; Bishop, C. The role of the dorsal raphe nucleus in the development, expression, and treatment of L-dopa-induced dyskinesia in hemiparkinsonian rats. Synapse 2009, 63, 610–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, J.W.; Wachtel, S.R.; Young, D.; Kang, U.J. Biochemical and anatomical characterization of forepaw adjusting steps in rat models of Parkinson’s disease: Studies on medial forebrain bundle and striatal lesions. Neuroscience 1999, 88, 617–628. [Google Scholar] [CrossRef]
- Ljungberg, T.; Ungerstedt, U. Sensory inattention produced by 6-hydroxydopamine-induced degeneration of ascending dopamine neurons in the brain. Exp. Neurol. 1976, 53, 585–600. [Google Scholar] [CrossRef]
- Marshall, J.F.; Richardson, J.S.; Teitelbaum, P. Nigrostriatal bundle damage and the lateral hypothalamic syndrome. J. Comp. Physiol. Psychol. 1974, 87, 808–830. [Google Scholar] [CrossRef] [PubMed]
- Gerfen, C.R.; Engber, T.M.; Mahan, L.C.; Susel, Z.; Chase, T.N.; Monsma, F.J., Jr.; Sibley, D.R. D1 and D2 dopamine receptor-regulated gene expression of striatonigral and striatopallidal neurons. Science 1990, 250, 1429–1432. [Google Scholar] [CrossRef] [PubMed]
- Steiner, H.; Gerfen, C.R. Dynorphin regulates D1 dopamine receptor-mediated responses in the striatum: Relative contributions of pre- and postsynaptic mechanisms in dorsal and ventral striatum demonstrated by altered immediate-early gene induction. J. Comp. Neurol. 1996, 376, 530–541. [Google Scholar] [CrossRef]
- Henry, B.; Crossman, A.R.; Brotchie, J.M. Effect of repeated L-DOPA, bromocriptine, or lisuride administration on preproenkephalin-A and preproenkephalin-B mRNA levels in the striatum of the 6-hydroxydopamine-lesioned rat. Exp. Neurol. 1999, 155, 204–220. [Google Scholar] [CrossRef]
- Westin, J.E.; Andersson, M.; Lundblad, M.; Cenci, M.A. Persistent changes in striatal gene expression induced by long-term L-DOPA treatment in a rat model of Parkinson’s disease. Eur. J. Neurosci. 2001, 14, 1171–1176. [Google Scholar] [CrossRef]
- Cenci, M.A.; Tranberg, A.; Andersson, M.; Hilbertson, A. Changes in the regional and compartmental distribution of FosB- and JunB-like immunoreactivity induced in the dopamine-denervated rat striatum by acute or chronic L-dopa treatment. Neuroscience 1999, 94, 515–527. [Google Scholar] [CrossRef]
- St-Hilaire, M.; Landry, E.; Lévesque, D.; Rouillard, C. Denervation and repeated L-DOPA induce complex regulatory changes in neurochemical phenotypes of striatal neurons: Implication of a dopamine D1-dependent mechanism. Neurobiol. Dis. 2005, 20, 450–460. [Google Scholar] [CrossRef]
- Sgambato-Faure, V.; Buggia, V.; Gilbert, F.; Lévesque, D.; Benabid, A.L.; Berger, F. Coordinated and spatial upregulation of arc in striatonigral neurons correlates with L-dopa-induced behavioral sensitization in dyskinetic rats. J. Neuropathol. Exp. Neurol. 2005, 64, 936–947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alcacer, C.; Andreoli, L.; Sebastianutto, I.; Jakobsson, J.; Fieblinger, T.; Cenci, M.A. Chemogenetic stimulation of striatal projection neurons modulates responses to Parkinson’s disease therapy. J. Clin. Invest. 2017, 127, 720–734. [Google Scholar] [CrossRef] [PubMed]
- Hernández, L.F.; Castela, I.; Ruiz-DeDiego, I.; Obeso, J.A.; Moratalla, R. Striatal activation by optogenetics induces dyskinesias in the 6-hydroxydopamine rat model of Parkinson disease. Mov. Disord. 2017, 32, 530–537. [Google Scholar] [CrossRef] [PubMed]
- Girasole, A.E.; Lum, M.Y.; Nathaniel, D.; Bair-Marshall, C.J.; Guenthner, C.J.; Luo, L.; Kreitzer, A.C.; Nelson, A.B. A subpopulation of striatal neurons mediates levodopa-induced dyskinesia. Neuron 2018, 97, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Keifman, E.; Ruiz-DeDiego, I.; Pafundo, D.E.; Paz, R.M.; Solís, O.; Murer, M.G.; Moratalla, R. Optostimulation of striatonigral terminals in substantia nigra induces dyskinesia that increases after L-DOPA in a mouse model of Parkinson’s disease. Br. J. Pharmacol. 2019, 176, 2146–2161. [Google Scholar] [CrossRef] [PubMed]
- Tamim, M.K.; Samadi, P.; Morissette, M.; Grégoire, L.; Ouattara, B.; Lévesque, D.; Rouillard, C.; Di Paolo, T. Effect of non-dopaminergic drug treatment on Levodopa induced dyskinesias in MPTP monkeys: Common implication of striatal neuropeptides. Neuropharmacology 2010, 58, 286–296. [Google Scholar] [CrossRef] [PubMed]
- Sgambato, V.; Tremblay, L. Pathophysiology of dyskinesia and behavioral disorders in non-human primates: The role of serotonergic fibers. J. Neural Transm. 2018, 125, 1145–1156. [Google Scholar] [CrossRef]
- Nisenbaum, L.K.; Kitai, S.T.; Crowley, W.R.; Gerfen, C.R. Temporal dissociation between changes in striatal enkephalin and substance P messenger RNAs following striatal dopamine depletion. Neuroscience 1994, 60, 927–937. [Google Scholar] [CrossRef]
- Morissette, M.; Goulet, M.; Soghomonian, J.J.; Blanchet, P.J.; Calon, F.; Bédard, P.J.; Di Paolo, T. Preproenkephalin mRNA expression in the caudate-putamen of MPTP monkeys after chronic treatment with the D2 agonist U91356A in continuous or intermittent mode of administration: Comparison with L-DOPA therapy. Mol. Brain Res. 1997, 49, 55–62. [Google Scholar] [CrossRef]
- Calon, F.; Birdi, S.; Rajput, A.H.; Hornykiewicz, O.; Bédard, P.J.; Di Paolo, T. Increase of preproenkephalin mRNA levels in the putamen of Parkinson disease patients with levodopa-induced dyskinesias. J. Neuropathol. Exp. Neurol. 2002, 61, 186–196. [Google Scholar] [CrossRef]
- Nisenbaum, L.K.; Kitai, S.T.; Gerfen, C.R. Dopaminergic and muscarinic regulation of striatal enkephalin and substance P messenger RNAs following striatal dopamine denervation: Effects of systemic and central administration of quinpirole and scopolamine. Neuroscience 1994, 63, 435–449. [Google Scholar] [CrossRef]
- Steiner, H. Psychostimulant-induced gene regulation in striatal circuits. In Handbook of Basal Ganglia Structure and Function; Steiner, H., Tseng, K.Y., Eds.; Academic Press/Elsevier: London, UK, 2017; Volume 24, pp. 639–672. [Google Scholar]
- Gerfen, C.R.; Keefe, K.A.; Gauda, E.B. D1 and D2 dopamine receptor function in the striatum: Coactivation of D1- and D2-dopamine receptors on separate populations of neurons results in potentiated immediate-early gene response in D1-containing neurons. J. Neurosci. 1995, 15, 8167–8176. [Google Scholar] [CrossRef] [PubMed]
- Kannari, K.; Tanaka, H.; Maeda, T.; Tomiyama, M.; Suda, T.; Matsunaga, M. Reserpine pretreatment prevents increases in extracellular striatal dopamine following L-DOPA administration in rats with nigrostriatal denervation. J. Neurochem. 2000, 74, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Ng, K.Y.; Chase, T.N.; Colburn, R.W.; Kopin, I.J. L-DOPA-induced release of cerebral monoamines. Science 1970, 170, 76–77. [Google Scholar] [CrossRef]
- Iravani, M.M.; Jackson, M.J.; Kuoppamäki, M.; Smith, L.A.; Jenner, P. 3, 4-methylenedioxymethamphetamine (ecstasy) inhibits dyskinesia expression and normalizes motor activity in 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine-treated primates. J. Neurosci. 2003, 23, 9107–9115. [Google Scholar] [CrossRef] [Green Version]
- Sellnow, R.C.; Newman, J.H.; Chambers, N.; West, A.R.; Steece-Collier, K.; Sandoval, I.M.; Benskey, M.J.; Bishop, C.; Manfredsson, F.P. Regulation of dopamine neurotransmission from serotonergic neurons by ectopic expression of the dopamine D2 autoreceptor blocks levodopa-induced dyskinesia. Acta Neuropathol. Commun. 2019, 7, 8. [Google Scholar] [CrossRef]
- Barnes, N.M.; Sharp, T. A review of central 5-HT receptors and their function. Neuropharmacology 1999, 38, 1083–1152. [Google Scholar] [CrossRef]
- Celada, P.; Puig, M.; Artigas, F. Serotonin modulation of cortical neurons and networks. Front. Integr. Neurosci. 2013, 7, 25. [Google Scholar] [CrossRef] [Green Version]
- Sgambato-Faure, V.; Cenci, M.A. Glutamatergic mechanisms in the dyskinesias induced by pharmacological dopamine replacement and deep brain stimulation for the treatment of Parkinson’s disease. Prog. Neurobiol. 2012, 96, 69–86. [Google Scholar] [CrossRef]
- Steiner, H.; Kitai, S.T. Regulation of rat cortex function by D1 dopamine receptors in the striatum. J. Neurosci. 2000, 20, 5449–5460. [Google Scholar] [CrossRef] [Green Version]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Altwal, F.; Moon, C.; West, A.R.; Steiner, H. The Multimodal Serotonergic Agent Vilazodone Inhibits L-DOPA-Induced Gene Regulation in Striatal Projection Neurons and Associated Dyskinesia in an Animal Model of Parkinson’s Disease. Cells 2020, 9, 2265. https://doi.org/10.3390/cells9102265
Altwal F, Moon C, West AR, Steiner H. The Multimodal Serotonergic Agent Vilazodone Inhibits L-DOPA-Induced Gene Regulation in Striatal Projection Neurons and Associated Dyskinesia in an Animal Model of Parkinson’s Disease. Cells. 2020; 9(10):2265. https://doi.org/10.3390/cells9102265
Chicago/Turabian StyleAltwal, Feras, Connor Moon, Anthony R. West, and Heinz Steiner. 2020. "The Multimodal Serotonergic Agent Vilazodone Inhibits L-DOPA-Induced Gene Regulation in Striatal Projection Neurons and Associated Dyskinesia in an Animal Model of Parkinson’s Disease" Cells 9, no. 10: 2265. https://doi.org/10.3390/cells9102265