Sex-Dependent Signaling Pathways Underlying Seizure Susceptibility and the Role of Chloride Cotransporters


Abstract
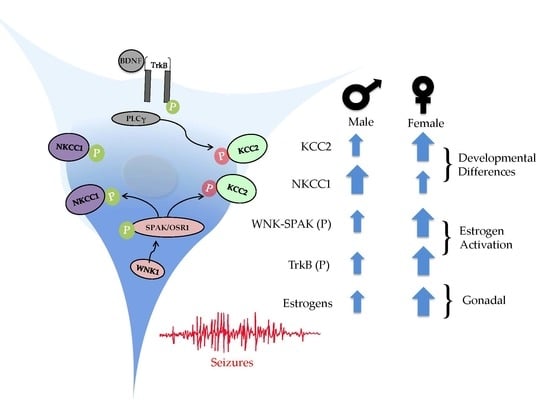
:
{kind=link}
{kind=link}
1. Introduction
2. KCC2-NKCC1
3. BDNF-TrkB Pathway Activation and Seizure Susceptibility
4. CCCs and Refractory Seizures
5. CCCs and Febrile Seizures
6. Sexual Dimorphism and Seizure Susceptibility
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Nardou, R.; Yamamoto, S.; Chazal, G.; Bhar, A.; Ferrand, N.; Dulac, O.; Ben-Ari, Y.; Khalilov, I. Neuronal chloride accumulation and excitatory GABA underlie aggravation of neonatal epileptiform activities by phenobarbital. Brain 2011, 134, 987–1002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nardou, R.; Ferrari, D.C.; Ben-Ari, Y. Mechanisms and effects of seizures in the immature brain. Semin Fetal Neonatal Med. 2013, 18, 175–184. [Google Scholar]
- Hollmann, M.; Heinemann, S. Cloned glutamate receptors. Annu. Rev. Neurosci. 1994, 17, 31–108. [Google Scholar] [CrossRef]
- Kumar, S.S.; Bacci, A.; Kharazia, V.; Huguenard, J.R. A developmental switch of AMPA receptor subunits in neocortical pyramidal neurons. J. Neurosci. 2002, 22, 3005–3015. [Google Scholar] [CrossRef]
- Volpe, J.; Inder, T.; Darras, B.; de Vries, L.; du Plessis, A.; Neill, J.; Perlman, J. Volpe’s Neurology of the Newborn, 6th ed.; Elsevier: Philadelphia, PA, USA, 2017. [Google Scholar]
- Kang, S.K.; Kadam, S.D. Neonatal Seizures: Impact on Neurodevelopmental Outcomes. Front. Pediatr. 2015, 3, 101. [Google Scholar] [CrossRef] [PubMed]
- Bassan, H.; Bental, Y.; Shany, E.; Berger, I.; Froom, P.; Levi, L.; Shiff, Y. Neonatal seizures: dilemmas in workup and management. Pediatr. Neurol. 2008, 38, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Kleen, J.K.; Scott, R.C.; Holmes, G.L.; Roberts, D.W.; Rundle, M.M.; Testorf, M.; Lenck-Santini, P.-P.; Jobst, B.C. Hippocampal interictal epileptiform activity disrupts cognition in humans. Neurology 2013, 81, 18–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lombroso, C.T. Neonatal seizures: gaps between the laboratory and the clinic. Epilepsia 2007, 48 (Suppl. 2), 83–106. [Google Scholar] [CrossRef]
- Walker, L.E.; Mirza, N.; Yip, V.L.M.; Marson, A.G.; Pirmohamed, M. Personalized medicine approaches in epilepsy. J. Int. Med. 2018, 218–234. [Google Scholar] [CrossRef]
- Gharaylou, Z.; Oghabian, M.A.; Azizi, Z.; Hadjighassem, M. Brain microstructural abnormalities correlate with KCC2 downregulation in refractory epilepsy. Neuroreport 2019, 30, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Huberfeld, G. Perturbed chloride homeostasis and GABAergic signaling in human temporal lobe epilepsy. J. Neurosci. 2007, 27, 9866–9873. [Google Scholar] [CrossRef]
- Carter, B.M.; Sullivan, B.J.; Landers, J.R.; Kadam, S.D. Dose-dependent reversal of KCC2 hypofunction and phenobarbital-resistant neonatal seizures by ANA12. Sci. Rep. 2018, 8, 11987. [Google Scholar] [CrossRef] [PubMed]
- He, X.P.; Pan, E.; Sciarretta, C.; Minichiello, L.; McNamara, J.O. Disruption of TrkB-Mediated Phospholipase Cγ Signaling Inhibits Limbic Epileptogenesis. J. Neurosci. 2010, 30, 6188–6196. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.K.; Ammanuel, S.; Thodupunuri, S.; Adler, D.A.; Johnston, M.V.; Kadam, S.D. Sleep dysfunction following neonatal ischemic seizures are differential by neonatal age of insult as determined by qEEG in a mouse model. Neurobiol. Dis. 2018, 116, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ben-Ari, Y. NKCC1 Chloride Importer Antagonists Attenuate Many Neurological and Psychiatric Disorders. Trends Neurosci. 2017, 40, 536–554. [Google Scholar] [CrossRef] [PubMed]
- Hadjikhani, N.; Asberg Johnels, J.; Lassalle, A.; Zurcher, N.R.; Hippolyte, L.; Gillberg, C.; Lemonnier, E.; Ben-Ari, Y. Bumetanide for autism: more eye contact, less amygdala activation. Sci. Rep. 2018, 8, 3602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sampath, D.; Valdez, R.; White, A.M.; Raol, Y.H. Anticonvulsant effect of flupirtine in an animal model of neonatal hypoxic-ischemic encephalopathy. Neuropharmacology 2017, 123, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Dunn, R.; Queenan, B.N.; Pak, D.T.S.; Forcelli, P.A. Divergent effects of levetiracetam and tiagabine against spontaneous seizures in adult rats following neonatal hypoxia. Epilepsy Res. 2018, 140, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Kadam, S. Pre-Clinical Models of Acquired Neonatal Seizures: Differential Effects of Injury on Function of Chloride Co-Transporters. Austin J. Cerebrovasc. Dis. Stroke 2014, 1. [Google Scholar]
- Greenfield, L.J. Molecular Mechanisms of Antiseizure Drug Activity at GABAA Receptors. Seizure 2013, 22, 589–600. [Google Scholar] [CrossRef]
- Boylan, G.B.; Pressler, R.M. Neonatal seizures: the journey so far. Semin. Fetal Neonatal Med. 2013, 18, 173–174. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, F.S.; Galanopoulou, A.S.; Moshé, S.L. Sex dimorphism in seizure-controlling networks. Neurobiol. Dis. 2014, 72 Pt B, 144–152. [Google Scholar] [CrossRef] [Green Version]
- Mirza, M.A.; Ritzel, R.; Xu, Y.; McCullough, L.D.; Liu, F. Sexually dimorphic outcomes and inflammatory responses in hypoxic-ischemic encephalopathy. J. Neuroinflammation 2015, 12, 32. [Google Scholar] [CrossRef] [PubMed]
- Johnston, M.V.; Hagberg, H. Sex and the pathogenesis of cerebral palsy. Dev. Med. Child Neurol. 2007, 49, 74–78. [Google Scholar]
- Velíšková, J.; DeSantis, K.A. Sex and hormonal influences on seizures and epilepsy. Horm. Behav. 2013, 63, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Murguía-Castillo, J.; Beas-Zárate, C.; Rivera-Cervantes, M.C.; Feria-Velasco, A.I.; Ureña-Guerrero, M.E. NKCC1 and KCC2 protein expression is sexually dimorphic in the hippocampus and entorhinal cortex of neonatal rats. Neurosci. Lett. 2013, 552, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Gamba, G. Molecular Physiology and Pathophysiology of Electroneutral Cation-Chloride Cotransporters. Physiol. Rev. 2005, 85, 423–493. [Google Scholar] [CrossRef] [Green Version]
- Tao, R.; Li, C.; Newburn, E.N.; Ye, T.; Lipska, B.K.; Herman, M.M.; Weinberger, D.R.; Kleinman, J.E.; Hyde, T.M. Transcript-specific associations of SLC12A5 (KCC2) in human prefrontal cortex with development, schizophrenia, and affective disorders. J. Neurosci. 2012, 32, 5216–5222. [Google Scholar] [CrossRef]
- Sedmak, G.; Jovanov-Milošević, N.; Puskarjov, M.; Ulamec, M.; Krušlin, B.; Kaila, K.; Judaš, M. Developmental Expression Patterns of KCC2 and Functionally Associated Molecules in the Human Brain. Cereb. Cortex 2016, 26, 4574–4589. [Google Scholar] [CrossRef]
- Kaila, K.; Price, T.J.; Payne, J.A.; Puskarjov, M.; Voipio, J. Cation-chloride cotransporters in neuronal development, plasticity and disease. Nat. Rev. Neurosci. 2014, 15, 637–654. [Google Scholar] [CrossRef] [Green Version]
- Markkanen, M.; Karhunen, T.; Llano, O.; Ludwig, A.; Rivera, C.; Uvarov, P.; Airaksinen, M.S. Distribution of neuronal KCC2a and KCC2b isoforms in mouse CNS. J. Comp. Neurol. 2014, 522, 1897–1914. [Google Scholar] [CrossRef] [PubMed]
- Uvarov, P.; Ludwig, A.; Markkanen, M.; Soni, S.; Hübner, C.A.; Rivera, C.; Airaksinen, M.S. Coexpression and Heteromerization of Two Neuronal K-Cl Cotransporter Isoforms in Neonatal Brain. J. Biol. Chem. 2009, 284, 13696–13704. [Google Scholar] [CrossRef]
- Dubois, C.J.; Cardoit, L.; Schwarz, V.; Markkanen, M.; Airaksinen, M.S.; Uvarov, P.; Simmers, J.; Thoby-Brisson, M. Role of the K+-Cl- Cotransporter KCC2a Isoform in Mammalian Respiration at Birth. eNeuro 2018, 5. [Google Scholar] [CrossRef] [PubMed]
- Kahle, K.T.; Deeb, T.Z.; Puskarjov, M.; Silayeva, L.; Liang, B.; Kaila, K.; Moss, S.J. Modulation of neuronal activity by phosphorylation of the K-Cl cotransporter KCC2. Trends Neurosci. 2013, 36, 726–737. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.H.C.; Walker, J.A.; Williams, J.R.; Goodier, R.J.; Payne, J.A.; Moss, S.J. Direct Protein Kinase C-dependent Phosphorylation Regulates the Cell Surface Stability and Activity of the Potassium Chloride Cotransporter KCC2. J. Biol. Chem. 2007, 282, 29777–29784. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.H.C.; Deeb, T.Z.; Walker, J.A.; Davies, P.A.; Moss, S.J. NMDA receptor activity downregulates KCC2 resulting in depolarizing GABAA receptor–mediated currents. Nat. Neurosci. 2011, 14, 736–743. [Google Scholar] [CrossRef] [PubMed]
- Alessi, D.R.; Zhang, J.; Khanna, A.; Hochdörfer, T.; Shang, Y.; Kahle, K.T. The WNK-SPAK/OSR1 pathway: master regulator of cation-chloride cotransporters. Sci. Signal. 2014, 7, re3. [Google Scholar] [CrossRef] [PubMed]
- de Los Heros, P.; Alessi, D.R.; Gourlay, R.; Campbell, D.G.; Deak, M.; Macartney, T.J.; Kahle, K.T.; Zhang, J. The WNK-regulated SPAK/OSR1 kinases directly phosphorylate and inhibit the K+-Cl- co-transporters. Biochem. J. 2014, 458, 559–573. [Google Scholar] [CrossRef]
- Rivera, C.; Voipio, J.; Payne, J.A.; Ruusuvuori, E.; Lahtinen, H.; Lamsa, K.; Pirvola, U.; Saarma, M.; Kaila, K. The K+/Cl− co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature 1999, 397, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Hubner, C.A. Disruption of KCC2 reveals an essential role of K-Cl cotransport already in early synaptic inhibition. Neuron 2001, 30, 515–524. [Google Scholar] [CrossRef]
- Woo, N.-S.; Lu, J.; England, R.; McClellan, R.; Dufour, S.; Mount, D.B.; Deutch, A.Y.; Lovinger, D.M.; Delpire, E. Hyperexcitability and epilepsy associated with disruption of the mouse neuronal-specific K–Cl cotransporter gene. Hippocampus 2002, 12, 258–268. [Google Scholar] [CrossRef] [PubMed]
- Succol, F.; Fiumelli, H.; Benfenati, F.; Cancedda, L.; Barberis, A. Intracellular chloride concentration influences the GABAA receptor subunit composition. Nat. Commun. 2012, 3, 738. [Google Scholar] [CrossRef] [PubMed]
- Magloire, V.; Cornford, J.; Lieb, A.; Kullmann, D.M.; Pavlov, I. KCC2 overexpression prevents the paradoxical seizure-promoting action of somatic inhibition. Nat. Commun. 2019, 10, 1225. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Khirug, S.; Cai, C.; Ludwig, A.; Blaesse, P.; Kolikova, J.; Afzalov, R.; Coleman, S.K.; Lauri, S.; Airaksinen, M.S.; et al. KCC2 interacts with the dendritic cytoskeleton to promote spine development. Neuron 2007, 56, 1019–1033. [Google Scholar] [CrossRef] [PubMed]
- Côme, E.; Heubl, M.; Schwartz, E.J.; Poncer, J.C.; Lévi, S. Reciprocal Regulation of KCC2 Trafficking and Synaptic Activity. Front. Cell. Neurosci. 2019, 13. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.; Guo, D. Dendritic spine pathology in epilepsy: Cause or consequence? Neuroscience 2013, 251, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Kasai, H.; Fukuda, M.; Watanabe, S.; Hayashi-Takagi, A.; Noguchi, J. Structural dynamics of dendritic spines in memory and cognition. Trends Neurosci. 2010, 33, 121–129. [Google Scholar] [CrossRef]
- Kharod, S.C.; Kang, S.K.; Kadam, S.D. Off-label use of bumetanide for brain disorders: An overview. Front. Neurosci. 2019, 13. [Google Scholar] [CrossRef]
- Galanopoulou, A.S.; Moshé, S.L. Role of sex hormones in the sexually dimorphic expression of KCC2 in rat substantia nigra. Exp. Neurol. 2003, 184, 1003–1009. [Google Scholar] [CrossRef]
- Boulenguez, P.; Liabeuf, S.; Bos, R.; Bras, H.; Jean-Xavier, C.; Brocard, C.; Stil, A.; Darbon, P.; Cattaert, D.; Delpire, E.; et al. Down-regulation of the potassium-chloride cotransporter KCC2 contributes to spasticity after spinal cord injury. Nat. Med. 2010, 16, 302–307. [Google Scholar] [CrossRef]
- Kang, S.K.; Markowitz, G.J.; Kim, S.T.; Johnston, M.V.; Kadam, S.D. Age- and sex-dependent susceptibility to phenobarbital-resistant neonatal seizures: role of chloride co-transporters. Front. Cell Neurosci. 2015, 9, 173. [Google Scholar] [CrossRef]
- Kharod, S.C.; Carter, B.M.; Kadam, S.D. Pharmaco-resistant neonatal seizures: critical mechanistic insights from a chemoconvulsant model. Dev. Neurobiol. 2018. [Google Scholar] [CrossRef]
- Yu, G.; Cheng, M.; Wang, W.; Zhao, R.; Liu, Z. Involvement of WNK1-mediated potassium channels in the sexual dimorphism of blood pressure. Biochem. Biophys. Res. Commun. 2017, 485, 255–260. [Google Scholar] [CrossRef]
- Nugent, B.; Valenzuela, C.; Simons, T.; McCarthy, M. Kinases SPAK and OSR1 are up regulated by estradiol and activate NKCC1 in the developing hypothalamus. J. Neurosci. 2012, 32, 593–598. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Song, X.; Shi, Y.; Shi, Z.; Niu, W.; Feng, X.; Gu, D.; Bao, H.-F.; Ma, H.-P.; Eaton, D.C.; et al. WNK1 Activates Large-Conductance Ca2+-Activated K+ Channels through Modulation of ERK1/2 Signaling. J. Am. Soc. Nephrol. 2015, 26, 844–854. [Google Scholar] [CrossRef]
- Chen, H.; Sun, D. The role of Na-K-Cl co-transporter in cerebral ischemia. Neurol. Res. 2005, 27, 280–286. [Google Scholar] [CrossRef]
- Hertz, L.; Peng, L.; Song, D. Ammonia, like K(+), stimulates the Na(+), K(+), 2 Cl(-) cotransporter NKCC1 and the Na(+),K(+)-ATPase and interacts with endogenous ouabain in astrocytes. Neurochem. Res. 2015, 40, 241–257. [Google Scholar] [CrossRef]
- Chan, C.B.; Ye, K. Sex Differences in Brain-Derived Neurotrophic Factor Signaling and Functions. J. Neurosci. Res. 2017, 95, 328–335. [Google Scholar] [CrossRef]
- Cunha, C.; Brambilla, R.; Thomas, K.L. A simple role for BDNF in learning and memory? Front. Mol. Neurosci. 2010, 3, 1. [Google Scholar] [CrossRef]
- Harward, S.C.; Hedrick, N.G.; Hall, C.E.; Parra-Bueno, P.; Milner, T.A.; Pan, E.; Laviv, T.; Hempstead, B.L.; Yasuda, R.; McNamara, J.O. Autocrine BDNF-TrkB signalling within a single dendritic spine. Nature 2016, 538, 99–103. [Google Scholar] [CrossRef] [Green Version]
- Lin, P.-Y.; Kavalali, E.T.; Monteggia, L.M. Genetic Dissection of Presynaptic and Postsynaptic BDNF-TrkB Signaling in Synaptic Efficacy of CA3-CA1 Synapses. Cell Rep. 2018, 24, 1550–1561. [Google Scholar] [CrossRef]
- Pan, E.; Zhao, Z.; McNamara, J.O. LTD at mossy fiber synapses onto stratum lucidum interneurons requires TrkB and retrograde endocannabinoid signaling. J. Neurophysiol. 2019, 121, 609–619. [Google Scholar] [CrossRef]
- Ferrini, F.; De Koninck, Y. Microglia control neuronal network excitability via BDNF signalling. Neural Plast. 2013, 2013, 429815. [Google Scholar] [CrossRef]
- Ulmann, L.; Hatcher, J.P.; Hughes, J.P.; Chaumont, S.; Green, P.J.; Conquet, F.; Buell, G.N.; Reeve, A.J.; Chessell, I.P.; Rassendren, F. Up-Regulation of P2X4 Receptors in Spinal Microglia after Peripheral Nerve Injury Mediates BDNF Release and Neuropathic Pain. J. Neurosci. 2008, 28, 11263–11268. [Google Scholar] [CrossRef] [Green Version]
- Trang, T.; Beggs, S.; Wan, X.; Salter, M.W. P2X4-Receptor-Mediated Synthesis and Release of Brain-Derived Neurotrophic Factor in Microglia Is Dependent on Calcium and p38-Mitogen-Activated Protein Kinase Activation. J. Neurosci. 2009, 29, 3518–3528. [Google Scholar] [CrossRef]
- Obuchowicz, E.; Nowacka, M.; Paul-Samojedny, M.; Bielecka-Wajdman, A.M.; Małecki, A. Sex differences in the effect of acute peripheral IL-1β administration on the brain and serum BDNF and VEGF expression in rats. Cytokine 2017, 90, 6–13. [Google Scholar] [CrossRef]
- Rivera, C.; Li, H.; Thomas-Crusells, J.; Lahtinen, H.; Viitanen, T.; Nanobashvili, A.; Kokaia, Z.; Airaksinen, M.S.; Voipio, J.; Kaila, K.; et al. BDNF-induced TrkB activation down-regulates the K+–Cl− cotransporter KCC2 and impairs neuronal Cl− extrusion. J. Cell Biol. 2002, 159, 747–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longo, F.M.; Massa, S.M. Small-molecule modulation of neurotrophin receptors: a strategy for the treatment of neurological disease. Nat. Rev. Drug Discov. 2013, 12, 507–525. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Johnston, M.; Kadam, S. Acute TrkB-inhibition rescues phenobarbital-resistant seizures in a mouse model of neonatal ischemia. Eur. J. Neurosci. 2015, 42, 2792–2804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, B.; Huang, Y.Z.; He, X.-P.; Joshi, R.B.; Jang, W.; McNamara, J.O. A Peptide Uncoupling BDNF Receptor TrkB from Phospholipase Cγ1 Prevents Epilepsy Induced by Status Epilepticus. Neuron 2015, 88, 484–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Zhu, Z.; Kalyani, M.; Janik, J.M.; Shi, H. Effects of energy status and diet on Bdnf expression in the ventromedial hypothalamus of male and female rats. Physiol. Behav. 2014, 130, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Snigdha, S.; Neill, J.C.; McLean, S.L.; Shemar, G.K.; Cruise, L.; Shahid, M.; Henry, B. Phencyclidine (PCP)-induced disruption in cognitive performance is gender-specific and associated with a reduction in brain-derived neurotrophic factor (BDNF) in specific regions of the female rat brain. J. Mol. Neurosci. 2011, 43, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Kight, K.E.; McCarthy, M.M. Sex differences and estrogen regulation of BDNF gene expression, but not propeptide content, in the developing hippocampus. J. Neurosci. Res. 2017, 95, 345–354. [Google Scholar] [CrossRef]
- Hill, R.A.; van den Buuse, M. Sex-dependent and region-specific changes in TrkB signaling in BDNF heterozygous mice. Brain Res. 2011, 1384, 51–60. [Google Scholar] [CrossRef]
- Spencer-Segal, J.L.; Waters, E.M.; Bath, K.G.; Chao, M.V.; McEwen, B.S.; Milner, T.A. Distribution of Phosphorylated TrkB Receptor in the Mouse Hippocampal Formation Depends on Sex and Estrous Cycle Stage. J. Neurosci. 2011, 31, 6780–6790. [Google Scholar] [CrossRef] [PubMed]
- Scharfman, H.E.; Mercurio, T.C.; Goodman, J.H.; Wilson, M.A.; MacLusky, N.J. Hippocampal excitability increases during the estrous cycle in the rat: a potential role for brain-derived neurotrophic factor. J. Neurosci. 2003, 23, 11641–11652. [Google Scholar] [CrossRef] [PubMed]
- Solum, D.T.; Handa, R.J. Estrogen regulates the development of brain-derived neurotrophic factor mRNA and protein in the rat hippocampus. J. Neurosci. 2002, 22, 2650–2659. [Google Scholar] [CrossRef]
- Moreno-Piovano, G.S.; Varayoud, J.; Luque, E.H.; Ramos, J.G. Long-term ovariectomy increases BDNF gene methylation status in mouse hippocampus. J. Steroid Biochem. Mol. Biol. 2014, 144 Pt B, 243–252. [Google Scholar] [CrossRef]
- Ottem, E.N.; Beck, L.A.; Jordan, C.L.; Breedlove, S.M. Androgen-dependent regulation of brain-derived neurotrophic factor and tyrosine kinase B in the sexually dimorphic spinal nucleus of the bulbocavernosus. Endocrinology 2007, 148, 3655–3665. [Google Scholar] [CrossRef]
- Li, M.; Masugi-Tokita, M.; Takanami, K.; Yamada, S.; Kawata, M. Testosterone has sublayer-specific effects on dendritic spine maturation mediated by BDNF and PSD-95 in pyramidal neurons in the hippocampus CA1 area. Brain Res. 2012, 1484, 76–84. [Google Scholar] [CrossRef]
- Allen, K.M.; Purves-Tyson, T.D.; Fung, S.J.; Shannon Weickert, C. The effect of adolescent testosterone on hippocampal BDNF and TrkB mRNA expression: relationship with cell proliferation. BMC Neurosci. 2015, 16, 4. [Google Scholar] [CrossRef]
- Purves-Tyson, T.D.; Allen, K.; Fung, S.; Rothmond, D.; Noble, P.L.; Handelsman, D.J.; Shannon Weickert, C. Adolescent testosterone influences BDNF and TrkB mRNA and neurotrophin-interneuron marker relationships in mammalian frontal cortex. Schizophr. Res. 2015, 168, 661–670. [Google Scholar] [CrossRef]
- Tekgul, H.; Gauvreau, K.; Soul, J.; Murphy, L.; Robertson, R.; Stewart, J.; Volpe, J.; Bourgeois, B.; Du Plessis, A.J. The current etiologic profile and neurodevelopmental outcome of seizures in term newborn infants. Pediatrics 2006, 117, 1270–1280. [Google Scholar] [CrossRef]
- Sankar, R.; Painter, M.J. Neonatal seizures: after all these years we still love what doesn’t work. Neurology 2005, 64, 776–777. [Google Scholar] [CrossRef]
- Boylan, G.B.; Stevenson, N.J.; Vanhatalo, S. Monitoring neonatal seizures. Semin. Fetal Neonatal Med. 2013, 18, 202–208. [Google Scholar] [CrossRef]
- Pressler, R.M.; Boylan, G.B.; Marlow, N.; Blennow, M.; Chiron, C.; Cross, J.H.; de Vries, L.S.; Hallberg, B.; Hellström-Westas, L.; Jullien, V.; et al. Bumetanide for the treatment of seizures in newborn babies with hypoxic ischaemic encephalopathy (NEMO): an open-label, dose finding, and feasibility phase 1/2 trial. Lancet Neurol. 2015, 14, 469–477. [Google Scholar] [CrossRef]
- Dzhala, V.I.; Talos, D.M.; Sdrulla, D.A.; Brumback, A.C.; Mathews, G.C.; Benke, T.A.; Delpire, E.; Jensen, F.E.; Staley, K.J. NKCC1 transporter facilitates seizures in the developing brain. Nat. Med. 2005, 11, 1205. [Google Scholar] [CrossRef]
- Bejot, Y.; Prigent-Tessier, A.; Cachia, C.; Giroud, M.; Mossiat, C.; Bertrand, N.; Garnier, P.; Marie, C. Time-dependent contribution of non neuronal cells to BDNF production after ischemic stroke in rats. Neurochem. Int. 2011, 58, 102–111. [Google Scholar] [CrossRef] [Green Version]
- Ruan, L.; Wang, B.; ZhuGe, Q.; Jin, K. Coupling of neurogenesis and angiogenesis after ischemic stroke. Brain Res. 2015, 1623, 166–173. [Google Scholar] [CrossRef]
- Rivera, C. Mechanism of activity-dependent downregulation of the neuron-specific K-Cl cotransporter KCC2. J. Neurosci. 2004, 24, 4683–4691. [Google Scholar] [CrossRef]
- Riffault, B.; Kourdougli, N.; Dumon, C.; Ferrand, N.; Buhler, E.; Schaller, F.; Chambon, C.; Rivera, C.; Gaiarsa, J.-L.; Porcher, C. Pro-Brain-Derived Neurotrophic Factor (proBDNF)-Mediated p75NTR Activation Promotes Depolarizing Actions of GABA and Increases Susceptibility to Epileptic Seizures. Cereb. Cortex 2018, 28, 510–527. [Google Scholar] [CrossRef]
- VonDran, M.W.; LaFrancois, J.; Padow, V.A.; Friedman, W.J.; Scharfman, H.E.; Milner, T.A.; Hempstead, B.L. p75NTR, but Not proNGF, Is Upregulated Following Status Epilepticus in Mice. ASN Neuro 2014, 6, 1759091414552185. [Google Scholar] [CrossRef]
- Chudotvorova, I.; Ivanov, A.; Rama, S.; H++bner, C.A.; Pellegrino, C.; Ben-Ari, Y.; Medina, I. Early expression of KCC2 in rat hippocampal cultures augments expression of functional GABA synapses. J. Physiol. 2005, 566, 671–679. [Google Scholar] [CrossRef] [Green Version]
- Chevy, Q.; Heubl, M.; Goutierre, M.; Backer, S.; Moutkine, I.; Eug+¿ne, E.; Bloch-Gallego, E.; L+¬vi, S.; Poncer, J.C. KCC2 Gates Activity-Driven AMPA Receptor Traffic through Cofilin Phosphorylation. J. Neurosci. 2015, 35, 15772–15786. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Gu, B.; He, X.-P.; Joshi, R.B.; Wackerle, H.D.; Rodriguiz, R.M.; Wetsel, W.C.; McNamara, J.O. Transient Inhibition of TrkB Kinase Following Status Epilepticus Prevents Development of Temporal Lobe Epilepsy. Neuron 2013, 79, 31–38. [Google Scholar] [CrossRef] [Green Version]
- Blauwblomme, T.; Dossi, E.; Pellegrino, C.; Goubert, E.; Iglesias, B.G.; Sainte-Rose, C.; Rouach, N.; Nabbout, R.; Huberfeld, G. Gamma-aminobutyric acidergic transmission underlies interictal epileptogenicity in pediatric focal cortical dysplasia. Ann. Neurol. 2018, 85, 204–217. [Google Scholar] [CrossRef]
- Karlócai, M.R.; Wittner, L.; Tóth, K.; Maglóczky, Z.; Katarova, Z.; Rásonyi, G.; Erőss, L.; Czirják, S.; Halász, P.; Szabó, G.; et al. Enhanced expression of potassium-chloride cotransporter KCC2 in human temporal lobe epilepsy. Brain Struct. Funct. 2016, 221, 3601–3615. [Google Scholar] [CrossRef]
- Steering Committee on Quality Improvement and Management, Subcommittee on Febrile Seizures American Academy of Pediatrics. Febrile Seizures: Clinical Practice Guideline for the Long-term Management of the Child With Simple Febrile Seizures. Pediatrics 2008, 121, 1281–1286. [Google Scholar] [CrossRef] [Green Version]
- Seinfeld, S.; Shinnar, S.; Sun, S.; Hesdorffer, D.C.; Deng, X.; Shinnar, R.C.; O’Hara, K.; Nordli, D.R.; Frank, L.M.; Gallentine, W.; et al. Emergency Management of Febrile Status Epilepticus: Results of the FEBSTAT study. Epilepsia 2014, 55, 388–395. [Google Scholar] [CrossRef] [Green Version]
- Puskarjov, M.; Seja, P.; Heron, S.E.; Williams, T.C.; Ahmad, F.; Iona, X.; Oliver, K.L.; Grinton, B.E.; Vutskits, L.; Scheffer, I.E.; et al. A variant of KCC2 from patients with febrile seizures impairs neuronal Cl− extrusion and dendritic spine formation. EMBO Rep. 2014, 15, 723–729. [Google Scholar] [CrossRef] [Green Version]
- Audenaert, D.; Schwartz, E.; Claeys, K.G.; Claes, L.; Deprez, L.; Suls, A.; Van Dyck, T.; Lagae, L.; Van Broeckhoven, C.; Macdonald, R.L.; et al. A novel GABRG2 mutation associated with febrile seizures. Neurology 2006, 67, 687–690. [Google Scholar] [CrossRef] [PubMed]
- Harkin, L.A.; Bowser, D.N.; Dibbens, L.M.; Singh, R.; Phillips, F.; Wallace, R.H.; Richards, M.C.; Williams, D.A.; Mulley, J.C.; Berkovic, S.F.; et al. Truncation of the GABAA-Receptor γ2 Subunit in a Family with Generalized Epilepsy with Febrile Seizures Plus. Am. J. Hum. Genet. 2002, 70, 530–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awad, P.N.; Sanon, N.T.; Chattopadhyaya, B.; Carri+oo, J.N.; Ouardouz, M.; Gagn+¬, J.; Duss, S.; Wolf, D.; Desgent, S.; Cancedda, L.; et al. Reducing premature KCC2 expression rescues seizure susceptibility and spine morphology in atypical febrile seizures. Neurobiol. Dis. 2016, 91, 10–20. [Google Scholar] [CrossRef]
- Hesdorffer, D.C.; Shinnar, S.; Lewis, D.V.; Nordli, D.R.; Pellock, J.M.; Moshé, S.L.; Shinnar, R.C.; Litherland, C.; Bagiella, E.; Frank, L.M.; et al. Risk factors for febrile status epilepticus: a case-control study. J. Pediatr. 2013, 163, 1147–1151. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.-J.; Xu, Z.-H.; Feng, B.; Xu, C.-L.; Zhao, H.-W.; Wu, D.-C.; Hu, W.-W.; Chen, Z. Gender difference in acquired seizure susceptibility in adult rats after early complex febrile seizures. Neurosci. Bull. 2014, 30, 913–922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadam, S.D.; White, A.M.; Staley, K.J.; Dudek, F.E. Continuous electroencephalographic monitoring with radio-telemetry in a rat model of perinatal hypoxia-ischemia reveals progressive post-stroke epilepsy. J. Neurosci. 2010, 30, 404–415. [Google Scholar] [CrossRef]
- Staley, K.J.; White, A.; Dudek, F.E. Interictal spikes: harbingers or causes of epilepsy? Neurosci. Lett. 2011, 497, 247–250. [Google Scholar] [CrossRef] [Green Version]
- McCarthny, C.R.; Du, X.; Wu, Y.C.; Hill, R.A. Investigating the Interactive Effects of Sex Steroid Hormones and Brain-Derived Neurotrophic Factor during Adolescence on Hippocampal NMDA Receptor Expression. Int. J. Endocrinol. 2018, 2018, 7231915. [Google Scholar] [CrossRef] [PubMed]
- Reddy, D.S.; Jian, K. The testosterone-derived neurosteroid androstanediol is a positive allosteric modulator of GABAA receptors. J. Pharmacol. Exp. Ther. 2010, 334, 1031–1041. [Google Scholar] [CrossRef] [PubMed]
- Sivaraaman, K.; Mintzer, S. Hormonal consequences of epilepsy and its treatment in men. Curr. Opin. Endocrinol. Diabetes Obes. 2011, 18, 204–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thagard, A.S.; Slack, J.L.; Estrada, S.M.; Kazanjian, A.A.; Chan, S.; Burd, I.; Napolitano, P.G.; Ieronimakis, N. Long-term impact of intrauterine neuroinflammation and treatment with magnesium sulphate and betamethasone: Sex-specific differences in a preterm labor murine model. Sci. Rep. 2017, 7, 17883. [Google Scholar] [CrossRef] [Green Version]
- Maxwell, J.R.; Denson, J.L.; Joste, N.E.; Robinson, S.; Jantzie, L.L. Combined in utero hypoxia-ischemia and lipopolysaccharide administration in rats induces chorioamnionitis and a fetal inflammatory response syndrome. Placenta 2015, 36, 1378–1384. [Google Scholar] [CrossRef]
- Koe, A.S.; Jones, N.C.; Salzberg, M.R. Early life stress as an influence on limbic epilepsy: an hypothesis whose time has come? Front. Behav. Neurosci. 2009, 3, 24. [Google Scholar] [CrossRef]
- Huang, L.-T. Early-life stress impacts the developing hippocampus and primes seizure occurrence: cellular, molecular, and epigenetic mechanisms. Front. Mol. Neurosci. 2014, 7. [Google Scholar] [CrossRef] [Green Version]
- Gholipoor, P.; Saboory, E.; Ghazavi, A.; Kiyani, A.; Roshan-Milani, S.; Mohammadi, S.; Javanmardi, E.; Rasmi, Y. Prenatal stress potentiates febrile seizure and leads to long-lasting increase in cortisol blood levels in children under 2years old. Epilepsy Behav. 2017, 72, 22–27. [Google Scholar] [CrossRef]
- Desgent, S.; Duss, S.; Sanon, N.T.; Lema, P.; Lévesque, M.; Hébert, D.; Rébillard, R.-M.; Bibeau, K.; Brochu, M.; Carmant, L. Early-life stress is associated with gender-based vulnerability to epileptogenesis in rat pups. PLoS ONE 2012, 7, e42622. [Google Scholar] [CrossRef]
- Verrotti, A.; D’Egidio, C.; Agostinelli, S.; Verrotti, C.; Pavone, P. Diagnosis and management of catamenial seizures: a review. Int. J. Womens Health 2012, 4, 535–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, D.S. The role of neurosteroids in the pathophysiology and treatment of catamenial epilepsy. Epilepsy Res. 2009, 85, 1–30. [Google Scholar] [CrossRef]
- Reddy, D.S.; Rogawski, M.A. Neurosteroid replacement therapy for catamenial epilepsy. Neurotherapeutics 2009, 6, 392–401. [Google Scholar] [CrossRef]
- Scharfman, H.E.; MacLusky, N.J. The Influence of Gonadal Hormones on Neuronal Excitability, Seizures, and Epilepsy in the Female. Epilepsia 2006, 47, 1423–1440. [Google Scholar] [CrossRef] [Green Version]
- Wickens, S.; Bowden, S.C.; D’Souza, W. Cognitive functioning in children with self-limited epilepsy with centrotemporal spikes: A systematic review and meta-analysis. Epilepsia 2017, 58, 1673–1685. [Google Scholar] [CrossRef] [PubMed]
- Beery, A.K.; Zucker, I. Sex bias in neuroscience and biomedical research. Neurosci. Biobehav. Rev. 2011, 35, 565–572. [Google Scholar]
- Ernst, L.D. Let’s Talk About Sex: Integrating Sex as a Biological Variable Into Epilepsy Research. Epilepsy Curr. 2018, 18, 292–294. [Google Scholar] [CrossRef] [PubMed]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kipnis, P.A.; Sullivan, B.J.; Kadam, S.D. Sex-Dependent Signaling Pathways Underlying Seizure Susceptibility and the Role of Chloride Cotransporters. Cells 2019, 8, 448. https://doi.org/10.3390/cells8050448
Kipnis PA, Sullivan BJ, Kadam SD. Sex-Dependent Signaling Pathways Underlying Seizure Susceptibility and the Role of Chloride Cotransporters. Cells. 2019; 8(5):448. https://doi.org/10.3390/cells8050448
Chicago/Turabian StyleKipnis, Pavel A., Brennan J. Sullivan, and Shilpa D. Kadam. 2019. "Sex-Dependent Signaling Pathways Underlying Seizure Susceptibility and the Role of Chloride Cotransporters" Cells 8, no. 5: 448. https://doi.org/10.3390/cells8050448