Mechanical Forces as Determinants of Disseminated Metastatic Cell Fate


Abstract
:1. Introduction
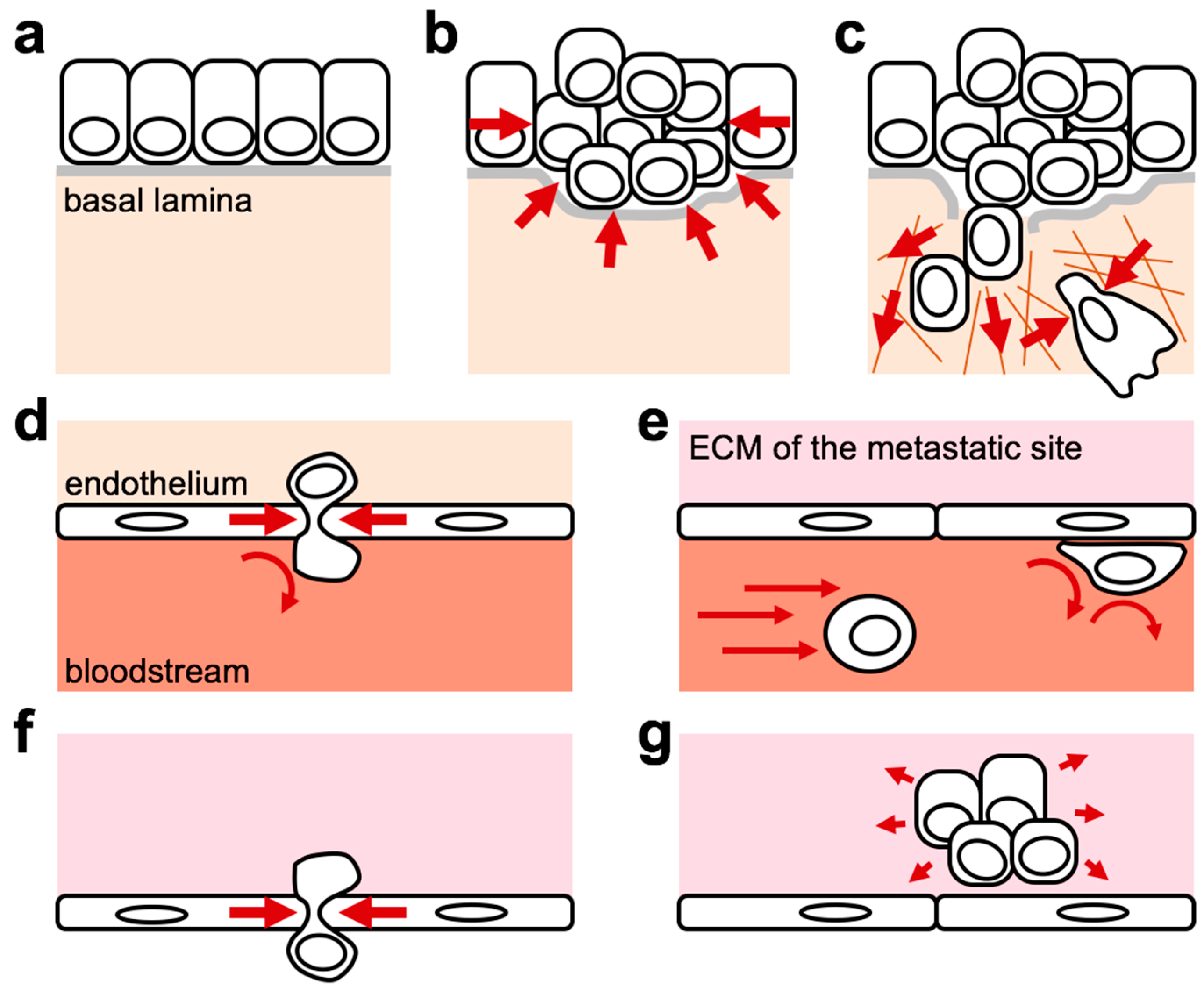
2. ECM Mechanical Forces in Cancer
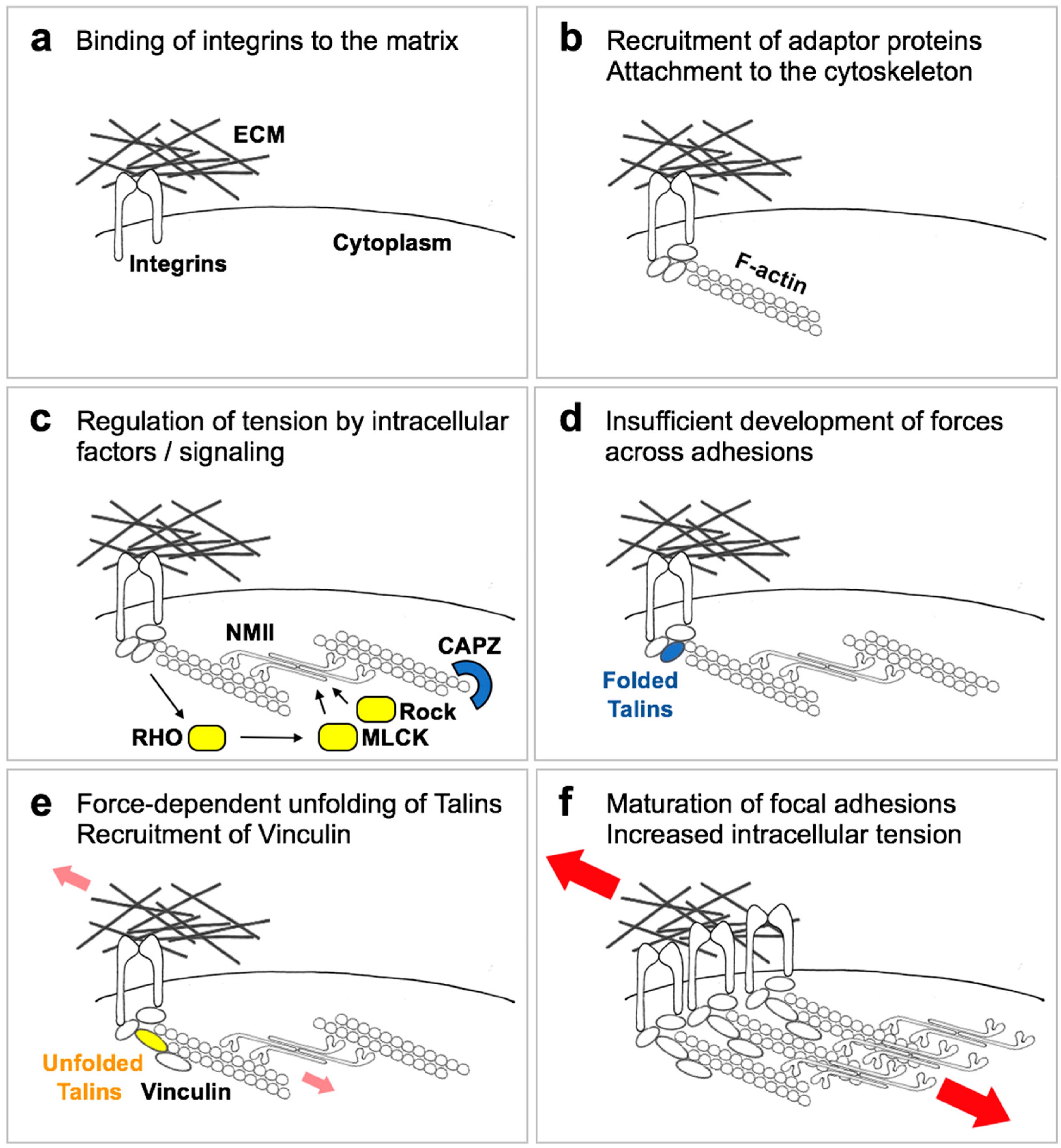
3. Cells Measure the Physical Properties of the ECM
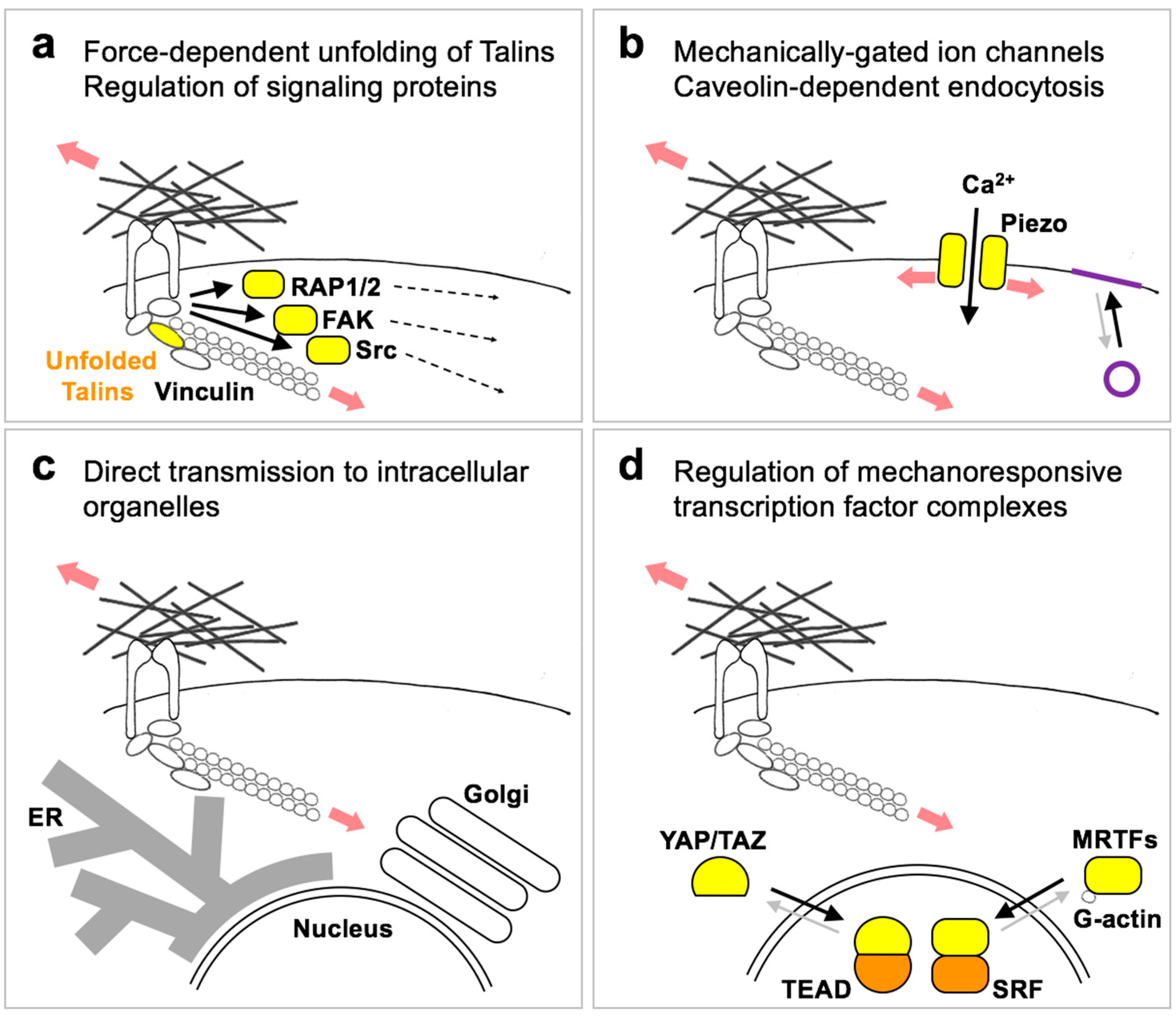
4. Downstream Pathways Modulated by Forces
5. Mechanical Forces as Regulators of Disseminated Cancer Cells’ Fate
6. Extracellular Matrix Proteins and Metastatic Outgrowth
7. Mechanosensing Receptors and Metastatic Fitness
8. Mechanosensing Pathways and Dormancy at the Metastatic Site
9. Conclusions
Funding
Conflicts of Interest
Abbreviations
| AKT | protein kinase B family |
| ARF1 | ADP ribosylation factor 1 |
| ARID1A | AT-rich interaction domain 1 |
| BMP | bone morphogenetic protein ligand family |
| BMP-1 | bone morphogenetic protein-1, astacin-like protease |
| CAPZ | capping actin protein of muscle z-line complex |
| COCO | DAN domain BMP antagonist family member 5 |
| CSC | Cancer Stem Cells |
| DDR | Discoidin Domain Receptor |
| ECM | Extracellular Matrix |
| EGF | Epidermal Growth Factor |
| EGFR | Epidermal Growth Factor Receptor |
| EMT | Epithelial to Mesenchymal Transition |
| ER | Endoplasmic Reticulum |
| ERK | Extracellular signal-Regulated Kinase |
| FA | Focal Adhesion |
| FAK | Focal Adhesion Kinase |
| FGF | Fibroblast Growth Factor |
| FLP | Filopodia-Like Protrusions |
| FN | Fibronectin |
| ILK | Integrin-Linked pseudokinase |
| JAK | Janus Kinase |
| KSR | Kinase Suppressor of Ras |
| L1CAM | L1 Cell Adhesion Molecule |
| LATS | Large Tumor Suppressor Kinase |
| LINK | Linker of Nucleoskeleton and Cytoskeleton Complex |
| LOX | Lysyl Oxidase |
| mDIA2 | Diaphanous-related formin 3 |
| MLCK | Myosin Light Chain Kinase |
| MMP-2 | Matrix Metallopeptidase 2 |
| MRTF | Myocardin-Related Transcription Factor Family |
| NET | Neutrophils Extracellular Trap |
| p130cas | BCAR1 scaffold protein, cas-family member |
| p190 | RHO GTPase activating protein 5 |
| p21 | Cyclin-Dependent Kinase inhibitor 1A |
| p27 | Cyclin-Dependent Kinase inhibitor 1B |
| p38 | Mitogen-Activated Protein Kinases |
| Par-1 | MARK, Microtubule-Affinity Regulating Kinases |
| PARD3 | Par-3 family Cell Polarity Regulator family |
| PARD6 | Par-6 family Cell Polarity Regulator family |
| PI3K | Phosphoinositide 3-Kinase |
| PKC | Protein Kinase C |
| POSTN | Periostin |
| RAP | RAP1A, RAP2A-C members of RAS oncogene family |
| Ras | Small GTPase |
| RHO | Ras Homology family member protein |
| SCP | Single Cell Population |
| SFRP2 | Secreted Frizzled-Related Protein 2 |
| Src | SRC proto-oncogene, non-receptor-tyrosine kinase |
| SREBP | Sterol-Regulatory Element Binding Proteins |
| SRF | Serum Response Factor |
| STAT3 | Signal Transducer and Activator of Transcription 3 |
| TAZ | WWTR1, WW-domain containing transcription regulator 1 |
| TEAD | TEA-Domain Transcription Factor family |
| TGFβ | Transforming Growth Factor β |
| TM4SF1 | Transmembrane 4 L Six Family member 1 |
| TNC | Tenascin C |
| TSP-1 | THBS-1, Thrombospondin 1 |
| VCAM-1 | Vascular Cell Adhesion Molecule 1 |
| vWF | Von Willebrand Factor |
| YAP | YES-Associated Protein 1 |
| ZEB | Zinc finger E-box Binding homeoboxes |
References
- Maman, S.; Witz, I.P. A history of exploring cancer in context. Nat. Rev. Cancer 2018, 18, 359–376. [Google Scholar] [CrossRef] [PubMed]
- LeGoff, L.; Lecuit, T. Mechanical Forces and Growth in Animal Tissues. Cold Spring Harb. Perspect. Biol. 2015, 8, a019232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petridou, N.I.; Spiró, Z.; Heisenberg, C.-P. Multiscale force sensing in development. Nat. Cell Biol. 2017, 19, 581–588. [Google Scholar] [CrossRef] [PubMed]
- Dupont, S. Role of YAP/TAZ in cell-matrix adhesion-mediated signalling and mechanotransduction. Exp. Cell Res. 2016, 343, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Iskratsch, T.; Wolfenson, H.; Sheetz, M.P. Appreciating force and shape—The rise of mechanotransduction in cell biology. Nat. Rev. Mol. Cell Biol. 2014, 15, 825–833. [Google Scholar] [CrossRef] [PubMed]
- Kirby, T.J.; Lammerding, J. Emerging views of the nucleus as a cellular mechanosensor. Nat. Cell Biol. 2018, 20, 373–381. [Google Scholar] [CrossRef]
- Vining, K.H.; Mooney, D.J. Mechanical forces direct stem cell behaviour in development and regeneration. Nat. Rev. Mol. Cell Biol. 2017, 18, 728–742. [Google Scholar] [CrossRef]
- Kai, F.; Drain, A.P.; Weaver, V.M. The Extracellular Matrix Modulates the Metastatic Journey. Dev. Cell 2019, 49, 332–346. [Google Scholar] [CrossRef]
- Wirtz, D.; Konstantopoulos, K.; Searson, P.C. The physics of cancer: The role of physical interactions and mechanical forces in metastasis. Nat. Rev. Cancer 2011, 11, 512. [Google Scholar] [CrossRef] [Green Version]
- Levental, K.R.; Yu, H.; Kass, L.; Lakins, J.N.; Egeblad, M.; Erler, J.T.; Fong, S.F.T.; Csiszar, K.; Giaccia, A.; Weninger, W.; et al. Matrix Crosslinking Forces Tumor Progression by Enhancing Integrin Signaling. Cell 2009, 139, 891–906. [Google Scholar] [CrossRef] [Green Version]
- Nelson, C.M.; Bissell, M.J. Of extracellular matrix, scaffolds, and signaling: Tissue architecture regulates development, homeostasis, and cancer. Annu. Rev. Cell Dev. Biol. 2006, 22, 287–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paszek, M.J.; Zahir, N.; Johnson, K.R.; Lakins, J.N.; Rozenberg, G.I.; Gefen, A.; Reinhart-King, C.A.; Margulies, S.S.; Dembo, M.; Boettiger, D.; et al. Tensional homeostasis and the malignant phenotype. Cancer Cell 2005, 8, 241–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weaver, V.M.; Petersen, O.W.; Wang, F.; Larabell, C.A.; Briand, P.; Damsky, C.; Bissell, M.J. Reversion of the malignant phenotype of human breast cells in three-dimensional culture and in vivo by integrin blocking antibodies. J. Cell Biol. 1997, 137, 231–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Follain, G.; Herrmann, D.; Harlepp, S.; Hyenne, V.; Osmani, N.; Warren, S.C.; Timpson, P.; Goetz, J.G. Fluids and their mechanics in tumour transit: Shaping metastasis. Nat. Rev. Cancer 2019. [Google Scholar] [CrossRef] [PubMed]
- Eyckmans, J.; Boudou, T.; Yu, X.; Chen, C.S. A hitchhiker’s guide to mechanobiology. Dev. Cell 2011, 21, 35–47. [Google Scholar] [CrossRef] [Green Version]
- Kechagia, J.Z.; Ivaska, J.; Roca-Cusachs, P. Integrins as biomechanical sensors of the microenvironment. Nat. Rev. Mol. Cell Biol. 2019, 20, 457–473. [Google Scholar] [CrossRef]
- Haupt, A.; Minc, N. How cells sense their own shape—Mechanisms to probe cell geometry and their implications in cellular organization and function. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef] [Green Version]
- Doyle, A.D.; Petrie, R.J.; Kutys, M.L.; Yamada, K.M. Dimensions in cell migration. Curr. Opin. Cell Biol. 2013, 25, 642–649. [Google Scholar] [CrossRef] [Green Version]
- van Helvert, S.; Storm, C.; Friedl, P. Mechanoreciprocity in cell migration. Nat. Cell Biol. 2018, 20, 8–20. [Google Scholar] [CrossRef]
- Pocaterra, A.; Santinon, G.; Romani, P.; Brian, I.; Dimitracopoulos, A.; Ghisleni, A.; Carnicer-Lombarte, A.; Forcato, M.; Braghetta, P.; Montagner, M.; et al. F-actin dynamics regulates mammalian organ growth and cell fate maintenance. J. Hepatol. 2019, 71, 130–142. [Google Scholar] [CrossRef] [Green Version]
- Samuel, M.S.; Lopez, J.I.; McGhee, E.J.; Croft, D.R.; Strachan, D.; Timpson, P.; Munro, J.; Schröder, E.; Zhou, J.; Brunton, V.G.; et al. Actomyosin-mediated cellular tension drives increased tissue stiffness and β-catenin activation to induce epidermal hyperplasia and tumor growth. Cancer Cell 2011, 19, 776–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sordella, R.; Jiang, W.; Chen, G.-C.; Curto, M.; Settleman, J. Modulation of Rho GTPase signaling regulates a switch between adipogenesis and myogenesis. Cell 2003, 113, 147–158. [Google Scholar] [CrossRef] [Green Version]
- Gjorevski, N.; Sachs, N.; Manfrin, A.; Giger, S.; Bragina, M.E.; Ordóñez-Morán, P.; Clevers, H.; Lutolf, M.P. Designer matrices for intestinal stem cell and organoid culture. Nature 2016, 539, 560–564. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Rowe, R.G.; Botvinick, E.L.; Kurup, A.; Putnam, A.J.; Seiki, M.; Weaver, V.M.; Keller, E.T.; Goldstein, S.; Dai, J.; et al. MT1-MMP-Dependent Control of Skeletal Stem Cell Commitment via a β1-Integrin/YAP/TAZ Signaling Axis. Dev. Cell 2013, 25, 402–416. [Google Scholar] [CrossRef] [Green Version]
- Charras, G.; Yap, A.S. Tensile Forces and Mechanotransduction at Cell-Cell Junctions. Curr. Biol. 2018, 28, R445–R457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yonemura, S.; Wada, Y.; Watanabe, T.; Nagafuchi, A.; Shibata, M. alpha-Catenin as a tension transducer that induces adherens junction development. Nat. Cell Biol. 2010, 12, 533–542. [Google Scholar] [CrossRef]
- Saini, K.; Discher, D.E. Forced Unfolding of Proteins Directs Biochemical Cascades. Biochemistry 2019, 58, 4893–4902. [Google Scholar] [CrossRef]
- Jiang, G.; Giannone, G.; Critchley, D.R.; Fukumoto, E.; Sheetz, M.P. Two-piconewton slip bond between fibronectin and the cytoskeleton depends on Talin. Nature 2003, 424, 334–337. [Google Scholar] [CrossRef]
- Sawada, Y.; Tamada, M.; Dubin-Thaler, B.J.; Cherniavskaya, O.; Sakai, R.; Tanaka, S.; Sheetz, M.P. Force sensing by mechanical extension of the Src family kinase substrate p130Cas. Cell 2006, 127, 1015–1026. [Google Scholar] [CrossRef] [Green Version]
- Klein, E.A.; Yin, L.; Kothapalli, D.; Castagnino, P.; Byfield, F.J.; Xu, T.; Levental, I.; Hawthorne, E.; Janmey, P.A.; Assoian, R.K. Cell-cycle control by physiological matrix elasticity and in vivo tissue stiffening. Curr. Biol. 2009, 19, 1511–1518. [Google Scholar] [CrossRef] [Green Version]
- Pirone, D.M.; Liu, W.F.; Ruiz, S.A.; Gao, L.; Raghavan, S.; Lemmon, C.A.; Romer, L.H.; Chen, C.S. An inhibitory role for FAK in regulating proliferation: A link between limited adhesion and RhoA-ROCK signaling. J. Cell Biol. 2006, 174, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Meng, Z.; Qiu, Y.; Lin, K.C.; Kumar, A.; Placone, J.K.; Fang, C.; Wang, K.-C.; Lu, S.; Pan, M.; Hong, A.W.; et al. RAP2 mediates mechanoresponses of the Hippo pathway. Nature 2018, 560, 655–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawada, Y.; Nakamura, K.; Doi, K.; Takeda, K.; Tobiume, K.; Saitoh, M.; Morita, K.; Komuro, I.; De Vos, K.; Sheetz, M.; et al. Rap1 is involved in cell stretching modulation of p38 but not ERK or JNK MAP kinase. J. Cell. Sci. 2001, 114, 1221–1227. [Google Scholar] [PubMed]
- Murthy, S.E.; Dubin, A.E.; Patapoutian, A. Piezos thrive under pressure: Mechanically activated ion channels in health and disease. Nat. Rev. Mol. Cell Biol. 2017, 18, 771–783. [Google Scholar] [CrossRef]
- Chen, X.; Wanggou, S.; Bodalia, A.; Zhu, M.; Dong, W.; Fan, J.J.; Yin, W.C.; Min, H.-K.; Hu, M.; Draghici, D.; et al. A Feedforward Mechanism Mediated by Mechanosensitive Ion Channel PIEZO1 and Tissue Mechanics Promotes Glioma Aggression. Neuron 2018, 100, 799–815. [Google Scholar] [CrossRef] [Green Version]
- Pathak, M.M.; Nourse, J.L.; Tran, T.; Hwe, J.; Arulmoli, J.; Le, D.T.T.; Bernardis, E.; Flanagan, L.A.; Tombola, F. Stretch-activated ion channel Piezo1 directs lineage choice in human neural stem cells. Proc. Natl. Acad. Sci. USA 2014, 111, 16148–16153. [Google Scholar] [CrossRef] [Green Version]
- Sinha, B.; Köster, D.; Ruez, R.; Gonnord, P.; Bastiani, M.; Abankwa, D.; Stan, R.V.; Butler-Browne, G.; Vedie, B.; Johannes, L.; et al. Cells respond to mechanical stress by rapid disassembly of caveolae. Cell 2011, 144, 402–413. [Google Scholar] [CrossRef] [Green Version]
- Goetz, J.G.; Minguet, S.; Navarro-Lérida, I.; Lazcano, J.J.; Samaniego, R.; Calvo, E.; Tello, M.; Osteso-Ibáñez, T.; Pellinen, T.; Echarri, A.; et al. Biomechanical remodeling of the microenvironment by stromal caveolin-1 favors tumor invasion and metastasis. Cell 2011, 146, 148–163. [Google Scholar] [CrossRef] [Green Version]
- Moreno-Vicente, R.; Pavón, D.M.; Martín-Padura, I.; Català-Montoro, M.; Díez-Sánchez, A.; Quílez-Álvarez, A.; López, J.A.; Sánchez-Álvarez, M.; Vázquez, J.; Strippoli, R.; et al. Caveolin-1 Modulates Mechanotransduction Responses to Substrate Stiffness through Actin-Dependent Control of YAP. Cell Rep. 2019, 26, 1679–1680. [Google Scholar] [CrossRef]
- Swift, J.; Ivanovska, I.L.; Buxboim, A.; Harada, T.; Dingal, P.C.D.P.; Pinter, J.; Pajerowski, J.D.; Spinler, K.R.; Shin, J.-W.; Tewari, M.; et al. Nuclear lamin-A scales with tissue stiffness and enhances matrix-directed differentiation. Science 2013, 341, 1240104. [Google Scholar] [CrossRef] [Green Version]
- Buxboim, A.; Swift, J.; Irianto, J.; Spinler, K.R.; Dingal, P.C.D.P.; Athirasala, A.; Kao, Y.-R.C.; Cho, S.; Harada, T.; Shin, J.-W.; et al. Matrix elasticity regulates lamin-A,C phosphorylation and turnover with feedback to actomyosin. Curr. Biol. 2014, 24, 1909–1917. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.; Vashisth, M.; Abbas, A.; Majkut, S.; Vogel, K.; Xia, Y.; Ivanovska, I.L.; Irianto, J.; Tewari, M.; Zhu, K.; et al. Mechanosensing by the Lamina Protects against Nuclear Rupture, DNA Damage, and Cell-Cycle Arrest. Dev. Cell 2019, 49, 920–935. [Google Scholar] [CrossRef] [PubMed]
- Raab, M.; Gentili, M.; de Belly, H.; Thiam, H.R.; Vargas, P.; Jimenez, A.J.; Lautenschlaeger, F.; Voituriez, R.; Lennon-Duménil, A.M.; Manel, N.; et al. ESCRT III repairs nuclear envelope ruptures during cell migration to limit DNA damage and cell death. Science 2016, 352, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Uhler, C.; Shivashankar, G.V. Regulation of genome organization and gene expression by nuclear mechanotransduction. Nat. Rev. Mol. Cell Biol. 2017, 18, 717–727. [Google Scholar] [CrossRef]
- Guet, D.; Mandal, K.; Pinot, M.; Hoffmann, J.; Abidine, Y.; Sigaut, W.; Bardin, S.; Schauer, K.; Goud, B.; Manneville, J.-B. Mechanical role of actin dynamics in the rheology of the Golgi complex and in Golgi-associated trafficking events. Curr. Biol. 2014, 24, 1700–1711. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.-J.; Joo, C.; Seong, J.; Vafabakhsh, R.; Botvinick, E.L.; Berns, M.W.; Palmer, A.E.; Wang, N.; Ha, T.; Jakobsson, E.; et al. Distinct mechanisms regulating mechanical force-induced Ca2+ signals at the plasma membrane and the ER in human MSCs. Elife 2015, 4, e04876. [Google Scholar] [CrossRef]
- Romani, P.; Brian, I.; Santinon, G.; Pocaterra, A.; Audano, M.; Pedretti, S.; Mathieu, S.; Forcato, M.; Bicciato, S.; Manneville, J.-B.; et al. Extracellular matrix mechanical cues regulate lipid metabolism through Lipin-1 and SREBP. Nat. Cell Biol. 2019, 21, 338–347. [Google Scholar] [CrossRef] [Green Version]
- Bertolio, R.; Napoletano, F.; Mano, M.; Maurer-Stroh, S.; Fantuz, M.; Zannini, A.; Bicciato, S.; Sorrentino, G.; Del Sal, G. Sterol regulatory element binding protein 1 couples mechanical cues and lipid metabolism. Nat. Commun. 2019, 10, 1326. [Google Scholar] [CrossRef] [Green Version]
- Porstmann, T.; Santos, C.R.; Griffiths, B.; Cully, M.; Wu, M.; Leevers, S.; Griffiths, J.R.; Chung, Y.-L.; Schulze, A. SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab. 2008, 8, 224–236. [Google Scholar] [CrossRef] [Green Version]
- Olson, E.N.; Nordheim, A. Linking actin dynamics and gene transcription to drive cellular motile functions. Nat. Rev. Mol. Cell Biol. 2010, 11, 353–365. [Google Scholar] [CrossRef] [Green Version]
- Miralles, F.; Posern, G.; Zaromytidou, A.-I.; Treisman, R. Actin dynamics control SRF activity by regulation of its coactivator MAL. Cell 2003, 113, 329–342. [Google Scholar] [CrossRef] [Green Version]
- McGee, K.M.; Vartiainen, M.K.; Khaw, P.T.; Treisman, R.; Bailly, M. Nuclear transport of the serum response factor coactivator MRTF-A is downregulated at tensional homeostasis. EMBO Rep. 2011, 12, 963–970. [Google Scholar] [CrossRef] [PubMed]
- Plessner, M.; Melak, M.; Chinchilla, P.; Baarlink, C.; Grosse, R. Nuclear F-actin formation and reorganization upon cell spreading. J. Biol. Chem. 2015, 290, 11209–11216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Somogyi, K.; Rørth, P. Evidence for tension-based regulation of Drosophila MAL and SRF during invasive cell migration. Dev. Cell 2004, 7, 85–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foster, C.T.; Gualdrini, F.; Treisman, R. Mutual dependence of the MRTF–SRF and YAP–TEAD pathways in cancer-associated fibroblasts is indirect and mediated by cytoskeletal dynamics. Genes Dev. 2017, 31, 2361–2375. [Google Scholar] [CrossRef] [Green Version]
- Pocaterra, A.; Romani, P.; Dupont, S. YAP/TAZ functions and regulation at a glance. J. Cell Sci. 2020, in press. [Google Scholar]
- Chang, L.; Azzolin, L.; Di Biagio, D.; Zanconato, F.; Battilana, G.; Lucon Xiccato, R.; Aragona, M.; Giulitti, S.; Panciera, T.; Gandin, A.; et al. The SWI/SNF complex is a mechanoregulated inhibitor of YAP and TAZ. Nature 2018, 563, 265–269. [Google Scholar] [CrossRef]
- Aureille, J.; Buffière-Ribot, V.; Harvey, B.E.; Boyault, C.; Pernet, L.; Andersen, T.; Bacola, G.; Balland, M.; Fraboulet, S.; Van Landeghem, L.; et al. Nuclear envelope deformation controls cell cycle progression in response to mechanical force. EMBO Rep. 2019, 20, e48084. [Google Scholar] [CrossRef]
- Elosegui-Artola, A.; Andreu, I.; Beedle, A.E.M.; Lezamiz, A.; Uroz, M.; Kosmalska, A.J.; Oria, R.; Kechagia, J.Z.; Rico-Lastres, P.; Le Roux, A.-L.; et al. Force Triggers YAP Nuclear Entry by Regulating Transport across Nuclear Pores. Cell 2017, 171, 1397–1410. [Google Scholar] [CrossRef]
- Dong, J.; Feldmann, G.; Huang, J.; Wu, S.; Zhang, N.; Comerford, S.A.; Gayyed, M.F.; Anders, R.A.; Maitra, A.; Pan, D. Elucidation of a Universal Size-Control Mechanism in Drosophila and Mammals. Cell 2007, 130, 1120–1133. [Google Scholar] [CrossRef] [Green Version]
- Santinon, G.; Brian, I.; Pocaterra, A.; Romani, P.; Franzolin, E.; Rampazzo, C.; Bicciato, S.; Dupont, S. dNTP metabolism links mechanical cues and YAP/TAZ to cell growth and oncogene-induced senescence. EMBO J. 2018, 37. [Google Scholar] [CrossRef]
- Zanconato, F.; Forcato, M.; Battilana, G.; Azzolin, L.; Quaranta, E.; Bodega, B.; Rosato, A.; Bicciato, S.; Cordenonsi, M.; Piccolo, S. Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nat. Cell Biol. 2015, 17, 1218–1227. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Ye, X.; Yu, J.; Li, L.; Li, W.; Li, S.; Yu, J.; Lin, J.D.; Wang, C.-Y.; Chinnaiyan, A.M.; et al. TEAD mediates YAP-dependent gene induction and growth control. Genes Dev. 2008, 22, 1962–1971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Totaro, A.; Castellan, M.; Di Biagio, D.; Piccolo, S. Crosstalk between YAP/TAZ and Notch Signaling. Trends Cell Biol. 2018, 28, 560–573. [Google Scholar] [CrossRef] [PubMed]
- Overholtzer, M.; Zhang, J.; Smolen, G.A.; Muir, B.; Li, W.; Sgroi, D.C.; Deng, C.-X.; Brugge, J.S.; Haber, D.A. Transforming properties of YAP, a candidate oncogene on the chromosome 11q22 amplicon. Proc. Natl. Acad. Sci. USA 2006, 103, 12405–12410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, Q.-Y.; Zhang, H.; Zhao, B.; Zha, Z.-Y.; Bai, F.; Pei, X.-H.; Zhao, S.; Xiong, Y.; Guan, K.-L. TAZ promotes cell proliferation and epithelial-mesenchymal transition and is inhibited by the hippo pathway. Mol. Cell. Biol. 2008, 28, 2426–2436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, S.C.; Fattet, L.; Tsai, J.H.; Guo, Y.; Pai, V.H.; Majeski, H.E.; Chen, A.C.; Sah, R.L.; Taylor, S.S.; Engler, A.J.; et al. Matrix stiffness drives epithelial–mesenchymal transition and tumour metastasis through a TWIST1–G3BP2 mechanotransduction pathway. Nat. Cell Biol. 2015, 17, 678–688. [Google Scholar] [CrossRef]
- Wang, L.; Luo, J.-Y.; Li, B.; Tian, X.Y.; Chen, L.-J.; Huang, Y.; Liu, J.; Deng, D.; Lau, C.W.; Wan, S.; et al. Integrin-YAP/TAZ-JNK cascade mediates atheroprotective effect of unidirectional shear flow. Nature 2016, 540, 579–582. [Google Scholar] [CrossRef]
- Wang, K.-C.; Yeh, Y.-T.; Nguyen, P.; Limqueco, E.; Lopez, J.; Thorossian, S.; Guan, K.-L.; Li, Y.-S.J.; Chien, S. Flow-dependent YAP/TAZ activities regulate endothelial phenotypes and atherosclerosis. Proc. Natl. Acad. Sci. USA 2016, 113, 11525–11530. [Google Scholar] [CrossRef] [Green Version]
- Zanconato, F.; Cordenonsi, M.; Piccolo, S. YAP/TAZ at the Roots of Cancer. Cancer Cell 2016, 29, 783–803. [Google Scholar] [CrossRef]
- Gupta, G.P.; Minn, A.J.; Kang, Y.; Siegel, P.M.; Serganova, I.; Cordón-Cardo, C.; Olshen, A.B.; Gerald, W.L.; Massagué, J. Identifying site-specific metastasis genes and functions. Cold Spring Harb. Symp. Quant. Biol. 2005, 70, 149–158. [Google Scholar] [CrossRef] [Green Version]
- Kang, Y.; Siegel, P.M.; Shu, W.; Drobnjak, M.; Kakonen, S.M.; Cordón-Cardo, C.; Guise, T.A.; Massagué, J. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell 2003, 3, 537–549. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.; Mu, E.; Wei, Y.; Riethdorf, S.; Yang, Q.; Yuan, M.; Yan, J.; Hua, Y.; Tiede, B.J.; Lu, X.; et al. VCAM-1 promotes osteolytic expansion of indolent bone micrometastasis of breast cancer by engaging α4β1-positive osteoclast progenitors. Cancer Cell 2011, 20, 701–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minn, A.J.; Kang, Y.; Serganova, I.; Gupta, G.P.; Giri, D.D.; Doubrovin, M.; Ponomarev, V.; Gerald, W.L.; Blasberg, R.; Massagué, J. Distinct organ-specific metastatic potential of individual breast cancer cells and primary tumors. J. Clin. Investig. 2005, 115, 44–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oskarsson, T.; Acharyya, S.; Zhang, X.H.-F.; Vanharanta, S.; Tavazoie, S.F.; Morris, P.G.; Downey, R.J.; Manova-Todorova, K.; Brogi, E.; Massagué, J. Breast cancer cells produce tenascin C as a metastatic niche component to colonize the lungs. Nat. Med. 2011, 17, 867–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kostic, A.; Lynch, C.D.; Sheetz, M.P. Differential Matrix Rigidity Response in Breast Cancer Cell Lines Correlates with the Tissue Tropism. PLoS ONE 2009, 4, e6361. [Google Scholar] [CrossRef] [PubMed]
- Barkan, D.; Kleinman, H.; Simmons, J.L.; Asmussen, H.; Kamaraju, A.K.; Hoenorhoff, M.J.; Liu, Z.; Costes, S.V.; Cho, E.H.; Lockett, S.; et al. Inhibition of metastatic outgrowth from single dormant tumor cells by targeting the cytoskeleton. Cancer Res. 2008, 68, 6241–6250. [Google Scholar] [CrossRef] [Green Version]
- Barkan, D.; El Touny, L.H.; Michalowski, A.M.; Smith, J.A.; Chu, I.; Davis, A.S.; Webster, J.D.; Hoover, S.; Simpson, R.M.; Gauldie, J.; et al. Metastatic growth from dormant cells induced by a col-I-enriched fibrotic environment. Cancer Res. 2010, 70, 5706–5716. [Google Scholar] [CrossRef] [Green Version]
- Naumov, G.N.; MacDonald, I.C.; Weinmeister, P.M.; Kerkvliet, N.; Nadkarni, K.V.; Wilson, S.M.; Morris, V.L.; Groom, A.C.; Chambers, A.F. Persistence of solitary mammary carcinoma cells in a secondary site: A possible contributor to dormancy. Cancer Res. 2002, 62, 2162–2168. [Google Scholar]
- Debnath, J.; Mills, K.R.; Collins, N.L.; Reginato, M.J.; Muthuswamy, S.K.; Brugge, J.S. The role of apoptosis in creating and maintaining luminal space within normal and oncogene-expressing mammary acini. Cell 2002, 111, 29–40. [Google Scholar] [CrossRef] [Green Version]
- Shibue, T.; Weinberg, R.A. Integrin beta1-focal adhesion kinase signaling directs the proliferation of metastatic cancer cells disseminated in the lungs. Proc. Natl. Acad. Sci. USA 2009, 106, 10290–10295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibue, T.; Brooks, M.W.; Inan, M.F.; Reinhardt, F.; Weinberg, R.A. The outgrowth of micrometastases is enabled by the formation of filopodium-like protrusions. Cancer Discov. 2012, 2, 706–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghajar, C.M.; Peinado, H.; Mori, H.; Matei, I.R.; Evason, K.J.; Brazier, H.; Almeida, D.; Koller, A.; Hajjar, K.A.; Stainier, D.Y.R.; et al. The perivascular niche regulates breast tumour dormancy. Nat. Cell Biol. 2013, 15, 807–817. [Google Scholar] [CrossRef] [PubMed]
- Lowy, C.M.; Oskarsson, T. Tenascin C in metastasis: A view from the invasive front. Cell Adh. Migr. 2015, 9, 112–124. [Google Scholar] [CrossRef] [Green Version]
- Malanchi, I.; Santamaria-Martínez, A.; Susanto, E.; Peng, H.; Lehr, H.-A.; Delaloye, J.-F.; Huelsken, J. Interactions between cancer stem cells and their niche govern metastatic colonization. Nature 2011, 481, 85–89. [Google Scholar] [CrossRef]
- Laklai, H.; Miroshnikova, Y.A.; Pickup, M.W.; Collisson, E.A.; Kim, G.E.; Barrett, A.S.; Hill, R.C.; Lakins, J.N.; Schlaepfer, D.D.; Mouw, J.K.; et al. Genotype tunes pancreatic ductal adenocarcinoma tissue tension to induce matricellular fibrosis and tumor progression. Nat. Med. 2016, 22, 497–505. [Google Scholar] [CrossRef] [Green Version]
- Northey, J.J.; Przybyla, L.; Weaver, V.M. Tissue force programs cell fate and tumor aggression. Cancer Discov. 2017, 7, 1224–1237. [Google Scholar] [CrossRef] [Green Version]
- Provenzano, P.P.; Eliceiri, K.W.; Campbell, J.M.; Inman, D.R.; White, J.G.; Keely, P.J. Collagen reorganization at the tumor-stromal interface facilitates local invasion. BMC Med. 2006, 4, 38. [Google Scholar] [CrossRef] [Green Version]
- Erler, J.T.; Bennewith, K.L.; Nicolau, M.; Dornhöfer, N.; Kong, C.; Le, Q.T.; Chi, J.T.A.; Jeffrey, S.S.; Giaccia, A.J. Lysyl oxidase is essential for hypoxia-induced metastasis. Nature 2006, 440, 1222–1226. [Google Scholar] [CrossRef]
- Erler, J.T.; Bennewith, K.L.; Cox, T.R.; Lang, G.; Bird, D.; Koong, A.; Le, Q.T.; Giaccia, A.J. Hypoxia-Induced Lysyl Oxidase Is a Critical Mediator of Bone Marrow Cell Recruitment to Form the Premetastatic Niche. Cancer Cell 2009, 15, 35–44. [Google Scholar] [CrossRef] [Green Version]
- Albrengues, J.; Shields, M.A.; Ng, D.; Park, C.G.; Ambrico, A.; Poindexter, M.E.; Upadhyay, P.; Uyeminami, D.L.; Pommier, A.; Küttner, V.; et al. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science 2018, 361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korah, R.; Boots, M.; Wieder, R. Integrin alpha5beta1 promotes survival of growth-arrested breast cancer cells: An in vitro paradigm for breast cancer dormancy in bone marrow. Cancer Res. 2004, 64, 4514–4522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barney, L.E.; Hall, C.L.; Schwartz, A.D.; Parks, A.N.; Sparages, C.; Galarza, S.; Platt, M.O.; Mercurio, A.M.; Peyton, S.R. Tumor cell-organized fibronectin is required to maintain a dormant breast cancer population. bioRxiv 2019, 686527. [Google Scholar] [CrossRef]
- Aguirre-Ghiso, J.A.; Liu, D.; Mignatti, A.; Kovalski, K.; Ossowski, L. Urokinase Receptor and Fibronectin Regulate the ERKMAPK to p38MAPK Activity Ratios That Determine Carcinoma Cell Proliferation or Dormancy In Vivo. Mol. Biol. Cell 2001, 12, 863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linde, N.; Fluegen, G.; Aguirre-Ghiso, J.A. The Relationship Between Dormant Cancer Cells and Their Microenvironment. Adv. Cancer Res. 2016, 132, 45–71. [Google Scholar] [PubMed] [Green Version]
- Provenzano, P.P.; Inman, D.R.; Eliceiri, K.W.; Keely, P.J. Matrix density-induced mechanoregulation of breast cell phenotype, signaling and gene expression through a FAK–ERK linkage. Oncogene 2009, 28, 4326–4343. [Google Scholar] [CrossRef] [Green Version]
- Paszek, M.J.; Dufort, C.C.; Rossier, O.; Bainer, R.; Mouw, J.K.; Godula, K.; Hudak, J.E.; Lakins, J.N.; Wijekoon, A.C.; Cassereau, L.; et al. The cancer glycocalyx mechanically primes integrin-mediated growth and survival. Nature 2014, 511, 319–325. [Google Scholar] [CrossRef] [Green Version]
- Woods, E.C.; Kai, F.; Barnes, J.M.; Pedram, K.; Pickup, M.W.; Hollander, M.J.; Weaver, V.M.; Bertozzi, C.R. A bulky glycocalyx fosters metastasis formation by promoting g1 cell cycle progression. eLife 2017, 6, e25752. [Google Scholar] [CrossRef]
- Midwood, K.S.; Chiquet, M.; Tucker, R.P.; Orend, G. Tenascin-C at a glance. J. Cell. Sci. 2016, 129, 4321–4327. [Google Scholar] [CrossRef] [Green Version]
- Huang, T.; Sun, L.; Yuan, X.; Qiu, H. Thrombospondin-1 is a multifaceted player in tumor progression. Oncotarget 2017, 8, 84546–84558. [Google Scholar] [CrossRef] [Green Version]
- González-González, L.; Alonso, J. Periostin: A Matricellular Protein with Multiple Functions in Cancer Development and Progression. Front. Oncol. 2018, 8, 225. [Google Scholar] [CrossRef] [PubMed]
- Francis, S.; Croft, D.; Schüttelkopf, A.W.; Parry, C.; Pugliese, A.; Cameron, K.; Claydon, S.; Drysdale, M.; Gardner, C.; Gohlke, A.; et al. Structure-based design, synthesis and biological evaluation of a novel series of isoquinolone and pyrazolo[4,3-c]pyridine inhibitors of fascin 1 as potential anti-metastatic agents. Bioorg. Med. Chem. Lett. 2019, 29, 1023–1029. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Huang, J.; Liu, B.; Xing, B.; Bordeleau, F.; Reinhart-King, C.A.; Li, W.; Zhang, J.J.; Huang, X.-Y. Improving fascin inhibitors to block tumor cell migration and metastasis. Mol. Oncol. 2016, 10, 966–980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGuire, S.; Kara, B.; Hart, P.C.; Montag, A.; Wroblewski, K.; Fazal, S.; Huang, X.-Y.; Lengyel, E.; Kenny, H.A. Inhibition of fascin in cancer and stromal cells blocks ovarian cancer metastasis. Gynecol. Oncol. 2019, 153, 405–415. [Google Scholar] [CrossRef] [PubMed]
- Aragona, M.; Panciera, T.; Manfrin, A.; Giulitti, S.; Michielin, F.; Elvassore, N.; Dupont, S.; Piccolo, S. A mechanical checkpoint controls multicellular growth through YAP/TAZ regulation by actin-processing factors. Cell 2013, 154, 1047–1059. [Google Scholar] [CrossRef] [Green Version]
- Carlson, P.; Dasgupta, A.; Grzelak, C.A.; Kim, J.; Barrett, A.; Coleman, I.M.; Shor, R.E.; Goddard, E.T.; Dai, J.; Schweitzer, E.M.; et al. Targeting the perivascular niche sensitizes disseminated tumour cells to chemotherapy. Nat. Cell Biol. 2019, 21, 238–250. [Google Scholar] [CrossRef]
- Er, E.E.; Valiente, M.; Ganesh, K.; Zou, Y.; Agrawal, S.; Hu, J.; Griscom, B.; Rosenblum, M.; Boire, A.; Brogi, E.; et al. Pericyte-like spreading by disseminated cancer cells activates YAP and MRTF for metastatic colonization. Nat. Cell Biol. 2018, 20, 966–978. [Google Scholar] [CrossRef]
- Gao, H.; Chakraborty, G.; Zhang, Z.; Akalay, I.; Gadiya, M.; Gao, Y.; Sinha, S.; Hu, J.; Jiang, C.; Akram, M.; et al. Multi-organ Site Metastatic Reactivation Mediated by Non-canonical Discoidin Domain Receptor 1 Signaling. Cell 2016, 166, 47–62. [Google Scholar] [CrossRef] [Green Version]
- Chute, C.; Yang, X.; Meyer, K.; Yang, N.; O’Neil, K.; Kasza, I.; Eliceiri, K.; Alexander, C.; Friedl, A. Syndecan-1 induction in lung microenvironment supports the establishment of breast tumor metastases. Breast Cancer Res. 2018, 20, 66. [Google Scholar] [CrossRef]
- Bragado, P.; Estrada, Y.; Parikh, F.; Krause, S.; Capobianco, C.; Farina, H.G.; Schewe, D.M.; Aguirre-Ghiso, J.A. TGF-β2 dictates disseminated tumour cell fate in target organs through TGF-β-RIII and p38α/β signalling. Nat. Cell Biol. 2013, 15, 1351–1361. [Google Scholar] [CrossRef] [Green Version]
- Yumoto, K.; Eber, M.R.; Wang, J.; Cackowski, F.C.; Decker, A.M.; Lee, E.; Nobre, A.R.; Aguirre-Ghiso, J.A.; Jung, Y.; Taichman, R.S. Axl is required for TGF-β2-induced dormancy of prostate cancer cells in the bone marrow. Sci. Rep. 2016, 6, 36520. [Google Scholar] [CrossRef] [PubMed]
- Yu-Lee, L.-Y.; Yu, G.; Lee, Y.-C.; Lin, S.-C.; Pan, J.; Pan, T.; Yu, K.-J.; Liu, B.; Creighton, C.J.; Rodriguez-Canales, J.; et al. Osteoblast-Secreted Factors Mediate Dormancy of Metastatic Prostate Cancer in the Bone via Activation of the TGFβRIII-p38MAPK-pS249/T252RB Pathway. Cancer Res. 2018, 78, 2911–2924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, H.; Chakraborty, G.; Lee-Lim, A.P.; Mo, Q.; Decker, M.; Vonica, A.; Shen, R.; Brogi, E.; Brivanlou, A.H.; Giancotti, F.G. The BMP inhibitor Coco reactivates breast cancer cells at lung metastatic sites. Cell 2012, 150, 764–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, A.; Okuda, H.; Xing, F.; Pandey, P.R.; Watabe, M.; Hirota, S.; Pai, S.K.; Liu, W.; Fukuda, K.; Chambers, C.; et al. Bone morphogenetic protein 7 in dormancy and metastasis of prostate cancer stem-like cells in bone. J. Exp. Med. 2011, 208, 2641–2655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derynck, R.; Weinberg, R.A. EMT and Cancer: More Than Meets the Eye. Dev. Cell 2019, 49, 313–316. [Google Scholar] [CrossRef]
- Xu, J.; Lamouille, S.; Derynck, R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 2009, 19, 156–172. [Google Scholar] [CrossRef]
- Weidenfeld, K.; Barkan, D. EMT and Stemness in Tumor Dormancy and Outgrowth: Are They Intertwined Processes? Front. Oncol. 2018, 8, 381. [Google Scholar] [CrossRef]
- De Cock, J.M.; Shibue, T.; Dongre, A.; Keckesova, Z.; Reinhardt, F.; Weinberg, R.A. Inflammation Triggers Zeb1-Dependent Escape from Tumor Latency. Cancer Res. 2016, 76, 6778–6784. [Google Scholar] [CrossRef] [Green Version]
- Wendt, M.K.; Schiemann, B.J.; Parvani, J.G.; Lee, Y.-H.; Kang, Y.; Schiemann, W.P. TGF-β stimulates Pyk2 expression as part of an epithelial-mesenchymal transition program required for metastatic outgrowth of breast cancer. Oncogene 2013, 32, 2005–2015. [Google Scholar] [CrossRef] [Green Version]
- Tran, D.D.; Corsa, C.A.S.; Biswas, H.; Aft, R.L.; Longmore, G.D. Temporal and spatial cooperation of Snail1 and Twist1 during epithelial-mesenchymal transition predicts for human breast cancer recurrence. Mol. Cancer Res. 2011, 9, 1644–1657. [Google Scholar] [CrossRef] [Green Version]
- Tsai, J.H.; Donaher, J.L.; Murphy, D.A.; Chau, S.; Yang, J. Spatiotemporal regulation of epithelial-mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell 2012, 22, 725–736. [Google Scholar] [CrossRef] [Green Version]
- Tran, H.D.; Luitel, K.; Kim, M.; Zhang, K.; Longmore, G.D.; Tran, D.D. Transient SNAIL1 expression is necessary for metastatic competence in breast cancer. Cancer Res. 2014, 74, 6330–6340. [Google Scholar] [CrossRef] [Green Version]
- Balic, M.; Lin, H.; Young, L.; Hawes, D.; Giuliano, A.; McNamara, G.; Datar, R.H.; Cote, R.J. Most early disseminated cancer cells detected in bone marrow of breast cancer patients have a putative breast cancer stem cell phenotype. Clin. Cancer Res. 2006, 12, 5615–5621. [Google Scholar] [CrossRef] [Green Version]
- Shiozawa, Y.; Berry, J.E.; Eber, M.R.; Jung, Y.; Yumoto, K.; Cackowski, F.C.; Yoon, H.J.; Parsana, P.; Mehra, R.; Wang, J.; et al. The marrow niche controls the cancer stem cell phenotype of disseminated prostate cancer. Oncotarget 2016, 7, 41217–41232. [Google Scholar] [CrossRef] [Green Version]
- Johnson, R.W.; Finger, E.C.; Olcina, M.M.; Vilalta, M.; Aguilera, T.; Miao, Y.; Merkel, A.R.; Johnson, J.R.; Sterling, J.A.; Wu, J.Y.; et al. Induction of LIFR confers a dormancy phenotype in breast cancer cells disseminated to the bone marrow. Nat. Cell Biol. 2016, 18, 1078–1089. [Google Scholar] [CrossRef] [Green Version]
- Sosa, M.S.; Parikh, F.; Maia, A.G.; Estrada, Y.; Bosch, A.; Bragado, P.; Ekpin, E.; George, A.; Zheng, Y.; Lam, H.-M.; et al. NR2F1 controls tumour cell dormancy via SOX9- and RARβ-driven quiescence programmes. Nat. Commun. 2015, 6, 6170. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, A.J.; Marengo, M.S.; Oltean, S.; Kemeny, G.; Bitting, R.L.; Turnbull, J.D.; Herold, C.I.; Marcom, P.K.; George, D.J.; Garcia-Blanco, M.A. Circulating Tumor Cells from Patients with Advanced Prostate and Breast Cancer Display Both Epithelial and Mesenchymal Markers. Mol. Cancer Res. 2011, 9, 997–1007. [Google Scholar] [CrossRef] [Green Version]
- Lecharpentier, A.; Vielh, P.; Perez-Moreno, P.; Planchard, D.; Soria, J.C.; Farace, F. Detection of circulating tumour cells with a hybrid (epithelial/mesenchymal) phenotype in patients with metastatic non-small cell lung cancer. Br. J. Cancer 2011, 105, 1338–1341. [Google Scholar] [CrossRef] [Green Version]
- Yu, M.; Bardia, A.; Wittner, B.S.; Stott, S.L.; Smas, M.E.; Ting, D.T.; Isakoff, S.J.; Ciciliano, J.C.; Wells, M.N.; Shah, A.M.; et al. Circulating Breast Tumor Cells Exhibit Dynamic Changes in Epithelial and Mesenchymal Composition. Science 2013, 339, 580–584. [Google Scholar] [CrossRef] [Green Version]
- Harper, K.L.; Sosa, M.S.; Entenberg, D.; Hosseini, H.; Cheung, J.F.; Nobre, R.; Avivar-Valderas, A.; Nagi, C.; Girnius, N.; Davis, R.J.; et al. Mechanism of early dissemination and metastasis in Her2+ mammary cancer. Nature 2016, 540, 588–592. [Google Scholar] [CrossRef]
- Keski-Oja, J.; Koli, K.; von Melchner, H. TGF-beta activation by traction? Trends Cell Biol. 2004, 14, 657–659. [Google Scholar] [CrossRef]
- Tschumperlin, D.J.; Shively, J.D.; Kikuchi, T.; Drazen, J.M. Mechanical Stress Triggers Selective Release of Fibrotic Mediators from Bronchial Epithelium. Am. J. Respir. Cell Mol. Biol. 2003, 28, 142–149. [Google Scholar] [CrossRef] [Green Version]
- Wipff, P.-J.; Rifkin, D.B.; Meister, J.-J.; Hinz, B. Myofibroblast contraction activates latent TGF-beta1 from the extracellular matrix. J. Cell Biol. 2007, 179, 1311–1323. [Google Scholar] [CrossRef] [Green Version]
- Leight, J.L.; Wozniak, M.A.; Chen, S.; Lynch, M.L.; Chen, C.S. Matrix rigidity regulates a switch between {TGF-β1-induced} apoptosis and epithelial-mesenchymal transition. Mol. Biol. Cell 2012, 23, 781–791. [Google Scholar] [CrossRef]
- Tschumperlin, D.J.; Dal, G.; Maly, I.V.; Kikuchi, T.; Lalho, L.H.; McVittie, A.K.; Haley, K.J.; Lilly, C.M.; So, P.T.C.; Lauffenburger, D.A.; et al. Mechanotransduction through growth-factor shedding into the extracellular space. Nature 2004, 429, 83–86. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.-H.; Asthagiri, A.R. Matrix stiffening sensitizes epithelial cells to EGF and enables the loss of contact inhibition of proliferation. J. Cell Sci. 2011, 124, 1280–1287. [Google Scholar] [CrossRef] [Green Version]
- Aguirre-Ghiso, J.A.; Sosa, M.S. Emerging Topics on Disseminated Cancer Cell Dormancy and the Paradigm of Metastasis. Annu. Rev. Cancer Biol. 2018, 2, 377–393. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Signaling Pathway | Biological Outcome |
|---|---|
| CollagenI (ECM stiffness)–Integrin–MLC–FAK/ERK/Src | Quiescence to proliferative switch at metastatic site |
| LOXL (Collagen crosslinking) | Metastatic efficiency |
| NETs–cleaved laminin–MLCK–YAP | Outgrowth of metastatic dormant cells |
| VWF/VCAM-1–Integrin | Resistance to chemotherapy of metastatic dormant cells |
| TNC – Notch/Wnt | Increased fitness of disseminated cells |
| TGFβ–fibrillar FN–Integrin–FAK/ERK | Metastatic cell survival |
| FN–ERK/p38 ratio | Quiescence to proliferative switch at primary site |
| Mucin-1–Integrin–PI3K/AKT | Increased fitness of disseminated cells |
| TSP-1 | Quiescence of disseminated cells |
| POSTN–BMP1/LOX and Wnt | Metastatic cell outgrowth |
| Rif/mDia2 + ILK/β-Parvin/Cofilin-FLPs | Metastatic cell outgrowth |
| Fascin–Filopodia | Metastatic efficiency |
| L1CAM–Integrin–ILK–YAP/MRTFs | Metastatic cell survival and outgrowth |
| Collagen–DDR1–JAK/STAT3 | Outgrowth of metastatic dormant cells |
| Syndecans–PAR polarity complexes-ERK | Metastatic cell quiescence |
| TGF-β superfamily | Ligand- and organ-dependent control of quiescence or proliferation of disseminated cells |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Montagner, M.; Dupont, S. Mechanical Forces as Determinants of Disseminated Metastatic Cell Fate. Cells 2020, 9, 250. https://doi.org/10.3390/cells9010250
Montagner M, Dupont S. Mechanical Forces as Determinants of Disseminated Metastatic Cell Fate. Cells. 2020; 9(1):250. https://doi.org/10.3390/cells9010250
Chicago/Turabian StyleMontagner, Marco, and Sirio Dupont. 2020. "Mechanical Forces as Determinants of Disseminated Metastatic Cell Fate" Cells 9, no. 1: 250. https://doi.org/10.3390/cells9010250