Applications of Genome Editing Technology in Research on Chromosome Aneuploidy Disorders


Abstract
:1. Introduction
1.1. Human Genetic Aneuploidy Disorders
1.2. Autosomal Chromosomes
1.2.1. Trisomy 21 (Down Syndrome)
1.2.2. Trisomy 18 (Edwards Syndrome)
1.2.3. Trisomy 13 (Patau Syndrome)
1.3. Sex Chromosomes
1.3.1. Turner Syndrome (45,X)
1.3.2. Klinefelter Syndrome
1.4. Mosaic Variegated Aneuploidy (MVA)
1.4.1. MVA1 or MVA1 Syndrome
1.4.2. MVA2
1.4.3. MVA3
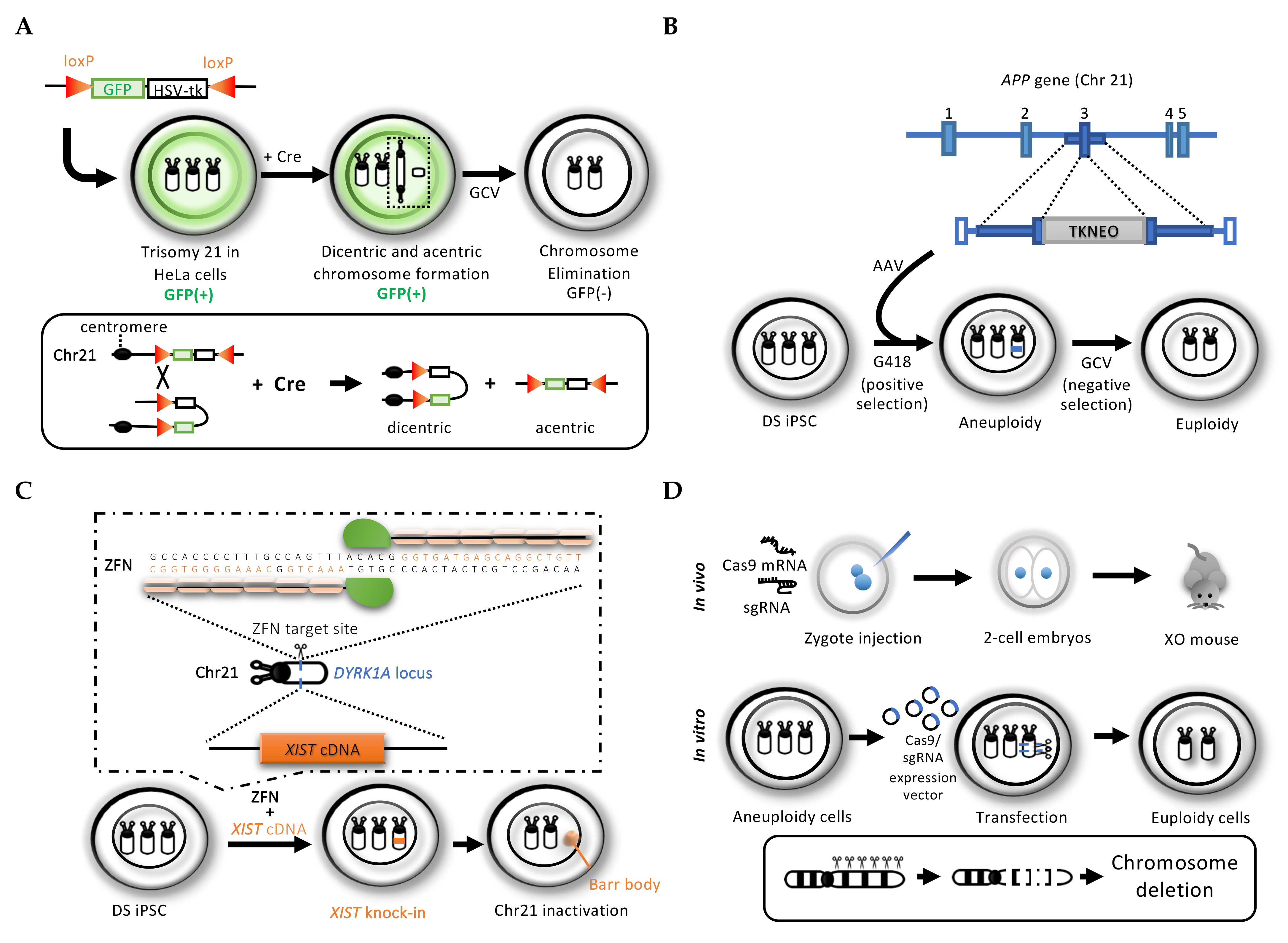
2. Gene Targeting-Mediated Chromosome Elimination
2.1. Cre/loxP System-Mediated Chromosome Elimination
2.2. Conventional Gene-Targeting Mediated Dual Drug Selection Cassette Knock-In
2.3. Genome Editing Technology for Rescuing Trisomy In Vitro and In Vivo
2.3.1. Zinc Finger Nuclease (ZFN) Mediated the XIST Gene Knock-In to Silence an Extra Chromosome 21 in Down Syndrome Patient Cells
2.3.2. CRISPR/Cas9 System to Introduce Multiple DNA Cleavages for Target Chromosome Elimination
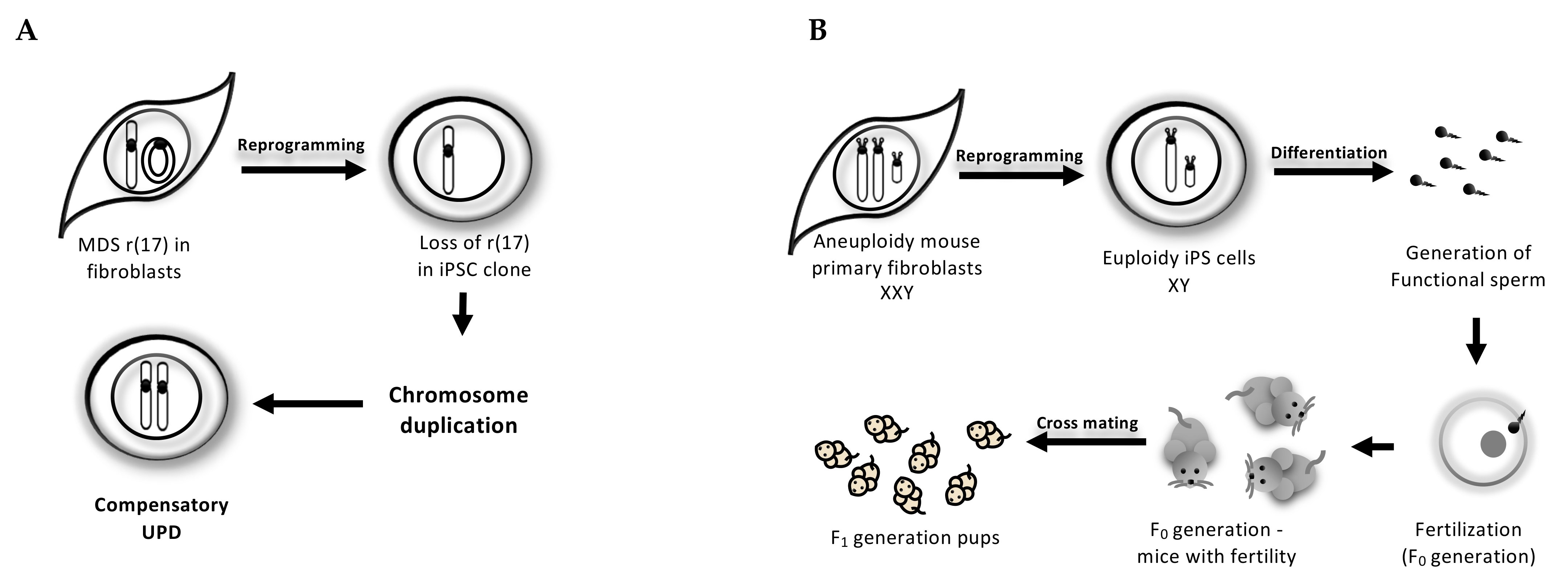
3. Reprogramming-Mediated Karyotype Correction
3.1. Cell-Autonomous Correction of Ring Chromosome During iPSC Reprogramming
3.2. Trisomy-Biased Chromosome Loss (TCL) to Convert the Trisomy into Disomy During iPSC Reprogramming
4. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Orr, B.; Godek, K.M.; Compton, D. Aneuploidy. Curr. Biol. 2015, 25, 538–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganmore, I.; Smooha, G.; Izraeli, S. Constitutional aneuploidy and cancer predisposition†. Hum. Mol. Genet. 2009, 18, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Spencer, K. Aneuploidy screening in the first trimester. Am. J. Med. Genet. C Semin. Med. Genet. 2007, 145, 18–32. [Google Scholar] [CrossRef] [PubMed]
- Thorsted, A.; Lauridsen, J.; Hoyer, B.; Arendt, L.; Bech, B.; Toft, G.; Hougaard, K.; Olsen, J.; Bonde, J.P.; Ramlau-Hansen, C. Birth weight for gestational age and the risk of infertility: A danish cohort study. Hum. Reprod. 2019. [Google Scholar] [CrossRef]
- Mantzouratou, A.; Mania, A.; Fragouli, E.; Xanthopoulou, L.; Tashkandi, S.; Fordham, K.; Ranieri, D.; Doshi, A.; Nuttall, S.; Harper, J.; et al. Variable aneuploidy mechanisms in embryos from couples with poor reproductive histories undergoing preimplantation genetic screening. Hum. Reprod. 2007, 22, 1844–1853. [Google Scholar] [CrossRef] [Green Version]
- Moorthie, S.; Congenital Disorders Expert Group; Blencowe, H.; Darlison, M.W.; Gibbons, S.; Lawn, J.E.; Mastroiacovo, P.; Morris, J.K.; Modell, B.; Bittles, A.H.; et al. Chromosomal disorders: Estimating baseline birth prevalence and pregnancy outcomes worldwide. J. Community Genet. 2017, 9, 377–386. [Google Scholar] [CrossRef]
- Hassold, T.; Hunt, P. Maternal age and chromosomally abnormal pregnancies: What we know and what we wish we knew. Curr. Opin. Pediatr. 2009, 21, 703–708. [Google Scholar] [CrossRef] [Green Version]
- Soler, A.; Morales, C.; Mademont-Soler, I.; Margarit, E.; Borrell, A.; Borobio, V.; Muñoz, M.; Sánchez, A. Overview of Chromosome Abnormalities in First Trimester Miscarriages: A Series of 1,011 Consecutive Chorionic Villi Sample Karyotypes. Cytogenet. Genome Res. 2017, 152, 81–89. [Google Scholar] [CrossRef]
- Allanson, J.E.; McGillivray, B.C.; Hall, J.G.; Shaw, D.; Kalousek, D.K. Cytogenetic findings in over 2000 amniocenteses. Can. Med. Assoc. J. 1983, 129, 846–850. [Google Scholar]
- Fragouli, E.; Alfarawati, S.; Spath, K.; Jaroudi, S.; Sarasa, J.; Enciso, M.; Wells, D. The origin and impact of embryonic aneuploidy. Hum. Genet. 2013, 132, 1001–1013. [Google Scholar] [CrossRef]
- Bianchi, D.W.; Parker, R.L.; Wentworth, J.; Madankumar, R.; Saffer, C.; Das, A.F.; Craig, J.A.; Chudova, D.I.; Devers, P.L.; Jones, K.W. DNA sequencing versus standard prenatal aneuploidy screening. N. Engl. J. Med. 2014, 370, 799–808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Driscoll, D.A.; Gross, S. Prenatal Screening for Aneuploidy. N. Engl. J. Med. 2009, 360, 2556–2562. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Ribas, I.; Diaz-Gimeno, P.; Sebastián-León, P.; Mercader, A.; Quiñonero, A.; Ballesteros, A.; Pellicer, A.; Domínguez, F. Transcriptomic behavior of genes associated with chromosome 21 aneuploidies in early embryo development. Fertil. Steril. 2019, 111, 991–1001. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, D.R. Transcriptional consequences of autosomal trisomy: Primary gene dosage with complex downstream effects. Trends. Genet. 2005, 21, 249–253. [Google Scholar] [CrossRef]
- Dierssen, M.; Herault, Y.; Estivill, X. Aneuploidy: From a Physiological Mechanism of Variance to Down Syndrome. Physiol. Rev. 2009, 89, 887–920. [Google Scholar] [CrossRef] [Green Version]
- Toufaily, M.H.; Westgate, M.N.; Lin, A.E.; Holmes, L.B. Causes of Congenital Malformations. Birth. Defects. Res. 2018, 110, 87–91. [Google Scholar] [CrossRef]
- Zhang, R.; Hao, L.; Wang, L.; Chen, M.; Li, W.; Li, R.; Yu, J.; Xiao, J.; Wu, J. Gene expression analysis of induced pluripotent stem cells from aneuploid chromosomal syndromes. BMC Genom. 2013. [Google Scholar] [CrossRef] [Green Version]
- Piovesan, A.; Pelleri, M.C.; Antonaros, F.; Strippoli, P.; Caracausi, M.; Vitale, L. On the length, weight and GC content of the human genome. BMC Res. Notes 2019. [Google Scholar] [CrossRef]
- Goel, N.; Morris, J.K.; Tucker, D.; De Walle, H.E.K.; Bakker, M.K.; Kancherla, V.; Marengo, L.; Canfield, M.A.; Kallen, K.; Lelong, N.; et al. Trisomy 13 and 18—Prevalence and mortality—A multi-registry population based analysis. Am. J. Med. Genet. A 2019, 179, 2382–2392. [Google Scholar] [CrossRef]
- Domingo, L.; Carey, J.C.; Eckhauser, A.; Wilkes, J.; Menon, S.C. Mortality and resource use following cardiac interventions in children with trisomy 13 and trisomy 18 and congenital heart disease. Pediatr. Cardiol. 2019, 40, 349–356. [Google Scholar] [CrossRef]
- Janvier, A.; Farlow, B.; Barrington, K.J.; Carey, J.C.; Kosho, T. Parental hopes, interventions, and survival of neonates with trisomy 13 and trisomy 18. Am. J. Med. Genet. C. Semin. Med. Genet. 2016, 172, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Hassold, T.J.; Jacobs, P.A. Trisomy in man. Ann. Rev. Genet. 1984, 18, 69–97. [Google Scholar] [CrossRef] [PubMed]
- Plaiasu, V. Down syndrome – genetics and cardiogenetics. Maedica 2017, 12, 208–213. [Google Scholar] [PubMed]
- Papavassiliou, P.; York, T.P.; Gursoy, N.; Hill, G.; Nicely, L.V.; Sundaram, U.; McClain, A.; Aggen, S.H.; Eaves, L.; Riley, B.; et al. The phenotype of persons having mosaicism for trisomy 21/Down syndrome reflects the percentage of trisomic cells present in different tissues. Am. J. Med. Genet. A. 2009, 149, 573–583. [Google Scholar] [CrossRef] [Green Version]
- Bray, I.; Wright, D.E.; Davies, C.; Hook, E.B. Joint estimation of down syndrome risk and ascertainment rates: A meta-analysis of nice published data sets. Prenat. Diagn. 1998, 18, 9–20. [Google Scholar] [CrossRef]
- Capone, G.T.; Chicoine, B.; Bulova, P.; Stephens, M.; Hart, S.; Crissman, B.; Videlefsky, A.; Myers, K.; Roizen, N.; Esbensen, A.; et al. Co-occurring medical conditions in adults with down syndrome: A systematic review toward the development of health care guidelines. Am. J. Med. Genet. A. 2018, 176, 116–133. [Google Scholar] [CrossRef]
- Li, W.; Wang, X.; Li, S. Investigation of copy number variations on chromosome 21 detected by comparative genomic hybridization (CGH) microarray in patients with congenital anomalies. Mol. Cytogenet. 2018. [Google Scholar] [CrossRef]
- Banno, K.; Omori, S.; Hirata, K.; Nawa, N.; Nakagawa, N.; Nishimura, K.; Ohtaka, M.; Nakanishi, M.; Sakuma, T.; Yamamoto, T.; et al. Systematic Cellular Disease Models Reveal Synergistic Interaction of Trisomy 21 and GATA1 Mutations in Hematopoietic Abnormalities. Cell Rep. 2016, 15, 1228–1241. [Google Scholar] [CrossRef] [Green Version]
- Bruns, D.A.; Martinez, A. An analysis of cardiac defects and surgical interventions in 84 cases with full trisomy 18. Am. J. Med. Genet. A. 2016, 170A, 337–343. [Google Scholar] [CrossRef]
- Cereda, A.; Carey, J.C. The trisomy 18 syndrome. Orphanet J. Rare Dis. 2012. [Google Scholar] [CrossRef] [Green Version]
- Gersen, S.L.; Keagle, M.B. The Principles of Clinical Cytogenetics, 2nd ed.; Humana Press Inc.: Totowa, NJ, USA, 2005; p. 616. [Google Scholar]
- Wyllie, J.P.; Wright, M.J.; Burn, J.; Hunter, S. Natural history of trisomy 13. Arch. Dis. Child. 1994, 71, 343–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crider, K.S.; Olney, R.S.; Cragan, J.D. Trisomies 13 and 18: Population prevalences, characteristics, and prenatal diagnosis, metropolitan Atlanta, 1994–2003. Am. J. Med. Genet. A. 2008, 146, 820–826. [Google Scholar] [CrossRef] [PubMed]
- Peroos, S.; Forsythe, E.; Pugh, J.H.; Arthur-Farraj, P.; Hodes, D. Longevity and Patau syndrome: What determines survival? BMJ Case Rep. 2012. [Google Scholar] [CrossRef] [PubMed]
- Plaiasu, V.; Ochiana, D.; Motei, G.; Anca, I.; Georgescu, A. Clinical relevance of cytogenetics to pediatric practice. Postnatal findings of Patau syndrome – Review of 5 cases. Maedica 2010, 5, 178–185. [Google Scholar]
- Heard, E.; Turner, J. Function of the Sex Chromosomes in Mammalian Fertility. Cold Spring Harb. Perspect. Boil. 2011. [Google Scholar] [CrossRef] [Green Version]
- Hassold, T.; Benham, F.; Leppert, M. Cytogenetic and molecular analysis of sex-chromosome monosomy. Am. J. Hum. Genet. 1988, 42, 534–541. [Google Scholar]
- Zhong, Q.; Layman, L.C. Genetic considerations in the patient with Turner syndrome--45,X with or without mosaicism. Fertil. Steril. 2012, 98, 775–779. [Google Scholar] [CrossRef] [Green Version]
- Pinsker, J.E. Clinical review: Turner syndrome: Updating the paradigm of clinical care. J. Clin. Endocrinol. Metab. 2012, 97, 994–1003. [Google Scholar] [CrossRef] [Green Version]
- Wikström, A.M.; Dunkel, L. Testicular Function in Klinefelter Syndrome. Horm. Res. Paediatr. 2008, 69, 317–326. [Google Scholar] [CrossRef]
- Ross, J.L.; Roeltgen, D.P.; Kushner, H.; Zinn, A.R.; Reiss, A.; Bardsley, M.Z.; McCauley, E.; Tartaglia, N. Behavioral and Social Phenotypes in Boys With 47,XYY Syndrome or 47,XXY Klinefelter Syndrome. Pediatrics 2012, 129, 769–778. [Google Scholar] [CrossRef] [Green Version]
- Giudice, M.G.; Vermeulen, M.; Wyns, C. Blood Testis Barrier and Somatic Cells Impairment in a Series of 35 Adult Klinefelter Syndrome Patients. Int. J. Mol. Sci. 2019, 5717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bojesen, A.; Juul, S.; Gravholt, C.H. Prenatal and Postnatal Prevalence of Klinefelter Syndrome: A National Registry Study. J. Clin. Endocrinol. Metab. 2003, 88, 622–626. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.; Telfer, M.A.; Richardson, C.E.; Clark, G.R. Chromosome Errors in Men With Antisocial Behavior. JAMA 1970, 214, 869–878. [Google Scholar] [CrossRef] [PubMed]
- Haslam, R.H.A.; Broske, S.P.; Moore, C.M.; Thomas, G.H.; Neill, C.A. Trisomy 9 Mosaicism with Multiple Congenital Anomalies. J. Med. Genet. 1973, 10, 180–184. [Google Scholar] [CrossRef] [Green Version]
- Francke, U.; Benirschke, K.; Jones, O.W. Prenatal diagnosis of trisomy 9. Qual. Life Res. 1975, 29, 243–250. [Google Scholar] [CrossRef]
- Crowe, C.A.; Schwartz, S.; Black, C.J.; Jaswaney, V. Mosaic trisomy 22: A case presentation and literature review of trisomy 22 phenotypes. Am. J. Med. Genet. 1997, 71, 406–413. [Google Scholar] [CrossRef]
- Hsu, L.Y.; Shapiro, L.R.; Gertner, M.; Lieber, E.; Hirschhorn, K. Trisomy 22: A clinical entity. J. Pediatr. 1971, 79, 12–19. [Google Scholar] [CrossRef]
- Hwang, S.; Williams, J.F.; Kneissig, M.; Lioudyno, M.; Rivera, I.; Helguera, P.; Busciglio, J.; Storchova, Z.; King, M.C.; Torres, E.M. Suppressing Aneuploidy-Associated Phenotypes Improves the Fitness of Trisomy 21 Cells. Cell Rep. 2019, 29, 2473–2488. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Chau, M.H.K.; Cao, Y.; Kwok, K.Y.; Choy, K.W. Chromosome copy number variants in fetuses with syndromic malformations. Birth Defects Res. 2017, 109, 725–733. [Google Scholar] [CrossRef]
- Trevisan, P.; Zen, T.D.; Rosa, R.F.M.; Da Silva, J.N.; Koshiyama, D.B.; Paskulin, G.A.; Zen, P.R.G. Chromosomal Abnormalities in Patients with Congenital Heart Disease. Arq. Bras. Cardiol. 2013, 101, 495–501. [Google Scholar] [CrossRef]
- Oliveira, P.H.A.; Souza, B.S.; Pacheco, E.N.; Menegazzo, M.S.; Corrêa, I.S.; Zen, P.R.G.; Rosa, R.F.M.; Cesa, C.C.; Pellanda, L.C.; Vilela, M.A.P. Genetic Syndromes Associated with Congenital Cardiac Defects and Ophthalmologic Changes - Systematization for Diagnosis in the Clinical Practice. Arq. Bras. Cardiol. 2018, 110, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.M. Genetic Syndromes associated with Congenital Heart Disease. Korean Circ. J. 2015, 45, 357–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahraman, S.; Benkhalifa, M.; Donmez, E.; Biricik, A.; Sertyel, S.; Findikli, N.; Berkil, H. The results of aneuploidy screening in 276 couples undergoing assisted reproductive techniques. Prenat. Diagn. 2004, 24, 307–311. [Google Scholar] [CrossRef] [PubMed]
- Callier, P.; Faivre, L.; Cusin, V.; Marle, N.; Thauvin-Robinet, C.; Sandre, D.; Rousseau, T.; Sagot, P.; Lacombe, E.; Faber, V.; et al. Microcephaly is not mandatory for the diagnosis of mosaic variegated aneuploidy syndrome. Am. J. Med. Genet. Part. A. 2005, 137, 204–207. [Google Scholar] [CrossRef] [PubMed]
- Kops, G.J.P.L.; Weaver, B.A.A.; Cleveland, D.W. On the road to cancer: Aneuploidy and the mitotic checkpoint. Nat. Rev. Cancer. 2005, 5, 773–785. [Google Scholar] [CrossRef] [PubMed]
- Vogt, E.; Kirsch-Volders, M.; Parry, J.; Eichenlaub-Ritter, U. Spindle formation, chromosome segregation and the spindle checkpoint in mammalian oocytes and susceptibility to meiotic error. Mutat. Res. Toxicol. Environ. Mutagen. 2008, 651, 14–29. [Google Scholar] [CrossRef]
- Försti, A.; Frank, C.; Smolkova, B.; Kazimirova, A.; Barancokova, M.; Vymetalkova, V.; Kroupa, M.; Naccarati, A.; Vodickova, L.; Buchancova, J.; et al. Genetic variation in the major mitotic checkpoint genes associated with chromosomal aberrations in healthy humans. Cancer Lett. 2016, 380, 442–446. [Google Scholar] [CrossRef]
- Sieben, C.J.; Jeganathan, K.B.; Nelson, G.G.; Sturmlechner, I.; Zhang, C.; Van Deursen, W.H.; Bakker, B.; Foijer, F.; Li, H.; Baker, D.J.; et al. BubR1 allelic effects drive phenotypic heterogeneity in mosaic-variegated aneuploidy progeria syndrome. J. Clin. Investig. 2019, 130, 171–188. [Google Scholar] [CrossRef]
- Kajii, T.; Ikeuchi, T.; Yang, Z.-Q.; Nakamura, Y.; Tsuji, Y.; Yokomori, K.; Kawamura, M.; Fukuda, S.; Horita, S.; Asamoto, A. Cancer-prone syndrome of mosaic variegated aneuploidy and total premature chromatid separation: Report of five infants. Am. J. Med. Genet. 2001, 104, 57–64. [Google Scholar] [CrossRef] [Green Version]
- Kajii, T.; Kawai, T.; Takumi, T.; Misu, H.; Mabuchi, O.; Takahashi, Y.; Tachino, M.; Nihei, F.; Ikeuchi, T. Mosaic variegated aneuploidy with multiple congenital abnormalities: Homozygosity for total premature chromatid separation trait. Am. J. Med. Genet. 1998, 78, 245–249. [Google Scholar] [CrossRef]
- Kajii, T.; Ikeuchi, T. Premature chromatid separation (PCS) vs. premature centromere division (PCD). Am. J. Med. Genet. 2004, 126, 433–434. [Google Scholar] [CrossRef] [PubMed]
- Ochiai, H.; Miyamoto, T.; Kanai, A.; Hosoba, K.; Sakuma, T.; Kudo, Y.; Asami, K.; Ogawa, A.; Watanabe, A.; Kajii, T.; et al. Talen-mediated single-base-pair editing identification of an intergenic mutation upstream of bub1b as causative of pcs (mva) syndrome. Proc. Natl. Acad. Sci. USA 2014, 111, 1461–1466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanks, S.; Coleman, K.; Reid, S.; Plaja, A.; Firth, H.; Fitzpatrick, D.R.; Kidd, A.; Méhes, K.; Nash, R.; Robin, N.; et al. Constitutional aneuploidy and cancer predisposition caused by biallelic mutations in BUB1B. Nat. Genet. 2004, 36, 1159–1161. [Google Scholar] [CrossRef] [PubMed]
- García-Castillo, H.; Vásquez-Velásquez, A.I.; Rivera, H.; Barros-Núñez, P. Clinical and genetic heterogeneity in patients with mosaic variegated aneuploidy: Delineation of clinical subtypes. Am. J. Med. Genet. A. 2008, 146, 1687–1695. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, S.; Ito, E.; Tauchi, H.; Komatsu, K.; Ikeuchi, T.; Kajii, T. Chromosomal Instability Syndrome of Total Premature Chromatid Separation with Mosaic Variegated Aneuploidy Is Defective in Mitotic-Spindle Checkpoint. Am. J. Hum. Genet. 2000, 67, 483–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Micale, M.A.; Schran, D.; Emch, S.; Kurczynski, T.W.; Rahman, N.; Van Dyke, D.L. Mosaic variegated aneuploidy without microcephaly: Implications for cytogenetic diagnosis. Am. J. Med. Genet. Part. A 2007, 143, 1890–1893. [Google Scholar] [CrossRef]
- Suijkerbuijk, S.J.; Van Osch, M.H.; Bos, F.L.; Hanks, S.; Rahman, N.; Kops, G.J. Molecular causes for BUBR1 dysfunction in the human cancer predisposition syndrome mosaic variegated aneuploidy. Cancer Res. 2010, 70, 4891–4900. [Google Scholar] [CrossRef] [Green Version]
- Kato, M.; Kato, T.; Hosoba, E.; Ohashi, M.; Fujisaki, M.; Ozaki, M.; Yamaguchi, M.; Sameshima, H.; Kurahashi, H. PCS/MVA syndrome caused by an Alu insertion in the BUB1B gene. Hum. Genome Var. 2017. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.; He, R.; Zhou, H.; Yu, A.C.; Zhang, B.; Teng, J.; Chen, J. Cep57, a NEDD1-binding pericentriolar material component, is essential for spindle pole integrity. Cell Res. 2012, 22, 1390–1401. [Google Scholar] [CrossRef] [Green Version]
- Brightman, D.S.; Ejaz, S.; Dauber, A. Mosaic variegated aneuploidy syndrome caused by a CEP57 mutation diagnosed by whole exome sequencing. Clin. Case Rep. 2018, 6, 1531–1534. [Google Scholar] [CrossRef]
- Aziz, K.; Sieben, C.J.; Jeganathan, K.B.; Hamada, M.; Davies, B.A.; Velasco, R.O.F.; Rahman, N.; Katzmann, D.J.; Van Deursen, J.M. Mosaic-variegated aneuploidy syndrome mutation or haploinsufficiency in Cep57 impairs tumor suppression. J. Clin. Investig. 2018, 128, 3517–3534. [Google Scholar] [CrossRef] [Green Version]
- De La Torre-García, O.; Mar-Aldama, R.; Salgado-Sangri, R.; Diaz-Gomez, N.; Bonilla-Arcaute, L.; Diaz-Ponce-Medrano, J.; Guevara-Yañez, R.; Córdova, E.J.; Monge-Cazares, T.; Orozco, L.; et al. A homozygous CEP57 c.915_925dupCAATGTTCAGC mutation in a patient with mosaic variegated aneuploidy syndrome with rhizomelic shortening in the upper and lower limbs and a narrow thorax. Eur. J. Med. Genet. 2019, 62, 195–197. [Google Scholar] [CrossRef]
- Snape, K.; Hanks, S.; Ruark, E.; Barros-Núñez, P.; Elliott, A.; Murray, A.; Lane, A.H.; Shannon, N.; Callier, P.; Chitayat, D.; et al. Mutations in CEP57 cause mosaic variegated aneuploidy syndrome. Nat. Genet. 2011, 43, 527–529. [Google Scholar] [CrossRef] [Green Version]
- Pinson, L.; Mannini, L.; Willems, M.; Cucco, F.; Sirvent, N.; Frebourg, T.; Quarantotti, V.; Collet, C.; Schneider, A.; Sarda, P.; et al. Cep57 mutation in a girl with mosaic variegated aneuploidy syndrome. Am. J. Med. Genet. A 2014, 164A, 177–181. [Google Scholar] [CrossRef] [PubMed]
- Yost, S.; De Wolf, B.; Hanks, S.; Zachariou, A.; Marcozzi, C.; Clarke, M.; De Voer, R.M.; Etemad, B.; Uijttewaal, E.; Ramsay, E.; et al. Biallelic TRIP13 mutations predispose to Wilms tumor and chromosome missegregation. Nat. Genet. 2017, 49, 1148–1151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alfieri, C.; Chang, L.; Barford, D. Mechanism for remodelling of the cell cycle checkpoint protein MAD2 by the ATPase TRIP13. Nature. 2018, 559, 274–278. [Google Scholar] [CrossRef] [PubMed]
- Marks, D.H.; Thomas, R.; Chin, Y.; Shah, R.; Khoo, C.; Benezra, R. Mad2 Overexpression Uncovers a Critical Role for TRIP13 in Mitotic Exit. Cell Rep. 2017, 19, 1832–1845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, R.; Marks, D.H.; Chin, Y.; Benezra, R. Whole chromosome loss and associated breakage-fusion-bridge cycles transform mouse tetraploid cells. EMBO J. 2018, 37, 201–218. [Google Scholar] [CrossRef] [PubMed]
- Tada, M.; Matsumura, H.; Kurose, Y.; Nakatsuji, N.; Tada, T. Target chromosomes of inducible deletion by a Cre/inverted loxP system in mouse embryonic stem cells. Chromosom. Res. 2009, 17, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.J.H.; Xian, J.; Richardson, M.; Johnstone, K.A.; Rabbitts, P.H. Cre-loxp chromosome engineering of a targeted deletion in the mouse corresponding to the 3p21.3 region of homozygous loss in human tumours. Oncogene 2002, 21, 4521–4529. [Google Scholar] [CrossRef] [Green Version]
- Sato, H.; Kato, H.; Yamaza, H.; Masuda, K.; Nguyen, H.T.N.; Pham, T.T.M.; Han, X.; Hirofuji, Y.; Nonaka, K. Engineering of Systematic Elimination of a Targeted Chromosome in Human Cells. BioMed. Res. Int. 2017, 2017, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Lewandoski, M.; Martin, G.R. Cre–mediated chromosome loss in mice. Nat. Genet. 1997, 17, 223–225. [Google Scholar] [CrossRef] [PubMed]
- Otsuji, T.; Matsumura, H.; Suzuki, T.; Nakatsuji, N.; Tada, T.; Tada, M. Rapid Induction of Large Chromosomal Deletions by a Cre/Inverted loxP System in Mouse ES Cell Hybrids. J. Mol. Biol. 2008, 378, 328–336. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, H.; Tada, M.; Otsuji, T.; Yasuchika, K.; Nakatsuji, N.; Surani, A.; Tada, T. Targeted chromosome elimination from es-somatic hybrid cells. Nat. Methods. 2007, 4, 23–25. [Google Scholar] [CrossRef]
- Li, L.B.; Chang, K.H.; Wang, P.R.; Hirata, R.K.; Papayannopoulou, T.; Russell, D.W. Trisomy correction in Down syndrome induced pluripotent stem cells. Cell. Stem. Cell. 2012, 11, 615–619. [Google Scholar] [CrossRef] [Green Version]
- Zuo, E.; Huo, X.; Yao, X.; Hu, X.; Sun, Y.; Yin, J.; He, B.; Wang, X.; Shi, L.; Ping, J.; et al. CRISPR/Cas9-mediated targeted chromosome elimination. Genome Boil. 2017. [Google Scholar] [CrossRef]
- Adikusuma, F.; Williams, N.; Grutzner, F.; Hughes, J.; Thomas, P. Targeted Deletion of an Entire Chromosome Using CRISPR/Cas9. Mol. Ther. 2017, 25, 1736–1738. [Google Scholar] [CrossRef]
- Jiang, J.; Jing, Y.; Cost, G.J.; Chiang, J.C.; Kolpa, H.J.; Cotton, A.M.; Carone, D.M.; Carone, B.R.; Shivak, D.A.; Guschin, D.Y.; et al. Translating dosage compensation to trisomy 21. Nature 2013, 500, 296–300. [Google Scholar] [CrossRef] [Green Version]
- Sauer, B.; Henderson, N. Cre-stimulated recombination at loxP -containing DNA sequences placed into the mammalian genome. Nucleic Acids Res. 1989, 17, 147–161. [Google Scholar] [CrossRef] [Green Version]
- Omori, S.; Tanabe, H.; Banno, K.; Tsuji, A.; Nawa, N.; Hirata, K.; Kawatani, K.; Kokubu, C.; Takeda, J.; Taniguchi, H.; et al. A Pair of Maternal Chromosomes Derived from Meiotic Nondisjunction in Trisomy 21 Affects Nuclear Architecture and Transcriptional Regulation. Sci. Rep. 2017. [Google Scholar] [CrossRef]
- Costa, V.; Sommese, L.; Casamassimi, A.; Colicchio, R.; Angelini, C.; Marchesano, V.; Milone, L.; Farzati, B.; Giovane, A.; Fiorito, C.; et al. Impairment of circulating endothelial progenitors in Down syndrome. BMC Med. Genom. 2010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sangrithi, M.N.; Turner, J.M.A. Mammalian X Chromosome Dosage Compensation: Perspectives From the Germ Line. BioEssays. 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, J.C.; Jiang, J.; Newburger, P.E.; Lawrence, J.B. Trisomy silencing by XIST normalizes Down syndrome cell pathogenesis demonstrated for hematopoietic defects in vitro. Nat. Commun. 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trevino, A.E.; Zhang, F. Genome Editing Using Cas9 Nickases. Methods Enzymol. 2014, 546, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Doudna, J.A.; Charpentier, E. The new frontier of genome engineering with CRISPR-Cas9. Science 2014. [Google Scholar] [CrossRef]
- Knott, G.J.; Doudna, J.A. CRISPR-Cas guides the future of genetic engineering. Science 2018, 361, 866–869. [Google Scholar] [CrossRef] [Green Version]
- Sternberg, S.H.; Redding, S.; Jinek, M.; Greene, E.C.; Doudna, J.A. DNA interrogation by the crispr rna-guided endonuclease cas9. Nature 2014, 507, 62–67. [Google Scholar] [CrossRef] [Green Version]
- O’Connell, M.R.; Oakes, B.L.; Sternberg, S.H.; East-Seletsky, A.; Kaplan, M.; Doudna, J.A. Programmable RNA recognition and cleavage by CRISPR/Cas9. Nature 2014, 516, 263–266. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Bershteyn, M.; Hayashi, Y.; Desachy, G.; Hsiao, E.C.; Sami, S.; Tsang, K.M.; Weiss, L.A.; Kriegstein, A.R.; Yamanaka, S.; Wynshaw-Boris, A. Cell-autonomous correction of ring chromosomes in human induced pluripotent stem cells. Nature 2014, 507, 99–103. [Google Scholar] [CrossRef] [Green Version]
- Hirota, T.; Ohta, H.; Powell, B.E.; Mahadevaiah, S.K.; Ojarikre, O.A.; Saitou, M.; Turner, J.M.A. Fertile offspring from sterile sex chromosome trisomic mice. Science 2017, 357, 932–935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gisselsson, D. Ring chromosomes: Vicious circles at the end and beginning of life. Atlas Genet. Cytogenet. Oncol. Haematol. 2011. [Google Scholar] [CrossRef] [Green Version]
- Dobyns, W.B.; Stratton, R.F.; Parke, J.T.; Greenberg, F.; Nussbaum, R.L.; Ledbetter, D.H. Miller-Dieker syndrome: Lissencephaly and monosomy 17p. J. Pediatr. 1983, 102, 552–558. [Google Scholar] [CrossRef]
- Plona, K.; Kim, T.; Halloran, K.; Wynshaw-Boris, A. Chromosome therapy: Potential strategies for the correction of severe chromosome aberrations. Am. J. Med. Genet. C Semin. Med. Genet. 2016, 172, 422–430. [Google Scholar] [CrossRef]
- Kim, T.; Plona, K.; Wynshaw-Boris, A. A novel system for correcting large-scale chromosomal aberrations: Ring chromosome correction via reprogramming into induced pluripotent stem cell (ipsc). Chromosoma 2017, 126, 457–463. [Google Scholar] [CrossRef]
- Hütter, G.; Nowak, D.; Mossner, M.; Ganepola, S.; Müßig, A.; Allers, K.; Schneider, T.; Hofmann, J.; Kücherer, C.; Blau, O.; et al. Long-Term Control of HIV byCCR5Delta32/Delta32 Stem-Cell Transplantation. New Engl. J. Med. 2009, 360, 692–698. [Google Scholar] [CrossRef] [Green Version]
- Couzin-Frankel, J. Cancer immunotherapy. Baby’s leukemia recedes after novel cell therapy. Science 2015. [Google Scholar] [CrossRef]
- Hamada, M.; Malureanu, L.A.; Wijshake, T.; Zhou, W.; Van Deursen, J.M. Reprogramming to Pluripotency Can Conceal Somatic Cell Chromosomal Instability. PLoS Genet. 2012. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Disease | Inheritance | Causative Gene (Gene Ontology) | Chromosome Imbalance | Frequency of Patients | Clinical Features | ||||
|---|---|---|---|---|---|---|---|---|---|
| Congenital Heart Defect | Microcephaly | Mental Retardation | Cancer Predisposition | Others | |||||
| Autosomal chromosome | |||||||||
| Down syndrome | IC | T 21 | 1/750 live births | + | − | + | + | upward-slanting palpebral fissures, epicanthal folds, single palm fold | |
| Edwards syndrome | IC | T 18 | 1/6000–1/8000 live births | + | + | unknown | unknown | prominent occiput, low-set malformed ear, micrognathia | |
| Patau syndrome | IC | T 13 | 1/20,000 live births | + | + | unknown | unknown | polydactyl, midline cleft lip, flexion of the fingers, polycystic kidneys | |
| Mosaic trisomy 8 | IC | Mosaic T 8 | >100 cases reported | + | − | + | + | morphological brain abnormalities, high arched or cleft palate, micrognathia, renal malformation | |
| Mosaic trisomy 9 | IC | Mosaic T 9 | >40 cases reported | + | − | + | + | morphological brain abnormalities, micrognathia, Dandy–Walker malformation, renal malformation | |
| Mosaic trisomy 22 | IC | Mosaic T 22 | >20 cases reported | + | + | + | unknown | hemi dystrophy, midfacial hypoplasia, cleft palate, micrognathia, renal hypoplasia | |
| Sex chromosome | |||||||||
| Turner syndrome | IC | M X | 1/2000–1/5000 live female births | + | + | + | − | posteriorly rotated ears, neck webbing, broad chest, short stature, micrognathia | |
| Klinefelter syndrome | IC | add chr X in male | 1/426–1/1000 live male births | − | − | + | − | tall stature, long limbs, hypogonadism, infertility | |
| XXX syndrome | IC | T X | 1/900 live female births | − | − | − | − | tall stature, normal fertility | |
| XYY syndrome | IC | add chr Y in male | 1/800–1/1000 live male births | − | − | − | − | tall stature, hyperactive behavior, distractibility, temper tantrums, low frustration tolerance | |
| Mosaic Variegated Aneuploidy (MVA)—autosomal and sex chromosome | |||||||||
| MVA1 or MVA | AR | BUB1B (mitotic SAC) | M, T, and double T | <1/1,000,000 live births | + | + | + | + | Dandy–Walker complex, cataracts, premature aging, multiple renal cysts |
| MVA2 | AR | Cep57 (spindle pole integrity) | M, T, and double T | 5 cases reported | + | + | + | − | rhizomelic shortening of the upper limbs, skull anomalies |
| MVA3 | AR | TRIP13 (mitotic SAC) | M, T, and double T | 6 cases reported | − | − | + | + | seizures, abnormal skin pigmentation, arthrogryposis |
| Used Genome Editing System | Aneuploidy Focused | Purpose | Cell Type | Target Gene Locus | Transgene | Selection Method | Initial → Final Genotype | Reference |
|---|---|---|---|---|---|---|---|---|
| Cre/inverted loxP | XY genotype | Chr del | Mouse zygotes | chr Y | Y-inverted loxP transgene | − | XY → XO | [83] |
| Tetraploid mESC | Chr del | mES somatic hybrid cells | chr 11, chr 12, chr 6 | CEC | Puro drug selection, sorting by FACS | 40,XY (2n) → 80,XXYY (4n)→ 79,XXYY (4n) | [85] | |
| Tetraploid mESC | Chr del | Hybrid cells from two CEC transgenic ESC lines (CEC-ESC) | chr 6, chr 11, chr 12, chr 17 | CEC | Puro and neo drug selection, sorting by FACS | 80,XXXY (4n) → 78,XXYY (4n) | [84] | |
| CEC-mESC | Chr del | Transgenic mESC containing a copy of CEC (CEC-ESC) | chr 5 (band F), chr 13 (band A) | CEC | Sorting by FACS | 40,XY → 39,XY | [80] | |
| Down syndrome | Chr del | HeLa cells with three copies of chr 21 | intergenic region between RCAN1 and CLIC6 genes | loxP-HSV-tk | GCV drug selection | 47,+21 → 46 | [82] | |
| Tetraploid MEF | Chr del | Tetraploid immortalized murine embryonic fibroblasts | chr 9, chr 10, chr 12, chr 14 | GFP-inverted loxP-hDC2 | Sorting by FACS | 40,XY (2n) → 80,XXYY (4n) → 79,XXYY (4n) | [79] | |
| Conventional gene targeting | Down syndrome | Knock-in | Down syndrome hiPSC | APP gene | TKNEO | Neo and GCV drug selection | 47,+21 → 46 | [86] |
| ZFNs | Down syndrome | Silencing the chr 21 | Down syndrome hiPSC | DYRK1A gene | XIST | Puro drug selection | 47,+21 → 47,+21(chr Barr) | [89] |
| CRISPR/Cas9 | XY genotype | Chr del | mESCs | SRE of centromere and long arm of chr Y | − | Puro drug selection | XY → XO | [88] |
| XY genotype | Chr del | mESCs | SRE of Rbmy1a1 and Ssty2 genes | − | Sorting by FACS | XY → XO | [87] | |
| XY genotype | Chr del | Mouse brain | SRE of Rbmy1a1 and Ssty2 genes | − | Sorting by FACS | XY → XO | [87] | |
| Turner syndrome | Chr del | Mouse zygotes | SRE of Rbmy1a1, Ssty1, and Ssty2 genes | − | − | XY → XO | [87] | |
| Turner syndrome | Chr del | Mouse zygotes | SRE of long arm of chr X | − | − | XX → XO | [87] | |
| mESC aneuploidy | Chr del | Stable mESC line with an extra human chr 14 established by chr transfer | SRE of long arm of chr 14 | − | Sorting by FACS | mChr14 = 1 → mChr14 = 0 | [87] | |
| Down syndrome | Chr del | mESCs with trisomy 21/hiPSCs with trisomy 21 | SRE of long arm of chr 21 | − | Sorting by FACS | 47,+21 → 46 | [87] | |
| Cancer | Chr del | Human cancer cell line HT-29 | SRE of short and long arm of chr 7 | − | Sorting by FACS | hChr7 = 4 →hChr7 = 3 | [87] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Akutsu, S.N.; Fujita, K.; Tomioka, K.; Miyamoto, T.; Matsuura, S. Applications of Genome Editing Technology in Research on Chromosome Aneuploidy Disorders. Cells 2020, 9, 239. https://doi.org/10.3390/cells9010239
Akutsu SN, Fujita K, Tomioka K, Miyamoto T, Matsuura S. Applications of Genome Editing Technology in Research on Chromosome Aneuploidy Disorders. Cells. 2020; 9(1):239. https://doi.org/10.3390/cells9010239
Chicago/Turabian StyleAkutsu, Silvia Natsuko, Kazumasa Fujita, Keita Tomioka, Tatsuo Miyamoto, and Shinya Matsuura. 2020. "Applications of Genome Editing Technology in Research on Chromosome Aneuploidy Disorders" Cells 9, no. 1: 239. https://doi.org/10.3390/cells9010239