Discovery of Small Molecules That Target Vascular Endothelial Growth Factor Receptor-2 Signalling Pathway Employing Molecular Modelling Studies


 ,
,  ,
,
Abstract
:
1. Introduction
2. Results
2.1. Structure-Based Pharmacophore Generation
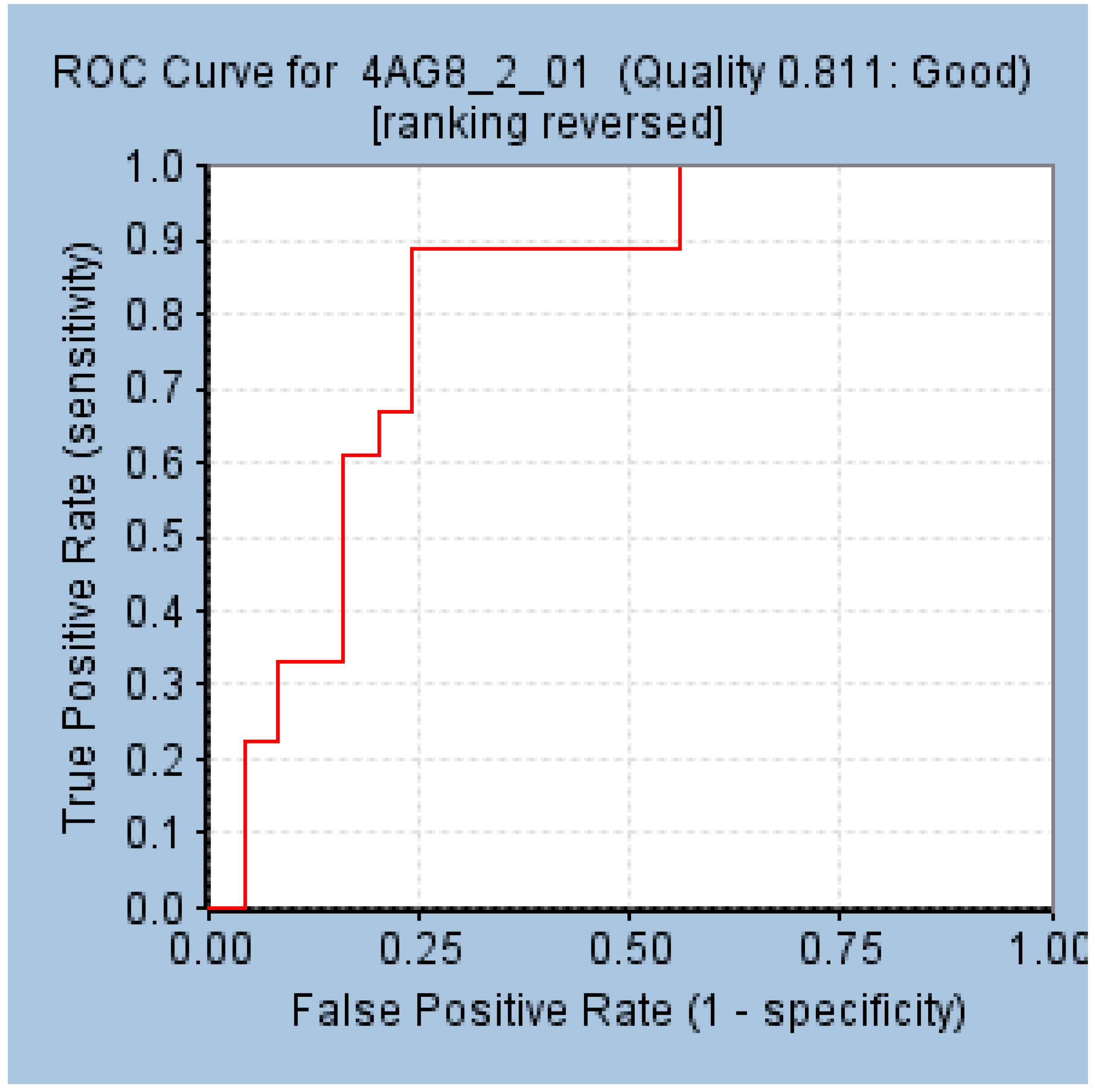
2.2. Validation of the Selected Pharmacophore Model
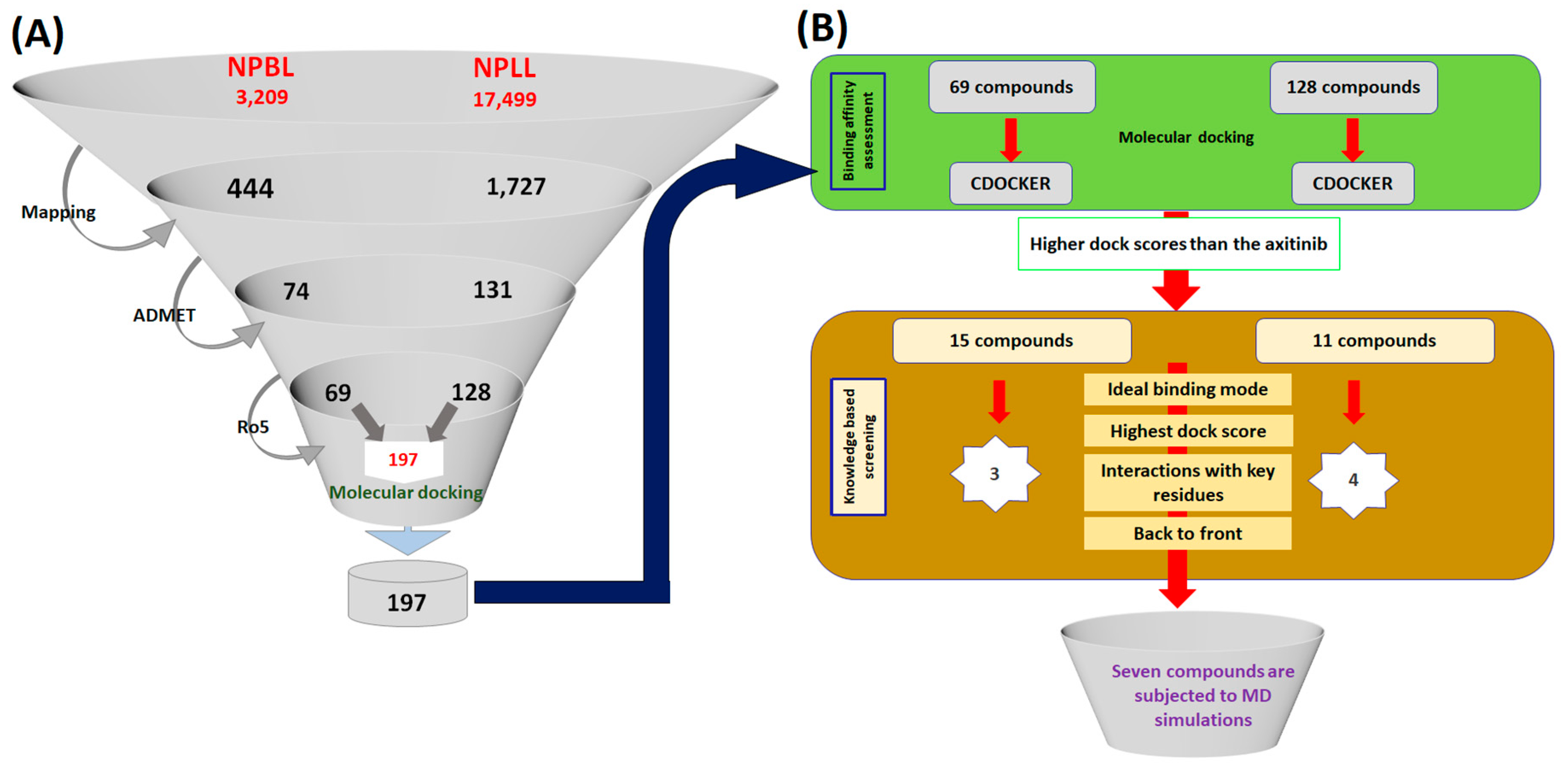
2.3. Virtual Screening for the Retrieval of Drug-Like Compounds
2.4. Molecular Docking-Based Screening
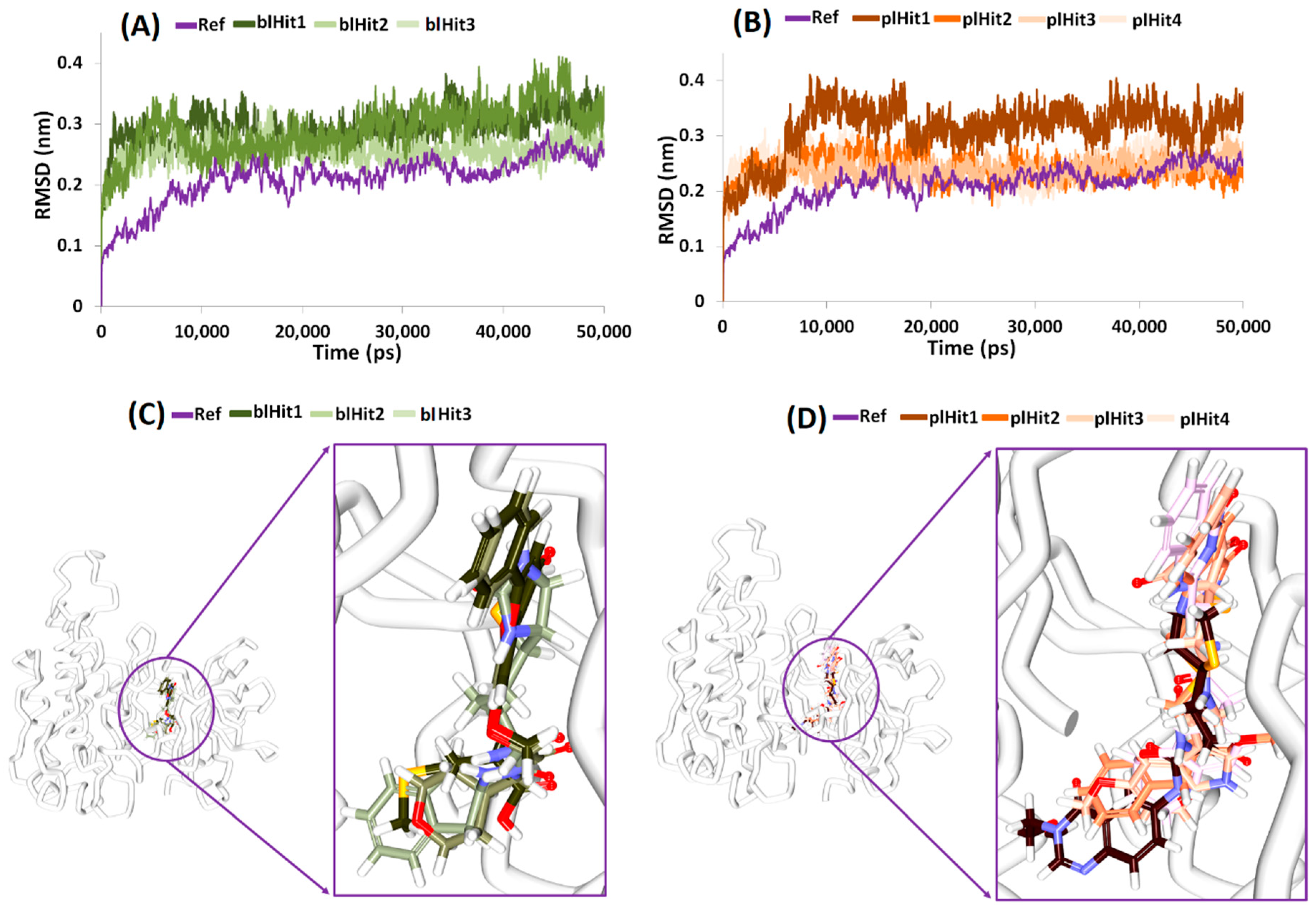
2.5. Molecular Dynamics Simulations Guided Binding Mode Analysis
2.6. Elucidation of Protein Stability and Binding Mode Analysis
2.7. Deciphering on Intermolecular Interactions
2.7.1. Protein-Reference
2.7.2. Protein-blHit1
2.7.3. Protein-blHit2
2.7.4. Protein-blHit3
2.7.5. Protein-plHit1
2.7.6. Protein-plHit2
2.7.7. Protein-plHit3
2.7.8. Protein-plHit4
3. Materials and Methods
3.1. Structure-Based Pharmacophore Generation
3.2. Pharmacophore Validation
3.3. Virtual Screening for the Retrieval of Drug-Like Compounds
3.4. Molecular Docking-Based Screening
3.5. Molecular Dynamics Simulation Studies
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gotink, K.J.; Verheul, H.M.W. Anti-angiogenic tyrosine kinase inhibitors: What is their mechanism of action? Angiogenesis 2010, 13, 1–14. [Google Scholar] [CrossRef]
- Yehya, A.H.S.; Asif, M.; Petersen, S.H.; Subramaniam, A.V.; Kono, K.; Majid, A.M.S.A.; Oon, C.E. Angiogenesis: Managing the culprits behind tumorigenesis and metastasis. Medicina 2018, 54, 8. [Google Scholar] [CrossRef]
- Bielenberg, D.R.; Zetter, B.R. The Contribution of Angiogenesis to the Process of Metastasis. Cancer J. 2015, 21, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Zetter, B.R. Angiogenesis and Tumor Metastasis. Annu. Rev. Med. 1998, 49, 407–424. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J. Tumor Angiogenesis: Therapeutic Implications. N. Engl. J. Med. 1971, 285, 1182–1186. [Google Scholar] [CrossRef]
- Kerbel, R.S. Tumor angiogenesis. N. Engl. J. Med. 2008, 358, 2039–2049. [Google Scholar] [CrossRef]
- Ishigami, S.I.; Arii, S.; Furutani, M.; Niwano, M.; Harada, T.; Mizumoto, M.; Mori, A.; Onodera, H.; Imamura, M. Predictive value of vascular endothelial growth factor (VEGF) in metastasis and prognosis of human colorectal cancer. Br. J. Cancer 1998, 78, 1379–1384. [Google Scholar] [CrossRef]
- Roberts, E.; Cossigny, D.A.F.; Quan, G.M.Y. The role of vascular endothelial growth factor in metastatic prostate cancer to the skeleton. Prostate Cancer 2013, 2013, 418340. [Google Scholar] [CrossRef]
- Holmes, D.I.R.; Zachary, I. The vascular endothelial growth factor (VEGF) family: Angiogenic factors in health and disease. Genome Biol. 2005, 6, 209. [Google Scholar] [CrossRef]
- Huang, L.; Huang, Z.; Bai, Z.; Xie, R.; Sun, L.; Lin, K. Development and strategies of VEGFR-2/KDR inhibitors. Future Med. Chem. 2012, 4, 1839–1852. [Google Scholar] [CrossRef] [PubMed]
- Hicklin, D.J.; Ellis, L.M. Role of the vascular endothelial growth factor pathway in tumor growth and angiogenesis. J. Clin. Oncol. 2005, 23, 1011–1027. [Google Scholar] [CrossRef]
- Meng, F. Molecular Dynamics Simulation of VEGFR2 with Sorafenib and Other Urea-Substituted Aryloxy Compounds. J. Theor. Chem. 2013, 2013, 739574. [Google Scholar] [CrossRef]
- Holmes, K.; Roberts, O.L.; Thomas, A.M.; Cross, M.J. Vascular endothelial growth factor receptor-2: Structure, function, intracellular signalling and therapeutic inhibition. Cell. Signal. 2007, 19, 2003–2012. [Google Scholar] [CrossRef]
- Aziz, M.A.; Serya, R.A.T.; Lasheen, D.S.; Abdel-Aziz, A.K.; Esmat, A.; Mansour, A.M.; Singab, A.N.B.; Abouzid, K.A.M. Discovery of Potent VEGFR-2 Inhibitors based on Furopyrimidine and Thienopyrimidne Scaffolds as Cancer Targeting Agents. Sci. Rep. 2016, 6, 24460. [Google Scholar] [CrossRef]
- Sanphanya, K.; Wattanapitayakul, S.K.; Phowichit, S.; Fokin, V.V.; Vajragupta, O. Novel VEGFR-2 kinase inhibitors identified by the back-to-front approach. Bioorg. Med. Chem. Lett. 2013, 23, 2962–2967. [Google Scholar] [CrossRef]
- Regan, J.; Breitfelder, S.; Cirillo, P.; Gilmore, T.; Graham, A.G.; Hickey, E.; Klaus, B.; Madwed, J.; Moriak, M.; Moss, N.; et al. Pyrazole Urea-Based Inhibitors of p38 MAP Kinase: From Lead Compound to Clinical Candidate. J. Med. Chem. 2002, 45, 2994–3008. [Google Scholar] [CrossRef]
- Baldwin, I.; Bamborough, P.; Haslam, C.G.; Hunjan, S.S.; Longstaff, T.; Mooney, C.J.; Patel, S.; Quinn, J.; Somers, D.O. Kinase array design, back to front: Biaryl amides. Bioorg. Med. Chem. Lett. 2008, 18, 5285–5289. [Google Scholar] [CrossRef]
- Iwata, H.; Oki, H.; Okada, K.; Takagi, T.; Tawada, M.; Miyazaki, Y.; Imamura, S.; Hori, A.; Lawson, J.D.; Hixon, M.S.; et al. A back-to-front fragment-based drug design search strategy targeting the DFG-out pocket of protein tyrosine kinases. ACS Med. Chem. Lett. 2012, 3, 342–346. [Google Scholar] [CrossRef]
- Al-Abd, A.M.; Abdulmohsin, J.A.; Ashraf, B.A.; Thikryat, A.N.; Osama, M.A. Anti-angiogenic agents for the treatment of solid tumors: Potential pathways, therapy and current strategies—A review. J. Adv. Res. 2017, 8, 591–605. [Google Scholar] [CrossRef]
- Cao, Y. Positive and negative modulation of angiogenesis by VEGFR1 ligands. Sci. Signal. 2009, 2, re1. [Google Scholar] [CrossRef]
- Verheul, H.M.W.; Pinedo, H.M. Possible molecular mechanisms involved in the toxicity of angiogenesis inhibition. Nat. Rev. Cancer 2007, 7, 475–485. [Google Scholar] [CrossRef] [PubMed]
- Bao, J.; Zhou, N.; Luo, K.; Zhang, W.; Li, X.; Wu, C.; Bao, J. In silico discovery of potential VEGFR-2 inhibitors from natural derivatives for anti-angiogenesis therapy. Int. J. Mol. Sci. 2014, 15, 15994–16001. [Google Scholar] [CrossRef]
- Harvey, A.L. Natural products in drug discovery. Drug Discov. Today 2008, 13, 894–901. [Google Scholar] [CrossRef]
- Cragg, G.M.; Newman, D.J. Natural products: A continuing source of novel drug leads. Biochim. Biophys. Acta Gen. Subj. 2013, 1830, 3670–3695. [Google Scholar] [CrossRef] [PubMed]
- Clardy, J.; Walsh, C. Lessons from natural molecules. Nature 2004, 432, 829–837. [Google Scholar] [CrossRef] [PubMed]
- Hong, J. Role of natural product diversity in chemical biology. Curr. Opin. Chem. Biol. 2011, 15, 350–354. [Google Scholar] [CrossRef]
- Kinghorn, A.D.; Chin, Y.-W.; Swanson, S.M. Discovery of natural product anticancer agents from biodiverse organisms. Curr. Opin. Drug Discov. Dev. 2009, 12, 189–196. [Google Scholar]
- Wang, J.; Jiang, Y.-F. Natural compounds as anticancer agents: Experimental evidence. World J. Exp. Med. 2012, 2, 45–57. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Sharma, B.; Kanwar, S.S.; Kumar, A. Lead Phytochemicals for Anticancer Drug Development. Front. Plant Sci. 2016, 7, 1667. [Google Scholar] [CrossRef]
- Kumar, R.; Deep, G.; Wempe, M.F.; Agarwal, R.; Agarwal, C. Procyanidin B2 3,3″-di-O-gallate Inhibits Endothelial Cells Growth and Motility by Targeting VEGFR2 and Integrin Signaling Pathways. Curr. Cancer Drug Targets 2015, 15, 14–26. [Google Scholar] [CrossRef]
- Jansen, R.J.; Robinson, D.P.; Stolzenberg-Solomon, R.Z.; Bamlet, W.R.; De Andrade, M.; Oberg, A.L.; Hammer, T.J.; Rabe, K.G.; Anderson, K.E.; Olson, J.E.; et al. Fruit and vegetable consumption is inversely associated with having pancreatic cancer. Cancer Causes Control 2011, 22, 1613–1625. [Google Scholar] [CrossRef]
- Detchokul, S.; Frauman, A.G. Recent developments in prostate cancer biomarker research: Therapeutic implications. Br. J. Clin. Pharmacol. 2011, 71, 157–174. [Google Scholar] [CrossRef] [PubMed]
- Gacche, R.N.; Meshram, R.J. Angiogenic factors as potential drug target: Efficacy and limitations of anti-angiogenic therapy. Biochim. Biophys. Acta Rev. Cancer 2014, 1846, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.J.; Koh, W.; Lee, E.O.; Lee, H.J.; Lee, H.J.; Bae, H.; Lü, J.; Kim, S.H. Antiangiogenic phytochemicals and medicinal herbs. Phyther. Res. 2011, 25, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Mirossay, L.; Varinská, L.; Mojžiš, J. Antiangiogenic effect of flavonoids and chalcones: An update. Int. J. Mol. Sci. 2018, 19, 27. [Google Scholar] [CrossRef]
- Rampogu, S.; Baek, A.; Zeb, A.; Lee, K.W. Exploration for novel inhibitors showing back-to-front approach against VEGFR-2 kinase domain (4AG8) employing molecular docking mechanism and molecular dynamics simulations. BMC Cancer 2018, 18, 264. [Google Scholar] [CrossRef]
- Rampogu, S.; Son, M.; Baek, A.; Park, C.; Rana, R.M.; Zeb, A.; Parameswaran, S.; Lee, K.W. Targeting natural compounds against HER2 kinase domain as potential anticancer drugs applying pharmacophore based molecular modelling approaches. Comput. Biol. Chem. 2018, 74, 327–338. [Google Scholar] [CrossRef]
- Zhu, X.; Lopes, P.E.M.; Mackerell, A.D. Recent developments and applications of the CHARMM force fields. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 167–185. [Google Scholar] [CrossRef]
- Zoete, V.; Cuendet, M.A.; Grosdidier, A.; Michielin, O. SwissParam: A fast force field generation tool for smallorganic molecules. J. Comput. Chem. 2011, 32, 2359–2368. [Google Scholar] [CrossRef] [PubMed]
- Rampogu, S.; Baek, A.; Son, M.; Zeb, A.; Park, C.; Kumar, R.; Lee, G.; Kim, D.; Choi, Y.; Cho, Y.; et al. Computational Exploration for Lead Compounds That Can Reverse the Nuclear Morphology in Progeria. BioMed Res. Int. 2017, 2017, 5270940. [Google Scholar] [CrossRef]
- Parrinello, M. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1998. [Google Scholar] [CrossRef]
- Miyamoto, S.; Kollman, P.A. Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Longatto Filho, A.; Lopes, J.M.; Schmitt, F.C. Angiogenesis and breast cancer. J. Oncol. 2010, 2010, 576384. [Google Scholar] [CrossRef]
- Wang, Z.; Dabrosin, C.; Yin, X.; Fuster, M.M.; Arreola, A.; Rathmell, W.K.; Generali, D.; Nagaraju, G.P.; El-Rayes, B.; Ribatti, D.; et al. Broad targeting of angiogenesis for cancer prevention and therapy. Semin. Cancer Biol. 2015, 35, S224–S243. [Google Scholar] [CrossRef]
- Ghorab, M.M.; Alsaid, M.S.; Soliman, A.M.; Ragab, F.A. VEGFR-2 inhibitors and apoptosis inducers: Synthesis and molecular design of new benzo[g]quinazolin bearing benzenesulfonamide moiety. J. Enzyme Inhib. Med. Chem. 2017, 32, 893–907. [Google Scholar] [CrossRef]
- Wu, P.; Nielsen, T.E.; Clausen, M.H. Small-molecule kinase inhibitors: An analysis of FDA-approved drugs. Drug Discov. Today 2016, 21, 5–10. [Google Scholar] [CrossRef]
- Noble, M.E.M.; Endicott, J.A.; Johnson, L.N. Protein Kinase Inhibitors: Insights into Drug Design from Structure. Science 2004, 303, 1800–1805. [Google Scholar] [CrossRef]
- Okamoto, K.; Ikemori-Kawada, M.; Jestel, A.; Von König, K.; Funahashi, Y.; Matsushima, T.; Tsuruoka, A.; Inoue, A.; Matsui, J. Distinct binding mode of multikinase inhibitor lenvatinib revealed by biochemical characterization. ACS Med. Chem. Lett. 2015, 6, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Eldehna, W.M.; Abou-Seri, S.M.; El Kerdawy, A.M.; Ayyad, R.R.; Hamdy, A.M.; Ghabbour, H.A.; Ali, M.M.; Abou El Ella, D.A. Increasing the binding affinity of VEGFR-2 inhibitors by extending their hydrophobic interaction with the active site: Design, synthesis and biological evaluation of 1-substituted-4-(4-methoxybenzyl)phthalazine derivatives. Eur. J. Med. Chem. 2016, 113, 50–62. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, C.; Adasme, F.; Alzate-Morales, J.H.; Vergara-Jaque, A.; Kniess, T.; Caballero, J. Study of differences in the VEGFR2 inhibitory activities between semaxanib and SU5205 using 3D-QSAR, docking, and molecular dynamics simulations. J. Mol. Graph. Model. 2012, 32, 39–48. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model No | Number of Features | Feature Set * | Selectivity |
|---|---|---|---|
| Model 1 | 5 | HBD, HBD, HyP, HyP, HBA | 9.84 |
| Model 2 | 5 | HBD, HyP, HyP, HBA, HBA | 8.93 |
| Model 3 | 5 | HBD, HyP, HyP, HyP, HBA | 8.93 |
| Model 4 | 4 | HBD, HBD, HyP, HBA | 8.33 |
| Model 5 | 4 | HBD, HBD, HyP, HBA | 8.33 |
| Model 6 | 4 | HBD, HBD, HyP, HyP | 8.33 |
| Model 7 | 5 | HBA, HBA, HyP, HyP, HyP | 8.01 |
| Model 8 | 4 | HBD, HBA, HyP, HyP | 7.41 |
| Model 9 | 4 | HBD, HBA, HyP, HyP | 7.41 |
| Model 10 | 4 | HBD, HBA, HyP, HyP | 7.41 |
| S. No | Parameters | Values |
|---|---|---|
| 1 | Total number of molecules in database (D) | 720 |
| 2 | Total number of actives in database (A) | 24 |
| 3 | Total number of hit molecules from the database (Ht) | 26 |
| 4 | Total number of active molecules in hit list (Ha) | 23 |
| 5 | % Yield of actives (Ha/Ht) | 88 |
| 6 | % Ratio of actives [(Ha/A) × 100] | 95.8 |
| 7 | Enrichment factor (EF) | 26.53 |
| 8 | False negatives (A-Ha) | 1 |
| 9 | False positives (Ht-Ha) | 3 |
| 10 | Goodness of fit score (GF) | 0.87 |
| Compound Name | Hydrogen Bond Interactions < 3 Å | π–π/π–alkyl Interactions | van der Waals Interactions |
|---|---|---|---|
| Reference | Glu885:OE2-N82 (2.6) Glu917:O-N15 (2.8) Cys919:N-N14 (2.9) Asp1046:N-O81 (2.9) | Leu840, Val848, Ala866, Lys868, Cys1045, Phe1047 | Val867, Leu889, Val899, Val914, Phe918, Lys920, Gly922 |
| blHit1 | Glu885:OE2-H40 (2.1) Glu917:O-H37 (1.9) Cys919:HN-O15 (1.9) Asp1046:O-H35 (2.0) | Leu840, Val848, Ala866, Leu1035, Cys1045 | Ile888, Leu889, Ile892, Val899, Val914, Val916, Lys920, Gly922, Asn923, Thr926, His1026, Ile1044, Phe1047, Ala1050 |
| blHit2 | Glu917:O-H35 (1.9) Cys919:HN-O12 (1.8) Asp1046:O-H34 (1.9) | Leu840,Val848, Ala866, Leu889, Leu1035, Cys1045 | Lys868, Glu885, Ile888, Ile892, Val899, Phe918, Lys920, Gly922, Val914, Asn923, Thr926, Ile1044, Ile1045, Phe1047 |
| blHit3 | Lys868:HZ3-O17 (1.9) Asp1046:O-H34 (2.1) Asn923:HN-O14 (2.8) Cys919:O-H27 (2.1) | His1026 | Val848, Ala866, Glu885, Ile888, Leu889, Ile892, Val899, Val916, Phe918, Gly922, Leu1019, Leu1035, Ile1044, Phe1047 |
| plHit1 | Glu885:OE2-H41 (2.8) Cys919:HN-O19 (2.1) Cys919:O-H33 (2.6) Asp1046:HN-O21 (2.6) | Leu840, His1026, Asp1046 | Gly841, Ala866, Leu889, His891, Val899, Val914, Val916, Glu917, Gly922, Asn923, Met1016, Leu1019, Arg1022, Cys1024, Ile1025, Arg1027, Leu1035, Cys1045, Phe1047, Ala1050, |
| plHit2 | Cys919:HN-O11 (2.6) Asp1046:O-H32 (2.0) | Leu840,Val848, Ala866, Lys868, Leu889, Val916 | Glu885, Ile892, Val899, Val914, Glu917, Phe918, Lys920, Gly922, Asn923, Leu1019, His1026, Leu1035, Ile1044, Phe1047 |
| plHit3 | Glu885:OE2-H34 (2.2) Cys919:O-H32 (2.4) Lys920:O-H32 (2.9) Asn923:HN-O14 (1.9) Asn923:HD21-O14 (1.9) | Leu840, Lys868, Leu889, Val916 | Leu840, Val848, Ala866, Ile888, Ile892, Val899, Val914, Glu917, Phe918, Gly922, Leu1091, His1026, Leu1035, Ile1044, Cys1045, Phe1047, Asp1046 |
| plHit4 | Cys919:O-H31 (2.5) Asp1046:HN-O12 (2.2) | Leu840, Lys868, Leu889, Val916, | Val848, Ala866, Glu885, Ile888, Ile892, Val914, Glu917, Phe918, Lys920, Gly922, Thr926, Leu1035, Phe1047 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rampogu, S.; Baek, A.; Park, C.; Son, M.; Parate, S.; Parameswaran, S.; Park, Y.; Shaik, B.; Kim, J.H.; Park, S.J.; et al. Discovery of Small Molecules That Target Vascular Endothelial Growth Factor Receptor-2 Signalling Pathway Employing Molecular Modelling Studies. Cells 2019, 8, 269. https://doi.org/10.3390/cells8030269
Rampogu S, Baek A, Park C, Son M, Parate S, Parameswaran S, Park Y, Shaik B, Kim JH, Park SJ, et al. Discovery of Small Molecules That Target Vascular Endothelial Growth Factor Receptor-2 Signalling Pathway Employing Molecular Modelling Studies. Cells. 2019; 8(3):269. https://doi.org/10.3390/cells8030269
Chicago/Turabian StyleRampogu, Shailima, Ayoung Baek, Chanin Park, Minky Son, Shraddha Parate, Saravanan Parameswaran, Yohan Park, Baji Shaik, Ju Hyun Kim, Seok Ju Park, and et al. 2019. "Discovery of Small Molecules That Target Vascular Endothelial Growth Factor Receptor-2 Signalling Pathway Employing Molecular Modelling Studies" Cells 8, no. 3: 269. https://doi.org/10.3390/cells8030269