The Interplay among PINK1/PARKIN/Dj-1 Network during Mitochondrial Quality Control in Cancer Biology: Protein Interaction Analysis


Abstract
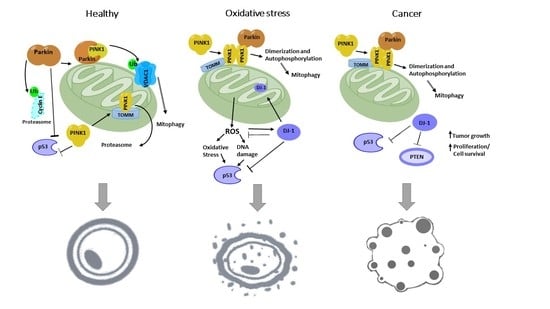
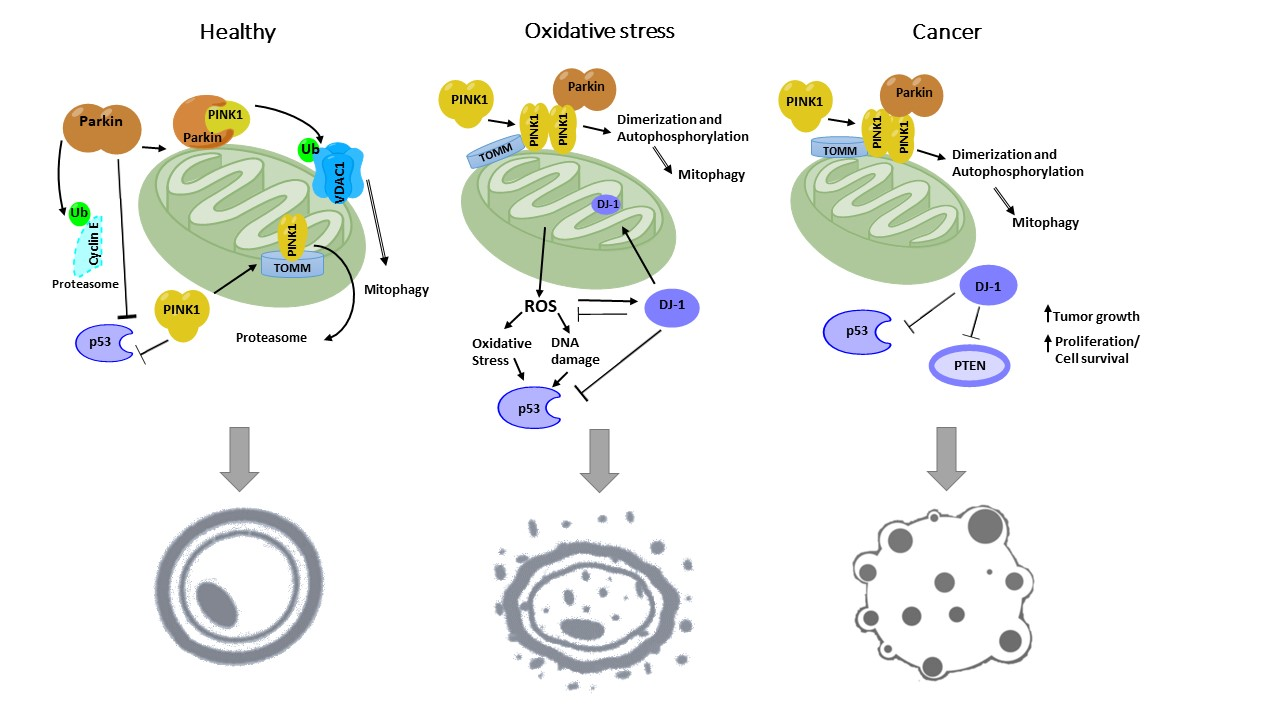
:
1. Biochemistry of Reactive Oxygen and Nitrogen Species (ROS/RNS)
2. How are Free Radicals Produced in the Mitochondria?
3. Mitochondrial Quality Control Systems
3.1. PINK1′s Role in Mitochondrial Quality Control
3.2. PARKIN’s Role in Mitochondrial Quality Control.
3.3. DJ-1 in Mitochondrial Quality Control
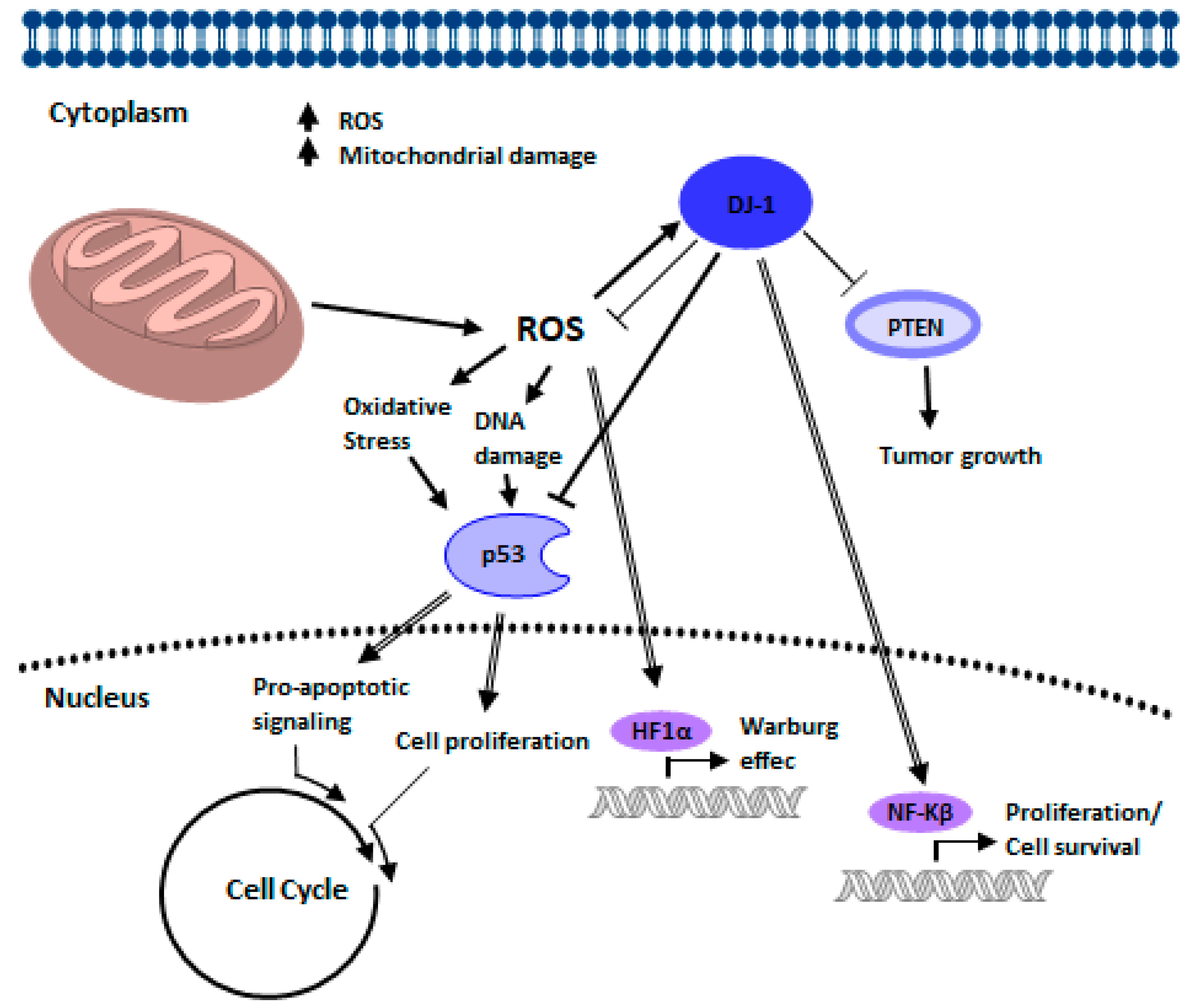
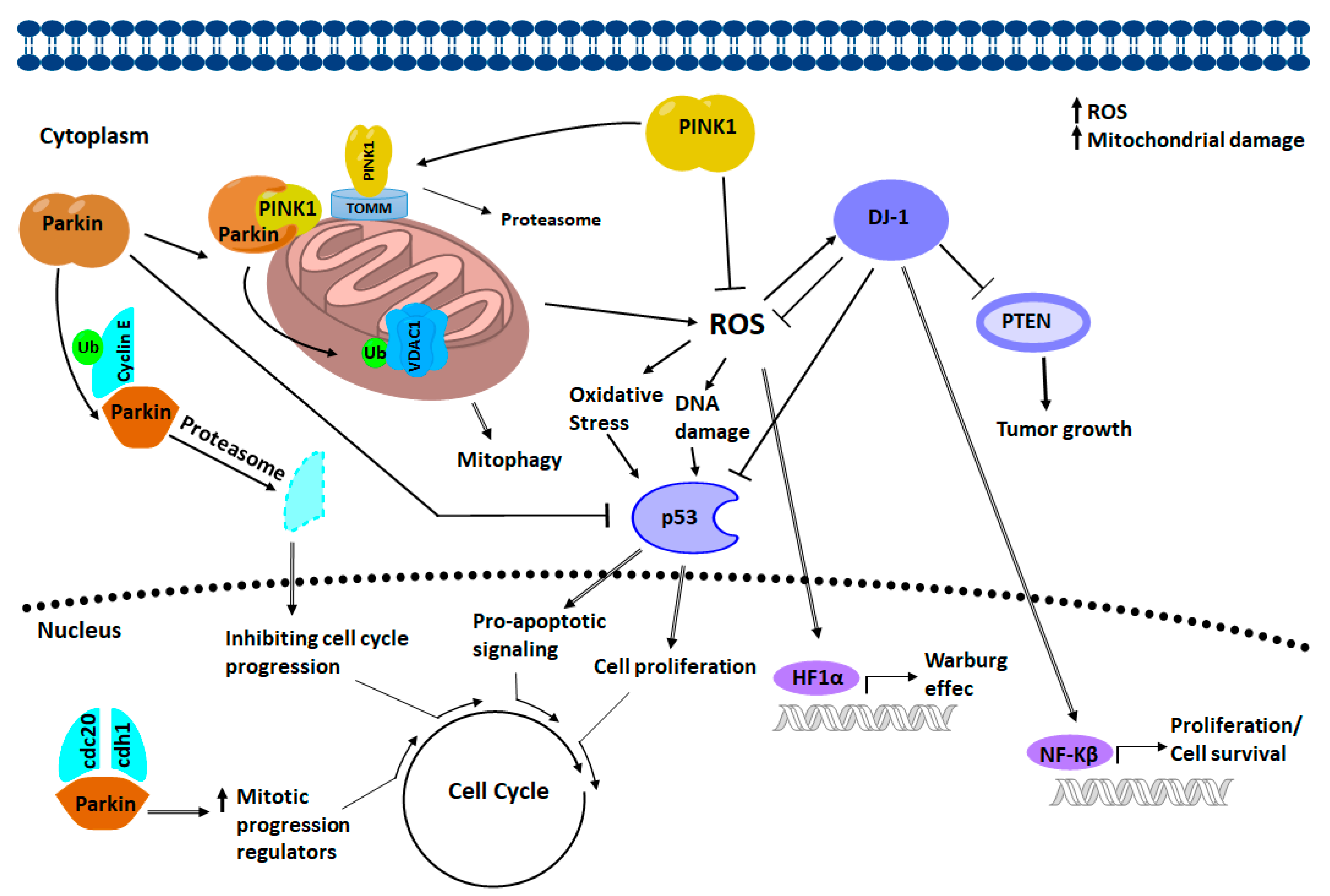
4. Relation of PINK-1/PARKIN/DJ-1 Network in Cancer Biology
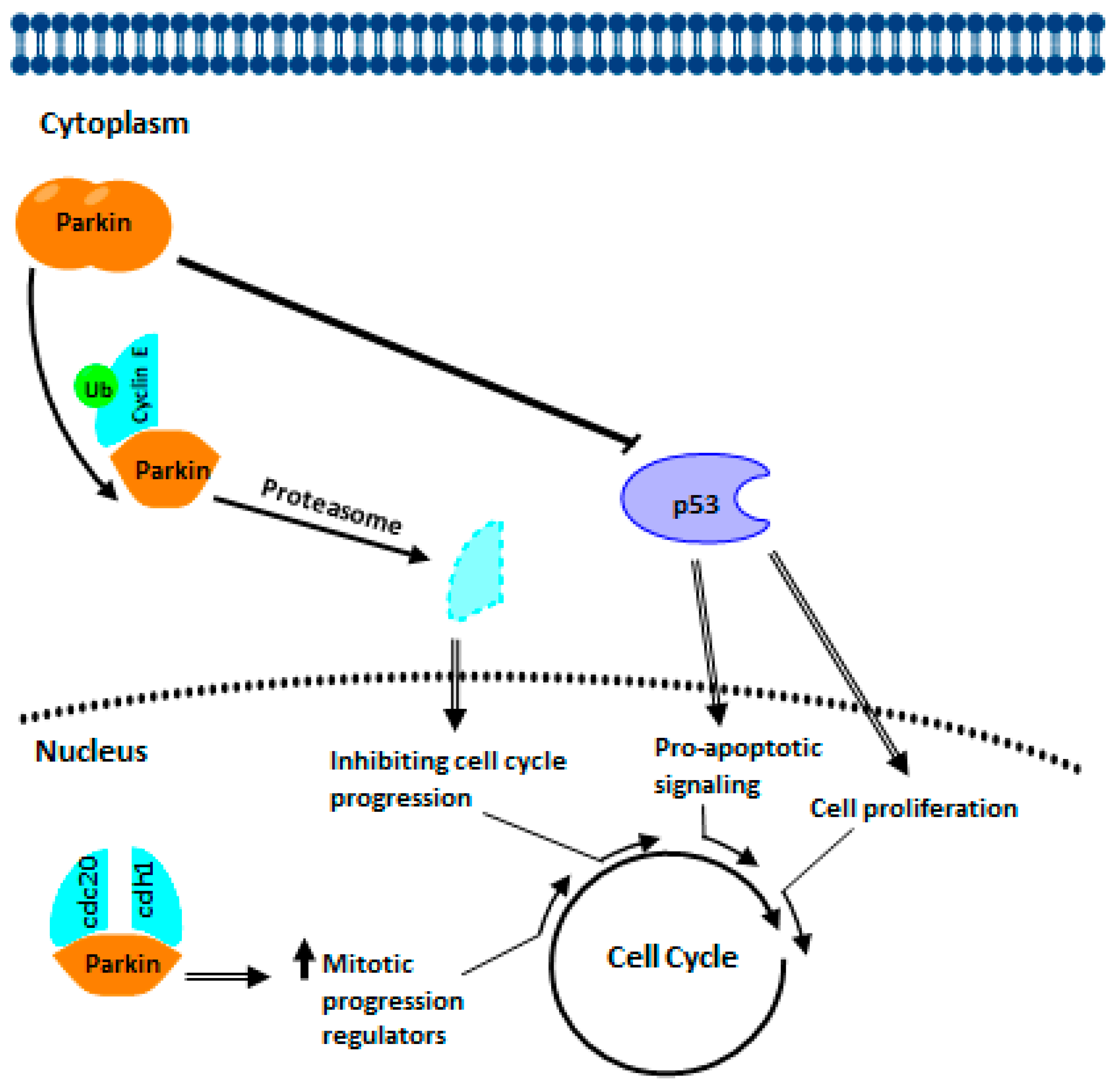
4.1. PARKIN Signaling in Cancer Biology
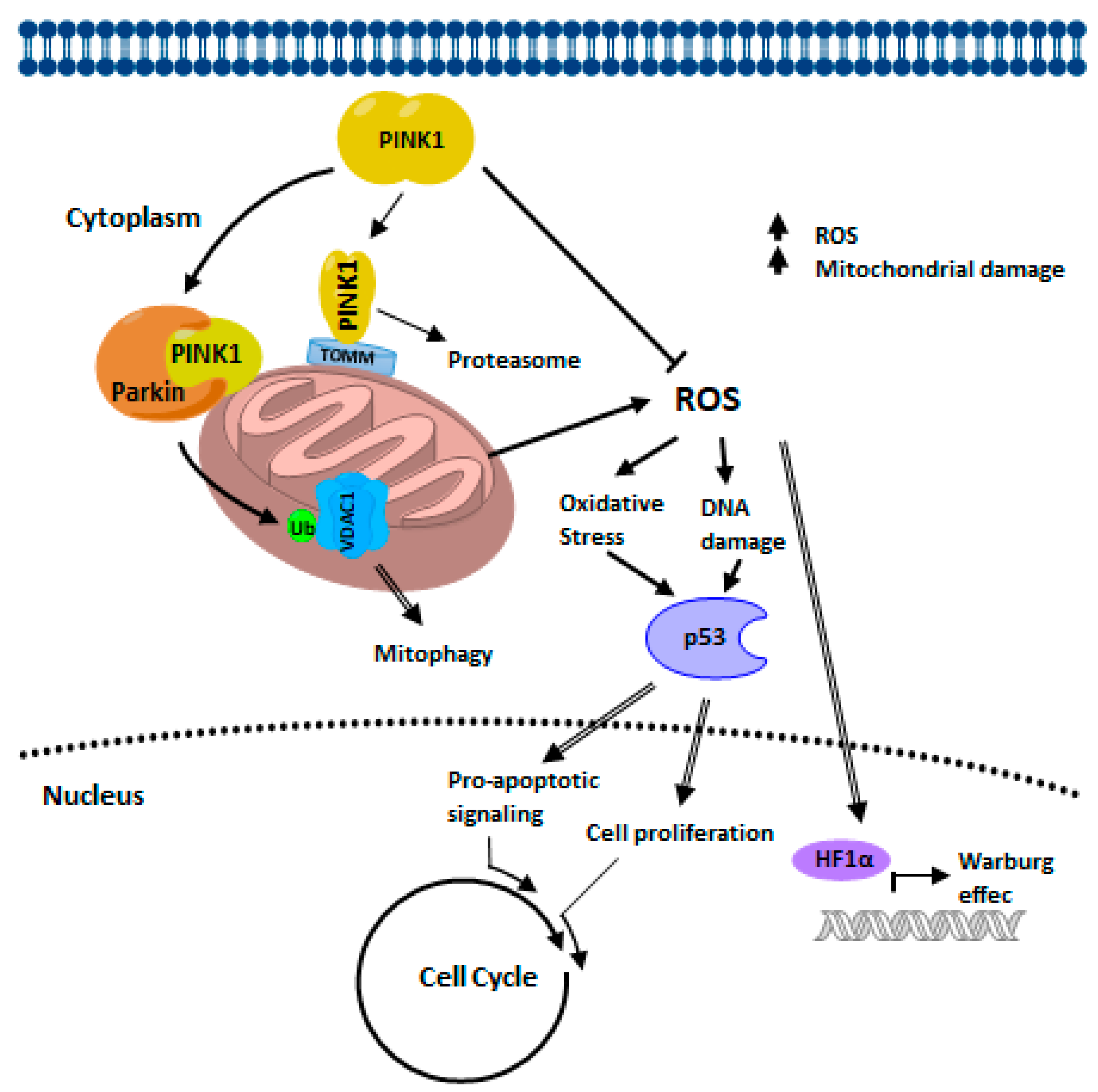
4.2. PINK1 Signaling in Cancer Biology
4.3. DJ-1 Signaling in Cancer Biology
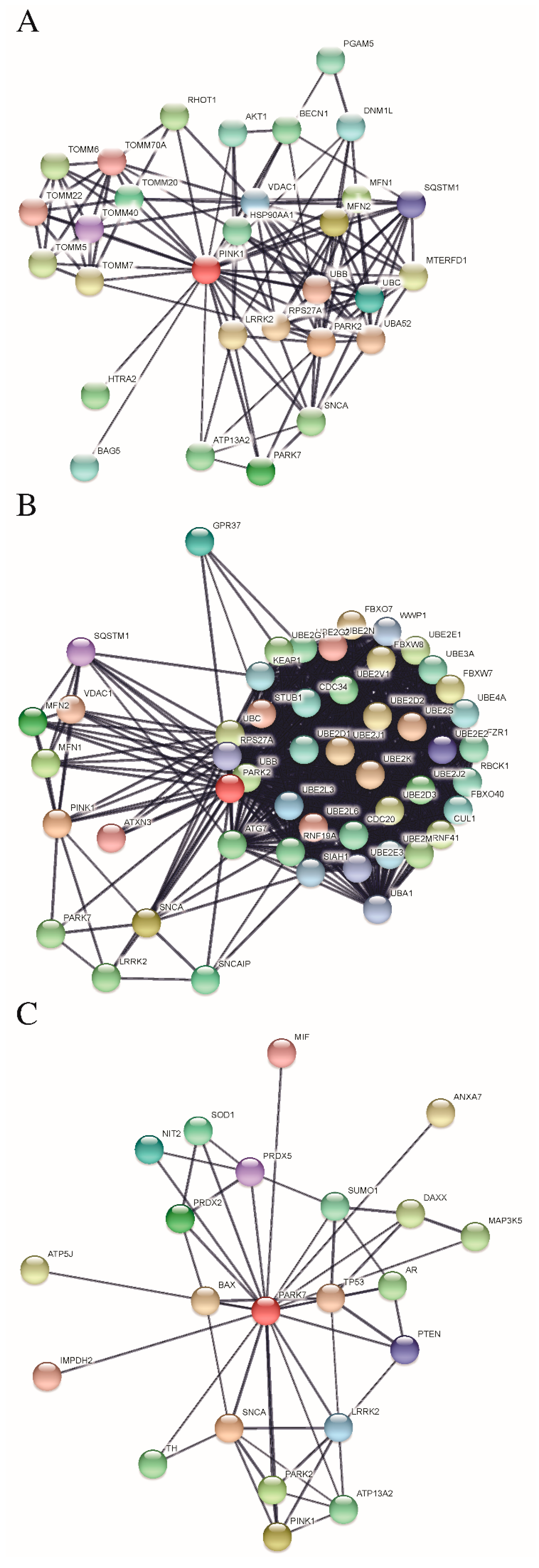
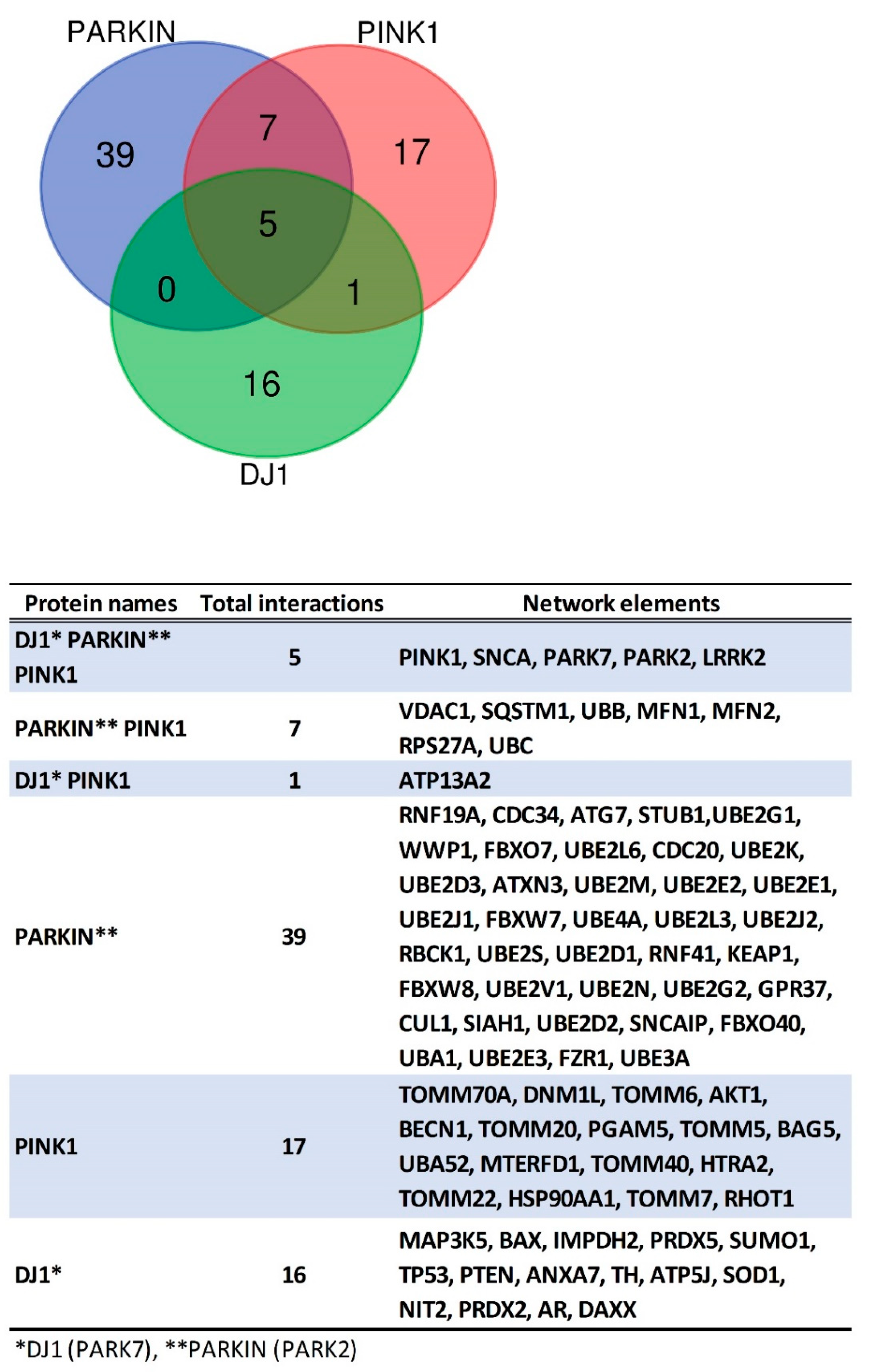
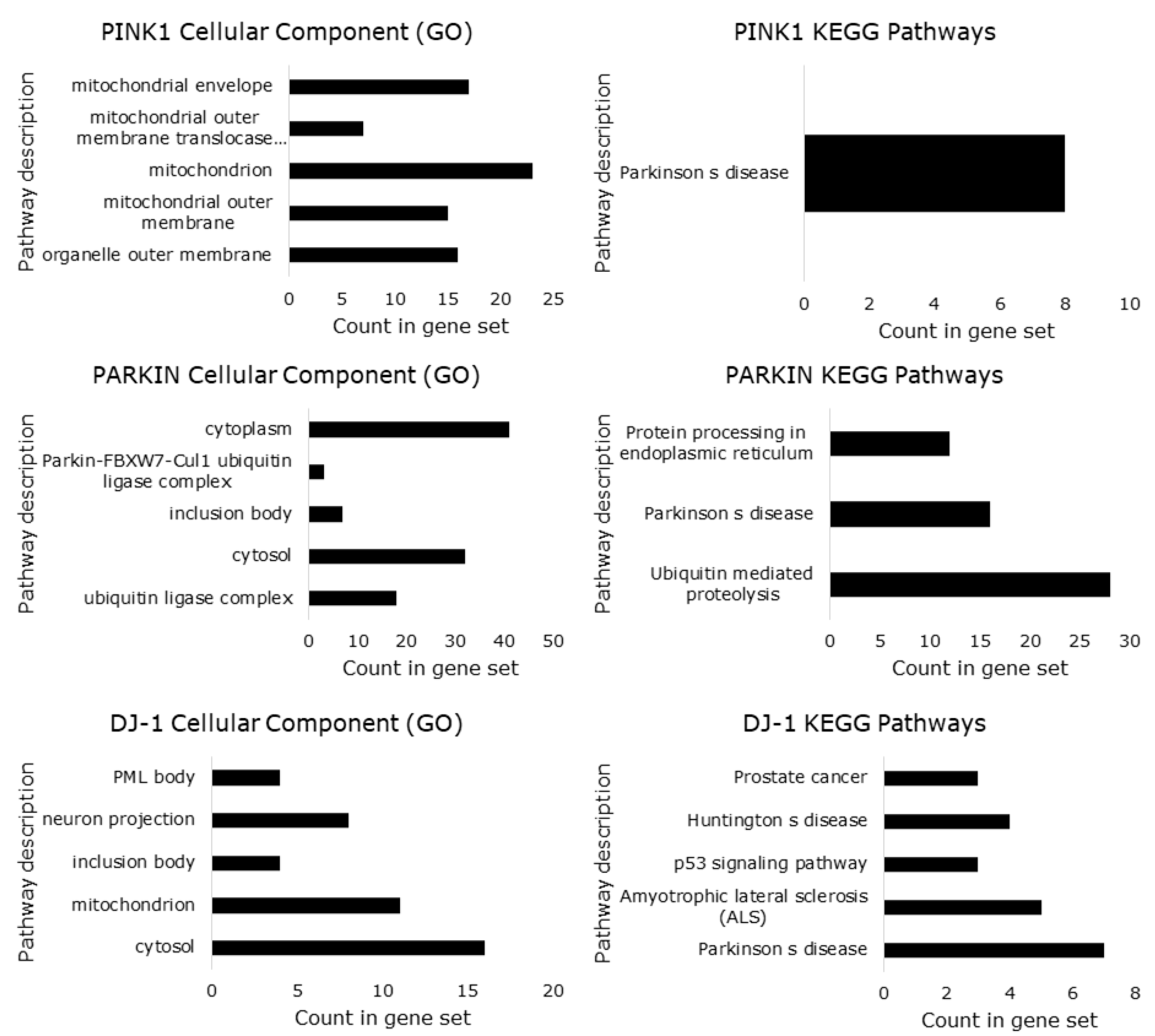
5. Protein-Protein Interactions Analysis of PINK1/PARKIN/DJ-1 Network
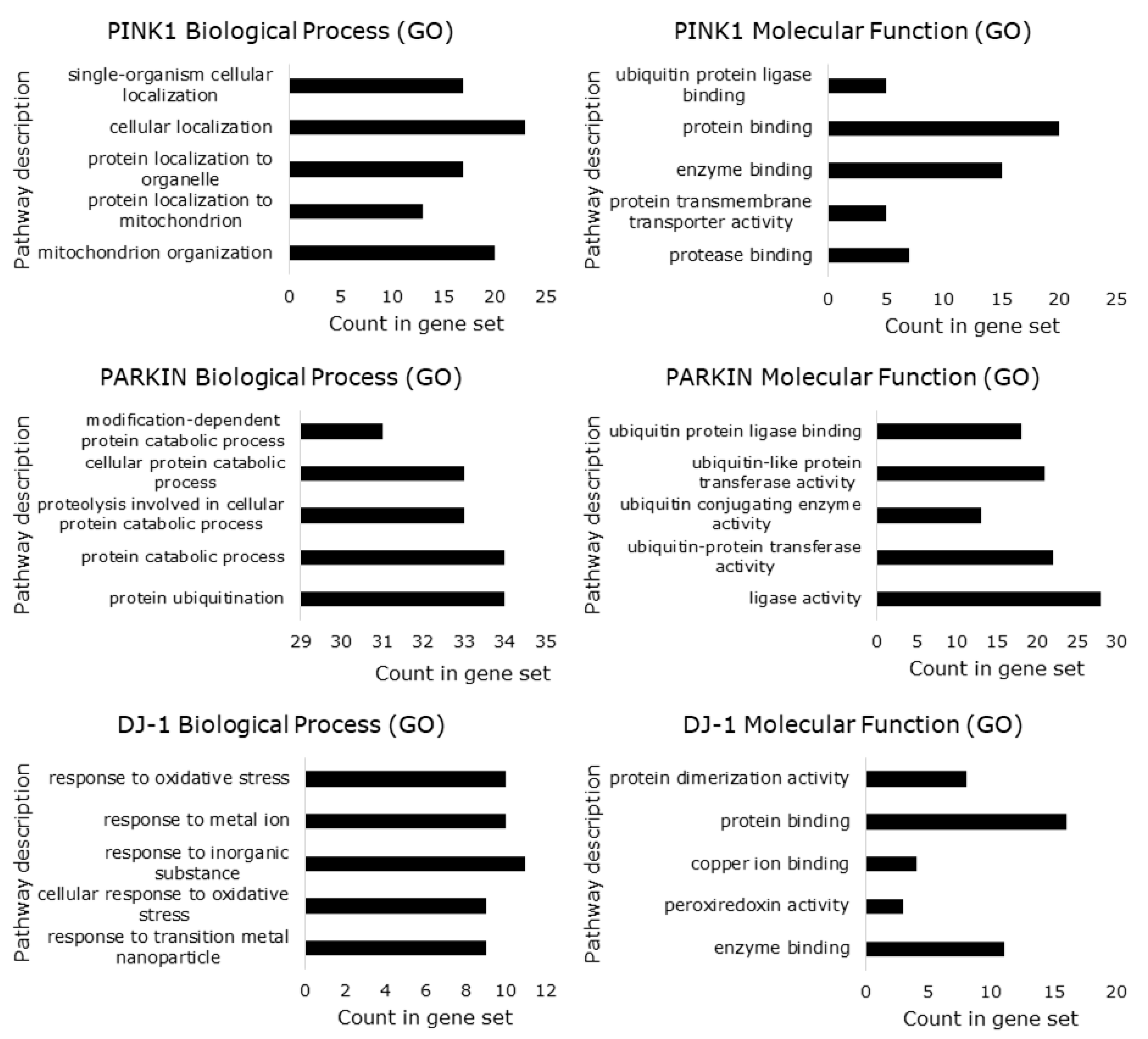
6. Predicted Transcription Factor Regulating PINK1, PARK2 and PARK7 Gene Expression
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Poyton, R.O.; Ball, K.A.; Castello, P.R. Mitochondrial generation of free radicals and hypoxic signaling. Trends Endocrinol. Metab. 2009, 20, 332–340. [Google Scholar] [CrossRef] [PubMed]
- Brand, M.D. The sites and topology of mitochondrial superoxide production. Exp. Gerontol. 2010, 45, 466–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brand, M.D. Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic. Biol. Med. 2016, 100, 14–31. [Google Scholar] [CrossRef] [PubMed]
- Fontmorin, J.M.; Burgos Castillo, R.C.; Tang, W.Z.; Sillanpää, M. Stability of 5,5-dimethyl-1-pyrroline-n-oxide as a spin-trap for quantification of hydroxyl radicals in processes based on fenton reaction. Water Res. 2016, 99, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Plecitá-Hlavatá, L.; Ježek, P. Integration of superoxide formation and cristae morphology for mitochondrial redox signaling. Int. J. Biochem. Cell Biol. 2016, 80, 31–50. [Google Scholar] [CrossRef] [PubMed]
- Ježek, P.; Hlavatá, L. Mitochondria in homeostasis of reactive oxygen species in cell, tissues, and organism. Int. J. Biochem. Cell Biol. 2005, 37, 2478–2503. [Google Scholar] [CrossRef] [PubMed]
- Kühlbrandt, W. Structure and function of mitochondrial membrane protein complexes. BMC Biol. 2015, 13, 89. [Google Scholar] [CrossRef] [PubMed]
- Schlattner, U.; Tokarska-Schlattner, M.; Rousseau, D.; Boissan, M.; Mannella, C.; Epand, R.; Lacombe, M.-L. Mitochondrial cardiolipin/phospholipid trafficking: The role of membrane contact site complexes and lipid transfer proteins. Chem. Phys. Lipids 2014, 179, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Huttemann, M.; Lee, I.; Pecinova, A.; Pecina, P.; Przyklenk, K.; Doan, J.W. Regulation of oxidative phosphorylation, the mitochondrial membrane potential, and their role in human disease. J. Bioenerg. Biomembr. 2008, 40, 445–456. [Google Scholar] [CrossRef] [PubMed]
- Wittig, I.; Carrozzo, R.; Santorelli, F.M.; Schägger, H. Supercomplexes and subcomplexes of mitochondrial oxidative phosphorylation. BBA Bioenerg. 2006, 1757, 1066–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schägger, H.; Pfeiffer, K. Supercomplexes in the respiratory chains of yeast and mammalian mitochondria. EMBO J. 2000, 19, 1777–1783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bornhövd, C.; Vogel, F.; Neupert, W.; Reichert, A.S. Mitochondrial membrane potential is dependent on the oligomeric state of f1f0-atp synthase supracomplexes. J. Biol. Chem. 2006, 281, 13990–13998. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, C.L.; Perevoshchikova, I.V.; Hey-Mogensen, M.; Orr, A.L.; Brand, M.D. Sites of reactive oxygen species generation by mitochondria oxidizing different substrates. Redox Biol. 2013, 1, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Lenaz, G. The mitochondrial production of reactive oxygen species: Mechanisms and implications in human pathology. IUBMB Life 2001, 52, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Andreyev, A.I.; Kushnareva, Y.E.; Starkov, A.A. Mitochondrial metabolism of reactive oxygen species. Biochemistry 2005, 70, 200–214. [Google Scholar] [CrossRef] [PubMed]
- Cadenas, E.; Davies, K.J.A. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic. Biol. Med. 2000, 29, 222–230. [Google Scholar] [CrossRef]
- Hirst, J.; Carroll, J.; Fearnley, I.M.; Shannon, R.J.; Walker, J.E. The nuclear encoded subunits of complex I from bovine heart mitochondria. BBA Bioenerg. 2003, 1604, 135–150. [Google Scholar] [CrossRef] [Green Version]
- Sazanov, L.A. Respiratory complex I: Mechanistic and structural insights provided by the crystal structure of the hydrophilic domain. Biochemistry 2007, 46, 2275–2288. [Google Scholar] [CrossRef] [PubMed]
- Lambert, A.J.; Brand, M.D. Inhibitors of the quinone-binding site allow rapid superoxide production from mitochondrial NADH: Ubiquinone oxidoreductase (complex I). J. Biol. Chem. 2004, 279, 39414–39420. [Google Scholar] [CrossRef] [PubMed]
- Santiago, A.P.S.A.; Chaves, E.A.; Oliveira, M.F.; Galina, A. Reactive oxygen species generation is modulated by mitochondrial kinases: Correlation with mitochondrial antioxidant peroxidases in rat tissues. Biochimie 2008, 90, 1566–1577. [Google Scholar] [CrossRef] [PubMed]
- Castello, P.R.; David, P.S.; McClure, T.; Crook, Z.; Poyton, R.O. Mitochondrial cytochrome oxidase produces nitric oxide under hypoxic conditions: Implications for oxygen sensing and hypoxic signaling in eukaryotes. Cell Metab. 2006, 3, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Rhee, H.-W.; Zou, P.; Udeshi, N.D.; Martell, J.D.; Mootha, V.K.; Carr, S.A.; Ting, A.Y. Proteomic mapping of mitochondria in living cells via spatially restricted enzymatic tagging. Science 2013, 339, 1328–1331. [Google Scholar] [CrossRef] [PubMed]
- Goard, C.A.; Schimmer, A.D. Mitochondrial matrix proteases as novel therapeutic targets in malignancy. Oncogene 2014, 33, 2690–2699. [Google Scholar] [CrossRef] [PubMed]
- Parra, V.; Verdejo, H.; del Campo, A.; Pennanen, C.; Kuzmicic, J.; Iglewski, M.; Hill, J.A.; Rothermel, B.A.; Lavandero, S. The complex interplay between mitochondrial dynamics and cardiac metabolism. J. Bioenerg. Biomembr. 2011, 43, 47–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westermann, B. Merging mitochondria matters: Cellular role and molecular machinery of mitochondrial fusion. EMBO Rep. 2002, 3, 527–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishra, P.; Chan, D.C. Mitochondrial dynamics and inheritance during cell division, development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 634. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.C. Fusion and fission: Interlinked processes critical for mitochondrial health. Annu. Rev. Genet. 2012, 46, 265–287. [Google Scholar] [CrossRef] [PubMed]
- Delettre, C.; Lenaers, G.; Griffoin, J.-M.; Gigarel, N.; Lorenzo, C.; Belenguer, P.; Pelloquin, L.; Grosgeorge, J.; Turc-Carel, C.; Perret, E.; et al. Nuclear gene opa1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat. Genet. 2000, 26, 207. [Google Scholar] [CrossRef] [PubMed]
- Santel, A.; Fuller, M.T. Control of mitochondrial morphology by a human mitofusin. J. Cell Sci. 2001, 114, 867–874. [Google Scholar] [PubMed]
- Richter, V.; Singh, A.P.; Kvansakul, M.; Ryan, M.T.; Osellame, L.D. Splitting up the powerhouse: Structural insights into the mechanism of mitochondrial fission. Cell. Mol. Life Sci. 2015, 72, 3695–3707. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Jeong, S.-Y.; Karbowski, M.; Smith, C.L.; Youle, R.J. Roles of the mammalian mitochondrial fission and fusion mediators fis1, drp1, and opa1 in apoptosis. Mol. Biol. Cell 2004, 15, 5001–5011. [Google Scholar] [CrossRef] [PubMed]
- Labrousse, A.M.; Zappaterra, M.D.; Rube, D.A.; van der Bliek, A.M. C. elegans dynamin-related protein drp-1 controls severing of the mitochondrial outer membrane. Mol. Cell 1999, 4, 815–826. [Google Scholar] [CrossRef]
- Lemasters, J.J. Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuv. Res. 2005, 8, 3–5. [Google Scholar] [CrossRef] [PubMed]
- Pickles, S.; Vigié, P.; Youle, R.J. Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr. Biol. 2018, 28, R170–R185. [Google Scholar] [CrossRef] [PubMed]
- Liesa, M.; Shirihai, O.S. Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab. 2013, 17, 491–506. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [PubMed]
- Bonifati, V.; Rizzu, P.; Squitieri, F.; Krieger, E.; Vanacore, N.; van Swieten, J.C.; Brice, A.; van Duijn, C.M.; Oostra, B.; Meco, G.; et al. Dj-1 (park7), a novel gene for autosomal recessive, early onset parkinsonism. Neurol. Sci. 2003, 24, 159–160. [Google Scholar] [CrossRef] [PubMed]
- Kitada, T.; Asakawa, S.; Matsumine, H.; Hattori, N.; Minoshima, S.; Shimizu, N.; Mizuno, Y. Positional cloning of the autosomal recessive juvenile parkinsonism (ar-jp) gene and its diversity in deletion mutations. Parkinsonism Relat. Disord. 1999, 5, 163–168. [Google Scholar] [CrossRef]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.K.; Harvey, K.; Gispert, S.; Ali, Z.; Del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary early-onset parkinson’s disease caused by mutations in pink1. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef] [PubMed]
- Valente, E.M.; Salvi, S.; Ialongo, T.; Marongiu, R.; Elia, A.E.; Caputo, V.; Romito, L.; Albanese, A.; Dallapiccola, B.; Bentivoglio, A.R. Pink1 mutations are associated with sporadic early-onset parkinsonism. Ann. Neurol. 2004, 56, 336–341. [Google Scholar] [CrossRef] [PubMed]
- Bernardini, J.P.; Lazarou, M.; Dewson, G. Parkin and mitophagy in cancer. Oncogene 2016, 36, 1315. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Lou, S.; Ying, M.; Yang, B. Dj-1 as a human oncogene and potential therapeutic target. Biochem. Pharmacol. 2015, 93, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Schumacher, S.E.; Wu, W.H.; Tang, F.; Beroukhim, R.; Chan, T.A. Pan-cancer analysis links park2 to bcl-xl-dependent control of apoptosis. Neoplasia 2017, 19, 75–83. [Google Scholar] [CrossRef] [PubMed]
- O’Flanagan, C.H.; O’Neill, C. Pink1 signalling in cancer biology. BBA Rev. Cancer 2014, 1846, 590–598. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Lin, D.; Yin, D.; Koeffler, H.P. An emerging role of park2 in cancer. J. Mol. Med. 2014, 92, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Mukherjee, S.; Fan, X.; Salameh, A.; Mujoo, K.; Huang, Z.; Li, L.; Salazar, G.T.; Zhang, N.; An, Z. Novel association of dj-1 with her3 potentiates her3 activation and signaling in cancer. Oncotarget 2016, 7, 65758–65769. [Google Scholar] [CrossRef] [PubMed]
- Cherra, S.J., III; Dagda, R.K.; Tandon, A.; Chu, C.T. Mitochondrial autophagy as a compensatory response to pink1 deficiency. Autophagy 2009, 5, 1213–1214. [Google Scholar] [CrossRef] [PubMed]
- Junn, E.; Jang, W.H.; Zhao, X.; Jeong, B.S.; Mouradian, M.M. Mitochondrial localization of dj-1 leads to enhanced neuroprotection. J. Neurosci. Res. 2009, 87, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Lev, N.; Ickowicz, D.; Melamed, E.; Offen, D. Oxidative insults induce dj-1 upregulation and redistribution: Implications for neuroprotection. Neurotoxicology 2008, 29, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Narendra, D.; Tanaka, A.; Suen, D.-F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narendra, D.; Tanaka, A.; Suen, D.-F.; Youle, R.J. Parkin-induced mitophagy in the pathogenesis of parkinson disease. Autophagy 2009, 5, 706–708. [Google Scholar] [CrossRef] [PubMed]
- Silvestri, L.; Caputo, V.; Bellacchio, E.; Atorino, L.; Dallapiccola, B.; Valente, E.M.; Casari, G. Mitochondrial import and enzymatic activity of pink1 mutants associated to recessive parkinsonism. Hum. Mol. Genet. 2005, 14, 3477–3492. [Google Scholar] [CrossRef] [PubMed]
- Vives-Bauza, C.; Zhou, C.; Huang, Y.; Cui, M.; de Vries, R.L.A.; Kim, J.; May, J.; Tocilescu, M.A.; Liu, W.; Ko, H.S.; et al. Pink1-dependent recruitment of parkin to mitochondria in mitophagy. Proc. Natl. Acad. Sci. USA 2010, 107, 378–383. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Huang, Y.; Shao, Y.; May, J.; Prou, D.; Perier, C.; Dauer, W.; Schon, E.A.; Przedborski, S. The kinase domain of mitochondrial pink1 faces the cytoplasm. Proc. Natl. Acad. Sci. USA 2008, 105, 12022–12027. [Google Scholar] [CrossRef] [PubMed]
- Narendra, D.P. Parkin/pink1 pathway for the selective isolation and degradation of impaired mitochondria. In Mitochondrial Mechanisms of Degeneration and Repair in Parkinson’s Disease; Buhlman, L.M., Ed.; Springer International Publishing: Cham, Switzerland, 2016; pp. 159–182. [Google Scholar]
- Gonzalez-Polo, R.A.; Niso-Santano, M.; Moran, J.M.; Ortiz-Ortiz, M.A.; Bravo-San Pedro, J.M.; Soler, G.; Fuentes, J.M. Silencing dj-1 reveals its contribution in paraquat-induced autophagy. J. Neurochem. 2009, 109, 889–898. [Google Scholar] [CrossRef] [PubMed]
- Vasseur, S.; Afzal, S.; Tardivel-Lacombe, J.; Park, D.S.; Iovanna, J.L.; Mak, T.W. Dj-1/park7 is an important mediator of hypoxia-induced cellular responses. Proc. Natl. Acad. Sci. USA 2009, 106, 1111–1116. [Google Scholar] [CrossRef] [PubMed]
- Clark, I.E.; Dodson, M.W.; Jiang, C.; Cao, J.H.; Huh, J.R.; Seol, J.H.; Yoo, S.J.; Hay, B.A.; Guo, M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 2006, 441, 1162–1166. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.; Dodson, M.W.; Huang, H.; Guo, M. The parkinson’s disease genes pink1 and parkin promote mitochondrial fission and/or inhibit fusion in drosophila. Proc. Natl. Acad. Sci. USA 2008, 105, 14503–14508. [Google Scholar] [CrossRef] [PubMed]
- Poole, A.C.; Thomas, R.E.; Andrews, L.A.; McBride, H.M.; Whitworth, A.J.; Pallanck, L.J. The pink1/parkin pathway regulates mitochondrial morphology. Proc. Natl. Acad. Sci. USA 2008, 105, 1638–1643. [Google Scholar] [CrossRef] [PubMed]
- Dagda, R.K.; Cherra, S.J., III; Kulich, S.M.; Tandon, A.; Park, D.; Chu, C.T. Loss of pink1 function promotes mitophagy through effects on oxidative stress and mitochondrial fission. J. Biol. Chem. 2009, 284, 13843–13855. [Google Scholar] [CrossRef] [PubMed]
- Dagda, R.K.; Zhu, J.; Chu, C.T. Mitochondrial kinases in parkinson’s disease: Converging insights from neurotoxin and genetic models. Mitochondrion 2009, 9, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Exner, N.; Treske, B.; Paquet, D.; Holmstrom, K.; Schiesling, C.; Gispert, S.; Carballo-Carbajal, I.; Berg, D.; Hoepken, H.-H.; Gasser, T.; et al. Loss-of-function of human pink1 results in mitochondrial pathology and can be rescued by parkin. J. Neurosci. 2007, 27, 12413–12418. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Ouyang, Y.; Yang, L.; Beal, M.F.; McQuibban, A.; Vogel, H.; Lu, B. Pink1 regulates mitochondrial dynamics through interaction with the fission/fusion machinery. Proc. Natl. Acad. Sci. USA 2008, 105, 7070–7075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamp, F.; Exner, N.; Lutz, A.K.; Wender, N.; Hegermann, J.; Brunner, B.; Nuscher, B.; Bartels, T.; Giese, A.; Beyer, K.; et al. Inhibition of mitochondrial fusion by α-synuclein is rescued by pink1, parkin and dj-1. EMBO J. 2010, 29, 3571–3589. [Google Scholar] [CrossRef] [PubMed]
- Lutz, A.K.; Exner, N.; Fett, M.E.; Schlehe, J.S.; Kloos, K.; Lämmermann, K.; Brunner, B.; Kurz-Drexler, A.; Vogel, F.; Reichert, A.S.; et al. Loss of parkin or pink1 function increases drp1-dependent mitochondrial fragmentation. J. Biol. Chem. 2009, 284, 22938–22951. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Dorn, G.W. Pink1-phosphorylated mitofusin 2 is a parkin receptor for culling damaged mitochondria. Science 2013, 340, 471–475. [Google Scholar] [CrossRef] [PubMed]
- Gong, G.; Song, M.; Csordas, G.; Kelly, D.P.; Matkovich, S.J.; Dorn, G.W. Parkin-mediated mitophagy directs perinatal cardiac metabolic maturation in mice. Science 2015, 350. [Google Scholar] [CrossRef] [PubMed]
- McCoy, M.K.; Cookson, M.R. Dj-1 regulation of mitochondrial function and autophagy through oxidative stress. Autophagy 2011, 7, 531–532. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.H.; Smith, P.D.; Aleyasin, H.; Hayley, S.; Mount, M.P.; Pownall, S.; Wakeham, A.; You-Ten, A.J.; Kalia, S.K.; Horne, P.; et al. Hypersensitivity of dj-1-deficient mice to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyrindine (MPTP) and oxidative stress. Proc. Natl. Acad. Sci. USA 2005, 102, 5215–5220. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Kim, S.Y.; Cha, G.H.; Lee, S.B.; Kim, S.; Chung, J. Drosophila dj-1 mutants show oxidative stress-sensitive locomotive dysfunction. Gene 2005, 361, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Unoki, M.; Nakamura, Y. Growth-suppressive effects of bpoz and egr2, two genes involved in the pten signaling pathway. Oncogene 2001, 20, 4457. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, S.; Muqit, M.M.K.; Stanyer, L.; Healy, D.G.; Abou-Sleiman, P.M.; Hargreaves, I.; Heales, S.; Ganguly, M.; Parsons, L.; Lees, A.J.; et al. Pink1 protein in normal human brain and parkinson’s disease. Brain 2006, 129, 1720–1731. [Google Scholar] [CrossRef] [PubMed]
- Petit, A.; Kawarai, T.; Paitel, E.; Sanjo, N.; Maj, M.; Scheid, M.; Chen, F.; Gu, Y.; Hasegawa, H.; Salehi-Rad, S.; et al. Wild-type pink1 prevents basal and induced neuronal apoptosis, a protective effect abrogated by parkinson disease-related mutations. J. Biol. Chem. 2005, 280, 34025–34032. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.-L.; Chou, A.-H.; Yeh, T.-H.; Li, A.H.; Chen, Y.-L.; Kuo, Y.-L.; Tsai, S.-R.; Yu, S.-T. Pink1 mutants associated with recessive parkinson’s disease are defective in inhibiting mitochondrial release of cytochrome c. Neurobiol. Dis. 2007, 28, 216–226. [Google Scholar] [CrossRef] [PubMed]
- Wood-Kaczmar, A.; Gandhi, S.; Yao, Z.; Abramov, A.S.Y.; Miljan, E.A.; Keen, G.; Stanyer, L.; Hargreaves, I.; Klupsch, K.; Deas, E.; et al. Pink1 is necessary for long term survival and mitochondrial function in human dopaminergic neurons. PLoS ONE 2008, 3, e2455. [Google Scholar] [CrossRef]
- Gautier, C.A.; Kitada, T.; Shen, J. Loss of pink1 causes mitochondrial functional defects and increased sensitivity to oxidative stress. Proc. Natl. Acad. Sci. USA 2008, 105, 11364–11369. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Lee, S.B.; Lee, S.; Kim, Y.; Song, S.; Kim, S.; Bae, E.; Kim, J.; Shong, M.H.; Kim, J.M.; et al. Mitochondrial dysfunction in drosophila pink1 mutants is complemented by parkin. Nature 2006, 441, 1157–1161. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Qian, L.; Xiong, H.; Liu, J.; Neckameyer, W.S.; Oldham, S.; Xia, K.; Wang, J.; Bodmer, R.; Zhang, Z. Antioxidants protect pink1-dependent dopaminergic neurons in drosophila. Proc. Natl. Acad. Sci. USA 2006, 103, 13520–13525. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Gehrke, S.; Imai, Y.; Huang, Z.; Ouyang, Y.; Wang, J.-W.; Yang, L.; Beal, M.F.; Vogel, H.; Lu, B. Mitochondrial pathology and muscle and dopaminergic neuron degeneration caused inactivation of drosophila pink1 is rescued by by parkin. Proc. Natl. Acad. Sci. USA 2006, 103, 10793–10798. [Google Scholar] [CrossRef] [PubMed]
- Amo, T.; Sato, S.; Saiki, S.; Wolf, A.M.; Toyomizu, M.; Gautier, C.A.; Shen, J.; Ohta, S.; Hattori, N. Mitochondrial membrane potential decrease caused by loss of pink1 is not due to proton leak, but to respiratory chain defects. Neurobiol. Dis. 2011, 41, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Gautier, C.A.; Giaime, E.; Caballero, E.; Núñez, L.; Song, Z.; Chan, D.; Villalobos, C.; Shen, J. Regulation of mitochondrial permeability transition pore by pink1. Mol. Neurodegener. 2012, 7, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morais, V.A.; Verstreken, P.; Roethig, A.; Smet, J.; Snellinx, A.; Vanbrabant, M.; Haddad, D.; Frezza, C.; Mandemakers, W.; Vogt-Weisenhorn, D.; et al. Parkinson’s disease mutations in pink1 result in decreased complex i activity and deficient synaptic function. EMBO Mol. Med. 2009, 1, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Heeman, B.; Van den Haute, C.; Aelvoet, S.-A.; Valsecchi, F.; Rodenburg, R.J.; Reumers, V.; Debyser, Z.; Callewaert, G.; Koopman, W.J.H.; Willems, P.H.G.M.; et al. Depletion of pink1 affects mitochondrial metabolism, calcium homeostasis and energy maintenance. J. Cell Sci. 2011, 124, 1115–1125. [Google Scholar] [CrossRef] [PubMed]
- Pridgeon, J.W.; Olzmann, J.A.; Chin, L.-S.; Li, L. Pink1 protects against oxidative stress by phosphorylating mitochondrial chaperone trap1. PLoS Biol. 2007, 5, e172. [Google Scholar] [CrossRef] [PubMed]
- Deas, E.; Wood, N.W.; Plun-Favreau, H. Mitophagy and parkinson’s disease: The pink1–parkin link. BBA Mol. Cell Res. 2011, 1813, 623–633. [Google Scholar] [CrossRef] [PubMed]
- Shiba-Fukushima, K.; Imai, Y.; Yoshida, S.; Ishihama, Y.; Kanao, T.; Sato, S.; Hattori, N. Pink1-mediated phosphorylation of the parkin ubiquitin-like domain primes mitochondrial translocation of parkin and regulates mitophagy. Sci. Rep. 2012, 2, 1002. [Google Scholar] [CrossRef] [PubMed]
- Nadtochiy, S.M.; Tompkins, A.J.; Brookes, P.S. Different mechanisms of mitochondrial proton leak in ischaemia/reperfusion injury and preconditioning: Implications for pathology and cardioprotection. Biochem. J. 2006, 395, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.M.; Youle, R.J. The accumulation of misfolded proteins in the mitochondrial matrix is sensed by pink1 to induce park2/parkin-mediated mitophagy of polarized mitochondria. Autophagy 2013, 9, 1750–1757. [Google Scholar] [CrossRef] [PubMed]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.-F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 is selectively stabilized on impaired mitochondria to activate parkin. PLoS Biol. 2010, 8, e100298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veeriah, S.; Taylor, B.S.; Meng, S.; Fang, F.; Yilmaz, E.; Vivanco, I.; Janakiraman, M.; Schultz, N.; Hanrahan, A.J.; Pao, W.; et al. Somatic mutations of the parkinson’s disease-associated gene park2 in glioblastoma and other human malignancies. Nat. Genet. 2010, 42, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Rankin, C.A.; Joazeiro, C.A.P.; Floor, E.; Hunter, T. E3 ubiquitin-protein ligase activity of parkin is dependent on cooperative interaction of ring finger (triad) elements. J. Biomed. Sci. 2001, 8, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Kazlauskaite, A.; Kondapalli, C.; Gourlay, R.; Campbell, D.G.; Ritorto, M.S.; Hofmann, K.; Alessi, D.R.; Knebel, A.; Trost, M.; Muqit, M.M.K. Parkin is activated by pink1-dependent phosphorylation of ubiquitin at ser65. Biochem. J. 2014, 460, 127–141. [Google Scholar] [CrossRef] [PubMed]
- Bouman, L.; Schlierf, A.; Lutz, A.K.; Shan, J.; Deinlein, A.; Kast, J.; Galehdar, Z.; Palmisano, V.; Patenge, N.; Berg, D.; et al. Parkin is transcriptionally regulated by ATF4: Evidence for an interconnection between mitochondrial stress and er stress. Cell Death Differ. 2011, 18, 769–782. [Google Scholar] [CrossRef] [PubMed]
- Millikin, D.; Meese, E.; Vogelstein, B.; Witkowski, C.; Trent, J. Loss of heterozygosity for loci on the long arm of chromosome-6 in human-malignant melanoma. Cancer Res. 1991, 51, 5449–5453. [Google Scholar] [PubMed]
- Cesari, R.; Martin, E.S.; Calin, G.A.; Pentimalli, F.; Bichi, R.; McAdams, H.; Trapasso, F.; Drusco, A.; Shimizu, M.; Mascillo, V.; et al. Parkin, a gene implicated in autosomal recessive juvenile parkinsonism, is a candidate tumor suppressor gene on chromosome 6q25-q27. Proc. Natl. Acad. Sci. USA 2003, 100, 5956–5961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondapalli, C.; Kazlauskaite, A.; Zhang, N.; Woodroof, H.I.; Campbell, D.G.; Gourlay, R.; Burchell, L.; Walden, H.; Macartney, T.J.; Deak, M.; et al. Pink1 is activated by mitochondrial membrane potential depolarization and stimulates parkin e3 ligase activity by phosphorylating serine 65. Open Biol. 2012, 2, 120080. [Google Scholar] [CrossRef] [PubMed]
- Koyano, F.; Okatsu, K.; Kosako, H.; Tamura, Y.; Go, E.; Kimura, M.; Kimura, Y.; Tsuchiya, H.; Yoshihara, H.; Hirokawa, T.; et al. Ubiquitin is phosphorylated by pink1 to activate parkin. Nature 2014, 510, 162. [Google Scholar] [CrossRef] [PubMed]
- Kane, L.A.; Lazarou, M.; Fogel, A.I.; Li, Y.; Yamano, K.; Sarraf, S.A.; Banerjee, S.; Youle, R.J. Pink1 phosphorylates ubiquitin to activate parkin e3 ubiquitin ligase activity. J. Cell Biol. 2014, 205, 143. [Google Scholar] [CrossRef] [PubMed]
- Wauer, T.; Komander, D. Structure of the human parkin ligase domain in an autoinhibited state. EMBO J. 2013, 32, 2099–2112. [Google Scholar] [CrossRef] [PubMed]
- Trempe, J.-F.; Sauvé, V.; Grenier, K.; Seirafi, M.; Tang, M.Y.; Ménade, M.; Al-Abdul-Wahid, S.; Krett, J.; Wong, K.; Kozlov, G.; et al. Structure of parkin reveals mechanisms for ubiquitin ligase activation. Science 2013, 340, 1451. [Google Scholar] [CrossRef] [PubMed]
- Gladkova, C.; Maslen, S.L.; Skehel, J.M.; Komander, D. Mechanism of parkin activation by pink1. Nature 2018, 559, 410–414. [Google Scholar] [CrossRef] [PubMed]
- Ordureau, A.; Sarraf, S.A.; Duda, D.M.; Heo, J.-M.; Jedrykowski, M.P.; Sviderskiy, V.; Olszewski, J.L.; Koerber, J.T.; Xie, T.; Beausoleil, S.A.; et al. Quantitative proteomics reveal a feed-forward model for mitochondrial parkin translocation and ub chain synthesis. Mol. Cell 2014, 56, 360–375. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.-W.; Hang, L.; Yao, T.-P.; Lim, K.-L. Parkin regulation and neurodegenerative disorders. Front. Aging Neurosci. 2015, 7, 248. [Google Scholar] [CrossRef] [PubMed]
- Vandiver, M.S.; Paul, B.D.; Xu, R.; Karuppagounder, S.; Rao, F.; Snowman, A.M.; Ko, H.S.; Lee, Y.I.; Dawson, V.L.; Dawson, T.M.; et al. Sulfhydration mediates neuroprotective actions of parkin. Nat. Commun. 2013, 4, 1626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, T.; Lipton, S.A. Emerging roles of s-nitrosylation in protein misfolding and neurodegenerative diseases. Antioxid. Redox Signal. 2007, 10, 87–102. [Google Scholar] [CrossRef] [PubMed]
- Sunico, C.; Nakamura, T.; Rockenstein, E.; Mante, M.; Adame, A.; Chan, S.; Newmeyer, T.; Masliah, E.; Nakanishi, N.; Lipton, S. S-nitrosylation of parkin as a novel regulator of p53-mediated neuronal cell death in sporadic parkinson’s disease. Mol. Neurodegener. 2013, 8, 29. [Google Scholar] [CrossRef] [PubMed]
- Joselin, A.P.; Hewitt, S.J.; Callaghan, S.M.; Kim, R.H.; Chung, Y.-H.; Mak, T.W.; Shen, J.; Slack, R.S.; Park, D.S. Ros-dependent regulation of parkin and dj-1 localization during oxidative stress in neurons. Hum. Mol. Genet. 2012, 21, 4888–4903. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, N.; Sato, S.; Shiba, K.; Okatsu, K.; Saisho, K.; Gautier, C.A.; Sou, Y.; Saiki, S.; Kawajiri, S.; Sato, F.; et al. Pink1 stabilized by mitochondrial depolarization recruits parkin to damaged mitochondria and activates latent parkin for mitophagy. J. Cell Biol. 2010, 189, 211–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.; Park, J.; Kim, S.; Song, S.; Won, S.-K.; Lee, S.-H.; Kitada, T.; Kim, J.-M.; Chung, J. Pink1 controls mitochondrial localization of parkin through direct phosphorylation. Biochem. Bioph. Res. Commun. 2008, 377, 975–980. [Google Scholar] [CrossRef] [PubMed]
- Geisler, S.; Holmstroem, K.M.; Skujat, D.; Fiesel, F.C.; Rothfuss, O.C.; Kahle, P.J.; Springer, W. Pink1/parkin-mediated mitophagy is dependent on vdac1 and p62/sqstm1. Nat. Cell Biol. 2010, 12, 119–U170. [Google Scholar] [CrossRef] [PubMed]
- Cummins, N.; Götz, J. Shedding light on mitophagy in neurons: What is the evidence for pink1/parkin mitophagy in vivo? Cell. Mol. Life Sci. 2018, 75, 1151–1162. [Google Scholar] [CrossRef] [PubMed]
- Ashrafi, G.; Schlehe, J.S.; LaVoie, M.J.; Schwarz, T.L. Mitophagy of damaged mitochondria occurs locally in distal neuronal axons and requires pink1 and parkin. J. Cell Biol. 2014, 206, 655–670. [Google Scholar] [CrossRef] [PubMed]
- Pickrell, A.M.; Huang, C.-H.; Kennedy, S.R.; Ordureau, A.; Sideris, D.P.; Hoekstra, J.G.; Harper, J.W.; Youle, R.J. Endogenous parkin preserves dopaminergic substantia nigral neurons following mitochondrial DNA mutagenic stress. Neuron 2015, 87, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Pan, T.; Li, X.; Jankovic, J. The association between parkinson’s disease and melanoma. Inter. J. Cancer 2011, 128, 2251–2260. [Google Scholar] [CrossRef] [PubMed]
- Maita, C.; Tsuji, S.; Yabe, I.; Hamada, S.; Ogata, A.; Maita, H.; Iguchi-Ariga, S.M.M.; Sasaki, H.; Ariga, H. Secretion of dj-1 into the serum of patients with parkinson’s disease. Neurosci. Lett. 2008, 431, 86–89. [Google Scholar] [CrossRef] [PubMed]
- Pei, X.; Wu, T.; Li, B.; Tian, X.; Li, Z.; Yang, Q. Increased expression of macrophage migration inhibitory factor and dj-1 contribute to cell invasion and metastasis of nasopharyngeal carcinoma. Int. J. Med. Sci. 2014, 11, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Aron, L.; Klein, P.; Pham, T.-T.; Kramer, E.R.; Wurst, W.; Klein, R. Pro-survival role for parkinson’s associated gene dj-1 revealed in trophically impaired dopaminergic neurons. PLoS Biol. 2010, 8, e1000349. [Google Scholar] [CrossRef] [PubMed]
- Cookson, M.R. Pathways to parkinsonism. Neuron 2003, 37, 7–10. [Google Scholar] [CrossRef]
- Usami, Y.; Hatano, T.; Imai, S.; Kubo, S.; Sato, S.; Saiki, S.; Fujioka, Y.; Ohba, Y.; Sato, F.; Funayama, M.; et al. Dj-1 associates with synaptic membranes. Neurobiol. Dis. 2011, 43, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Ren, H.; Jia, N.; Fei, E.; Zhou, T.; Jiang, P.; Wu, M.; Wang, G. Dj-1 decreases bax expression through repressing p53 transcriptional activity. J. Biol. Chem. 2008, 283, 4022–4030. [Google Scholar] [CrossRef] [PubMed]
- Clements, C.M.; McNally, R.S.; Conti, B.J.; Mak, T.W.; Ting, J.P.-Y. Dj-1, a cancer- and parkinson’s disease-associated protein, stabilizes the antioxidant transcriptional master regulator nrf2. Proc. Natl. Acad. Sci. USA 2006, 103, 15091–15096. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-J.; Park, Y.-J.; Hwang, I.-Y.; Youdim, M.B.H.; Park, K.-S.; Oh, Y.J. Nuclear translocation of dj-1 during oxidative stress-induced neuronal cell death. Free Radic. Biol. Med. 2012, 53, 936–950. [Google Scholar] [CrossRef] [PubMed]
- Nagakubo, D.; Taira, T.; Kitaura, H.; Ikeda, M.; Tamai, K.; Iguchi-Ariga, S.M.M.; Ariga, H. Dj-1, a novel oncogene which transforms mouse nih3t3 cells in cooperation withras. Biochem. Biophys. Res. Commun. 1997, 231, 509–513. [Google Scholar] [CrossRef] [PubMed]
- Aleyasin, H.; Rousseaux, M.W.C.; Phillips, M.; Kim, R.H.; Bland, R.J.; Callaghan, S.; Slack, R.S.; During, M.J.; Mak, T.W.; Park, D.S. The parkinson’s disease gene dj-1 is also a key regulator of stroke-induced damage. Proc. Natl. Acad. Sci. USA 2007, 104, 18748–18753. [Google Scholar] [CrossRef] [PubMed]
- Andres-Mateos, E.; Perier, C.; Zhang, L.; Blanchard-Fillion, B.; Greco, T.M.; Thomas, B.; Ko, H.S.; Sasaki, M.; Ischiropoulos, H.; Przedborski, S.; et al. Dj-1 gene deletion reveals that dj-1 is an atypical peroxiredoxin-like peroxidase. Proc. Natl. Acad. Sci. USA 2007, 104, 14807–14812. [Google Scholar] [CrossRef] [PubMed]
- Taira, T.; Saito, Y.; Niki, T.; Iguchi-Ariga, S.M.M.; Takahashi, K.; Ariga, H. Dj-1 has a role in antioxidative stress to prevent cell death. EMBO Rep. 2004, 5, 213–218. [Google Scholar] [CrossRef] [PubMed]
- Irrcher, I.; Aleyasin, H.; Seifert, E.L.; Hewitt, S.J.; Chhabra, S.; Phillips, M.; Lutz, A.K.; Rousseaux, M.W.C.; Bevilacqua, L.; Jahani-Asl, A.; et al. Loss of the parkinson’s disease-linked gene dj-1 perturbs mitochondrial dynamics. Hum. Mol. Genet. 2010, 19, 3734–3746. [Google Scholar] [CrossRef] [PubMed]
- Behrens, M.I.; Lendon, C.; Roe, C.M. A common biological mechanism in cancer and alzheimer’s disease? Curr. Alzheimer Res. 2009, 6, 196–204. [Google Scholar] [CrossRef] [PubMed]
- Plun-Favreau, H.; Lewis, P.A.; Hardy, J.; Martins, L.M.; Wood, N.W. Cancer and neurodegeneration: Between the devil and the deep blue sea. PLoS Genet. 2010, 6, e1001257. [Google Scholar] [CrossRef] [PubMed]
- Doshay, L.J. Problem situations in the treatment of paralysis agitans. J. Am. Med. Assoc. 1954, 156, 680–684. [Google Scholar] [CrossRef] [PubMed]
- Driver, J.A.; Beiser, A.; Au, R.; Kreger, B.E.; Splansky, G.L.; Kurth, T.; Kiel, D.P.; Lu, K.P.; Seshadri, S.; Wolf, P.A. Inverse association between cancer and alzheimer’s disease: Results from the framingham heart study. BMJ 2012, 344, e1442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Driver, J.A.; Kurth, T.; Buring, J.E.; Gaziano, J.M.; Logroscino, G. Prospective case–control study of nonfatal cancer preceding the diagnosis of parkinson’s disease. Cancer Causes Control 2007, 18, 705–711. [Google Scholar] [CrossRef] [PubMed]
- Elbaz, A.; Peterson, B.J.; Bower, J.H.; Yang, P.; Maraganore, D.M.; McDonnell, S.K.; Ahlskog, J.E.; Rocca, W.A. Risk of cancer after the diagnosis of parkinson’s disease: A historical cohort study. Mov. Disord. 2005, 20, 719–725. [Google Scholar] [CrossRef] [PubMed]
- Elbaz, A.; Peterson, B.J.; Yang, P.; Van Gerpen, J.A.; Bower, J.H.; Maraganore, D.M.; McDonnell, S.K.; Eric Ahlskog, J.; Rocca, W.A. Nonfatal cancer preceding parkinson’s disease: A case-control study. Epidemiology 2002, 13, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Fois, A.F.; Wotton, C.J.; Yeates, D.; Turner, M.R.; Goldacre, M.J. Cancer in patients with motor neuron disease, multiple sclerosis, and parkinson’s disease: Record-linkage studies. J. Neurol. Neurosurg. Psychiatry 2009, 81, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Olsen, J.H.; Friis, S.; Frederiksen, K. Malignant melanoma and other types of cancer preceding parkinson disease. Epidemiology 2006, 17, 582–587. [Google Scholar] [CrossRef] [PubMed]
- Olsen, J.H.; Friis, S.; Frederiksen, K.; McLaughlin, J.K.; Mellemkjaer, L.; Møller, H. Atypical cancer pattern in patients with parkinson‘s disease. Br. J. Cancer 2004, 92, 201. [Google Scholar] [CrossRef] [PubMed]
- Barnett, K.; Mercer, S.W.; Norbury, M.; Watt, G.; Wyke, S.; Guthrie, B. Epidemiology of multimorbidity and implications for health care, research, and medical education: A cross-sectional study. Lancet 2012, 380, 37–43. [Google Scholar] [CrossRef]
- Valderas, J.M.; Starfield, B.; Sibbald, B.; Salisbury, C.; Roland, M. Defining comorbidity: Implications for understanding health and health services. Ann. Fam. Med. 2009, 7, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Bajaj, A.; Driver, J.A.; Schernhammer, E.S. Parkinson’s disease and cancer risk: A systematic review and meta-analysis. Cancer Causes Control 2010, 21, 697–707. [Google Scholar] [CrossRef] [PubMed]
- Catts, V.S.; Catts, S.V.; O’Toole, B.I.; Frost, A.D.J. Cancer incidence in patients with schizophrenia and their first-degree relatives—A meta-analysis. Acta Psychiatr. Scand. 2008, 117, 323–336. [Google Scholar] [CrossRef] [PubMed]
- Tabarés-Seisdedos, R.; Rubenstein, J.L. Inverse cancer comorbidity: A serendipitous opportunity to gain insight into cns disorders. Nat. Rev. Neurosci. 2013, 14, 293. [Google Scholar] [CrossRef] [PubMed]
- Catalá-López, F.; Suárez-Pinilla, M.; Suárez-Pinilla, P.; Valderas, J.M.; Gómez-Beneyto, M.; Martinez, S.; Balanzá-Martínez, V.; Climent, J.; Valencia, A.; McGrath, J.; et al. Inverse and direct cancer comorbidity in people with central nervous system disorders: A meta-analysis of cancer incidence in 577,013 participants of 50 observational studies. Psychother. Psychosom. 2014, 83, 89–105. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, A.K.; Ho, H.; Bodemann, B.; Petersen, S.; Aruri, J.; Koshy, S.; Richardson, Z.; Le, L.Q.; Krasieva, T.; Roth, M.G.; et al. Genome-wide sirna-based functional genomics of pigmentation identifies novel genes and pathways that impact melanogenesis in human cells. PLoS Genet. 2008, 4, e1000298. [Google Scholar] [CrossRef] [PubMed]
- Olzmann, J.A.; Chin, L.-S. Parkin-mediated k63-linked polyubiquitination: A signal for targeting misfolded proteins to the aggresome-autophagy pathway. Autophagy 2008, 4, 85–87. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Erviti, L.; Rodriguez-Oroz, M.C.; Cooper, J.M.; Caballero, C.; Ferrer, I.; Obeso, J.A.; Schapira, A.H.V. Chaperone-mediated autophagy markers in parkinson disease brains. Arch. Neurol. 2010, 67, 1464–1472. [Google Scholar] [CrossRef] [PubMed]
- Cheung, Z.H.; Ip, N.Y. The emerging role of autophagy in parkinson’s disease. Mol. Brain 2009, 2, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, T.; Kondo, S.; Le, W.; Jankovic, J. The role of autophagy-lysosome pathway in neurodegeneration associated with parkinson’s disease. Brain 2008, 131, 1969–1978. [Google Scholar] [CrossRef] [PubMed]
- Rubinsztein, D.C. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 2006, 443, 780–786. [Google Scholar] [CrossRef] [PubMed]
- Pimkina, J.; Murphy, M.E. Arf, autophagy and tumor suppression. Autophagy 2009, 5, 397–399. [Google Scholar] [CrossRef] [PubMed]
- Miracco, C.; Cevenini, G.; Franchi, A.; Luzi, P.; Cosci, E.; Mourmouras, V.; Monciatti, I.; Mannucci, S.; Biagioli, M.; Toscano, M.; et al. Beclin 1 and lc3 autophagic gene expression in cutaneous melanocytic lesions. Hum. Pathol. 2010, 41, 503–512. [Google Scholar] [CrossRef] [PubMed]
- Lazova, R.; Klump, V.; Pawelek, J. Autophagy in cutaneous malignant melanoma. J. Cutan. Pathol. 2010, 37, 256–268. [Google Scholar] [CrossRef] [PubMed]
- Denison, S.R.; Wang, F.; Becker, N.A.; Schüle, B.; Kock, N.; Phillips, L.A.; Klein, C.; Smith, D.I. Alterations in the common fragile site gene parkin in ovarian and other cancers. Oncogene 2003, 22, 8370. [Google Scholar] [CrossRef] [PubMed]
- Tay, S.-P.; Yeo, C.W.S.; Chai, C.; Chua, P.-J.; Tan, H.-M.; Ang, A.X.Y.; Yip, D.L.H.; Sung, J.-X.; Tan, P.H.; Bay, B.-H.; et al. Parkin enhances the expression of cyclin-dependent kinase 6 and negatively regulates the proliferation of breast cancer cells. J. Biol. Chem. 2010, 285, 29231–29238. [Google Scholar] [CrossRef] [PubMed]
- Poulogiannis, G.; McIntyre, R.E.; Dimitriadi, M.; Apps, J.R.; Wilson, C.H.; Ichimura, K.; Luo, F.; Cantley, L.C.; Wyllie, A.H.; Adams, D.J.; et al. PARK2 deletions occur frequently in sporadic colorectal cancer and accelerate adenoma development in Apc mutant mice. Proc. Natl. Acad. Sci. USA 2010, 107, 15145–15150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staropoli, J.F.; McDermott, C.; Martinat, C.; Schulman, B.; Demireva, E.; Abeliovich, A. Parkin is a component of an scf-like ubiquitin ligase complex and protects postmitotic neurons from kainate excitotoxicity. Neuron 2003, 37, 735–749. [Google Scholar] [CrossRef]
- Ikeuchi, K.; Marusawa, H.; Fujiwara, M.; Matsumoto, Y.; Endo, Y.; Watanabe, T.; Iwai, A.; Sakai, Y.; Takahashi, R.; Chiba, T. Attenuation of proteolysis-mediated cyclin e regulation by alternatively spliced parkin in human colorectal cancers. Int. J. Cancer 2009, 125, 2029–2035. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.B.; Kim, J.J.; Nam, H.-J.; Gao, B.; Yin, P.; Qin, B.; Yi, S.-Y.; Ham, H.; Evans, D.; Kim, S.-H.; et al. Parkin regulates mitosis and genomic stability through cdc20/cdh1. Mol. Cell 2015, 60, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Lucas, J.J.; Domenico, J.; Gelfand, E.W. Cyclin-dependent kinase 6 inhibits proliferation of human mammary epithelial cells. Mol. Cancer Res. 2004, 2, 105–114. [Google Scholar] [PubMed]
- Wang, H.; Liu, B.; Zhang, C.; Peng, G.; Liu, M.; Li, D.; Gu, F.; Chen, Q.; Dong, J.-T.; Fu, L.; et al. Parkin regulates paclitaxel sensitivity in breast cancer via a microtubule-dependent mechanism. J. Pathol. 2009, 218, 76–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.; Lee, M.H.; Kang, Y.W.; Rhee, K.-J.; Kim, T.U.; Kim, Y.S. Parkin induces apoptotic cell death in tnf-α-treated cervical cancer cells. BMB Rep. 2012, 45, 526–531. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Anjomani-Virmouni, S.; Koundouros, N.; Dimitriadi, M.; Choo-Wing, R.; Valle, A.; Zheng, Y.; Chiu, Y.-H.; Agnihotri, S.; Zadeh, G.; et al. Park2 depletion connects energy and oxidative stress to pi3k/akt activation via pten s-nitrosylation. Mol. Cell 2017, 65, 999–1013. [Google Scholar] [CrossRef] [PubMed]
- Gegg, M.E.; Cooper, J.M.; Schapira, A.H.V.; Taanman, J.-W. Silencing of pink1 expression affects mitochondrial DNA and oxidative phosphorylation in dopaminergic cells. PLoS ONE 2009, 4, e4756. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Sawada, T.; Lee, S.; Yu, W.; Silverio, G.; Alapatt, P.; Millan, I.; Shen, A.; Saxton, W.; Kanao, T.; et al. Parkinson’s disease–associated kinase pink1 regulates miro protein level and axonal transport of mitochondria. PLoS Genet. 2012, 8, e1002537. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, A.; Kataoka, K.; Hong, M.; Sakaguchi, M.; Huh, N. BRPK, a novel protein kinase showing increased expression in mouse cancer cell lines with higher metastatic potential. Cancer Lett. 2003, 201, 195–201. [Google Scholar] [CrossRef]
- Liang, H.; He, S.; Yang, J.; Jia, X.; Wang, P.; Chen, X.; Zhang, Z.; Zou, X.; McNutt, M.A.; Shen, W.H.; et al. Ptenα, a pten isoform translated through alternative initiation, regulates mitochondrial function and energy metabolism. Cell Metab. 2014, 19, 836–848. [Google Scholar] [CrossRef] [PubMed]
- O’Flanagan, C.H.; Morais, V.A.; Wurst, W.; De Strooper, B.; O’Neill, C. The parkinson’s gene pink1 regulates cell cycle progression and promotes cancer-associated phenotypes. Oncogene 2015, 34, 1363. [Google Scholar] [CrossRef] [PubMed]
- Agnihotri, S.; Golbourn, B.; Huang, X.; Remke, M.; Younger, S.; Cairns, R.A.; Chalil, A.; Smith, C.A.; Krumholtz, S.-L.; Mackenzie, D.; et al. Pink1 is a negative regulator of growth and the warburg effect in glioblastoma. Cancer Res. 2016, 76, 4708–4719. [Google Scholar] [CrossRef] [PubMed]
- Ariga, H. Common mechanisms of onset of cancer and neurodegenerative diseases. Biol. Pharm. Bull. 2015, 38, 795–808. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.C.; Kitaura, H.; Iguchi-Ariga, S.M.M.; Ariga, H. Dj-1, an oncogene and causative gene for familial parkinson’s disease, is essential for sv40 transformation in mouse fibroblasts through up-regulation of c-myc. FEBS Lett. 2010, 584, 3891–3895. [Google Scholar] [CrossRef] [PubMed]
- Bandopadhyay, R.; Kingsbury, A.E.; Cookson, M.R.; Reid, A.R.; Evans, I.M.; Hope, A.D.; Pittman, A.M.; Lashley, T.; Canet-Aviles, R.; Miller, D.W.; et al. The expression of dj-1 (park7) in normal human cns and idiopathic parkinson’s disease. Brain 2004, 127, 420–430. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Cui, J.; Zhang, C.; Yang, D.; Chen, J.; Zan, W.; Li, B.; Li, Z.; He, Y. High-expression of dj-1 and loss of pten associated with tumor metastasis and correlated with poor prognosis of gastric carcinoma. Int. J. Med. Sci. 2013, 10, 1689–1697. [Google Scholar] [CrossRef] [PubMed]
- Rizzu, P.; Hinkle, D.A.; Zhukareva, V.; Bonifati, V.; Severijnen, L.A.; Martinez, D.; Ravid, R.; Kamphorst, W.; Eberwine, J.H.; Lee, V.M.Y.; et al. Dj-1 colocalizes with tau inclusions: A link between parkinsonism and dementia. Ann. Neurol. 2004, 55, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Davidson, B.; Hadar, R.; Schlossberg, A.; Sternlicht, T.; Slipicevic, A.; Skrede, M.; Risberg, B.; Flørenes, V.A.; Kopolovic, J.; Reich, R. Expression and clinical role of dj-1, a negative regulator of pten, in ovarian carcinoma. Hum. Pathol. 2008, 39, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Hod, Y. Differential control of apoptosis by dj-1 in prostate benign and cancer cells. J. Cell. Biochem. 2004, 92, 1221–1233. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.H.; Peters, M.; Jang, Y.; Shi, W.; Pintilie, M.; Fletcher, G.C.; DeLuca, C.; Liepa, J.; Zhou, L.; Snow, B.; et al. Dj-1, a novel regulator of the tumor suppressor pten. Cancer Cell 2005, 7, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Pan, B.; Li, B.; Li, Y.; Tian, X.; Li, Z. Dj-1 is activated in medulloblastoma and is associated with cell proliferation and differentiation. World J. Surg. Oncol. 2014, 12, 373. [Google Scholar] [CrossRef] [PubMed]
- Yarden, Y.; Pines, G. The erbb network: At last, cancer therapy meets systems biology. Nat. Rev. Cancer 2012, 12, 553. [Google Scholar] [CrossRef] [PubMed]
- Prigent, S.A.; Gullick, W.J. Identification of c-erbb-3 binding sites for phosphatidylinositol 3′-kinase and shc using an egf receptor/c-erbb-3 chimera. EMBO J. 1994, 13, 2831–2841. [Google Scholar] [CrossRef] [PubMed]
- Soltoff, S.P.; Carraway, K.L.; Prigent, S.A.; Gullick, W.G.; Cantley, L.C. Erbb3 is involved in activation of phosphatidylinositol 3-kinase by epidermal growth factor. Mol. Cell. Biol. 1994, 14, 3550–3558. [Google Scholar] [CrossRef] [PubMed]
- Hellyer, N.J.; Cheng, K.; Koland, J.G. Erbb3 (her3) interaction with the p85 regulatory subunit of phosphoinositide 3-kinase. Biochem. J. 1998, 333, 757–763. [Google Scholar] [CrossRef] [PubMed]
- Mujoo, K.; Choi, B.-K.; Huang, Z.; Zhang, N.; An, Z. Regulation of erbb3/her3 signaling in cancer. Oncotarget 2014, 5, 10222–10236. [Google Scholar] [CrossRef] [PubMed]
- Vijapurkar, U.; Cheng, K.; Koland, J.G. Mutation of a shc binding site tyrosine residue in erbb3/her3 blocks heregulin-dependent activation of mitogen-activated protein kinase. J. Biol. Chem. 1998, 273, 20996–21002. [Google Scholar] [CrossRef] [PubMed]
- McNally, R.S.; Davis, B.K.; Clements, C.M.; Accavitti-Loper, M.A.; Mak, T.W.; Ting, J.P.-Y. Dj-1 enhances cell survival through the binding of cezanne, a negative regulator of nf-κb. J. Biol. Chem. 2011, 286, 4098–4106. [Google Scholar] [CrossRef] [PubMed]
- Bargou, R.C.; Emmerich, F.; Krappmann, D.; Bommert, K.; Mapara, M.Y.; Arnold, W.; Royer, H.D.; Grinstein, E.; Greiner, A.; Scheidereit, C.; et al. Constitutive nuclear factor-kappab-rela activation is required for proliferation and survival of hodgkin’s disease tumor cells. J. Clin. Investig. 1997, 100, 2961–2969. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.; Cohen, J.; Arun, P.; Chen, Z.; Van Waes, C. Nf-κb in carcinoma therapy and prevention. Expert Opin. Ther. Targets 2008, 12, 1109–1122. [Google Scholar] [CrossRef] [PubMed]
- Landberg, G.; Tan, E.M. Characterization of a DNA-binding nuclear autoantigen mainly associated with s phase and g2 cells. Exp. Cell Res. 1994, 212, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Muro, Y.; Chan, E.K.L.; Landberg, G.; Tan, E.M. A cell-cycle nuclear autoantigen containing WD-40 motifs expressed mainly in S and G2 phase cells. Biochem. Biophys. Res. Commun. 1995, 207, 1029–1037. [Google Scholar] [CrossRef] [PubMed]
- Tanti, G.K.; Goswami, S.K. Sg2na recruits dj-1 and akt into the mitochondria and membrane to protect cells from oxidative damage. Free Radic. Biol. Med. 2014, 75, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Kawate, T.; Iwaya, K.; Koshikawa, K.; Moriya, T.; Yamasaki, T.; Hasegawa, S.; Kaise, H.; Fujita, T.; Matsuo, H.; Nakamura, T.; et al. High levels of dj-1 protein and isoelectric point 6.3 isoform in sera of breast cancer patients. Cancer Sci. 2015, 106, 938–943. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.-Q.; He, C.; Tao, L.; Liu, F. Role of dj-1 sirna in reverse sensitivity of breast cancer cells to chemotherapy and its possible mechanism. Inter. J. Clin. Exp. Pathol. 2015, 8, 6944–6951. [Google Scholar]
- Szklarczyk, D.; Franceschini, A.; Kuhn, M.; Simonovic, M.; Roth, A.; Minguez, P.; Doerks, T.; Stark, M.; Muller, J.; Bork, P.; et al. The string database in 2011: Functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2011, 39, D561–D568. [Google Scholar] [CrossRef] [PubMed]
- STRING. Protein-Protein Interactions Network. Available online: https://string-db.org/ (accessed on 18 July 2018).
- Bioinformatics-and-Evolutionary-Genomics. Calculate and Draw Custom Venn Diagrams. Available online: http://bioinformatics.psb.ugent.be/webtools/Venn/ (accessed on 18 July 2018).










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metal-Catalyzed Haber-Weiss Reaction |
|---|
| (1) |
| (2) |
| (3) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salazar, C.; Ruiz-Hincapie, P.; Ruiz, L.M. The Interplay among PINK1/PARKIN/Dj-1 Network during Mitochondrial Quality Control in Cancer Biology: Protein Interaction Analysis. Cells 2018, 7, 154. https://doi.org/10.3390/cells7100154
Salazar C, Ruiz-Hincapie P, Ruiz LM. The Interplay among PINK1/PARKIN/Dj-1 Network during Mitochondrial Quality Control in Cancer Biology: Protein Interaction Analysis. Cells. 2018; 7(10):154. https://doi.org/10.3390/cells7100154
Chicago/Turabian StyleSalazar, Celia, Paula Ruiz-Hincapie, and Lina María Ruiz. 2018. "The Interplay among PINK1/PARKIN/Dj-1 Network during Mitochondrial Quality Control in Cancer Biology: Protein Interaction Analysis" Cells 7, no. 10: 154. https://doi.org/10.3390/cells7100154