TRP Channel Involvement in Salivary Glands—Some Good, Some Bad

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Historical Overview of Transient Receptor Potential (TRP) Channels
3. TRPC Channel Regulation and Function
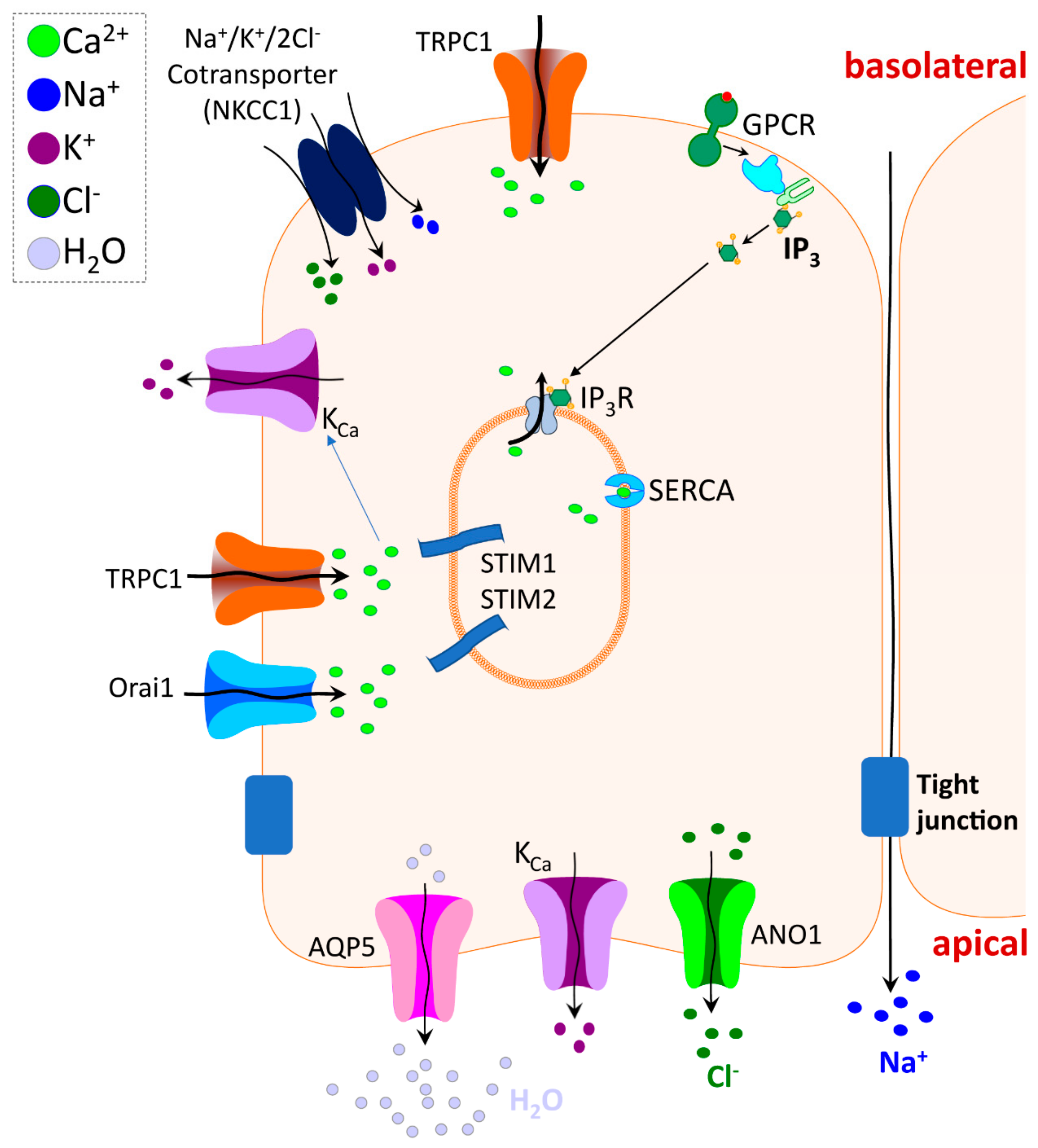
4. TRPC Channel Function in Exocrine Glands
5. TRPV4 and Other TRP Channel Function in Salivary Glands
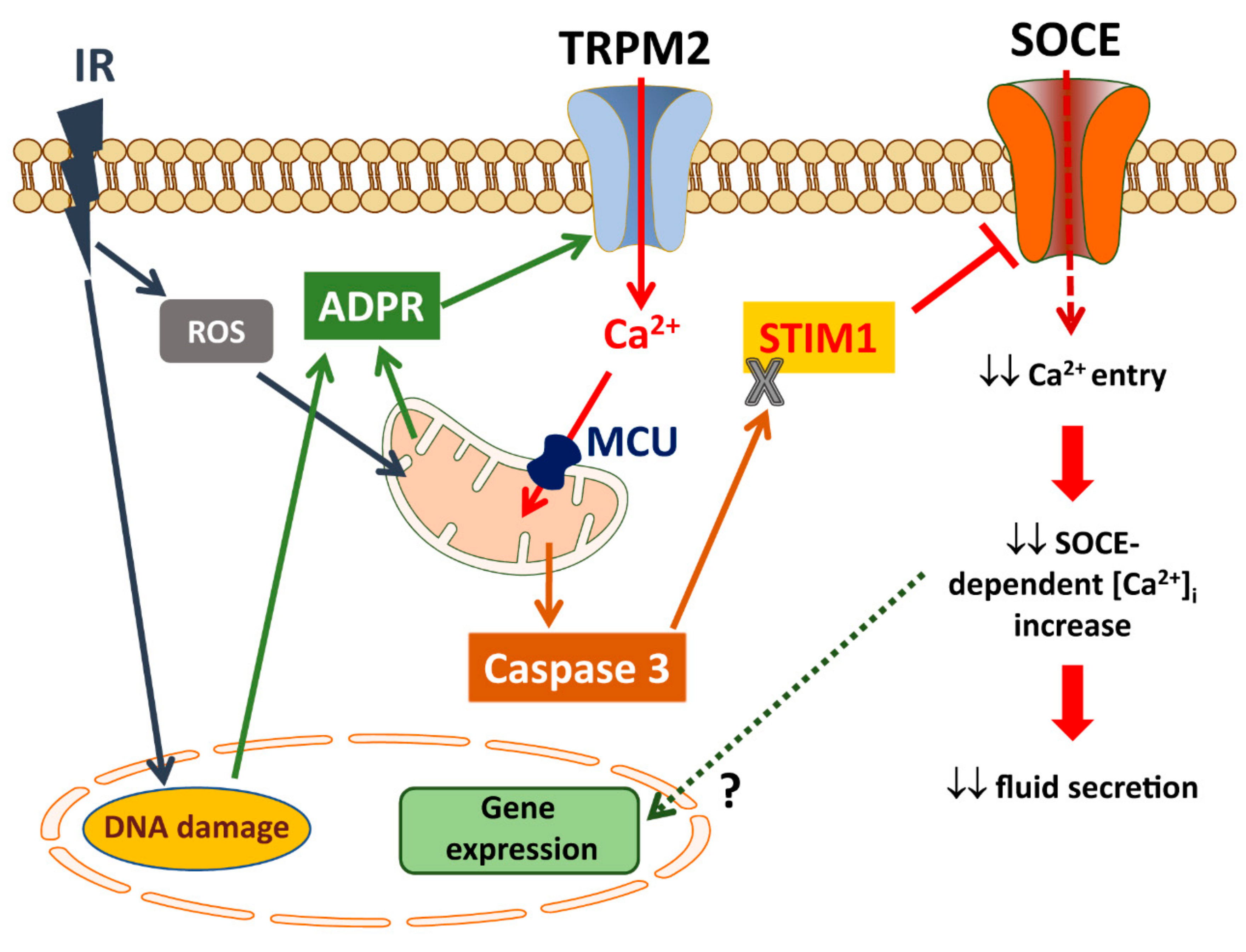
6. Role of TRPM2 in Salivary Gland Dysfunction
6.1. Regulation and Activation of TRPM2
6.2. TRPM2 and Radiation-Induced Loss of Salivary Flow
6.3. TRPM2 in Inflammatory Disorders
7. Conclusions
Funding
Conflicts of Interest
References
- Ambudkar, I.S. Dissection of calcium signaling events in exocrine secretion. Neurochem. Res. 2011, 36, 1212–1221. [Google Scholar] [CrossRef] [PubMed]
- Ambudkar, I.S. Polarization of calcium signaling and fluid secretion in salivary gland cells. Curr. Med. Chem. 2012, 19, 5774–5781. [Google Scholar] [CrossRef] [PubMed]
- Melvin, J.E.; Yule, D.; Shuttleworth, T.; Begenisich, T. Regulation of fluid and electrolyte secretion in salivary gland acinar cells. Annu. Rev. Physiol. 2005, 67, 445–469. [Google Scholar] [CrossRef] [PubMed]
- Futatsugi, A.; Nakamura, T.; Yamada, M.K.; Ebisui, E.; Nakamura, K.; Uchida, K.; Kitaguchi, T.; Takahashi-Iwanaga, H.; Noda, T.; Aruga, J.; et al. IP3 receptor types 2 and 3 mediate exocrine secretion underlying energy metabolism. Science 2005, 309, 2232–2234. [Google Scholar] [CrossRef] [PubMed]
- Yule, D.I. Subtype-specific regulation of inositol 1,4,5-trisphosphate receptors: Controlling calcium signals in time and space. J. Gen. Physiol. 2001, 117, 431–434. [Google Scholar] [CrossRef] [PubMed]
- Ambudkar, I.S. Ca2+ signaling and regulation of fluid secretion in salivary gland acinar cells. Cell Calcium 2014, 55, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Ambudkar, I.S. Calcium signalling in salivary gland physiology and dysfunction. J. Physiol. 2016, 594, 2813–2824. [Google Scholar] [CrossRef] [PubMed]
- Putney, J.W., Jr. Capacitative calcium entry revisited. Cell Calcium 1990, 11, 611–624. [Google Scholar] [CrossRef]
- Cheng, K.T.; Ong, H.L.; Liu, X.; Ambudkar, I.S. Contribution and regulation of TRPC channels in store-operated Ca2+ Entry. In Store-Operated Calcium Channels; Prakriya, M., Ed.; Elsevier: Amsterdam, The Netherlands, 2013; Volume 71, pp. 149–179. [Google Scholar]
- Hogan, P.G.; Lewis, R.S.; Rao, A. Molecular basis of calcium signaling in lymphocytes: Stim and Orai. Ann. Rev. Immunol. 2010, 28, 491–533. [Google Scholar] [CrossRef] [PubMed]
- Liou, J.; Kim, M.L.; Heo, W.D.; Jones, J.T.; Myers, J.W.; Ferrell, J.E., Jr.; Meyer, T. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr. Biol. 2005, 15, 1235–1241. [Google Scholar] [CrossRef] [PubMed]
- Prakriya, M. Store-operated Orai channels: Structure and function. Curr. Top. Membr. 2013, 71, 1–32. [Google Scholar] [PubMed]
- Yuan, J.P.; Zeng, W.; Dorwart, M.R.; Choi, Y.J.; Worley, P.F.; Muallem, S. SOAR and the polybasic STIM1 domains gate and regulate Orai channels. Nat. Cell Biol. 2009, 11, 337–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, W.; Yuan, J.P.; Kim, M.S.; Choi, Y.J.; Huang, G.N.; Worley, P.F.; Muallem, S. STIM1 gates TRPC channels, but not Orai1, by electrostatic interaction. Mol. Cell 2008, 32, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.L.; Yu, Y.; Roos, J.; Kozak, J.A.; Deerinck, T.J.; Ellisman, M.H.; Stauderman, K.A.; Cahalan, M.D. STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature 2005, 437, 902–905. [Google Scholar] [CrossRef] [PubMed]
- Ong, H.L.; de Souza, L.B.; Zheng, C.; Cheng, K.T.; Liu, X.; Goldsmith, C.M.; Feske, S.; Ambudkar, I.S. STIM2 enhances receptor-stimulated Ca2+ signaling by promoting recruitment of STIM1 to the endoplasmic reticulum-plasma membrane junctions. Sci. Signal. 2015, 8, ra3. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.H.; Li, Q.; Kim, M.S.; Shin, D.M.; Feske, S.; Birnbaumer, L.; Cheng, K.T.; Ambudkar, I.S.; Muallem, S. Polarized but differential localization and recruitment of STIM1, Orai1 and TRPC channels in secretory cells. Traffic 2011, 12, 232–245. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Cheng, K.T.; Bandyopadhyay, B.C.; Pani, B.; Dietrich, A.; Paria, B.C.; Swaim, W.D.; Beech, D.; Yildrim, E.; Singh, B.B.; et al. Attenuation of store-operated Ca2+ current impairs salivary gland fluid secretion in TRPC1−/− mice. Proc. Natl. Acad. Sci. USA 2007, 104, 17542–17547. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, W.; Singh, B.B.; Lockwich, T.; Jadlowiec, J.; O’Connell, B.; Wellner, R.; Zhu, M.X.; Ambudkar, I.S. Trp1, a candidate protein for the store-operated Ca2+ influx mechanism in salivary gland cells. J. Biol. Chem. 2000, 275, 3403–3411. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.T.; Liu, X.; Ong, H.L.; Ambudkar, I.S. Functional requirement for Orai1 in store-operated TRPC1-STIM1 channels. J. Biol. Chem. 2008, 283, 12935–12940. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.T.; Liu, X.; Ong, H.L.; Swaim, W.; Ambudkar, I.S. Local Ca2+ entry via Orai1 regulates plasma membrane recruitment of TRPC1 and controls cytosolic Ca2+ signals required for specific cell functions. PLoS Biol. 2011, 9, e1001025. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.; Maleth, J.; Jha, A.; Lee, K.P.; Kim, M.S.; So, I.; Ahuja, M.; Muallem, S. The TRPCs-STIM1-Orai interaction. Handbook Exp. Pharmacol. 2014, 223, 1035–1054. [Google Scholar]
- Lee, K.P.; Yuan, J.P.; Hong, J.H.; So, I.; Worley, P.F.; Muallem, S. An endoplasmic reticulum/plasma membrane junction: STIM1/Orai1/TRPCs. FEBS Lett. 2010, 584, 2022–2027. [Google Scholar] [CrossRef] [PubMed]
- Delaleu, N.; Jonsson, R.; Koller, M.M. Sjögren’s syndrome. Eur. J. Oral. Sci. 2005, 113, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Mavragani, C.P.; Moutsopoulos, H.M. Sjögren’s syndrome. Ann. Rev. Path. 2014, 9, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Delli, K.; Spijkervet, F.K.; Kroese, F.G.; Bootsma, H.; Vissink, A. Xerostomia. Monogr. Oral. Sci. 2014, 24, 109–125. [Google Scholar] [PubMed]
- Vissink, A.; Jansma, J.; Spijkervet, F.K.; Burlage, F.R.; Coppes, R.P. Oral sequelae of head and neck radiotherapy. Crit. Rev. Oral. Biol. Med. 2003, 14, 199–212. [Google Scholar] [CrossRef] [PubMed]
- Montell, C.; Rubin, G.M. Molecular characterization of the Drosophila trp locus: A putative integral membrane protein required for phototransduction. Neuron 1989, 2, 1313–1323. [Google Scholar] [CrossRef]
- Montell, C.; Birnbaumer, L.; Flockerzi, V. The TRP channels, a remarkably functional family. Cell 2002, 108, 595–598. [Google Scholar] [CrossRef]
- Schlingmann, K.P.; Gudermann, T. A critical role of TRPM channel-kinase for human magnesium transport. J. Physiol. 2005, 566, 301–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bargal, R.; Avidan, N.; Ben-Asher, E.; Olender, Z.; Zeigler, M.; Frumkin, A.; Raas-Rothschild, A.; Glusman, G.; Lancet, D.; Bach, G. Identification of the gene causing mucolipidosis type IV. Nat. Genet. 2000, 26, 118–123. [Google Scholar] [PubMed]
- Bassi, M.T.; Manzoni, M.; Monti, E.; Pizzo, M.T.; Ballabio, A.; Borsani, G. Cloning of the gene encoding a novel integral membrane protein, mucolipidin-and identification of the two major founder mutations causing mucolipidosis type IV. Am. J. Hum. Genet. 2000, 67, 1110–1120. [Google Scholar] [CrossRef]
- Mochizuki, T.; Wu, G.; Hayashi, T.; Xenophontos, S.L.; Veldhuisen, B.; Saris, J.J.; Reynolds, D.M.; Cai, Y.; Gabow, P.A.; Pierides, A.; et al. Pkd2, a gene for polycystic kidney disease that encodes an integral membrane protein. Science 1996, 272, 1339–1342. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Goldin, E.; Stahl, S.; Falardeau, J.L.; Kennedy, J.C.; Acierno, J.S., Jr.; Bove, C.; Kaneski, C.R.; Nagle, J.; Bromley, M.C.; et al. Mucolipidosis type IV is caused by mutations in a gene encoding a novel transient receptor potential channel. Hum. Mol. Genet. 2000, 9, 2471–2478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaneko, Y.; Szallasi, A. TRP channels as therapeutic targets. Curr. Top. Med. Chem. 2013, 13, 241–243. [Google Scholar] [CrossRef] [PubMed]
- Nilius, B.; Owsianik, G. Transient receptor potential channelopathies. Pflugers Arch. 2010, 460, 437–450. [Google Scholar] [CrossRef] [PubMed]
- Nilius, B.; Szallasi, A. Transient receptor potential channels as drug targets: From the science of basic research to the art of medicine. Pharmacol. Rev. 2014, 66, 676–814. [Google Scholar] [CrossRef] [PubMed]
- Tóth, B.I.; Nilius, B. Transient receptor potential dysfunctions in hereditary diseases. In TRP Channels as Therapeutic Targets; Szallasi, A., Ed.; Academic Press: Amsterdam, The Netherlands, 2015; pp. 13–33. [Google Scholar]
- Kaneko, Y.; Szallasi, A. Transient receptor potential (TRP) channels: A clinical perspective. Br. J. Pharmacol. 2014, 171, 2474–2507. [Google Scholar] [CrossRef] [PubMed]
- Smani, T.; Shapovalov, G.; Skryma, R.; Prevarskaya, N.; Rosado, J.A. Functional and physiopathological implications of TRP channels. Biochim. Biophys. Acta 2015, 1853, 1772–1782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nilius, B.; Owsianik, G. The transient receptor potential family of ion channels. Genome Biol. 2011, 12, 218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bandell, M.; Macpherson, L.J.; Patapoutian, A. From chills to chilis: Mechanisms for thermosensation and chemesthesis via thermotrps. Curr. Opin. Neurobiol. 2007, 17, 490–497. [Google Scholar] [CrossRef] [PubMed]
- Patapoutian, A.; Peier, A.M.; Story, G.M.; Viswanath, V. ThermoTRP channels and beyond: Mechanisms of temperature sensation. Nat. Rev. Neurosci. 2003, 4, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Tominaga, M. The role of TRP channels in thermosensation. In TRP Ion Channel Function in Sensory Transduction and Cellular Signaling Cascades; Liedtke, W.B., Heller, S., Eds.; CRC Press: Boca Raton, FL, USA, 2007. [Google Scholar]
- Caterina, M.J.; Rosen, T.A.; Tominaga, M.; Brake, A.J.; Julius, D. A capsaicin-receptor homologue with a high threshold for noxious heat. Nature 1999, 398, 436–441. [Google Scholar] [CrossRef] [PubMed]
- Caterina, M.J.; Schumacher, M.A.; Tominaga, M.; Rosen, T.A.; Levine, J.D.; Julius, D. The capsaicin receptor: A heat-activated ion channel in the pain pathway. Nature 1997, 389, 816–824. [Google Scholar] [PubMed]
- McKemy, D.D.; Neuhausser, W.M.; Julius, D. Identification of a cold receptor reveals a general role for TRP channels in thermosensation. Nature 2002, 416, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Peier, A.M.; Moqrich, A.; Hargarden, A.C.; Reeve, A.J.; Andersson, D.A.; Story, G.M.; Earley, T.J.; Dragoni, I.; MaIntyre, P.; Bevan, S.; et al. A TRP channel that senses cold stimuli and menthol. Cell 2002, 108, 705–715. [Google Scholar] [CrossRef]
- Vriens, J.; Owsianik, G.; Hofmann, T.; Philipp, S.E.; Stab, J.; Chen, X.; Benoit, M.; Xue, F.; Janssens, A.; Kerselaers, S.; et al. TRPM3 is a nociceptor channel involved in the detection of noxious heat. Neuron 2011, 70, 482–494. [Google Scholar] [CrossRef] [PubMed]
- Story, G.M.; Peier, A.M.; Reeve, A.J.; Eid, S.R.; Mosbacher, J.; Hricik, T.R.; Earley, T.J.; Hergarden, A.C.; Andersson, D.A.; Hwang, S.W.; et al. ANKTM1, a trp-like channel expressed in nociceptive neurons, is activated by cold temperatures. Cell 2003, 112, 819–829. [Google Scholar] [CrossRef]
- Bandell, M.; Story, G.M.; Hwang, S.W.; Viswanath, V.; Eid, S.R.; Petrus, M.J.; Earley, T.J.; Patapoutian, A. Noxious cold ion channel TRPA1 is activated by pungent compounds and bradykinin. Neuron 2004, 41, 849–857. [Google Scholar] [CrossRef]
- Chen, J.; Kang, D.; Xu, J.; Lake, M.; Hogan, J.O.; Sun, C.; Walter, K.; Yao, B.; Kim, D. Species differences and molecular determinant of TRPA1 cold sensitivity. Nat. Commun. 2013, 4, 2501. [Google Scholar] [CrossRef] [PubMed]
- Jordt, S.E.; Bautista, D.M.; Chuang, H.H.; McKemy, D.D.; Zygmunt, P.M.; Hogestatt, E.D.; Meng, I.D.; Julius, D. Mustard oils and cannabinoids excite sensory nerve fibres through the TRP channel ANKTM1. Nature 2004, 427, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Bautista, D.M.; Pellegrino, M.; Tsunozaki, M. TRPA1: A gatekeeper for inflammation. Annu. Rev. Physiol. 2013, 75, 181–200. [Google Scholar] [CrossRef] [PubMed]
- Caspani, O.; Heppenstall, P.A. TRPA1 and cold transduction: An unresolved issue? J. Gen. Physiol. 2009, 133, 245–249. [Google Scholar] [CrossRef] [PubMed]
- Doerner, J.F.; Gisselmann, G.; Hatt, H.; Wetzel, C.H. Transient receptor potential channel A1 is directly gated by calcium ions. J. Biol. Chem. 2007, 282, 13180–13189. [Google Scholar] [CrossRef] [PubMed]
- Fujita, F.; Uchida, K.; Moriyama, T.; Shima, A.; Shibasaki, K.; Inada, H.; Sokabe, T.; Tominaga, M. Intracellular alkalization causes pain sensation through activation of TRPA1 in mice. J. Clin. Investig. 2008, 118, 4049–4057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nilius, B.; Appendino, G.; Owsianik, G. The transient receptor potential channel TRPA1: From gene to pathophysiology. Pflugers Arch. 2012, 464, 425–458. [Google Scholar] [CrossRef] [PubMed]
- Nilius, B.; Prenen, J.; Owsianik, G. Irritating channels: The case of TRPA1. J. Physiol. 2011, 589, 1543–1549. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.F.; Chen, J.; Faltynek, C.R.; Moreland, R.B.; Neelands, T.R. Transient receptor potential A1 mediates an osmotically activated ion channel. Eur. J. Neurosci. 2008, 27, 605–611. [Google Scholar] [CrossRef] [PubMed]
- Liedtke, W.; Choe, Y.; Marti-Renom, M.A.; Bell, A.M.; Denis, C.S.; Sali, A.; Hudspeth, A.J.; Friendman, J.M.; Heller, S. Vanilloid receptor-related osmotically activated channel (VR-OAC), a candidate vertebrate osmoreceptor. Cell 2000, 103, 525–535. [Google Scholar] [CrossRef]
- Strotmann, R.; Harteneck, C.; Nennenmacher, K.; Schultz, G.; Plant, T.D. OTRPC4, a nonselective cation channel that confers sensitivity to extracellular osmolarity. Nat. Cell Biol. 2000, 2, 695–702. [Google Scholar] [CrossRef] [PubMed]
- Tominaga, M.; Caterina, M.J.; Malmberg, A.B.; Rosen, T.A.; Gilbert, H.; Skinner, K.; Raumann, B.E.; Basbaum, A.I.; Julius, D. The cloned capsaicin receptor integrates multiple pain-producing stimuli. Neuron 1998, 21, 531–543. [Google Scholar] [CrossRef]
- Bautista, D.M.; Jordt, S.E.; Nikai, T.; Tsuruda, P.R.; Read, A.J.; Poblete, J.; Yamoah, E.N.; Basbaum, A.I.; Julius, D. TRPA1 mediates the inflammatory actions of environmental irritants and proalgesic agents. Cell 2006, 124, 1269–1282. [Google Scholar] [CrossRef] [PubMed]
- Chuang, H.H.; Prescott, E.D.; Kong, H.; Shields, S.; Jordt, S.E.; Basbaum, A.I.; Chao, M.V.; Julius, D. Bradykinin and nerve growth factor release the capsaicin receptor from Ptdins(4,5)p2-mediated inhibition. Nature 2001, 411, 957–962. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Qin, F. Functional control of cold- and menthol-sensitive TRPM8 ion channels by phosphatidylinositol 4,5-bisphosphate. J. Neurosci. 2005, 25, 1674–1681. [Google Scholar] [CrossRef] [PubMed]
- Prescott, E.D.; Julius, D. A modular PIP2 binding site as a determinant of capsaicin receptor sensitivity. Science 2003, 300, 1284–1288. [Google Scholar] [CrossRef] [PubMed]
- Rohacs, T.; Lopes, C.M.; Michailidis, I.; Logothetis, D.E. PI(4,5)P2 regulates the activation and desensitization of TRPM8 channels through the TRP domain. Nat. Neurosci. 2005, 8, 626–634. [Google Scholar] [CrossRef] [PubMed]
- Romanovsky, A.A.; Almeida, M.C.; Garami, A.; Steiner, A.A.; Norman, M.H.; Morrison, S.F.; Nakamura, K.; Burmeister, J.J.; Nucci, T.B. The transient receptor potential vanilloid-1 channel in thermoregulation: A thermosensor it is not. Pharmacol. Rev. 2009, 61, 228–261. [Google Scholar] [CrossRef] [PubMed]
- Szolcsanyi, J. Effect of capsaicin on thermoregulation: An update with new aspects. Temperature (Austin) 2015, 2, 277–296. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Siemens, J. TRP ion channels in thermosensation, thermoregulation and metabolism. Temperature (Austin) 2015, 2, 178–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minke, H.; Cook, B. TRP channel proteins and signal transduction. Physiol. Rev. 2002, 82, 429–472. [Google Scholar] [CrossRef] [PubMed]
- Montell, C. The TRP superfamily of cation channels. Sci. STKE 2005, 272, re3. [Google Scholar] [CrossRef] [PubMed]
- Venkatachalam, K.; Montell, C. TRp channels. Annu. Rev. Biochem. 2007, 76, 387–417. [Google Scholar] [CrossRef] [PubMed]
- Ong, H.L.; de Souza, L.B.; Cheng, K.T.; Ambudkar, I.S. Physiological functions and regulation of TRPC channels. Handbook Exp. Pharmacol. 2014, 223, 1005–1034. [Google Scholar]
- Nesin, V.; Tsiokas, L. TRPC1. Handbook Exp. Pharmacol. 2014, 222, 15–51. [Google Scholar]
- Miller, B.A. TRPC2. Handbook Exp. Pharmacol. 2014, 222, 53–65. [Google Scholar]
- Lichtenegger, M.; Groschner, K. TRPC3: A multifunctional signaling molecule. Handbook Exp. Pharmacol. 2014, 222, 67–84. [Google Scholar]
- Freichel, M.; Tsvilovskyy, V.; Camacho-Londono, J.E. TRPC4- and TRPC4-containing channels. Handbook Exp. Pharmacol. 2014, 222, 85–128. [Google Scholar]
- Zholos, A.V. TRPC5. Handbook Exp. Pharmacol. 2014, 222, 129–156. [Google Scholar]
- Dietrich, A.; Gudermann, T. TRPC6: Physiological function and pathophysiological relevance. Handbook Exp. Pharmacol. 2014, 222, 157–188. [Google Scholar]
- Zhang, X.; Trebak, M. Transient receptor potential canonical 7: A diacylglycerol-activated non-selective cation channel. Handbook Exp. Pharmacol. 2014, 222, 189–204. [Google Scholar]
- Wes, P.D.; Chevesich, J.; Jeromin, A.; Rosenberg, C.; Stetten, S.; Montell, C. TRPC1, a human homolog of a drosophila store-operated channel. Proc. Natl. Acad. Sci. USA 1995, 92, 9652–9656. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Chu, P.B.; Peyton, M.; Birnbaumer, L. Molecular cloning of a widely expressed human homologue for the drosophila trp gene. FEBS Lett. 1995, 373, 193–198. [Google Scholar] [CrossRef]
- Liu, X.; Singh, B.B.; Ambudkar, I.S. TRPC1 is required for functional store-operated Ca2+ channels. Role of acidic amino acid residues in the s5-s6 region. J. Biol. Chem. 2003, 278, 11337–11343. [Google Scholar] [CrossRef] [PubMed]
- Hoth, M. Depletion of intracellular calcium stores activates an outward potassium current in mast and RBL-1 cells that is correlated with CRAC channel activation. FEBS Lett. 1996, 390, 285–288. [Google Scholar] [CrossRef] [Green Version]
- Ambudkar, I.S. TRPC1: A core component of store-operated calcium channels. Biochem. Soc. Trans. 2007, 35, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Beech, D.J. TRPC1: Store-operated channel and more. Pflugers Arch. 2005, 451, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Birnbaumer, L.; Singh, B.B. TRPC1 regulates calcium-activated chloride channels in salivary gland cells. J. Cell. Physiol. 2015, 230, 2848–2856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pani, B.; Ong, H.L.; Brazer, S.C.; Liu, X.; Rauser, K.; Singh, B.B.; Ambudkar, I.S. Activation of TRPC1 by STIM1 in ER-PM microdomains involves release of the channel from its scaffold caveolin-1. Proc. Natl. Acad. Sci. USA 2009, 106, 20087–20092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pani, B.; Liu, X.; Bollimuntha, S.; Cheng, K.T.; Niesman, I.R.; Zheng, C.; Achen, V.R.; Patel, H.H.; Ambudkar, I.S.; Singh, B.B. Impairment of TRPC1-STIM1 channel assembly and aqp5 translocation compromise agonist-stimulated fluid secretion in mice lacking caveolin1. J. Cell. Sci. 2013, 126, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Hong, J.H.; Li, Q.; Shin, D.M.; Abramowitz, J.; Birnbaumer, L.; Muallem, S. Deletion of TRPC3 in mice reduces store-operated Ca2+ influx and the severity of acute pancreatitis. Gastroenterology 2009, 137, 1509–1517. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.P.; Choi, S.; Hong, J.H.; Ahuja, M.; Graham, S.; Ma, R.; So, I.; Shin, D.M.; Muallem, S.; Yuan, J.P. Molecular determinants mediating gating of transient receptor potential canonical (TRPC) channels by stromal interaction molecule 1 (STIM1). J. Biol. Chem. 2014, 289, 6372–6382. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.P.; Zeng, W.; Huang, G.N.; Worley, P.F.; Muallem, S. STIM1 heteromultimerizes TRPC channels to determine their function as store-operated channels. Nat. Cell Biol. 2007, 9, 636–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.S.; Lee, K.P.; Yang, D.; Shin, D.M.; Abramowitz, J.; Kiyonaka, S.; Birnbaumer, L.; Mori, Y.; Muallem, S. Genetic and pharmacologic inhibition of the Ca2+ influx channel TRPC3 protects secretory epithelia from Ca2+-dependent toxicity. Gastroenterology 2011, 140, 2107–2115. [Google Scholar] [CrossRef] [PubMed]
- Xing, J.; Petranka, J.G.; Davis, F.M.; Desai, P.N.; Putney, J.W.; Bird, G.S. Role of Orai1 and store-operated calcium entry in mouse lacrimal gland signalling and function. J. Physiol. 2014, 592, 927–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahuja, M.; Jha, A.; Maleth, J.; Park, S.; Muallem, S. cAMP and Ca2+ signaling in secretory epithelia: Crosstalk and synergism. Cell Calcium 2014, 55, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Bandyopadhyay, B.; Nakamoto, T.; Singh, B.; Liedtke, W.; Melvin, J.E.; Ambudkar, I. A role for AQP5 in activation of trpv4 by hypotonicity: Concerted involvement of AQP5 and TRPC4 in regulation of cell volume recovery. J. Biol. Chem. 2006, 281, 15485–15495. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Catalan, M.A.; Melvin, J.E. TRPV4 activation in mouse submandibular gland modulates Ca2+ influx and salivation. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 303, G1365–G1372. [Google Scholar] [CrossRef] [PubMed]
- Derouiche, S.; Takayama, Y.; Murakami, M.; Tominaga, M. TRPV4 heats up ANO1-dependent exocrine gland fluid secretion. FASEB J. 2018, 32, 1841–1854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sumoza-Toledo, A.; Penner, R. TRPM2: A multifunctional ion channel for calcium signalling. J. Physiol. 2011, 589, 1515–1525. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.H.; Yang, W.; Zou, J.; Beech, D.J. TRPM2 channel properties, functions and therapeutic potentials. Expert Opin. Ther. Targets 2010, 14, 973–988. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Cotrim, A.; Teos, L.; Zheng, C.; Swaim, W.; Mitchell, J.; Mori, Y.; Ambudkar, I. Loss of TRPM2 function protects against irradiation-induced salivary gland dysfunction. Nat. Commun. 2013, 4, 1515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perraud, A.L.; Shen, B.; Dunn, C.A.; Rippe, K.; Smith, M.K.; Bessman, M.J.; Stoddard, B.L.; Scharenberg, A.M. NUDT9, a member of the nudix hydrolase family, is an evolutionarily conserved mitochondrial ADP-ribose pyrophosphatase. J. Biol. Chem. 2003, 278, 1794–1801. [Google Scholar] [CrossRef] [PubMed]
- Knowles, H.; Li, Y.; Perraud, A.L. The TRPM2 ion channel, an oxidative stress and metabolic sensor regulating innate immunity and inflammation. Immunol. Res. 2013, 55, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Gao, Y.; Bao, X.; Li, F.; Yao, W.; Feng, Z.; Yin, Y. TRPM2: A potential drug target to retard oxidative stress. Front. Biosci. (Landmark Ed.) 2017, 22, 1427–1438. [Google Scholar] [PubMed]
- Perraud, A.L.; Takanishi, C.L.; Shen, B.; Kang, S.; Smith, M.K.; Schmitz, C.; Knowles, H.M.; Ferraris, D.; Li, W.; Zhang, J.; et al. Accumulation of free ADP-ribose from mitochondria mediates oxidative stress-induced gating of TRPM2 cation channels. J. Biol. Chem. 2005, 280, 6138–6148. [Google Scholar] [CrossRef] [PubMed]
- Toth, B.; Iordanov, I.; Csanady, L. Ruling out pyridine dinucleotides as true TRPM2 channel activators reveals novel direct agonist ADP-ribose-2’-Phosphate. J. Gen. Physiol. 2015, 145, 419–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faouzi, M.; Penner, R. TRPM2. Handbook Exp. Pharmacol. 2014, 222, 403–426. [Google Scholar]
- Konings, A.W.; Coppes, R.P.; Vissink, A. On the mechanism of salivary gland radiosensitivity. Int. J. Rad. Oncol. Biol. Phys. 2005, 62, 1187–1194. [Google Scholar] [CrossRef] [PubMed]
- Stephens, L.C.; Schultheiss, T.E.; Price, R.E.; Ang, K.K.; Peters, L.J. Radiation apoptosis of serous acinar cells of salivary and lacrimal glands. Cancer 1991, 67, 1539–1543. [Google Scholar] [CrossRef] [Green Version]
- Zeilstra, L.J.; Vissink, A.; Konings, A.W.; Coppes, R.P. Radiation induced cell loss in rat submandibular gland and its relation to gland function. Int. J. Rad. Oncol. Biol. Phys. 2000, 76, 419–429. [Google Scholar]
- Liu, X.; Gong, B.; de Souza, L.B.; Ong, H.L.; Subedi, K.P.; Cheng, K.T.; Swaim, W.; Zheng, C.; Mori, Y.; Ambudkar, I.S. Radiation inhibits salivary gland function by promoting STIM1 cleavage by caspase-3 and loss of SOCE through a TRPM2-dependent pathway. Sci. Signal. 2017, 10, eaal4064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teos, L.Y.; Zheng, C.Y.; Liu, X.; Swaim, W.D.; Goldsmith, C.M.; Cotrim, A.P.; Baum, B.J.; Ambudkar, I.S. Adenovirus-mediated hAQP1 expression in irradiated mouse salivary glands causes recovery of saliva secretion by enhancing acinar cell volume decrease. Gene Ther. 2016, 23, 572–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Citrin, D.; Cotrim, A.P.; Hyodo, F.; Baum, B.J.; Krishna, M.C.; Mitchell, J.B. Radioprotectors and mitigators of radiation-induced normal tissue injury. Oncologist 2010, 15, 360–371. [Google Scholar] [CrossRef] [PubMed]
- Prins, D.; Groenendyk, J.; Touret, N.; Michalak, M. Modulation of STIM1 and capacitative Ca2+ entry by the endoplasmic reticulum luminal oxidoreductase ERP57. EMBO Rep. 2011, 12, 1182–1188. [Google Scholar] [CrossRef] [PubMed]
- Tong, B.C.; Lee, C.S.; Cheng, W.H.; Lai, K.O.; Foskett, J.K.; Cheung, K.H. Familial Alzheimer’s disease-associated presenilin 1 mutants promote γ-secretase cleavage of STIM1 to impair store-operated Ca2+ entry. Sci. Signal. 2016, 9, ra89. [Google Scholar] [CrossRef] [PubMed]
- Keil, J.M.; Shen, Z.; Briggs, S.P.; Patrick, G.N. Regulation of STIM1 and SOCE by the ubiquitin-proteasome system (UPS). PLoS ONE 2010, 5, e13465. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.I.; Ong, H.L.; Liu, X.; Alevizos, I.; Ambudkar, I.S. Up-regulation of store-operated Ca2+ entry and nuclear factor of activated T cells promote the acinar phenotype of the primary human salivary gland cells. J. Biol. Chem. 2016, 291, 8709–8720. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, N.; Kozai, D.; Kobayashi, R.; Ebert, M.; Mori, Y. Roles of TRPM2 in oxidative stress. Cell Calcium 2011, 50, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Gasser, A.; Glassmeier, G.; Fliegert, R.; Langhorst, M.F.; Meinke, S.; Hein, D.; Kruger, S.; Weber, K.; Heiner, I.; Oppenheimer, N.; et al. Activation of T cell calcium influx by the second messenger ADP-ribose. J. Biol. Chem. 2006, 281, 2489–2496. [Google Scholar] [CrossRef] [PubMed]
- Dammermann, W.; Zhang, B.; Nebel, M.; Cordiglieri, C.; Odoardi, F.; Kirchberger, T.; Kawakami, N.; Dowden, J.; Schmid, F.; Dornmair, K.; et al. NAADP-mediated Ca2+ signaling via type 1 ryanodine receptor in T cells revealed by a synthetic NAADP antagonist. Proc. Natl. Acad. Sci. USA 2009, 106, 10678–10683. [Google Scholar] [CrossRef] [PubMed]
- Bari, M.R.; Akbar, S.; Eweida, M.; Kuhn, F.J.; Gustafsson, A.J.; Luckhoff, A.; Islam, M.S. H2O2-induced Ca2+ influx and its inhibition by n-(p-amylcinnamoyl) anthranilic acid in the β-cells: Involvement of TRPM2 channels. J. Cell. Mol. Med. 2009, 13, 3260–3267. [Google Scholar] [CrossRef] [PubMed]
- Kraft, R.; Grimm, C.; Frenzel, H.; Harteneck, C. Inhibition of TRPM2 cation channels by n-(p-amylcinnamoyl)anthranilic acid. Br. J. Pharmacol. 2006, 148, 264–273. [Google Scholar] [CrossRef] [PubMed]
- Sumoza-Toledo, A.; Lange, I.; Cortado, H.; Bhagat, H.; Mori, Y.; Fleig, A.; Penner, R.; Partida-Sanchez, S. Dendritic cell maturation and chemotaxis is regulated by TRPM2-mediated lysosomal Ca2+ release. FASEB J. 2011, 25, 3529–3542. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Shimizu, S.; Kiyonaka, S.; Takahashi, N.; Wajima, T.; Hara, Y.; Negoro, T.; Hiroi, T.; Kiuchi, Y.; Okada, T.; et al. TRPM2-mediated Ca2+influx induces chemokine production in monocytes that aggravates inflammatory neutrophil infiltration. Nat. Med. 2008, 14, 738–747. [Google Scholar] [CrossRef] [PubMed]
- Haraguchi, K.; Kawamoto, A.; Isami, K.; Maeda, S.; Kusano, A.; Asakura, K.; Shirakawa, H.; Mori, Y.; Nakagawa, T.; Kaneko, S. TRPM2 contributes to inflammatory and neuropathic pain through the aggravation of pronociceptive inflammatory responses in mice. J. Neurosci. 2012, 32, 3931–3941. [Google Scholar] [CrossRef] [PubMed]
- Melzer, N.; Hicking, G.; Gobel, K.; Wiendl, H. TRPM2 cation channels modulate T cell effector functions and contribute to autoimmune CNS inflammation. PLoS ONE 2012, 7, e47617. [Google Scholar] [CrossRef] [PubMed]
- Beceiro, S.; Radin, J.N.; Chatuvedi, R.; Piazuelo, M.B.; Horvarth, D.J.; Cortado, H.; Gu, Y.; Dixon, B.; Gu, C.; Lange, I.; et al. TRPM2 ion channels regulate macrophage polarization and gastric inflammation during helicobacter pylori infection. Mucosal Immunol. 2017, 10, 493–507. [Google Scholar] [CrossRef] [PubMed]
- Yonezawa, R.; Yamamoto, S.; Takenaka, M.; Kage, Y.; Negoro, T.; Toda, T.; Ohbayashi, M.; Numata, T.; Nakano, Y.; Yamamoto, T.; et al. TRPM2 channels in alveolar epithelial cells mediate bleomycin-induced lung inflammation. Free Radic. Biol. Med. 2016, 90, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Oda, S.; Uchida, K.; Wang, X.; Lee, J.; Shimada, Y.; Tominaga, M.; Kadowaki, M. TRPM2 contributes to antigen-stimulated Ca2+ influx in mucosal mast cells. Pflugers Arch. 2013, 465, 1023–1030. [Google Scholar] [CrossRef] [PubMed]
- Ham, H.Y.; Hong, C.W.; Lee, S.N.; Kwon, M.S.; Kim, Y.J.; Song, D.K. Sulfur mustard primes human neutrophils for increased degranulation and stimulates cytokine release via TRPM2/p38 MAPK signaling. Toxicol. Appl. Pharmacol. 2012, 258, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Di, A.; Gao, X.P.; Qian, F.; Kawamura, T.; Han, J.; Hecquet, C.; Ye, R.D.; Vogel, S.M.; Malik, A.B. The redox-sensitive cation channel TRPM2 modulates phagocyte ROS production and inflammation. Nat. Immunol. 2011, 13, 29–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knowles, H.; Heizer, J.W.; Li, Y.; Chapman, K.; Ogden, C.A.; Andreasen, K.; Shapland, E.; Kucera, G.; Mogan, J.; Humann, J.; et al. Transient receptor potential melastatin 2 (TRPM2) ion channel is required for innate immunity against listeria monocytogenes. Proc. Natl. Acad. Sci. USA 2011, 108, 11578–11583. [Google Scholar] [CrossRef] [PubMed]
- Miyake, T.; Shirakawa, H.; Kusano, A.; Sakimoto, S.; Konno, M.; Nakagawa, T.; Mori, Y.; Kaneko, S. TRPM2 contributes to lPS/IFNγ-induced production of nitric oxide via the p38/JNK pathway in microglia. Biochem. Biophys. Res. Commun. 2014, 444, 212–217. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Mayor, A.; Tschopp, J. The inflammasomes: Guardians of the body. Annu. Rev. Immunol. 2009, 27, 229–265. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.; Ainscough, J.F.; Yang, W.; Sedo, A.; Yu, S.P.; Mei, Z.Z.; Sivaprasadarao, A.; Beech, D.J.; Jiang, L.H. A differential role of macrophage TRPM2 channels in Ca2+ signaling and cell death in early responses to H2O2. Am. J. Physiol. Cell. Physiol. 2013, 305, C61–C69. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.Y.; Zhai, Y.G.; Liang, S.; Mori, Y.S.; Han, R.Z.; Sutterwala, F.S.; Qiao, L. TRPM2 links oxidative stress to NLRP3 inflammasome activation. Nat. Commun. 2013, 4, 1611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bikker, A.; van Woerkom, J.M.; Kruize, A.A.; Wenting-van Wijk, M.; de Jager, W.; Bijlsma, J.W.; Lafeber, F.P.; van Roon, J.A. Increased expression of interleukin-7 in labial salivary glands of patients with primary Sjögren’s Syndrome correlates with increased inflammation. Arthritis Rheum. 2010, 62, 969–977. [Google Scholar] [CrossRef] [PubMed]
- Baturone, R.; Soto, M.J.; Marquez, M.; Macias, I.; de Oca, M.M.; Medina, F.; Chozas, N.; Garcia-Perez, S.; Giron-Gonzalez, J.A. Health-related quality of life in patients with primary Sjögren’s Syndrome: Relationship with serum levels of proinflammatory cytokines. Scand. J. Rheumatol. 2009, 38, 386–389. [Google Scholar] [CrossRef] [PubMed]
- Sakai, A.; Sugawara, Y.; Kuroishi, T.; Sasano, T.; Sugawara, S. Identification of il-18 and th17 cells in salivary glands of patients with sjogren’s syndrome, and amplification of il-17-mediated secretion of inflammatory cytokines from salivary gland cells by il-18. J. Immunol. 2008, 181, 2898–2906. [Google Scholar] [CrossRef] [PubMed]
- Lindholm, C.; Acheva, A.; Salomaa, S. Clastogenic plasma factors: A short overview. Radiat. Environ. Biophys. 2010, 49, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Obrador, E.; Navarro, J.; Mompo, J.; Asensi, M.; Pellicer, J.A.; Estrela, J.M. Regulation of tumour cell sensitivity to TNF-induced oxidative stress and cytotoxicity: Role of glutathione. Biofactors 1998, 8, 23–26. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.Y.; Lee, E.K.; Choi, Y.J.; Kim, J.M.; Kim, D.H.; Zou, Y.; Kim, C.H.; Lee, J.; Kim, H.S.; Kim, N.D.; et al. Molecular inflammation as an underlying mechanism of the aging process and age-related diseases. J. Dent. Res. 2011, 90, 830–840. [Google Scholar] [CrossRef] [PubMed]
- Reuter, S.; Gupta, S.C.; Chaturvedi, M.M.; Aggarwal, B.B. Oxidative stress, inflammation, and cancer: How are they linked? Free Radic. Biol. Med. 2010, 49, 1603–1616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elmarakby, A.A.; Sullivan, J.C. Relationship between oxidative stress and inflammatory cytokines in diabetic nephropathy. Cardiovasc. Ther. 2012, 30, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Cejkova, J.; Ardan, T.; Simonova, Z.; Cejka, C.; Malec, J.; Dotrelova, D.; Brunova, B. Decreased expression of antioxidant enzymes in the conjunctival epithelium of dry eye (Sjögren’s Syndrome) and its possible contribution to the development of ocular surface oxidative injuries. Histol. Histopathol. 2008, 23, 1477–1483. [Google Scholar] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, X.; Ong, H.L.; Ambudkar, I. TRP Channel Involvement in Salivary Glands—Some Good, Some Bad. Cells 2018, 7, 74. https://doi.org/10.3390/cells7070074
Liu X, Ong HL, Ambudkar I. TRP Channel Involvement in Salivary Glands—Some Good, Some Bad. Cells. 2018; 7(7):74. https://doi.org/10.3390/cells7070074
Chicago/Turabian StyleLiu, Xibao, Hwei Ling Ong, and Indu Ambudkar. 2018. "TRP Channel Involvement in Salivary Glands—Some Good, Some Bad" Cells 7, no. 7: 74. https://doi.org/10.3390/cells7070074