1. Introduction
Doxorubicin (DOX) is a widely used anticancer drug belonging to the anthracyclines family and used in the treatment of carcinomas, sarcomas and hematologic tumors. Breast cancer is one of the most carcinomas treated with DOX, especially when there is no indication of a targeted therapy. Nowadays, the main limitation of DOX treatment comes from its problematic cardiotoxicity [
1,
2]. The severity and irreversibility of the DOX-induced cardiotoxicity depends on the cumulated DOX doses and range from subclinical myopathy to severe heart failure, leading to the patient’s death in the worst cases [
3]. Cardiotoxicity is believed to result from excessive oxidative stress induced by DOX in cardiomyocytes through the overproduction of reactive oxygen species (ROS) [
1,
4,
5], which impair the mitochondrial function and membrane integrity [
4]. Eventually, cardiomyocytes undergo increasing apoptosis and necrosis, clinically expressed by progressive heart failure [
6].
These findings led to clinical trials evaluating the protective effects of some antioxidants to prevent DOX-induced cardiotoxicity. However, only dexrazoxane (DEX) demonstrated some significant protective effects in patients and was therefore approved by the FDA as a cardioprotective drug. DEX acts by reducing ROS production through iron chelation. DEX is intravenously injected into patients 10 to 30 min prior to the onset of DOX treatment at a recommended DEX/DOX dose ratio of 10:1 [
4]. Clinically, the DEX cardioprotective effect has been studied for more than 20 years. There are evidences that the incidence of heart failure is significantly reduced in patients pretreated with DEX [
7]. Despite its clear ability to reduce the incidence of heart complications in DOX-treated cancerous patients, DEX can be responsible for different adverse effects, particularly in pediatric patients. These findings led to a restriction of the indication of DEX for cardioprotection [
4,
8]. Therefore, the need for new cardioprotective strategies is obvious, hopefully safer and more efficacious than DEX.
Although the increased oxidative stress is considered as the main toxicological mechanism responsible of DOX cardiotoxicity, it remains unclear why, among all the antioxidants clinically tested, only DEX showed some substantial benefice. These unexpected findings led to the conclusion that the oxidative stress was most likely not the only toxic mechanism, and other causes should be investigated. Besides the beneficial effects of DEX on DOX-induced oxidative stress, some authors reported that DEX is also able to prevent its inhibition on the mitochondrial topoisomerase IIβ [
9], another proposed cardiotoxicological mechanism [
10]. This revived the search for other mechanisms of DOX-induced cardiotoxicity.
Besides their well-known roles in cell respiration [
11], some Krebs cycle intermediates are suspected to play an active role in crucial cell activities. For instance, it was proposed that succinate could accumulate in the interstitial space in the case of ischemia [
12]. In 2004, succinate was also pointed as the endogenous ligand of GPR91, a previously orphan G-protein-coupled receptor. Succinate was therefore considered to have unexpected signaling functions as an “alarming” signal able to trigger GPR91 [
13]. Since then, the physiological functions and pathological implications of GPR91 have been studied. The receptor expression was found in many tissues and organs, including the heart [
14]. However, its roles in cardiac physiology and pathophysiology are still unclear. While some evidences link it to cardiac hypertrophy through intracellular pathways involving cytoplasmic Ca
2+, RAS proteins, mitogen-activated protein kinases (MAPK) and inositol 3,4,5-triphosphate (IP3) generated by phospholipase C [
15], other findings associate GPR91 to the induction of apoptosis in cardiomyocytes through the activation of protein kinase A (PKA) [
16]. Such opposite effects suggest that GPR91 stimulation could regulate the cardiomyocytes’ fate, promoting either cell death or cell growth and survival on purpose. Clearly, should GPR91 be considered as a pharmacological target in drug-induced cardiotoxicity, a better understanding of how GPR91 functions and how it is regulated is needed. The discovery of a GPR91-succinate interaction brings new perspectives for the development of new treatments to counteract different cardiomyopathies involving either a loss of cardiomyocytes or an excess of cardiomyocyte growth. We previously studied the effects of DOX and DEX on H9C2 cardiomyoblasts metabolism by
1H-NMR metabonomics. We highlighted that exposing cells to DEX prior to DOX caused an oversecretion of succinate in the culture medium. This led us to hypothesize that the release of succinate in DEX-exposed cells could protect against DOX-induced cardiotoxicity in H9C2 cells by an autocrine activation of the GPR91 receptor and the subsequent triggering of cell survival pathways [
17].
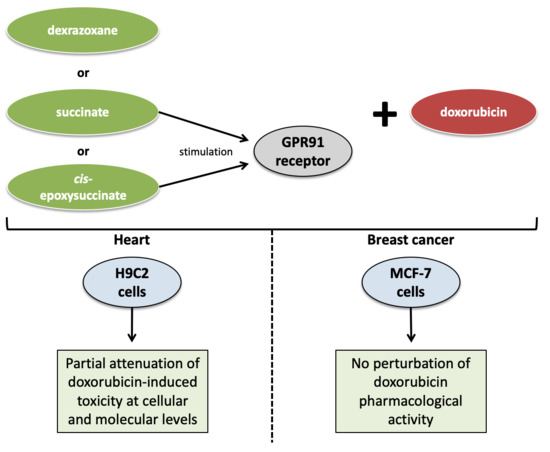
In this study, we investigated the putative protective role of GPR91 against DOX-induced cardiotoxicity in rat H9C2 cells. In parallel, the effect of stimulating GPR91 on DOX anticancer efficacy was checked in breast cancer-derived MCF-7 cells.
2. Materials and Methods
2.1. Materials
The H9C2 (2-1) (ECACC 88092904) cell line was purchased from the European Collection of Authenticated Cell Cultures (Salisbury, United Kingdom). The MCF-7 cell line was obtained from ATCC (Manassas, VA, USA). Dulbecco’s modified Eagle’s medium (DMEM), trypsin-EDTA 0.05%, penicillin/streptomycin, L-glutamine, cis-epoxysuccinate (cis-ES) and Alexa Fluor® 488 goat anti-rabbit immunoglobulin G (IgG) secondary antibody (AB) were acquired from Gibco (Thermo Fisher Scientific, Waltham, MA, USA). Dulbecco’s phosphate-buffered solution (D-PBS), phosphate-buffered solution (PBS), fetal bovine serum (FBS), doxorubicin hydrochloride (DOX), dexrazoxane (DEX), dichloro-dihydro-fluorescein diacetate (DCFH-DA), glutaraldehyde, triton, crystal violet, succinate and 4′,6-diamidino-2-phénylindole (DAPI) mounting solution were obtained from Sigma-Aldrich (Saint-Louis, MO, USA). Rabbit anti-GPR91 AB (BS2961) was purchased from BioWorld Technology (Bloomington, IN, USA).
2.2. Cell Culture
H9C2 cells were cultured in DMEM high glucose supplemented with L-Glutamine 2 mM, heat-inactivated FBS 10% and antibiotics (100 µg/mL of streptomycin and 100 unit/mL of penicillin), incubated at 5% CO2 and 37 °C, in a humidified atmosphere. Culture medium was replaced every 2 to 3 days, and cells were split when they reached 80% of confluence using trypsin-EDTA 0.05%. For viability and ROS quantification procedures, 30,000 cells were first seeded in 96-well plates and were kept growing during 24 h before any exposure. For the caspase 3 quantification assay, cells were seeded at a density of 30,000 cells/cm2 and were kept growing during 48 h before any exposure.
MCF-7 breast cancer cells were cultured in DMEM high glucose supplemented with L-glutamine 4 mM, heat-inactivated FBS 10% and antibiotics (100 µg/mL of streptomycin and 100 unit/mL of penicillin), incubated at 5% CO2 and 37 °C, in a humidified atmosphere. Culture medium was replaced every 2 to 3 days, and cells were split when they reached 100% of confluence using trypsin-EDTA 0.05%. For the proliferation assay, 30,000 cells were first seeded in 96-well plates and were kept growing during 24 h before any exposure.
2.3. Immunofluorescence Staining of GPR91
For immunostaining purposes, H9C2 and MCF-7 cells were grown on lamellae in 6-well plates. Cells were first washed with PBS and fixed with paraformaldehyde for 20 min. Cells were permeabilized using triton 0.05% for 15 min and then incubated during 20 min with 0.05% casein for blocking. After rinsing with PBS, cells were labeled with a specific rabbit primary anti-GPR91 AB diluted in a blocking solution (1:100) for 2 h at room temperature. Cells were washed with PBS and exposed to Alexa Fluor® goat anti-rabbit IgG (secondary AB) during 1 h in the dark at room temperature. Cells were finally washed, and lamellae were mounted on slides with DAPI mounting solution and sealed with nail polish. A control without primary AB was made for both cell types. Coverslips were analyzed using a fluorescent microscope Olympic Fluoview FV1000 (Olympus, Berchem, Belgium). All images were obtained at a magnification of 60× by Z-projection.
2.4. Cell Viability Assay
The impacts of DOX, DEX, succinate (SUC) and
cis-ES on the H9C2 cell viability were assessed by a crystal violet procedure in 96-well plates, as previously mentioned [
17]. After cell exposure, the culture medium was removed, and cells were washed twice with PBS and incubated with 100 µL of glutaraldehyde 1% for 15 min at room temperature (RT). Glutaraldehyde was then removed, and 100 µL of crystal violet 1% was added for 30 min at RT. The plate was profusely washed with water and then dried at RT. The plate was incubated with 100 µL of triton 0.2% and agitated for 60 min. The absorbance was read at 570 nm with a VersaMax plate reader (Molecular Devices, Wokingham, England). Relative mean cellular viability was determined. All exposures were performed by diluting the compounds into a fresh culture medium. Cells were randomly assigned to different conditions in 96-well plates: a control condition receiving the vehicle; cells receiving DOX (0.3 µM); cells receiving either DEX, SUC or
cis-ES (3 µM) and cells receiving either DEX, SUC or
cis-ES 30 min prior to DOX exposure. Viability was measured after 24, 48 and 72 h of exposure. Moreover, increasing the doses of SUC or
cis-ES (3–6–30–100 µM) and the combined exposures of either SUC or
cis-ES with DEX were evaluated for their possible protective roles against DOX-induced cell mortality after 48 h of exposure.
2.5. Caspase 3 Activity Assay
The ability of DOX, DEX, SUC and
cis-ES to induce or prevent apoptosis was indirectly assessed using the caspase 3 enzyme activity, as previously described [
17]. H9C2 cells were randomly split in different conditions and were exposed to DOX 5 µM, DEX 50 µM, SUC 50 µM and
cis-ES 50 µM in 96-well plates. Cells were also exposed once to each cardioprotective compound 30 min before DOX exposure, keeping a protective compound/DOX dose ratio of 10/1. After 4 h of exposure, the culture medium was removed. Cells were washed twice with PBS and then collected in PBS by scraping. A centrifugation at 260×
g during 5 min was performed at 4 °C. The cell pellet was stored at −80 °C before further analysis. The activity of caspase 3 was evaluated using EnzChek Caspase-3 Assay Kit 1 (Thermo Fisher Scientific, Waltham, MA, USA), according to the manufacturer’s instructions. The intensity of fluorescence was read with a Glomax Explorer (Promega, Leiden, The Netherlands) plate reader at 342/441 nm (excitation/emission). Data were expressed as the relative mean of caspase 3 activity.
2.6. Oxidative Stress Quantification Assay
The effects of DOX, DEX, SUC and
cis-ES on the redox state of H9C2 cells were indirectly assessed by a quantification of ROS production using a DCFH-DA probe [
18]. Cells were randomly exposed in PBS to DOX 5 µM, DEX 50 µM, SUC 50 µM and
cis-ES 50 µM in 96-well plates for 2 h. Cells were also exposed once to each cardioprotective compound 30 min before DOX exposure, keeping a protective compound/DOX dose ratio of 10/1. A positive control was performed by a 10-mM H
2O
2 exposure, and a negative control was carried out with cells only receiving PBS. After incubation, a DCFH-DA solution was added in each well to reach a final concentration of 100 µM for 30 min at 37 °C and in darkness. Intensity of the fluorescence was read with a Glomax Explorer (Promega, Leiden, The Netherlands) plate reader at 490/510 nm (excitation/emission). Data were expressed as the relative means of the ROS level.
2.7. Cells Exposure and Samples Collection for Metabonomic Study
H9C2 cells were randomly exposed to DOX 0.3 µM, DEX 3 µM, SUC 3 µM and
cis-ES 3 µM in T-175 flasks for 24 h. Cells were also exposed once to each cardioprotective compound 30 min before DOX exposure, keeping a protective compound/DOX dose ratio of 10/1. After exposure, extracellular media samples were collected and stored at −80 °C. Then, the cell mat was washed twice with PBS and collected in 6 mL of cold methanol by scraping. Samples were then rapidly frozen in liquid nitrogen and stored at −80 °C [
19].
2.8. Samples Preparation for 1H-NMR, Data Acquisition and Treatment
A chloroform-methanol-water extraction procedure was carried out on cells to extract intracellular metabolites [
20]. Briefly, a cell lysis by sonication was performed, and the 3 solvents were added to separate the cell contents into an aqueous phase containing hydrophilic metabolites and a chloroform phase containing lipophilic metabolites. The methanol-water phases were collected and evaporated with a speed vacuum concentrator. As previously described, 700 µL of a phosphate-buffered solution (0.2-M Na
2HPO
4/0.04-M NaH
2PO
4, pH 7.4) were used to dissolve the hydrophilic metabolites. Samples were centrifuged at 10,000×
g during 10 min. Fifty microliters of trimethylsilylpropanoic acid (TSP) 3.5 mM was added to the supernatant (650 µL) in NMR tubes. Extracellular media samples (500 µL) were mixed with a phosphate-buffered solution (250 µL). Samples were centrifuged at 10,000×
g during 10 min. Fifty microliters of trimethylsilylpropanoic acid (TSP) 14 mM was added to the supernatant (650 µL) in NMR tubes [
17].
1H-NMR spectra of both intracellular and extracellular compartments were acquired by a Bruker Advance 600 MHz spectrometer (Bruker BioSpin GmbH, Kontich, Belgium). The employed sequence was NOESYPRESAT-1D, with a number of 256 scans. The acquired free induction decay signals were Fourier-transformed to obtain spectra. Baseline and phase corrections were performed using MestReNova 10.0.2 (Mestrelab Research, Santiago de Compostela, Spain). Spectra were arbitrarily calibrated by setting the TSP-arising resonance to 0.00 ppm. Region from 0.08 to 10 ppm was divided into subareas of 0.04-ppm widths that were further integrated. The water peak (from 4.50 to 5.00 ppm) was suppressed. A total area normalization was carried out for each subarea integral using Excel functionalities (Microsoft Office® 16.36, Redmond, WA, USA).
2.9. Multivariate Data Analysis, Metabolites Identification and Statistical Tests
Metabonomic data were analyzed by a projection to a latent structure discriminant analysis (PLS-DA), where the experimental groups were defined as classes. SIMCA P+ 12 (Umetrics, Umeå, Sweden) was used for this purpose. R
2cum and Q
2cum parameters, as well as the
p-value of ANOVA of the cross-validated residuals (CV-ANOVA), were worked out. A cross-validation using 200 permutations was performed to ensure the suitability of the model. The variables characterized by a variable importance in projection (VIP) value > 0.8 were selected as the most discriminant ones, according to recommendations in [
21]. Corresponding metabolites were identified with several databases: the Human Metabolome Database (HMDB) [
22], Chenomx Profiler software 8.3 (Edmonton, Canada) [
23] and “in-house” databases.
Due to the descriptor size of 0.04 ppm that could contain several metabolite chemical shifts but, also, the low signal-to-noise ratio of some metabolites, a semi-quantification comparison of the spectra was then processed to specify the metabolic changes. Statistical significance of the discriminant metabolites was assessed by integrating the
1H-NMR peaks of each metabolite with a VIP value > 0.8. Each integral was normalized to the spectral total area, as previously mentioned [
17]. The most appropriate statistical tests to compare variables between the different conditions were chosen to respect the two restrictive hypotheses, allowing the use of the parametric tests: the normal distribution of the variable and the equality of the variances. Normality of data was evaluated by a Shapiro-Wilk test [
24]. Homoscedasticity of variances was evaluated by a Bartlett’s test [
25]. For variables characterized by a normal distribution and a variance homoscedasticity, statistical significance was determined using one-way ANOVA. For other variables, significance was determined using the Dunn’s test. For proportion comparisons, a two-proportion z-test was applied. The significance was determined at
p-value * < 0.05, **
p-value < 0.01 and
p-value *** < 0.001. Heatmaps for both intracellular and extracellular compartments were established from the mean normalized integrals of the discriminant metabolites, as previously described [
17].
To highlight the most relevant metabolic pathways, an enrichment analysis was carried out on the discriminant metabolites. The MetaboAnalyst 4.0. online software was used for this purpose. A metabolite set enrichment analysis (MSEA) was performed [
26]. The MSEA is a tool designed to help the interpretation of metabonomic data by highlighting the most relevant biological pathways linked to the signature identified through our analysis. The analysis provides a classification of suggested pathways depending on the number of imputed metabolites found in the identified pathways. A depending
p-value was worked out and compared to the 0.05 alpha value threshold.
2.10. MCF-7 Cell Proliferation Assay
The effects of DOX and protective compounds DEX, SUC and cis-ES alone or in combination with DOX on MCF-7 cell proliferation were evaluated by the crystal violet assay. For this purpose, the living cells population was measured at different time points after exposure (days 1, 3, 6, 8 and 9). Exposure medium was renewed every 3 days. Relative mean of the living cells population was determined for each condition.
4. Discussion
H9C2 cells are derived from a rat cardiomyoblast cell line similar to differentiated cardiomyocytes but without any cardiac functional characteristics. This cellular model was employed due to the constraints imposed by
1H-NMR spectroscopy that require more than 10 million cells per sample to ensure a correct signal-to-noise ratio. For this purpose, the ability of H9C2 cells to reproduce the main features of the DOX toxic mode of action, such as oxidative stress [
27,
28], apoptosis [
29,
30], topoisomerases inhibition and sarcoplasmic reticulum stress [
31,
32], were ensured through a literature review and our own experimental data. Despite some obvious drawbacks, the H9C2 cell line remains a suitable model for studying DOX-induced cardiotoxicity and cardioprotective strategies. However, a further investigation of the GPR91 agonist-induced protective effect should be carried out on more human extrapolated in vitro/in vivo models for validation.
In a previous work, we noticed an oversecretion of SUC in the extracellular medium when H9C2 cells were co-exposed to DEX and DOX during 24 h. We hypothesized that DEX could possibly protect against DOX-induced cardiotoxicity through an auto/paracrine stimulation of the GPR91 receptor by SUC, triggering the cell survival pathways [
17]. In this study, the protective effects of SUC and
cis-ES, two GPR91 agonists, against DOX-induced cardiotoxicity were investigated on H9C2 cells. For long periods of exposure (24 to 72 h), DOX caused a decrease of cell viability, an effect partially counteracted by both SUC and
cis-ES pre-exposures. Interestingly, the protection obtained with both agonists was close to the one already seen for DEX pre-exposure, the reference cardioprotective compound. Moreover, on a short time of exposure (4 h), DOX significantly triggered the apoptotic process by an enhanced activity of the caspase 3 enzyme, as already reported [
33]. SUC pre-exposure was able to decrease the activity of caspase 3 to a similar extent as DEX pre-exposure, whereas
cis-ES pre-exposure provoked an even more pronounced effect. The important elevation of ROS levels caused by DOX was partially prevented by SUC and DEX to a similar extent, whereas
cis-ES showed a more drastic effect. These results suggest that agonism of the GPR91 receptor protects H9C2 cells against DOX-induced toxicity, especially when using
cis-ES as the agonist.
cis-ES was used in this study because of its higher specificity and affinity to GPR91 than SUC [
34] and to avoid any possible additional effects of SUC that are also involved in cell respiration.
In our conditions, a pre-exposure to GPR91 agonists inhibited apoptosis induced by DOX. There are some evidences linking GPR91 to both apoptosis and cell growth. Indeed, some authors reported that GPR91 induces cardiac hypertrophy [
15,
35], while others observed that a prolonged incubation of cardiomyocytes with high concentrations of succinate (10 mM) promoted apoptosis [
16]. Similar observations were made in the context of an ischemic injury, where the release of succinate and the triggering of the GPR91-associated signaling pathway were responsible for mitochondrial dysfunction and apoptosis in cardiomyocytes [
36]. In the present study, although both agonists significantly reduced the DOX-induced overactivity of caspase 3, SUC alone induced an increased activity of caspase 3, while no such effect was found for
cis-ES alone. Those findings suggest that the proapoptotic or the pro-growth effect mediated by GPR91 could depend on the exposure context, and exposure conditions and should be further investigated.
It has been reported that succinate dehydrogenase (SDH), forming complex II of the respiratory chain in mitochondria, has an important role in redox regulation in the heart as an enhancer or suppressor of ROS. Some authors claim that SUC is an important regulator of SDH-induced ROS production and that a low level of SUC is associated with a higher ROS production by SDH [
37,
38,
39]. The SUC level seems to be a critical factor for the ROS regulation performed by SDH. In our study, we observed a decreased ROS level associated with a higher level of SUC, as detected by
1H-NMR, when H9C2 cells were pre-exposed to SUC or
cis-ES before adding DOX, as compared to DOX alone. Those results suggest that the decrease of the ROS level may be caused by the higher level of SUC promoting the ROS suppression activity of SDH. The link between GPR91 activation by agonists and the regulation of the ROS level by SDH is still unclear. Additional investigations are needed to demonstrate if SUC and
cis-ES reduce the ROS level trough a GPR91-dependent or -independent manner.
The expression of GPR91 was validated in H9C2 cells by immunofluorescence staining, in accordance to similar observations by other authors [
36], suggesting that this receptor may be strongly implicated in the protective effects of
cis-ES and SUC. However, to ensure and validate the crucial role of the GPR91 receptor in the observed protective effects, a receptor knockout should be performed, as already reported [
40,
41].
In a previous study, we characterized the metabolic changes induced by DOX and DEX in H9C2 cells [
17]. In this study, we focused on the metabolic effects of a 30-min preincubation with SUC or
cis-ES before 24 h-DOX exposure on H9C2 cells using a
1H-NMR-based metabonomic approach. Both the intra- and extracellular compartments of H9C2 cells were investigated for this purpose. The metabonomic profiles obtained under such conditions pointed out that the main metabolic differences were due to cardioprotective compounds rather than DOX exposure. SUC and
cis-ES exposures and pre-exposure had similar metabolic profiles, which were different from the DEX-induced profiles. The enrichment analysis using MetaboAnalyst software highlighted many pathways that are related to energy metabolism and anabolism. Identified metabolite variations strongly suggest that both SUC and
cis-ES stimulate aerobic metabolism mainly through glycolysis and the Krebs cycle. The extracellular level of glucose is decreased in all SUC and
cis-ES conditions, suggesting a higher uptake of glucose from the culture medium. A strong increase in extracellular pyruvate and a small increase in intra- and extracellular lactate concentrations are also observed in all SUC and
cis-ES conditions, but the extracellular L:P remains higher than those of the CTR and DOX conditions. The L:P ratio is commonly used in serums to detect mitochondrial disorders, as it reflects the equilibrium between the product and substrate of the reaction catalyzed by lactate dehydrogenase and indirectly reflects the NADH:NAD
+ redox status of the intracellular compartment [
42,
43]. A high L:P ratio is associated with dysfunction of the mitochondrial respiratory chain, where there is an increase in reducing equivalents (excess of NADH and absence of NAD
+) [
44]. Therefore, this ratio can be used as an indicator of the cell aerobic or anaerobic status. Here, we transposed the L:P ratio to the culture fluid that mimics the blood compartment. As previously observed, DOX induced a metabolic switch to anaerobic glycolysis [
17], characterized by a high L:P ratio. All protective compounds induced a decrease of the L:P ratio compared to the DOX and CTR conditions. This strongly suggests that SUC,
cis-ES and DEX stimulate the mitochondrial respiratory chain, resulting in a stronger aerobic metabolism. Besides glycolysis, the Krebs cycle can also use other substrates for adenosine triphosphate (ATP) production. The metabonomic results highlight that glycolysis was used as the main pathway to supply the Krebs cycle when the cells were exposed to the GPR91 agonists. Indeed, the intracellular content of carnitine was lowered in such conditions, suggesting that fatty acid oxidation is poorly used for ATP production. Moreover, the intracellular detection of glutamine and glutamate reflects that glutamine present in the culture medium entered into cells where it was converted into glutamate by a glutaminase enzyme. Glutamate can be used as fuel for the Krebs cycle. The intracellular Gln:Glu ratio was calculated to evaluate the activity of glutaminase. This ratio was higher in all SUC and
cis-ES conditions, assuming that glutamine is poorly converted into glutamate, as a consequence of a lower need for glutamate to supply ATP production. The Gln:Glu ratio was already used to evaluate the dependence of cancer cells to glutamine for energy purposes and as an indicator of tissue oxygenation [
45,
46]. All SUC and
cis-ES conditions were characterized by a lower intracellular level of proline, a precursor of α-ketoglutarate, to supply the citric cycle. Thus, a lower level of proline may highlight that this amino acid is less-produced, highlighting a lower need to form α-ketoglutarate. The results also indicate a higher level of isoleucine in the extracellular compartment of cells exposed to GPR91 agonists, as compared to the CTR. Isoleucine is an essential amino acid that cannot be produced by cells and must be supplied by the culture medium. A higher extracellular concentration reflects a lower consumption by cells. Despite the fact that isoleucine can be used as a “non-glucose” substrate for the Krebs cycle, its lower consumption is also an argument that SUC and
cis-ES stimulate the aerobic energy metabolism using glucose as the main fuel. An increase in the SUC level was observed in the extracellular compartment of cells exposed to GPR91 agonists. When SUC was used as the agonist (at a nondetectable concentration by
1H-NMR), this increase was bigger than the one observed with DEX exposure. An enhancement of the SUC level strongly indicates a stimulation of the Krebs cycle in the mitochondria for energy production that is induced by GPR91 agonists. Moreover, the observations suggest that SUC is secreted in the extracellular compartment and could stimulate the GPR91 receptor to sustain the primary effect of SUC and
cis-ES on this receptor. This hypothesis could explain why an exposure with a very low concentration of SUC and
cis-ES (3 µM) is able to ensure a cardioprotective effect that is not enhanced with a higher exposure concentration as a “plateau effect”. Some evidences suggest that SUC may impair the pyruvate dehydrogenase (PDH) activity through the GPR91-dependent and -independent signaling pathways in cardiomyocytes suffering from an ischemia/reperfusion injury [
47]. Hence, the association between high extracellular levels of SUC and pyruvate might be linked to this phenomenon. However, as previously discussed, the low L:P ratio observed in cells exposed to either GPR91 agonists or DEX strongly suggests an aerobic mitochondrial metabolism for energy production and a lower production of lactate from pyruvate. Therefore, the effect of the GPR91 agonists on the PDH activity should be further investigated to elucidate this phenomenon in the context of DOX-induced cardiotoxicity.
The metabonomic data also highlighted an important SUC and
cis-ES-induced stimulation of phospholipids synthesis, although to different extents, as seen with DEX. First, both the phosphocholine (PCho) and glycerophosphocholine (GPC) levels were enhanced in cells exposed to GPR91 agonists but to a lower extent than DEX, indicating an ongoing activation of the Kennedy’s pathway responsible for phosphatidylcholine synthesis, a major phospholipid found in biological membranes [
48]. Moreover, the levels of choline and its precursor glycine were decreased in such conditions compared to the DEX pre-exposure effect, also supporting that the activation of the Kennedy’s pathway by SUC and
cis-ES remains less intense in DEX conditions. A similar observation was made about phosphatidylserine synthesis. Indeed, the serine level, the precursor of this phospholipid, was elevated in SUC and
cis-ES conditions, again to a lower extent than in DEX conditions. However, SUC seems to more actively stimulate phosphatidylinositol synthesis than DEX. Indeed, the intracellular level of
myo-inositol, the precursor of phosphatidylinositol, was higher with the agonists than with DEX. Therefore, our results strongly suggest that both GPR91 agonists were able to stimulate phospholipids biosynthesis to counteract DOX-induced cardiotoxicity, although to a lower extent than DEX does.
Glutathione is a tripeptide composed of glutamate, glycine and cysteine. It is found in all mammal tissues and is a powerful agent for redox homeostasis maintenance, especially in conditions where cells could be damaged by oxidative stress [
49]. DOX induced a harmful oxidative stress in H9C2 that can be partially prevented by pre-exposing cells to either SUC or
cis-ES. The metabonomic results pointed out a decrease of glutathione and precursors (glycine and glutamate) induced by the GPR91 agonists. Moreover, our results showed a decrease of the intracellular taurine and carnitine contents. Considering that those compounds can be used as antioxidants to counteract DOX-induced oxidative stress [
50,
51], their decreased concentration could indicate a lower cell need for redox maintenance. It sounds as if both GPR91 agonists induced a decrease of oxidative stress using other mechanisms than glutathione, taurine and carnitine synthesis. As already discussed, the increase of the SUC level is suspected to reduce the ROS production induced by SDH in mitochondria and should be responsible for a lower need of antioxidant compounds such as glutathione, carnitine and taurine for redox maintenance. Oxidative stress was considered for long as the main DOX-induced toxicological mechanism causing mitochondrial dysfunction and cell death. Oxidative stress was therefore seen as the “starting point” of all cell damages induced by DOX in the heart. However, both SUC and
cis-ES GPR91 agonists were able to reduce DOX-induced oxidative stress, while those compounds have no known antioxidant properties. This observation, together with the lack of protective effect reported for many antioxidants but DEX, contributes to questioning the role of oxidative stress in DOX-induced cardiotoxicity. Indeed, among all the tested antioxidants, only DEX was able to exhibit a cardioprotective effect in the clinical trial. Thereafter, some authors reported that DEX was also able to prevent the DOX-induced inhibition of the mitochondrial topoisomerase IIβ [
9]. Considering these evidences, it appears that DOX-induced oxidative stress should be considered as a “consequence” rather than a “cause” in the cardiotoxicological process. Our metabonomic data strongly suggest that the synthesis of endogenous antioxidants is decreased in cells pre-exposed to GPR91 agonists. Both agonists may act on the cause(s) of the oxidative stress rather than on the oxidative stress itself, which could then be targeted for the development of cardioprotective strategies.
So far, there have been no clinical trial evaluating the benefit of a GPR91 agonist or antagonist against any particular pathology, as well as there being no drug candidate involving SUC or cis-ES as an active ingredient. All discovered GPR91 agonists and antagonists are still experimental molecules only available for research purposes. Considering that more than 50% currently approved drugs target G-protein coupled receptors, there are many hopes that drug candidates targeting GPR91 will be developed. In our in vitro study, we highlighted that targeting GPR91 with an agonist could partially counteract DOX-induced cardiotoxicity. However, further investigations are needed for studying GPR91 agonists’ efficacy and safety in the relevant animal models before considering clinical studies. As GPR91 agonists are expected to activate GPR91 in all cells expressing it, some optimization would be necessary to ensure the heart-selective delivery and to avoid adverse effects due to interactions on other GPR91-expressing organs. In this context, tissue-selective drug delivery systems such as liposomes or specific vectors may be developed and evaluated in rigorous preclinical studies.
As breast cancer is one of the main indications for DOX treatment, human breast cancer-derived MCF-7 cells were selected to assess the effect of a cardioprotective strategy targeting the GPR91 receptor on cancer cell growth and on DOX pharmacological action. Our results demonstrate that such agonists do not interfere with the DOX anticancer effects on MCF-7 cells, alone or in combination. GPR91 expression was also undetected in MCF-7 cells. Based on those findings, one can reasonably assume that targeting the GPR91 receptor for cardioprotective purposes should not interfere with the DOX pharmacological effects on cancer cells that do not express the GPR91 receptor. Although the possible involvement of GPR91 and SUC in tumor development is not clearly established, some works suggest that SUC and its receptor may be tumor promoters. It has been reported that the intracellular concentration of SUC, as well as other metabolites such as fumarate, aspartate and 2-hydroxyglutarate, is increased in some cancers and is responsible for the epigenetic changes associated with carcinogenesis and impacting the activity of proteins involved in cell signaling and metabolism [
52,
53]. The accumulation of SUC in cancer cells is secondary to mutations in SDH causing a loss-of-function of the enzyme, which no longer converts SUC to fumarate. These mutations are sufficient to promote the development of some cancers, including renal carcinoma [
54]. There is currently no evidence of a direct link between the GPR91 receptor and carcinogenesis. Nevertheless, the known implications of the receptor in processes found during tumor development such as angiogenesis [
55], fibrosis [
56] and inflammation [
57] raise questions about its possible participation in the development of some cancers. In addition, a recent study pointed out that some cancer cells, whose SDH activity was reduced, were able to secrete SUC in the tumor microenvironment, causing the activation of macrophages to “tumor-associated macrophages” (TAM) that play a role in tumor progression and metastases developments [
58]. The presence of TAM in the tumor microenvironment is indeed associated with a poor prognosis in breast cancer, ovarian cancer and in some types of gliomas and lymphomas [
59]. For the development of a cardioprotective therapy targeting the GPR91 receptor to counteract the adverse effects of anticancer drugs, special attention should be paid to a possible adverse effect promoting the development of cancers. Further studies should determine whether the expression of the GPR91 receptor in cancer cells, the secretion of succinate by the tumor and the presence of TAM in the tumor microenvironment could be exclusive criteria or not for a cardioprotective strategy targeting this receptor.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}