Targeting the Oncogenic FGF-FGFR Axis in Gastric Carcinogenesis

 and
and
Abstract
:1. Introduction
2. Emerging Role of FGF-FGFR in Solid Tumors
2.1. FGF Family Induces Tumor Growth
2.2. FGFR Family Drives Oncogenesis
2.2.1. Functional Structures of FGFR
2.2.2. Mechanisms of FGFR in Driving Cancer
2.3. Partner Proteins Mediate FGF-FGFR Signal Transduction
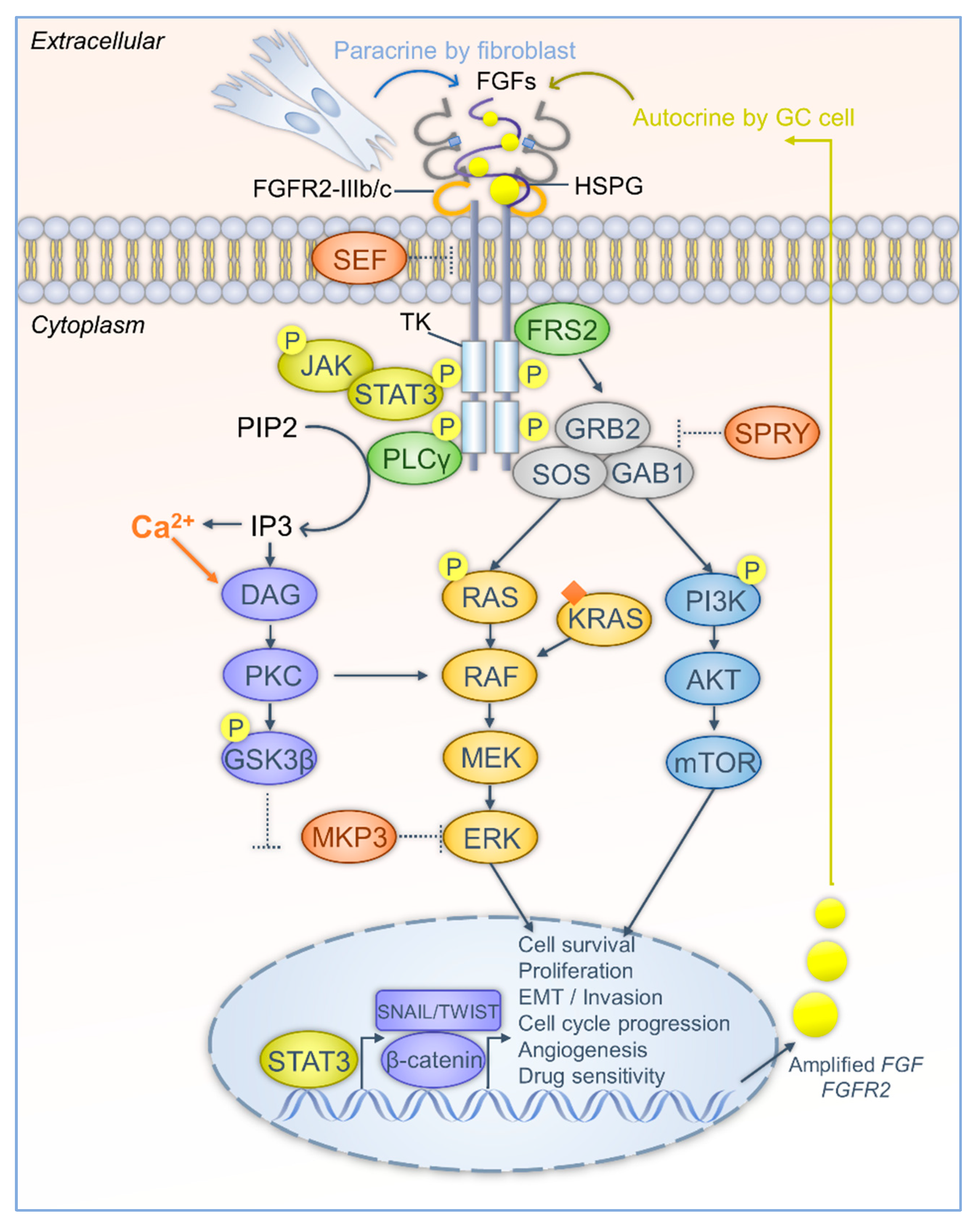
2.4. Signaling Pathways Respond to FGF-FGFR Activation
3. Deregulation of the FGF-FGFR Signaling in Gastric Carcinogenesis
3.1. Significance of FGFR2 in Gastric Tissues
3.2. Aberrant FGF-FGFRs Advance Gastric Tumorigenesis
3.3. FGFR2 Crosstalk in GC
4. Targeting Aberrant FGF-FGFR Activation in GC by Specific Antibodies or Small Molecules
4.1. Specific Antibodies and FGF Traps
4.2. Small Molecules: Non-Selective and Selective FGFR Inhibitors
5. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Correa, P. Human gastric carcinogenesis: A multistep and multifactorial process—First American Cancer Society Award Lecture on Cancer Epidemiology and Prevention. Cancer Res. 1992, 52, 6735–6740. [Google Scholar] [PubMed]
- Uemura, N.; Okamoto, S.; Yamamoto, S.; Matsumura, N.; Yamaguchi, S.; Yamakido, M.; Taniyama, K.; Sasaki, N.; Schlemper, R.J. Helicobacter pylori infection and the development of gastric cancer. N. Engl. J. Med. 2001, 345, 784–789. [Google Scholar] [CrossRef] [PubMed]
- Pharoah, P.D.; Guilford, P.; Caldas, C. International Gastric Cancer Linkage C: Incidence of gastric cancer and breast cancer in CDH1 (E-cadherin) mutation carriers from hereditary diffuse gastric cancer families. Gastroenterology 2001, 121, 1348–1353. [Google Scholar] [CrossRef] [PubMed]
- Hansford, S.; Kaurah, P.; Li-Chang, H.; Woo, M.; Senz, J.; Pinheiro, H.; Schrader, K.A.; Schaeffer, D.F.; Shumansky, K.; Zogopoulos, G.; et al. Hereditary Diffuse Gastric Cancer Syndrome: CDH1 Mutations and Beyond. JAMA Oncol. 2015, 1, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Yanai, K.; Nakamura, M.; Akiyoshi, T.; Nagai, S.; Wada, J.; Koga, K.; Noshiro, H.; Nagai, E.; Tsuneyoshi, M.; Tanaka, M.; et al. Crosstalk of hedgehog and Wnt pathways in gastric cancer. Cancer Lett. 2008, 263, 145–156. [Google Scholar] [CrossRef]
- Corso, S.; Ghiso, E.; Cepero, V.; Sierra, J.R.; Migliore, C.; Bertotti, A.; Trusolino, L.; Comoglio, P.M.; Giordano, S. Activation of HER family members in gastric carcinoma cells mediates resistance to MET inhibition. Mol. Cancer 2010, 9, 121. [Google Scholar] [CrossRef]
- Fu, Y.F.; Gui, R.; Liu, J. HER-2-induced PI3K signaling pathway was involved in the pathogenesis of gastric cancer. Cancer Gene Ther. 2015, 22, 145–153. [Google Scholar] [CrossRef]
- Riquelme, I.; Saavedra, K.; Espinoza, J.A.; Weber, H.; García, P.; Nervi, B.; Garrido, M.; Corvalán, A.H.; Roa, J.C.; Bizama, C. Molecular classification of gastric cancer: Towards a pathway-driven targeted therapy. Oncotarget 2015, 6, 24750–24779. [Google Scholar] [CrossRef] [Green Version]
- Cui, Z.-L.; Han, F.-F.; Peng, X.-H.; Chen, X.; Luan, C.-Y.; Han, R.-C.; Xu, W.-G.; Guo, X.-J. Yes-Associated Protein 1 Promotes Adenocarcinoma Growth and Metastasis through Activation of the Receptor Tyrosine Kinase Axl. Int. J. Immunopathol. Pharmacol. 2012, 25, 989–1001. [Google Scholar] [CrossRef]
- Kang, W.; Cheng, A.S.; Yu, J.; To, K.F. Emerging role of Hippo pathway in gastric and other gastrointestinal cancers. World J. Gastroenterol. 2016, 22, 1279–1288. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Huang, T.; Zhang, J.; Wong, C.C.; Zhang, B.; Dong, Y.; Wu, F.; Tong, J.H.M.; Wu, W.K.K.; Cheng, A.S.L.; et al. TEAD1/4 exerts oncogenic role and is negatively regulated by miR-4269 in gastric tumorigenesis. Oncogene 2017, 36, 6518–6530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014, 513, 202–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friesel, R.; Neilson, K.M. Ligand-independent Activation of Fibroblast Growth Factor Receptors by Point Mutations in the Extracellular, Transmembrane, and Kinase Domains. J. Boil. Chem. 1996, 271, 25049–25057. [Google Scholar] [Green Version]
- Thisse, B.; Thisse, C. Functions and regulations of fibroblast growth factor signaling during embryonic development. Dev. Biol. 2005, 287, 390–402. [Google Scholar] [CrossRef] [PubMed]
- Dieci, M.V.; Arnedos, M.; Andre, F.; Soria, J.C. Fibroblast Growth Factor Receptor Inhibitors as a Cancer Treatment: From a Biologic Rationale to Medical Perspectives. Cancer Discov. 2013, 3, 264–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, L.; Su, X.; Zhang, L.; Yin, X.; Tang, L.; Zhang, X.; Xu, Y.; Gao, Z.; Liu, K.; Zhou, M.; et al. FGFR2 gene amplification in gastric cancer predicts sensitivity to the selective FGFR inhibitor AZD4547. Clin. Cancer Res. 2013, 19, 2572–2583. [Google Scholar] [CrossRef]
- Su, X.; Zhan, P.; Gavine, P.R.; Morgan, S.; Womack, C.; Ni, X.; Shen, D.; Bang, Y.-J.; Im, S.-A.; Kim, W.H.; et al. FGFR2 amplification has prognostic significance in gastric cancer: Results from a large international multicentre study. Br. J. Cancer 2014, 110, 967–975. [Google Scholar] [CrossRef]
- Ahn, S.; Lee, J.; Hong, M.; Kim, S.T.; Park, S.H.; Choi, M.G.; Lee, J.-H.; Sohn, T.S.; Bae, J.M.; Kim, S.; et al. FGFR2 in gastric cancer: Protein overexpression predicts gene amplification and high H-index predicts poor survival. Mod. Pathol. 2016, 29, 1095–1103. [Google Scholar] [CrossRef]
- Katoh, M.; Nakagama, H. FGF receptors: Cancer biology and therapeutics. Med. Res. Rev. 2014, 34, 280–300. [Google Scholar] [CrossRef]
- Itoh, N.; Ornitz, D.M. Fibroblast growth factors: From molecular evolution to roles in development, metabolism and disease. J. Biochem. 2011, 149, 121–130. [Google Scholar] [CrossRef]
- Ghedini, G.C.; Ronca, R.; Presta, M.; Giacomini, A. Future applications of FGF/FGFR inhibitors in cancer. Expert Rev. Anticancer. Ther. 2018, 18, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.; Wilhelm, K.; Dubrac, A.; Tung, J.K.; Alves, T.C.; Fang, J.S.; Xie, Y.; Zhu, J.; Chen, Z.; De Smet, F.; et al. FGF-dependent metabolic control of vascular development. Nature 2017, 545, 224–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maddaluno, L.; Urwyler, C.; Werner, S. Fibroblast growth factors: Key players in regeneration and tissue repair. Development 2017, 144, 4047–4060. [Google Scholar] [CrossRef]
- Hsu, P.I.; Chow, N.H.; Lai, K.H.; Yang, H.B.; Chan, S.H.; Lin, X.Z.; Cheng, J.S.; Huang, J.S.; Ger, L.P.; Huang, S.M.; et al. Implications of serum basic fibroblast growth factor levels in chronic liver diseases and hepatocellular carcinoma. Anticancer. Res. 1997, 17, 2803–2809. [Google Scholar] [PubMed]
- Poon, R.T.-P.; Ng, I.O.-L.; Lau, C.; Yu, W.-C.; Fan, S.-T.; Wong, J. Correlation of serum basic fibroblast growth factor levels with clinicopathologic features and postoperative recurrence in hepatocellular carcinoma. Am. J. Surg. 2001, 182, 298–304. [Google Scholar] [CrossRef]
- Jibiki, N.; Saito, N.; Kameoka, S.; Kobayashi, M. Clinical Significance of Fibroblast Growth Factor (FGF) Expression in Colorectal Cancer. Int. Surg. 2014, 99, 493–499. [Google Scholar] [CrossRef]
- Helsten, T.; Schwaederle, M.; Kurzrock, R. Fibroblast growth factor receptor signaling in hereditary and neoplastic disease: Biologic and clinical implications. Cancer Metastasis Rev. 2015, 34, 479–496. [Google Scholar] [CrossRef]
- Korc, M.; Friesel, R. The Role of Fibroblast Growth Factors in Tumor Growth. Curr. Cancer Drug Targets 2009, 9, 639–651. [Google Scholar] [CrossRef]
- Lee, P.; Johnson, D.; Cousens, L.; Fried, V.; Williams, L. Purification and complementary DNA cloning of a receptor for basic fibroblast growth factor. Sci. 1989, 245, 57–60. [Google Scholar] [CrossRef]
- Johnson, D.E.; Williams, L.T. Structural and functional diversity in the FGF receptor multigene family. Adv. Cancer Res. 1993, 60, 1–41. [Google Scholar]
- Haugsten, E.M.; Wiedlocha, A.; Olsnes, S.; Wesche, J. Roles of Fibroblast Growth Factor Receptors in Carcinogenesis. Mol. Cancer Res. 2010, 8, 1439–1452. [Google Scholar] [CrossRef] [Green Version]
- Holzmann, K.; Grunt, T.; Heinzle, C.; Sampl, S.; Steinhoff, H.; Reichmann, N.; Kleiter, M.; Hauck, M.; Marian, B. Alternative Splicing of Fibroblast Growth Factor Receptor IgIII Loops in Cancer. J. Nucleic Acids 2012, 2012, 950508. [Google Scholar] [CrossRef]
- Yeh, B.K.; Igarashi, M.; Eliseenkova, A.V.; Plotnikov, A.N.; Sher, I.; Ron, D.; Aaronson, S.A.; Mohammadi, M. Structural basis by which alternative splicing confers specificity in fibroblast growth factor receptors. Proc. Natl. Acad. Sci. USA 2003, 100, 2266–2271. [Google Scholar] [CrossRef] [Green Version]
- Babina, I.S.; Turner, N.C. Advances and challenges in targeting FGFR signalling in cancer. Nat. Rev. Cancer 2017, 17, 318–332. [Google Scholar] [CrossRef]
- Trueb, B. Biology of FGFRL1, the fifth fibroblast growth factor receptor. Cell. Mol. Life Sci. 2011, 68, 951–964. [Google Scholar] [CrossRef]
- Helsten, T.; Elkin, S.; Arthur, E.; Tomson, B.N.; Carter, J.; Kurzrock, R. The FGFR Landscape in Cancer: Analysis of 4,853 Tumors by Next-Generation Sequencing. Clin. Cancer Res. 2016, 22, 259–267. [Google Scholar] [CrossRef]
- Latko, M.; Czyrek, A.; Porebska, N.; Kucinska, M.; Otlewski, J.; Zakrzewska, M.; Opalinski, L. Cross-Talk between Fibroblast Growth Factor Receptors and Other Cell Surface Proteins. Cells 2019, 8, 455. [Google Scholar] [CrossRef]
- Mulloy, B.; Linhardt, R.J. Order out of complexity – protein structures that interact with heparin. Curr. Opin. Struct. Boil. 2001, 11, 623–628. [Google Scholar] [CrossRef]
- Beenken, A.; Mohammadi, M. The FGF family: Biology, pathophysiology and therapy. Nat. Rev. Drug Discov. 2009, 8, 235–253. [Google Scholar] [CrossRef]
- Venero Galanternik, M.; Kramer, K.L.; Piotrowski, T. Heparan Sulfate Proteoglycans Regulate Fgf Signaling and Cell Polarity during Collective Cell Migration. Cell Rep. 2015, 10, 414–428. [Google Scholar] [CrossRef] [Green Version]
- Goetz, R.; Mohammadi, M. Exploring mechanisms of FGF signalling through the lens of structural biology. Nat. Rev. Mol. Cell Biol. 2013, 14, 166–180. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Lee, K.W.; Goldfarb, M. Novel Recognition Motif on Fibroblast Growth Factor Receptor Mediates Direct Association and Activation of SNT Adapter Proteins. J. Boil. Chem. 1998, 273, 17987–17990. [Google Scholar] [CrossRef] [Green Version]
- Hoch, R.V.; Soriano, P. Context-specific requirements for Fgfr1 signaling through Frs2 and Frs3 during mouse development. Development 2006, 133, 663–673. [Google Scholar] [CrossRef] [Green Version]
- Gotoh, N.; Laks, S.; Nakashima, M.; Lax, I.; Schlessinger, J. FRS2 family docking proteins with overlapping roles in activation of MAP kinase have distinct spatial-temporal patterns of expression of their transcripts. FEBS Lett. 2004, 564, 14–18. [Google Scholar] [CrossRef] [Green Version]
- Mohammadi, M.; Olsen, S.K.; Ibrahimi, O.A. Structural basis for fibroblast growth factor receptor activation. Cytokine Growth Factor Rev. 2005, 16, 107–137. [Google Scholar] [CrossRef]
- Zhang, Y.; McKeehan, K.; Lin, Y.; Zhang, J.; Wang, F. Fibroblast growth factor receptor 1 (FGFR1) tyrosine phosphorylation regulates binding of FGFR substrate 2alpha (FRS2alpha) but not FRS2 to the receptor. Mol. Endocrinol. 2008, 22, 167–175. [Google Scholar] [CrossRef]
- Fürthauer, M.; Lin, W.; Ang, S.-L.; Thisse, B.; Thisse, C. Sef is a feedback-induced antagonist of Ras/MAPK-mediated FGF signalling. Nat. Cell Biol. 2002, 4, 170–174. [Google Scholar] [CrossRef]
- Tsang, M.; Friesel, R.; Kudoh, T.; Dawid, I.B. Identification of Sef, a novel modulator of FGF signalling. Nat. Cell Biol. 2002, 4, 165–169. [Google Scholar] [CrossRef]
- Tsang, M.; Dawid, I.B. Promotion and attenuation of FGF signaling through the Ras-MAPK pathway. Sci. Stke 2004, 2004, pe17. [Google Scholar] [CrossRef]
- Dey, J.H.; Bianchi, F.; Voshol, J.; Bonenfant, D.; Oakeley, E.J.; Hynes, N.E. Targeting Fibroblast Growth Factor Receptors Blocks PI3K/AKT Signaling, Induces Apoptosis, and Impairs Mammary Tumor Outgrowth and Metastasis. Cancer Res. 2010, 70, 4151–4162. [Google Scholar] [CrossRef]
- Eswarakumar, V.; Lax, I.; Schlessinger, J.; Eswarakumar, J. Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev. 2005, 16, 139–149. [Google Scholar] [CrossRef]
- Turner, N.; Grose, R. Fibroblast growth factor signalling: From development to cancer. Nat. Rev. Cancer 2010, 10, 116–129. [Google Scholar] [CrossRef]
- Kamata, T. Keratinocyte growth factor regulates proliferation and differentiation of hematopoietic cells expressing the receptor gene K-sam. Exp. Hematol. 2002, 30, 297–305. [Google Scholar] [CrossRef]
- Cailliau, K.; Browaeys-Poly, E.; Vilain, J.P. Fibroblast growth factors 1 and 2 differently activate MAP kinase in Xenopus oocytes expressing fibroblast growth factor receptors 1 and 4. Biochim. Biophys. Acta 2001, 1538, 228–233. [Google Scholar] [CrossRef] [Green Version]
- Peters, K.G.; Marie, J.; Wilson, E.; Ives, H.E.; Escobedo, J.; Del Rosario, M.; Mirda, D.; Williams, L.T. Point mutation of an FGF receptor abolishes phosphatidylinositol turnover and Ca2+ flux but not mitogenesis. Nature 1992, 358, 678–681. [Google Scholar] [CrossRef]
- Dudka, A.A.; Sweet, S.M.; Heath, J.K. Signal transducers and activators of transcription-3 binding to the fibroblast growth factor receptor is activated by receptor amplification. Cancer Res. 2010, 70, 3391–3401. [Google Scholar] [CrossRef]
- Candi, E.; Rufini, A.; Terrinoni, A.; Giamboi-Miraglia, A.; Lena, A.M.; Mantovani, R.; Knight, R.; Melino, G. DeltaNp63 regulates thymic development through enhanced expression of FgfR2 and Jag2. Proc. Natl. Acad. Sci. USA 2007, 104, 11999–12004. [Google Scholar] [CrossRef]
- Katoh, M.; Katoh, M. Cross-talk of WNT and FGF signaling pathways at GSK3beta to regulate beta-catenin and SNAIL signaling cascades. Cancer Biol. Ther. 2006, 5, 1059–1064. [Google Scholar] [CrossRef]
- Fogarty, M.P.; Emmenegger, B.A.; Grasfeder, L.L.; Oliver, T.G.; Wechsler-Reya, R.J. Fibroblast growth factor blocks Sonic hedgehog signaling in neuronal precursors and tumor cells. Proc. Natl. Acad. Sci. USA 2007, 104, 2973–2978. [Google Scholar] [CrossRef] [Green Version]
- Dudley, A.T.; Godin, R.E.; Robertson, E.J. Interaction between FGF and BMP signaling pathways regulates development of metanephric mesenchyme. Genes Dev. 1999, 13, 1601–1613. [Google Scholar] [CrossRef] [Green Version]
- Peters, K.G.; Werner, S.; Chen, G.; Williams, L.T. Two FGF receptor genes are differentially expressed in epithelial and mesenchymal tissues during limb formation and organogenesis in the mouse. Development 1992, 114, 233–243. [Google Scholar]
- Orr-Urtreger, A.; Bedford, M.T.; Burakova, T.; Arman, E.; Zimmer, Y.; Yayon, A.; Givol, D.; Lonai, P. Developmental Localization of the Splicing Alternatives of Fibroblast Growth Factor Receptor-2 (FGFR2). Dev. Boil. 1993, 158, 475–486. [Google Scholar] [CrossRef]
- Eswarakumar, V.P.; Monsonego-Ornan, E.; Pines, M.; Antonopoulou, I.; Morriss-Kay, G.M.; Lonai, P. The IIIc alternative of Fgfr2 is a positive regulator of bone formation. Development 2002, 129, 3783–3793. [Google Scholar]
- Zhang, X.; Ibrahimi, O.A.; Olsen, S.K.; Umemori, H.; Mohammadi, M.; Ornitz, D.M. Receptor specificity of the fibroblast growth factor family. The complete mammalian FGF family. J. Boil. Chem. 2006, 281, 15694–15700. [Google Scholar] [CrossRef]
- Ornitz, D.M.; Xu, J.; Colvin, J.S.; McEwen, D.G.; MacArthur, C.A.; Coulier, F.; Gao, G.; Goldfarb, M. Receptor specificity of the fibroblast growth factor family. J. Biol. Chem. 1996, 271, 15292–15297. [Google Scholar] [CrossRef]
- Nyeng, P.; Norgaard, G.A.; Kobberup, S.; Jensen, J. FGF10 signaling controls stomach morphogenesis. Dev. Boil. 2007, 303, 295–310. [Google Scholar] [CrossRef] [Green Version]
- Hattori, Y.; Odagiri, H.; Nakatani, H.; Miyagawa, K.; Naito, K.; Sakamoto, H.; Katoh, O.; Yoshida, T.; Sugimura, T.; Terada, M. K-sam, an amplified gene in stomach cancer, is a member of the heparin-binding growth factor receptor genes. Proc. Natl. Acad. Sci. USA 1990, 87, 5983–5987. [Google Scholar] [CrossRef]
- Nakatani, H.; Sakamoto, H.; Yoshida, T.; Yokota, J.; Tahara, E.; Sugimura, T.; Terada, M. Isolation of an Amplified DNA Sequence in Stomach Cancer. Jpn. J. Cancer Res. 1990, 81, 707–710. [Google Scholar] [CrossRef]
- Cristescu, R.; Lee, J.; Nebozhyn, M.; Kim, K.-M.; Ting, J.C.; Wong, S.S.; Liu, J.; Yue, Y.G.; Wang, J.; Yu, K.; et al. Molecular analysis of gastric cancer identifies subtypes associated with distinct clinical outcomes. Nat. Med. 2015, 21, 449–456. [Google Scholar] [CrossRef]
- Deng, N.; Goh, L.K.; Wang, H.; Das, K.; Tao, J.; Tan, I.B.; Zhang, S.; Lee, M.; Wu, J.; Lim, K.H.; et al. A comprehensive survey of genomic alterations in gastric cancer reveals systematic patterns of molecular exclusivity and co-occurrence among distinct therapeutic targets. Gut 2012, 61, 673–684. [Google Scholar] [CrossRef]
- Jung, E.-J.; Jung, E.-J.; Min, S.Y.; Kim, M.A.; Kim, W.H. Fibroblast growth factor receptor 2 gene amplification status and its clinicopathologic significance in gastric carcinoma. Hum. Pathol. 2012, 43, 1559–1566. [Google Scholar] [CrossRef]
- Matsumoto, K.; Arao, T.; Hamaguchi, T.; Shimada, Y.; Kato, K.; Oda, I.; Taniguchi, H.; Koizumi, F.; Yanagihara, K.; Sasaki, H.; et al. FGFR2 gene amplification and clinicopathological features in gastric cancer. Br. J. Cancer 2012, 106, 727–732. [Google Scholar] [CrossRef]
- Park, Y.S.; Na, Y.-S.; Ryu, M.-H.; Lee, C.-W.; Park, H.J.; Lee, J.-K.; Park, S.R.; Ryoo, B.-Y.; Kang, Y.-K. FGFR2 Assessment in Gastric Cancer Using Quantitative Real-Time Polymerase Chain Reaction, Fluorescent In Situ Hybridization, and Immunohistochemistry. Am. J. Clin. Pathol. 2015, 143, 865–872. [Google Scholar] [CrossRef] [Green Version]
- Das, K.; Gunasegaran, B.; Tan, I.B.; Deng, N.; Lim, K.H.; Tan, P. Mutually exclusive FGFR2, HER2, and KRAS gene amplifications in gastric cancer revealed by multicolour FISH. Cancer Lett. 2014, 353, 167–175. [Google Scholar] [CrossRef]
- Kuboki, Y.; Schatz, C.A.; Koechert, K.; Schubert, S.; Feng, J.; Wittemer-Rump, S.; Ziegelbauer, K.; Krahn, T.; Nagatsuma, A.K.; Ochiai, A. In situ analysis of FGFR2 mRNA and comparison with FGFR2 gene copy number by dual-color in situ hybridization in a large cohort of gastric cancer patients. Gastric Cancer 2018, 21, 401–412. [Google Scholar] [CrossRef]
- Hosoda, K.; Yamashita, K.; Ushiku, H.; Ema, A.; Moriya, H.; Mieno, H.; Washio, M.; Watanabe, M. Prognostic relevance of FGFR2 expression in stage II/III gastric cancer with curative resection and S-1 chemotherapy. Oncol. Lett. 2018, 15, 1853–1860. [Google Scholar]
- Van Cutsem, E.; Bang, Y.J.; Mansoor, W.; Petty, R.D.; Chao, Y.; Cunningham, D.; Ferry, D.R.; Smith, N.R.; Frewer, P.; Ratnayake, J.; et al. A randomized, open-label study of the efficacy and safety of AZD4547 monotherapy versus paclitaxel for the treatment of advanced gastric adenocarcinoma with FGFR2 polysomy or gene amplification. Ann. Oncol. 2017, 28, 1316–1324. [Google Scholar] [CrossRef]
- Nakazawa, K.; Yashiro, M.; Hirakawa, K. Keratinocyte growth factor produced by gastric fibroblasts specifically stimulates proliferation of cancer cells from scirrhous gastric carcinoma. Cancer Res. 2003, 63, 8848–8852. [Google Scholar]
- Huang, T.; Wang, L.; Liu, D.; Li, P.; Xiong, H.; Zhuang, L.; Sun, L.; Yuan, X.; Qiu, H. FGF7/FGFR2 signal promotes invasion and migration in human gastric cancer through upregulation of thrombospondin-1. Int. J. Oncol. 2017, 50, 1501–1512. [Google Scholar] [CrossRef] [Green Version]
- Ren, C.; Chen, H.; Han, C.; Fu, D.; Wang, F.; Wang, D.; Ma, L.; Zhou, L.; Han, D. The anti-apoptotic and prognostic value of fibroblast growth factor 9 in gastric cancer. Oncotarget 2016, 7, 36655–36665. [Google Scholar] [CrossRef]
- Ooi, A.; Oyama, T.; Nakamura, R.; Tajiri, R.; Ikeda, H.; Fushida, S.; Nakamura, H.; Dobashi, Y. Semi-comprehensive analysis of gene amplification in gastric cancers using multiplex ligation-dependent probe amplification and fluorescence in situ hybridization. Mod. Pathol. 2015, 28, 861–871. [Google Scholar] [CrossRef] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Sun, Q.; Lin, P.; Zhang, J.; Li, X.; Yang, L.; Huang, J.; Zhou, Z.; Liu, P.; Liu, N. Expression of Fibroblast Growth Factor 10 Is Correlated with Poor Prognosis in Gastric Adenocarcinoma. Tohoku J. Exp. Med. 2015, 236, 311–318. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Zhou, Y.; Huang, T.; Wu, F.; Pan, Y.; Dong, Y.; Wang, Y.; Chan, A.K.Y.; Liu, L.; Kwan, J.S.H.; et al. FGF18, a prominent player in FGF signaling, promotes gastric tumorigenesis through autocrine manner and is negatively regulated by miR-590-5p. Oncogene 2019, 38, 33–46. [Google Scholar] [CrossRef]
- Chang, J.; Wang, S.; Zhang, Z.; Liu, X.; Wu, Z.; Geng, R.; Ge, X.; Dai, C.; Liu, R.; Zhang, Q.; et al. Multiple receptor tyrosine kinase activation attenuates therapeutic efficacy of the fibroblast growth factor receptor 2 inhibitor AZD4547 in FGFR2 amplified gastric cancer. Oncotarget 2015, 6, 2009–2022. [Google Scholar] [CrossRef]
- Liu, Y.J.; Shen, D.; Yin, X.; Gavine, P.; Zhang, T.; Su, X.; Zhan, P.; Xu, Y.; Lv, J.; Qian, J.; et al. HER2, MET and FGFR2 oncogenic driver alterations define distinct molecular segments for targeted therapies in gastric carcinoma. Br. J. Cancer 2014, 110, 1169–1178. [Google Scholar] [CrossRef]
- Lau, W.M.; Teng, E.; Huang, K.K.; Tan, J.W.; Das, K.; Zang, Z.; Chia, T.; The, M.; Kono, K.; Yong, W.P.; et al. Acquired Resistance to FGFR Inhibitor in Diffuse-Type Gastric Cancer through an AKT-Independent PKC-Mediated Phosphorylation of GSK3beta. Mol. Cancer Ther. 2018, 17, 232–242. [Google Scholar] [CrossRef]
- Huang, T.; Liu, D.; Wang, Y.; Li, P.; Sun, L.; Xiong, H.; Dai, Y.; Zou, M.; Yuan, X.; Qiu, H. FGFR2 Promotes Gastric Cancer Progression by Inhibiting the Expression of Thrombospondin4 via PI3K-Akt-Mtor Pathway. Cell. Physiol. Biochem. 2018, 50, 1332–1345. [Google Scholar] [CrossRef]
- Zhu, D.-Y.; Guo, Q.-S.; Li, Y.-L.; Cui, B.; Guo, J.; Liu, J.-X.; Li, P. Twist1 correlates with poor differentiation and progression in gastric adenocarcinoma via elevation of FGFR2 expression. World J. Gastroenterol. 2014, 20, 18306–18315. [Google Scholar] [CrossRef]
- Grygielewicz, P.; Dymek, B.; Bujak, A.; Gunerka, P.; Stanczak, A.; Lamparska-Przybysz, M.; Wieczorek, M.; Dzwonek, K.; Zdzalik, D. Epithelial-mesenchymal transition confers resistance to selective FGFR inhibitors in SNU-16 gastric cancer cells. Gastric Cancer 2016, 19, 53–62. [Google Scholar] [CrossRef]
- Hallinan, N.; Finn, S.; Cuffe, S.; Rafee, S.; O’Byrne, K.; Gately, K. Targeting the fibroblast growth factor receptor family in cancer. Cancer Treat. Rev. 2016, 46, 51–62. [Google Scholar] [CrossRef] [Green Version]
- Hui, Q.; Jin, Z.; Li, X.; Liu, C.; Wang, X. FGF Family: From Drug Development to Clinical Application. Int. J. Mol. Sci. 2018, 19, 1875. [Google Scholar] [CrossRef]
- Zhao, W.-M.; Wang, L.; Park, H.; Chhim, S.; Tanphanich, M.; Yashiro, M.; Kim, K.J. Monoclonal Antibodies to Fibroblast Growth Factor Receptor 2 Effectively Inhibit Growth of Gastric Tumor Xenografts. Clin. Cancer Res. 2010, 16, 5750–5758. [Google Scholar] [CrossRef]
- Sommer, A.; Kopitz, C.; Schatz, C.A.; Nising, C.F.; Mahlert, C.; Lerchen, H.G.; Stelte-Ludwig, B.; Hammer, S.; Greven, S.; Schuhmacher, J.; et al. Preclinical Efficacy of the Auristatin-Based Antibody-Drug Conjugate BAY 1187982 for the Treatment of FGFR2-Positive Solid Tumors. Cancer Res. 2016, 76, 6331–6339. [Google Scholar] [CrossRef]
- Katoh, M. Therapeutics Targeting FGF Signaling Network in Human Diseases. Trends Pharmacol. Sci. 2016, 37, 1081–1096. [Google Scholar] [CrossRef]
- Ronca, R.; Giacomini, A.; Di Salle, E.; Coltrini, D.; Pagano, K.; Ragona, L.; Matarazzo, S.; Rezzola, S.; Maiolo, D.; Torella, R.; et al. Long-Pentraxin 3 Derivative as a Small-Molecule FGF Trap for Cancer Therapy. Cancer Cell 2015, 28, 225–239. [Google Scholar] [CrossRef] [Green Version]
- Pierce, K.L.; Deshpande, A.M.; Stohr, B.A.; Gemo, A.T.; Patil, N.S.; Brennan, T.J.; Bellovin, D.I.; Palencia, S.; Giese, T.; Huang, C.; et al. FPA144, a humanized monoclonal antibody for both FGFR2-amplified and nonamplified, FGFR2b-overexpressing gastric cancer patients. J. Clin. Oncol. 2014, 32, e15074. [Google Scholar] [CrossRef]
- Jiang, X.F.; Dai, Y.; Peng, X.; Shen, Y.Y.; Su, Y.; Wei, M.M.; Liu, W.R.; Ding, Z.B.; Zhang, A.; Shi, Y.H.; et al. SOMCL-085, a novel multi-targeted FGFR inhibitor, displays potent anticancer activity in FGFR-addicted human cancer models. Acta Pharmacol. Sin. 2018, 39, 243–250. [Google Scholar] [CrossRef]
- Hilberg, F.; Tontsch-Grunt, U.; Baum, A.; Le, A.T.; Doebele, R.C.; Lieb, S.; Gianni, D.; Voss, T.; Garin-Chesa, P.; Haslinger, C.; et al. Triple Angiokinase Inhibitor Nintedanib Directly Inhibits Tumor Cell Growth and Induces Tumor Shrinkage via Blocking Oncogenic Receptor Tyrosine Kinases. J. Pharmacol. Exp. Ther. 2018, 364, 494–503. [Google Scholar] [CrossRef]
- Cha, Y.; Kim, H.-P.; Lim, Y.; Han, S.-W.; Song, S.-H.; Kim, T.-Y. FGFR2 amplification is predictive of sensitivity to regorafenib in gastric and colorectal cancers in vitro. Mol. Oncol. 2018, 12, 993–1003. [Google Scholar] [CrossRef]
- Burbridge, M.F.; Bossard, C.J.; Saunier, C.; Fejes, I.; Bruno, A.; Leonce, S.; Ferry, G.; Da Violante, G.; Bouzom, F.; Cattan, V.; et al. S49076 is a novel kinase inhibitor of MET, AXL, and FGFR with strong preclinical activity alone and in association with bevacizumab. Mol. Cancer Ther. 2013, 12, 1749–1762. [Google Scholar] [CrossRef]
- Gozgit, J.M.; Wong, M.J.; Moran, L.; Wardwell, S.; Mohemmad, Q.K.; Narasimhan, N.I.; Shakespeare, W.C.; Wang, F.; Clackson, T.; Rivera, V.M. Ponatinib (AP24534), a multitargeted pan-FGFR inhibitor with activity in multiple FGFR-amplified or mutated cancer models. Mol. Cancer Ther. 2012, 11, 690–699. [Google Scholar] [CrossRef]
- Jang, J.; Kim, H.K.; Bang, H.; Kim, S.T.; Kim, S.Y.; Park, S.H.; Lim, H.Y.; Kang, W.K.; Lee, J.; Kim, K.-M. Antitumor Effect of AZD4547 in a Fibroblast Growth Factor Receptor 2–Amplified Gastric Cancer Patient–Derived Cell Model1. Transl. Oncol. 2017, 10, 469–475. [Google Scholar] [CrossRef]
- Michael, M.; Bang, Y.J.; Park, Y.S.; Kang, Y.K.; Kim, T.M.; Hamid, O.; Thornton, D.; Tate, S.C.; Raddad, E.; Tie, J. A Phase 1 Study of LY2874455, an Oral Selective pan-FGFR Inhibitor, in Patients with Advanced Cancer. Target. Oncol. 2017, 12, 463–474. [Google Scholar] [CrossRef]
- Kim, S.Y.; Ahn, T.; Bang, H.; Ham, J.S.; Kim, J.; Kim, S.T.; Jang, J.; Shim, M.; Kang, S.Y.; Park, S.H.; et al. Acquired resistance to LY2874455 in FGFR2-amplified gastric cancer through an emergence of novel FGFR2-ACSL5 fusion. Oncotarget 2017, 8, 15014–15022. [Google Scholar] [CrossRef]
- Brameld, K.A.; Owens, T.D.; Verner, E.; Venetsanakos, E.; Bradshaw, J.M.; Phan, V.T.; Tam, D.; Leung, K.; Shu, J.; LaStant, J.; et al. Discovery of the Irreversible Covalent FGFR Inhibitor 8-(3-(4-Acryloylpiperazin-1-yl) propyl)-6-(2,6-dichloro-3,5-dimethoxyphenyl)-2-(me thylamino) pyrido [2,3-d] pyrimidin-7 (8 H)-one (PRN1371) for the Treatment of Solid Tumors. J. Med. Chem. 2017, 60, 6516–6527. [Google Scholar] [CrossRef]
- Tsimafeyeu, I.; Daeyaert, F.; Joos, J.B.; Aken, K.V.; Ludes-Meyers, J.; Byakhov, M.; Tjulandin, S. Molecular Modeling, de novo Design and Synthesis of a Novel, Extracellular Binding Fibroblast Growth Factor Receptor 2 Inhibitor Alofanib (RPT835). Med. Chem. 2016, 12, 303–317. [Google Scholar] [CrossRef]
- Hall, T.G.; Yu, Y.; Eathiraj, S.; Wang, Y.; Savage, R.E.; Lapierre, J.M.; Schwartz, B.; Abbadessa, G. Preclinical Activity of ARQ 087, a Novel Inhibitor Targeting FGFR Dysregulation. PLoS ONE 2016, 11, e0162594. [Google Scholar] [CrossRef]
- Schmidt, K.; Moser, C.; Hellerbrand, C.; Zieker, D.; Wagner, C.; Redekopf, J.; Schlitt, H.J.; Geissler, E.K.; Lang, S.A. Targeting Fibroblast Growth Factor Receptor (FGFR) with BGJ398 in a Gastric Cancer Model. Anticancer Res. 2015, 35, 6655–6665. [Google Scholar]
- Inokuchi, M.; Murase, H.; Otsuki, S.; Kawano, T.; Kojima, K. Different clinical significance of FGFR1-4 expression between diffuse-type and intestinal-type gastric cancer. World J. Surg. Oncol. 2017, 15, 2. [Google Scholar] [CrossRef]
- Pearson, A.; Smyth, E.; Babina, I.S.; Herrera-Abreu, M.T.; Tarazona, N.; Peckitt, C.; Kilgour, E.; Smith, N.R.; Geh, C.; Rooney, C.; et al. High-Level Clonal FGFR Amplification and Response to FGFR Inhibition in a Translational Clinical Trial. Cancer Discov. 2016, 6, 838–851. [Google Scholar] [CrossRef] [Green Version]
- Hierro, C.; Alsina, M.; Sanchez, M.; Serra, V.; Rodon, J.; Tabernero, J. Targeting the fibroblast growth factor receptor 2 in gastric cancer: Promise or pitfall? Ann. Oncol. 2017, 28, 1207–1216. [Google Scholar] [CrossRef]


{kind=link}
| Monoclonal Antibodies | Targets | References |
|---|---|---|
| GAL-FR21 and GAL-FR22 | FGFR2 | [94] |
| FPA144 (Bemarituzumab) | FGFR2 amplification or overexpression | [98] |
| BAY 1179470 | FGFR2 amplification or overexpression | [95] |
| FGF traps | ||
| GSK3052230 | FGFs | [93,96] |
| NSC12 | FGFs | [97] |
| Non-Selective FGFR Inhibitors | Main Targets | References |
|---|---|---|
| SOMCL-085 | FGFR, VEGFR, and PDGFR | [99] |
| Nintedanib | FGFR, VEGFR, and PDGFR | [100] |
| Regorafenib | FGFR2, VEGFR1-3, PDGFR, c-Kit, and RET | [101] |
| S49076 | MET, AXL/MER, and FGFR1-3 | [102] |
| Ponatinib | BCR-ABL, VEGFR2-3, and FGFR1-4 | [103] |
| Selective FGFR inhibitors | ||
| AZD4547 | FGFR1, FGFR2 and FGFR3 | [17,78] |
| LY2874455 | FGFR1, FGFR2, FGFR3 and FGFR4 | [105] |
| PRN1371 | FGFR1, FGFR2, FGFR3 and FGFR4 | [107] |
| RPT835 | FGFR2 | [108] |
| ARQ 087 | FGFR1, FGFR2 and FGFR3 | [109] |
| BGJ398 | FGFR1, FGFR2 and FGFR3 | [110] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, J.; Tang, P.M.K.; Zhou, Y.; Cheng, A.S.L.; Yu, J.; Kang, W.; To, K.F. Targeting the Oncogenic FGF-FGFR Axis in Gastric Carcinogenesis. Cells 2019, 8, 637. https://doi.org/10.3390/cells8060637
Zhang J, Tang PMK, Zhou Y, Cheng ASL, Yu J, Kang W, To KF. Targeting the Oncogenic FGF-FGFR Axis in Gastric Carcinogenesis. Cells. 2019; 8(6):637. https://doi.org/10.3390/cells8060637
Chicago/Turabian StyleZhang, Jinglin, Patrick M. K. Tang, Yuhang Zhou, Alfred S. L. Cheng, Jun Yu, Wei Kang, and Ka Fai To. 2019. "Targeting the Oncogenic FGF-FGFR Axis in Gastric Carcinogenesis" Cells 8, no. 6: 637. https://doi.org/10.3390/cells8060637