C-Met as a Key Factor Responsible for Sustaining Undifferentiated Phenotype and Therapy Resistance in Renal Carcinomas

 , , , ,
, , , ,
Abstract
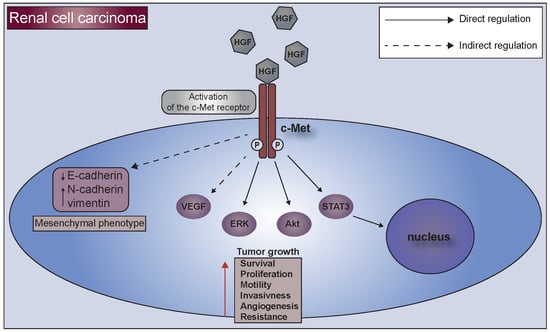
:
1. Introduction
2. C-Met Receptor
3. C-Met Receptor and Kidney Tumors
3.1. C-Met is Associated with an Advanced Stage and Poor Survival of Patients
3.2. Germline and Somatic Mutations of C-Met Receptor in Renal Carcinomas
3.3. Phosphorylation of C-Met in Renal Carcinomas
4. Resistance to Anti-Cancer Therapies in RCC
4.1. C-Met Receptor and Therapy Resistance
4.2. Epithelial to Mesenchymal Transition in the Acquisition of Therapeutic Resistance
5. C-Met Receptor as a Marker of Undifferentiated Cells
6. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Uehara, Y.; Minowa, O.; Mori, C.; Shiota, K.; Kuno, J.; Noda, T.; Kitamura, N. Placental defect and embryonic lethality in mice lacking hepatocyte growth factor/scatter factor. Nature 1995, 373, 702–705. [Google Scholar] [CrossRef] [PubMed]
- Bladt, F.; Riethmacher, D.; Isenmann, S.; Aguzzi, A.; Birchmeier, C. Essential role for the c-Met receptor in the migration of myogenic precursor cells into the limb bud. Nature 1995, 376, 768–771. [Google Scholar] [CrossRef] [PubMed]
- Huh, C.G.; Factor, V.M.; Sánchez, A.; Uchida, K.; Conner, E.A.; Thorgeirsson, S.S. Hepatocyte growth factor/c-Met signaling pathway is required for efficient liver regeneration and repair. Proc. Natl. Acad. Sci. USA 2004, 101, 4477–4482. [Google Scholar] [CrossRef]
- Trusolino, L.; Bertotti, A.; Comoglio, P.M. MET signalling: Principles and functions in development, organ regeneration and cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 834–848. [Google Scholar] [CrossRef] [PubMed]
- Comoglio, P.M.; Giordano, S.; Trusolino, L. Drug development of MET inhibitors: Targeting oncogene addiction and expedience. Nat. Rev. Drug Discov. 2008, 7, 504–516. [Google Scholar] [CrossRef] [PubMed]
- Paumelle, R.; Tulashe, D.; Kherrouche, Z.; Plaza, S.; Leroy, C.; Reveneau, S.; Vandenbunder, B.; Fafeur, V. Hepatocyte growth factor/scatter factor activates the ETS1 transcription factor by a RAS-RAF-MEK-ERK signaling pathway. Oncogene 2002, 21, 2309–2319. [Google Scholar] [CrossRef] [Green Version]
- Furge, K.A.; Kiewlich, D.; Le, P.; Vo, M.N.; Faure, M.; Howlett, A.R.; Lipson, K.E.; Vande Woude, G.F.; Webb, C.P. Suppression of Ras-mediated tumorigenicity and metastasis through inhibition of the Met receptor tyrosine kinase. Proc. Natl. Acad. Sci. USA 2001, 98, 10722–10727. [Google Scholar] [CrossRef] [Green Version]
- Pennacchietti, S.; Michieli, P.; Galluzzo, M.; Mazzone, M.; Giordano, S.; Comoglio, P.M. Hypoxia promotes invasive growth by transcriptional activation of the met oncogene. Cancer Cell 2003, 3, 347–361. [Google Scholar] [CrossRef]
- Szturz, P.; Raymond, E.; Abitbol, C.; Albert, S.; de Gramont, A.; Faivre, S. Understanding c-MET signalling in squamous cell carcinoma of the head & neck. Crit. Rev. Oncol. Hematol. 2017, 111, 39–51. [Google Scholar]
- Kim, B.; Jung, N.; Lee, S.; Sohng, J.K.; Jung, H.J. Apigenin inhibits cancer stem cell-like phenotypes in human Glioblastoma cells via suppression of c-Met Signaling. Phytother. Res. 2016, 30, 1833–1840. [Google Scholar] [CrossRef]
- Bender, S.; Gronych, J.; Warnatz, H.J.; Hutter, B.; Gröbner, S.; Ryzhova, M.; Pfaff, E.; Hovestadt, V.; Weinberg, F.; Halbach, S.; et al. Recurrent MET fusion genes represent a drug target in pediatric glioblastoma. Nat. Med. 2016, 22, 1314–1320. [Google Scholar]
- Di Renzo, M.F.; Narsimhan, R.; Olivero, M.; Bretti, S.; Giordano, S.; Medico, E.; Gaglia, P.; Zara, P.; Comoglio, P.M. Expression of the Met/HGF receptor in normal and neoplastic human tissues. Oncogene 1991, 6, 1997–2003. [Google Scholar]
- Prat, M.; Crepaldi, T.; Gandino, L.; Giordano, S.; Longati, P.; Comoglio, P. C-terminal truncated forms of Met, the hepatocyte growth factor receptor. Mol. Cell. Biol. 1991, 11, 5954–5962. [Google Scholar] [CrossRef]
- Sonnenberg, E.; Meyer, D.; Weidner, K.M.; Birchmeier, C. Scatter factor/hepatocyte growth factor and its receptor, the c-met tyrosine kinase, can mediate a signal exchange between mesenchyme and epithelial during mouse development. J. Cell Biol. 1993, 123, 223–235. [Google Scholar] [CrossRef]
- Igawa, T.; Kanda, S.; Kanetake, H.; Saitoh, Y.; Ichihara, A.; Tomita, Y.; Nakamura, T. Hepatocyte growth factor is a potent mitogen for cultured rabbit renal tubular epithelial cells. Biochem. Biophys. Res. Commun. 1991, 174, 831–838. [Google Scholar] [CrossRef]
- Kawaida, K.; Matsumoto, K.; Shimazu, H.; Nakamura, T. Hepatocyte growth factor prevents acute renal failure and accelerates renal regeneration in mice. Proc. Natl. Acad. Sci. USA 1994, 91, 4357–4361. [Google Scholar] [CrossRef]
- Nagaike, M.; Hirao, S.; Tajima, H.; Noji, S.; Taniguchi, S.; Matsumoto, K.; Nakamura, T. Renotropic functions of hepatocyte growth factor in renal regeneration after unilateral nephrectomy. J. Biol. Chem. 1991, 266, 22781–22784. [Google Scholar]
- Montesano, R.; Matsumoto, K.; Nakamura, T.; Orci, L. Identification of a fibroblast-derived epithelial morphogen as hepatocyte growth factor. Cell 1991, 67, 901–908. [Google Scholar] [CrossRef]
- Bajorin, D.F. Tumors of the kidney, bladder, ureters, and renal pelvis. In Goldman’s Cecil Medicine, 24th ed.; Goldman, L., Schafer, A.I., Eds.; Elsevier Saunders: Philadelphia, PA, USA, 2012; pp. 1303–1308. [Google Scholar]
- Gibney, G.T.; Aziz, S.A.; Camp, R.L.; Conrad, P.; Schwartz, B.E.; Chen, C.R.; Kelly, W.K.; Kluger, H.M. c-Met is a prognostic marker and potential therapeutic target in clear cell renal cell carcinoma. Ann. Oncol. 2013, 24, 343–349. [Google Scholar] [CrossRef]
- Betsunoh, H.; Mukai, S.; Akiyama, Y.; Fukushima, T.; Minamiguchi, N.; Hasui, Y.; Osada, Y.; Kataoka, H. Clinical relevance of hepsin and hepatocyte growth factor activator inhibitor type 2 expression in renal cell carcinoma. Cancer Sci. 2007, 98, 491–498. [Google Scholar] [CrossRef] [Green Version]
- Natali, P.G.; Prat, M.; Nicotra, M.R.; Bigotti, A.; Olivero, M.; Comoglio, P.M.; Di Renzo, M.F. Overexpression of the met/HGF receptor in renal cell carcinomas. Int. J. Cancer 1996, 69, 212–217. [Google Scholar] [CrossRef] [Green Version]
- Miyata, Y.; Kanetake, H.; Kanda, S. Presence of phosphorylated hepatocyte growth factor receptor/c-Met is associated with tumor progression and survival in patients with conventional renal cell carcinoma. Clin. Cancer Res. 2006, 12, 4876–4881. [Google Scholar] [CrossRef]
- Maxwell, P.H.; Wiesener, M.S.; Chang, G.; Clifford, S.C.; Vaux, E.C.; Cockman, M.E.; Wykoff, C.C.; Pugh, C.W.; Maher, E.R.; Ratcliffe, P.J. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999, 399, 271–275. [Google Scholar] [CrossRef]
- Linehan, W.M.; Rubin, J.S.; Bottaro, D.P. VHL loss of function and its impact on oncogenic signaling networks in clear cell renal cell carcinoma. Int. J. Biochem. Cell Biol. 2009, 41, 753–756. [Google Scholar] [CrossRef] [Green Version]
- Matsumura, A.; Kubota, T.; Taiyoh, H.; Fujiwara, H.; Okamoto, K.; Ichikawa, D.; Shiozaki, A.; Komatsu, S.; Nakanishi, M.; Kuriu, Y.; et al. HGF regulates VEGF expression via the c-Met receptor downstream pathways, PI3K/Akt, MAPK and STAT3, in CT26 murine cells. Int. J. Oncol. 2013, 42, 535–542. [Google Scholar] [CrossRef]
- Nakaigawa, N.; Yao, M.; Baba, M.; Kato, S.; Kishida, T.; Hattori, K.; Nagashima, Y.; Kubota, Y. Inactivation of von Hippel-Lindau gene induces constitutive phosphorylation of MET protein in clear cell renal carcinoma. Cancer Res. 2006, 66, 3699–3705. [Google Scholar] [CrossRef]
- Oh, R.R.; Park, J.Y.; Lee, J.H.; Shin, M.S.; Kim, H.S.; Lee, S.K.; Kim, Y.S.; Lee, S.H.; Lee, S.N.; Yang, Y.M.; et al. Expression of HGF/SF and Met protein is associated with genetic alterations of VHL gene in primary renal cell carcinomas. APMIS 2002, 110, 229–238. [Google Scholar] [CrossRef]
- Semenza, G.L. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 2003, 3, 721–732. [Google Scholar] [CrossRef]
- Pisters, L.L.; el-Naggar, A.K.; Luo, W.; Malpica, A.; Lin, S.H. C-met proto-oncogene expression in benign and malignant human renal tissues. J. Urol. 1997, 158, 724–728. [Google Scholar] [CrossRef]
- Fuhrman, S.A.; Lasky, L.C.; Limas, C. Prognostic significance of morphologic parameters in renal cell carcinoma. Am. J. Surg. Pathol. 1982, 6, 655–663. [Google Scholar] [CrossRef]
- Choi, J.S.; Kim, M.K.; Seo, J.W.; Choi, Y.L.; Kim, D.H.; Chun, Y.K.; Ko, Y.H. MET expression in sporadic renal cell carcinomas. J. Korean Med. Sci. 2006, 21, 672–677. [Google Scholar] [CrossRef]
- Ma, P.C.; Tretiakova, M.S.; MacKinnon, A.C.; Ramnath, N.; Johnson, C.; Dietrich, S.; Seiwert, T.; Christensen, J.G.; Jagadeeswaran, R.; Krausz, T.; et al. Expression and mutational analysis of MET in human solid cancers. Genes Chromosomes Cancer 2008, 47, 1025–1037. [Google Scholar] [CrossRef]
- Macher-Goeppinger, S.; Keith, M.; Endris, V.; Penzel, R.; Tagscherer, K.E.; Pahernik, S.; Hohenfellner, M.; Gardner, H.; Grüllich, C.; Schirmacher, P.; et al. MET expression and copy number status in clear-cell renal cell carcinoma: Prognostic value and potential predictive marker. Oncotarget 2016, 8, 1046–1057. [Google Scholar] [CrossRef]
- Kim, J.H.; Kim, B.J.; Kim, H.S. Clinicopathological impacts of high c-Met expression in head and neck squamous cell carcinoma: A meta-analysis and review. Oncotarget 2017, 8, 113120–113128. [Google Scholar] [CrossRef]
- Shuch, B.; Falbo, R.; Parisi, F.; Adeniran, A.; Kluger, Y.; Kluger, H.M.; Jilaveanu, L.B. MET expression in primary and metastatic Clear Cell Renal Cell Carcinoma: Implications of correlative biomarker assessment to MET pathway inhibitors. Biomed Res. Int. 2015, 2015, 192406. [Google Scholar] [CrossRef]
- Pavlovich, C.P.; Walther, M.M.; Eyler, R.A.; Hewitt, S.M.; Zbar, B.; Linehan, W.M.; Merino, M.J. Renal tumors in the Birt-Hogg-Dubé syndrome. Am. J. Surg. Pathol. 2002, 26, 1542–1552. [Google Scholar] [CrossRef]
- Dean, M.; Park, M.; Le Beau, M.M.; Robins, T.S.; Diaz, M.O.; Rowley, J.D.; Blair, D.G.; Vande Woude, G.F. The human met oncogene is related to the tyrosine kinase oncogenes. Nature 1985, 318, 385–388. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, L.; Duh, F.M.; Chen, F.; Kishida, T.; Glenn, G.; Choyke, P.; Scherer, S.W.; Zhuang, Z.; Lubensky, I.; Dean, M.; et al. Germline and somatic mutations in the tyrosine kinase domain of the MET proto-oncogene in papillary renal carcinomas. Nat. Genet. 1997, 16, 68–73. [Google Scholar] [CrossRef]
- Schmidt, L.; Junker, K.; Nakaigawa, N.; Kinjerski, T.; Weirich, G.; Miller, M.; Lubensky, I.; Neumann, H.P.; Brauch, H.; Decker, J.; et al. Novel mutations of the MET proto-oncogene in papillary renal carcinomas. Oncogene 1999, 18, 2343–2350. [Google Scholar] [CrossRef] [Green Version]
- Stransky NCerami, E.; Schalm, S.; Kim, J.L.; Lengauer, C. The landscape of kinase fusions in cancer. Nat. Commun. 2014, 5, 4846. [Google Scholar] [CrossRef] [PubMed]
- Olivero, M.; Valente, G.; Bardelli, A.; Longati, P.; Ferrero, N.; Cracco, C.; Terrone, C.; Rocca-Rossetti, S.; Comoglio, P.M.; Di Renzo, M.F. Novel mutation in the ATP-binding site of the MET oncogene tyrosine kinase in a HPRCC family. Int. J. Cancer 1999, 82, 640–643. [Google Scholar] [CrossRef]
- Marona, P.; Górka, J.; Mazurek, Z.; Wilk, W.; Rys, J.; Majka, M.; Jura, J.; Miekus, K. MCPIP1 downregulation in clear cell renall cell carcinoma promotes vascularization and metastatic progression. Cancer Res. 2017, 77, 4905–4920. [Google Scholar] [PubMed]
- Sweeney, P.; El-Naggar, A.K.; Lin, S.H.; Pisters, L.L. Biological significance of c-met over expression in papillary renal cell carcinoma. J. Urol. 2002, 168, 51–55. [Google Scholar] [CrossRef]
- Gontero, P.; Ceratti, G.; Gugliemetti, S.; Andorno, A.; Terrone, C.; Bonvini, D.; Faggiano, F.; Tizzani, A.; Frea, B.; Valente, G. Prognostic factors in a prospective series of papillary renal cell carcinoma. Bju Int. 2008, 102, 697–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukai, S.; Yorita, K.; Kawagoe, Y.; Katayama, Y.; Nakahara, K.; Kamibeppu, T.; Sugie, S.; Tukino, H.; Kamoto, T.; Kataoka, H. Matriptase and MET are prominently expressed at the site of bone metastasis in renal cell carcinoma: Immunohistochemical analysis. Hum. Cell 2015, 28, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Sierra, J.R.; Cepero, V.; Giordano, S. Molecular mechanisms of acquired resistance to tyrosine kinase targeted therapy. Mol. Cancer 2010, 9, 75. [Google Scholar] [CrossRef] [PubMed]
- Rini, B.I.; Atkins, M.B. Resistance to targeted therapy in renal cell carcinoma. Lancet Oncol. 2009, 10, 992–1000. [Google Scholar] [CrossRef]
- Gottesman, M.M. Mechanisms of cancer drug resistance. Annu. Rev. Med. 2002, 53, 615–627. [Google Scholar] [CrossRef]
- Foo, J.; Michor, F. Evolution of acquired resistance to anti-cancer therapy. J. Theor. Biol. 2014, 355, 10–20. [Google Scholar] [CrossRef] [Green Version]
- Bielecka, Z.F.; Czarnecka, A.M.; Solarek, W.; Kornakiewicz, A.; Szczylik, C. Mechanisms of acquired resistance to tyrosine kinase inhibitors in clear-cell Renal Cell Carcinoma (ccRCC). Curr. Signal Transduct. Ther. 2014, 8, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Hammers, H.J.; Verheul, H.M.; Salumbides, B.; Sharma, R.; Rudek, M.; Jaspers, J.; Shah, P.; Ellis, L.; Shen, L.; Paesante, S.; et al. Reversible epithelial to mesenchymal transition and acquired resistance to sunitinib in patients with renal cell carcinoma: Evidence from a xenograft study. Mol. Cancer Ther. 2010, 9, 1525–1535. [Google Scholar] [CrossRef] [PubMed]
- Hamid, O.; Carvajal, R.D. Anti-programmed death-1 and anti-programmed death-ligand 1 antibodies in cancer therapy. Expert Opin. Biol. Ther. 2013, 13, 847–861. [Google Scholar] [CrossRef] [PubMed]
- Escudier, B.; Pluzanska, A.; Koralewski, P.; Ravaud, A.; Bracarda, S.; Szczylik, C.; Chevreau, C.; Filipek, M.; Melichar, B.; Bajetta, E.; et al. Bevacizumab plus interferon alfa-2a for treatment of metastatic renal cell carcinoma: A randomised, doubleblind phase III trial. Lancet 2007, 370, 2103–2111. [Google Scholar] [CrossRef]
- Escudier, B.; Eisen, T.; Stadler, W.M.; Szczylik, C.; Oudard, S.; Siebels, M.; Negrier, S.; Chevreau, C.; Solska, E.; Desai, A.A.; et al. Sorafenib in advanced clear-cell renal-cell carcinoma. N. Engl. J. Med. 2007, 356, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Hutson, T.E.; Tomczak, P.; Michaelson, M.D.; Bukowski, R.M.; Rixe, O.; Oudard, S.; Negrier, S.; Szczylik, C.; Kim, S.T.; et al. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N. Engl. J. Med. 2007, 356, 115–124. [Google Scholar] [CrossRef]
- Sternberg, C.N.; Davis, I.D.; Mardiak, J.; Szczylik, C.; Lee, E.; Wagstaff, J.; Barrios, C.H.; Salman, P.; Gladkov, O.A.; Kavina, A.; et al. Pazopanib in locally advanced or metastatic renal cell carcinoma: Results of a randomized phase III trial. J. Clin. Oncol. 2010, 28, 1061–1068. [Google Scholar] [CrossRef]
- Rini, B.I.; Escudier, B.; Tomczak, P.; Kaprin, A.; Szczylik, C.; Hutson, T.E.; Michaelson, M.D.; Gorbunova, V.A.; Gore, M.E.; Rusakov, I.G.; et al. Comparative effectiveness of axitinib versus sorafenib in advanced renal cell carcinoma (AXIS): A randomised phase 3 trial. Lancet 2011, 378, 1931–1939. [Google Scholar] [CrossRef]
- Motzer, R.J.; Hutson, T.E.; Glen, H.; Michaelson, M.D.; Molina, A.; Eisen, T.; Jassem, J.; Zolnierek, J.; Maroto, J.P.; Mellado, B.; et al. Lenvatinib, everolimus, and the combination in patients with metastatic renal cell carcinoma: A randomised, phase 2, openlabel, multicentre trial. Lancet Oncol. 2015, 16, 1473–1482. [Google Scholar] [CrossRef]
- Bhargava, P.; Robinson, M.O. Development of second-generation VEGFR tyrosine kinase inhibitors: Current status. Curr. Oncol. Rep. 2011, 13, 103–111. [Google Scholar] [CrossRef]
- Duran, I.; Lambea, J.; Maroto, P.; González-Larriba, L.; Flores, L.; Granados-Principal, S.; Graupera, M.; Sáez, B.; Vivancos, A.; Casanovas, O. Resistance to targeted therapies in renal cancer: The importance of changing the mechanism of action. Target Oncol. 2017, 12, 19–35. [Google Scholar] [CrossRef] [PubMed]
- Adelaiye, R.; Ciamporcero, E.; Miles, K.M.; Sotomayor, P.; Bard, J.; Tsompana, M.; Conroy, D.; Shen, L.; Ramakrishnan, S.; Ku, S.Y.; et al. Sunitinib dose escalation overcomes transient resistance in clear cell renal cell carcinoma and is associated with epigenetic modifications. Mol. Cancer Ther. 2015, 14, 513–522. [Google Scholar] [CrossRef]
- Shojaei, F.; Lee, J.H.; Simmons, B.H.; Wong, A.; Esparza, C.O.; Plumlee, P.A.; Feng, J.; Stewart, A.E.; Hu-Lowe, D.D.; Christensen, J.G. HGF/c-Met acts as an alternative angiogenic pathway in sunitinib-resistant tumors. Cancer Res. 2010, 70, 10090–10100. [Google Scholar] [CrossRef] [PubMed]
- Sennino, B.; Ishiguro-Oonuma, T.; Wei, Y.; Naylor, R.; Williamson, C.; Bhagwandin, V.; Tabruyn, S.P.; You, W.K.; Chapman, H.A.; Christensen, J.G.; et al. Suppression of tumor invasion and metastasis by concurrent inhibition of c-Met and VEGF signaling in pancreatic neuroendocrine tumors. Cancer Discov. 2012, 2, 270–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, L.; Liu, X.; Sun, M.; Zhang, X.; German, P.; Bai, S.; Ding, Z.; Tannir, N.; Wood, C.G.; Matin, S.F.; et al. Targeting MET and AXL overcomes resistance to sunitinib therapy in renal cell carcinoma. Oncogene 2016, 35, 2687–2697. [Google Scholar] [CrossRef]
- Folberg, R.; Hendrix, M.J.; Maniotis, A.J. Vasculogenic mimicry and tumor angiogenesis. Am. J. Pathol. 2000, 156, 361–381. [Google Scholar] [CrossRef]
- Maniotis, A.J.; Folberg, R.; Hess, A.; Seftor, E.A.; Gardner, L.M.G.; Pe’er, J.; Trent, J.M.; Meltzer, P.S.; Hendrix, M.J. Vascular channel formation by human melanoma cells in vivo and in vitro: Vasculogenic mimicry. Am. J. Pathol. 1999, 155, 739–752. [Google Scholar] [CrossRef]
- Huang, D.; Ding, Y.; Zhou, M.; Rini, B.; Petillo, D.; Qian, C.; Kahnoski, R.; Futreal, P.A.; Furge, K.A.; Teh, B.T. Interleukin-8 mediates resistance to antiangiogenic agent sunitinib in renal cell carcinoma. Cancer Res. 2010, 70, 1063–1071. [Google Scholar] [CrossRef]
- Ciamporcero, E.; Miles, K.; Adelaiye, R.; Ramakrishanan, S.; Shen, L.; Ku, S.; Pizzimenti, S.; Sennino, B.; Barrera, G.; Pili, R. Combination strategy targeting VEGF and HGF/c-Met in human renal cell carcinoma models. Mol. Cancer Ther. 2015, 14, 101–110. [Google Scholar] [CrossRef]
- Rankin, E.B.; Fuh, K.C.; Castellini, L.; Viswanathan, K.; Finger, E.C.; Diep, A.N.; LaGory, E.L.; Kariolis, M.S.; Chan, A.; Lindgren, D.; et al. Direct regulation of GAS6/AXL signaling by HIF promotes renal metastasis through SRC and MET. Proc. Natl. Acad. Sci. USA 2014, 111, 13373–13378. [Google Scholar] [CrossRef] [Green Version]
- Peltola, K.; Penttilä, P.; Rautiola, J.; Joensuu, H.; Hänninen, E.; Ristimäki, A.; Bono, P. Correlation of c-Met expression and outcome in patients with renal cell carcinoma (RCC) treated with sunitinib. Clin. Genitourin. Cancer 2017, 15, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Lacy, S.A.; Miles, D.R.; Nguyen, L.T. Clinical pharmacokinetics and pharmacodynamics of cabozantinib. Clin. Pharmacokinet. 2017, 56, 477–491. [Google Scholar] [CrossRef] [PubMed]
- FDA. Available online: https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm497483.htm (accessed on 8 January 2019).
- Choueiri, T.K.; Pal, S.K.; McDermott, D.F.; Morrissey s Ferguson, K.C.; Holland, J.; Kaelin, W.G.; Dutcher, J.P. A phase I study of cabozantinib (XL184) in patients with renal cell cancer. Ann. Oncol. 2014, 25, 1603–1608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choueiri, T.K.; Halabi, S.; Sanford, B.L.; Hahn, O.; Michaelson, M.D.; Walsh, M.K.; Feldman, D.R.; Olencki, T.; Picus, J.; Small, E.J.; et al. Cabozantinib versus sunitinib as initial targeted therapy for patients with metastatic renal cell carcinoma of poor or intermediate risk: The Alliance A031203 CABOSUN Trial. J. Clin. Oncol. 2017, 35, 591–597. [Google Scholar] [CrossRef]
- Choueiri, T.K.; Escudier, B.; Powles, T.; Tannir, N.M.; Mainwaring, P.N.; Rini, B.I.; Donskov, F.; Hammers, H.; Hutson, T.E.; Lee, J.L.; et al. Cabozantinib versus everolimus in advanced renal cell carcinoma (METEOR): Final results from a randomised, open-label, phase 3 trial. Lancet Oncol. 2016, 17, 917–927. [Google Scholar] [CrossRef]
- Choueiri, T.K.; Vaishampayan, U.; Rosenberg JELogan, T.F.; Harzstark, A.L.; Bukowski, R.M.; Rini, B.I.; Srinivas, S.; Stein, M.N.; Adams, L.M.; Ottesen, L.H.; et al. Phase II and biomarker study of the dual MET/VEGFR2 inhibitor foretinib in patients with papillary renal cell carcinoma. J. Clin. Oncol. 2013, 31, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Shook, D.; Keller, R. Mechanisms, mechanics and function of epithelial-mesenchymal transitions in early development. Mech. Dev. 2003, 120, 1351–1383. [Google Scholar] [CrossRef]
- Thiery, J.P.; Sleeman, J.P. Complex networks orchestrate epithelial-mesenchymal transitions. Nat. Rev. Mol. Cell Biol. 2006, 7, 131–142. [Google Scholar] [CrossRef]
- Lee, M.Y.; Chou, C.Y.; Tang, M.J.; Shen, M.R. Epithelial-mesenchymal transition in cervical cancer: Correlation with tumor progression, epidermal growth factor receptor overexpression, and snail up-regulation. Clin. Cancer Res. 2008, 14, 4743–4750. [Google Scholar] [CrossRef]
- Polyak, K.; Weinberg, R.A. Transitions between epithelial and mesenchymal states: Acquisition of malignant and stem cell traits. Nat. Rev. Cancer 2009, 9, 265–273. [Google Scholar] [CrossRef]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Lewis, M.T.; Huang, J.; Gutierrez, C.; Osborne, C.K.; Wu, M.F.; Hilsenbeck, S.G.; Pavlick, A.; Zhang, X.; Chamness, G.C.; et al. Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J. Natl. Cancer Inst. 2008, 100, 672–679. [Google Scholar] [CrossRef] [PubMed]
- Miekus, K. The Met tyrosine kinase receptor as a therapeutic target and a potential cancer stem cell factor responsible for therapy resistance (Review). Oncol. Rep. 2017, 37, 647–656. [Google Scholar] [CrossRef] [PubMed]
- Taulli, R.; Scuoppo, C.; Bersani, F.; Accornero, P.; Forni, P.E.; Miretti, S.; Grinza, A.; Allegra, P.; Schmitt-Ney, M.; Crepaldi, T.; et al. Validation of met as a therapeutic target in alveolar and embryonal rhabdomyosarcoma. Cancer Res. 2006, 66, 4742–4749. [Google Scholar] [CrossRef] [PubMed]
- Jankowski, K.; Kucia, M.; Wysoczynski, M.; Reca, R.; Zhao, D.; Trzyna, E.; Trent, J.; Peiper, S.; Zembala, M.; Ratajczak, J.; et al. Both hepatocyte growth factor (HGF) and stromal-derived factor-1 regulate the metastatic behavior of human rhabdomyosarcoma cells, but only HGF enhances their resistance to radiochemotherapy. Cancer Res. 2003, 63, 7926–7935. [Google Scholar] [PubMed]
- Skrzypek, K.; Kusienicka, A.; Szewczyk, B.; Adamus, T.; Lukasiewicz, E.; Miekus, K.; Majka, M. Constitutive activation of MET signaling impairs myogenic differentiation of rhabdomyosarcoma and promotes its development and progression. Oncotarget 2015, 6, 31378–31398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miekus, K.; Lukasiewicz, E.; Jarocha, D.; Sekula, M.; Drabik, G.; Majka, M. The decreased metastatic potential of rhabdomyosarcoma cells obtained through MET receptor downregulation and the induction of differentiation. Cell Death Dis. 2013, 4, e459. [Google Scholar] [CrossRef]
- Charytonowicz, E.; Cordon-Cardo, C.; Matushansky, I.; Ziman, M. Alveolar rhabdomyosarcoma: Is the cell of origin a mesenchymal stem cell? Cancer Lett. 2009, 279, 126–136. [Google Scholar] [CrossRef]
- Tatsumi, R.; Anderson, J.E.; Nevoret, C.J.; Halevy, O.; Allen, R.E. HGF/SF is present in normal adult skeletal muscle and is capable of activating satellite cells. Dev. Biol. 1998, 194, 114–128. [Google Scholar] [CrossRef]
- Miekus, K.; Pawlowska, M.; Sekuła, M.; Drabik, G.; Madeja, Z.; Adamek, D.; Majka, M. MET receptor is a potential therapeutic target in high grade cervical cancer. Oncotarget 2015, 6, 10086–10101. [Google Scholar] [CrossRef] [Green Version]
- Bolós, V.; Peinado, H.; Pérez-Moreno, M.A.; Fraga, M.F.; Esteller, M.; Cano, A. The transcription factor Slug represses E-cadherin expression and induces epithelial to mesenchymal transitions: A comparison with Snail and E47 repressors. J. Cell Sci. 2003, 116, 499–511. [Google Scholar] [CrossRef]
- Witta, S.E.; Gemmill, R.M.; Hirsch, F.R.; Coldren, C.D.; Hedman, K.; Ravdel, L.; Helfrich, B.; Dziadziuszko, R.; Chan, D.C.; Sugita, M.; et al. Restoring E-cadherin expression increases sensitivity to epi- dermal growth factor receptor inhibitors in lung cancer cell lines. Cancer Res. 2006, 66, 944–950. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Study (Year) | Type | c-Met Detection Method | Expression c-Met (Number Positive/Number Tested) | c-Met Expression in Percent | Major Conclusion |
|---|---|---|---|---|---|
| Natali et al. (1996) [23] | RCC | Immunohist-ochemical (IHC) | 39/45 | 87% | - increased expression at various levels in kidney cells of tumors with different histological and cytological properties; - c-Met may be involved in the formation and progression of renal cancer cells; |
| Pisters et al. (1997) [31] | Papillary cell | IHC | 3/3 | 68% | - high expression of c-Met in renal cell carcinomas; - correlation between the higher level of c-Met and higher nuclear grade renal cancers; |
| Mixed clear and granular cell | 24/37 | ||||
| Rhabdoid/sarcomatoid | 1/1 | ||||
| Oncocytoma | IHC | 8/8 | 100% | ||
| Sweeney et al. (2002) [45] | Papillary renal carcinoma | IHC | 40/50 | 80% | 20% of papillary cancers did not express c-Met—another possible mechanism responsible for tumorigenesis; - correlation of the growing stage of cancer with c-Met expression; - a tendency for higher overall survival in patients with c-Met -negative tumors; |
| Jong Sun Choi et al. (2006) [33] | Conventional renal carcinoma | IHC | 43/96 | 45% | - significant relationship between c-Met expression and high nuclear level, as well as several clinical–pathological parameters, which indicates tumor invasion or aggressiveness; - the ability to distinguish between RCC subtypes by diffuse and strong immunoexpression of c-Met in RCC and collecting duct carcinomas as compared to subtypes with tubulo-papillary growth; |
| Papillary renal carcinoma | 18/20 | 90% | |||
| Chromophobe renal carcinoma | 2/24 | 8% | |||
| Collecting duct carcinoma | 5/5 | 100% | |||
| Urothelial carcinoma of renal pelvis | 23/25 | 92% | |||
| Oncocytoma | 0/12 | 0% | |||
| Miyata et al. (2006) [24] | RCC | IHC | 73/114 | 64% | - detection of pY1349 c-Met expression is an excellent predictor for the prognosis of patients with sporadic conventional RCC because it is positively associated with tumor grade, stage and size as well as cancer cell proliferation in patients; |
| Gontero et al. (2008) [46] | Papillary renal carcinoma | IHC | 13/46 | 29% | - positive c-Met expression was significantly more common in papillary RCC (PRCC) type 2 (p < 0.001), however lack of correlation with multifocal kidney disease (p = 0.86) and occurrence of metachronous tumors (p = 0.93); - c-Met expression had no prognostic value; |
| Mukai et al. (2015) [47] | RCC | IHC | 8/17 | 47% | - high c-Met and matriptase expression was found in RCC cells that had metastasized to bone and was accompanied by matriptase expression in osteoclasts, which indicates a significant role for these molecules in bone metastasis; |
| Macher-Goeppinger et al. (2017) [35] | ccRCC | IHC and CISH analyses | 476/572 | 83% | - high expression of c-Met and MET copy number gains was associated with clinical and pathological features in the primary tumor, an aggressive phenotype and an unfavorable patient outcome; - c-Met expression and MET copy number should be used in relapses or metastases to target anti c-Met therapy in patients with ccRCC; |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marona, P.; Górka, J.; Kotlinowski, J.; Majka, M.; Jura, J.; Miekus, K. C-Met as a Key Factor Responsible for Sustaining Undifferentiated Phenotype and Therapy Resistance in Renal Carcinomas. Cells 2019, 8, 272. https://doi.org/10.3390/cells8030272
Marona P, Górka J, Kotlinowski J, Majka M, Jura J, Miekus K. C-Met as a Key Factor Responsible for Sustaining Undifferentiated Phenotype and Therapy Resistance in Renal Carcinomas. Cells. 2019; 8(3):272. https://doi.org/10.3390/cells8030272
Chicago/Turabian StyleMarona, Paulina, Judyta Górka, Jerzy Kotlinowski, Marcin Majka, Jolanta Jura, and Katarzyna Miekus. 2019. "C-Met as a Key Factor Responsible for Sustaining Undifferentiated Phenotype and Therapy Resistance in Renal Carcinomas" Cells 8, no. 3: 272. https://doi.org/10.3390/cells8030272