
Structural Mechanisms and Drug Discovery Prospects of Rho GTPases

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
1.1. Ras Family and Rho Subfamily
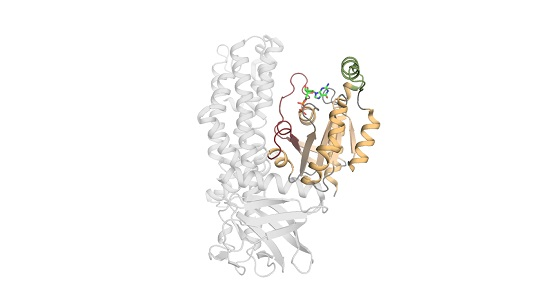
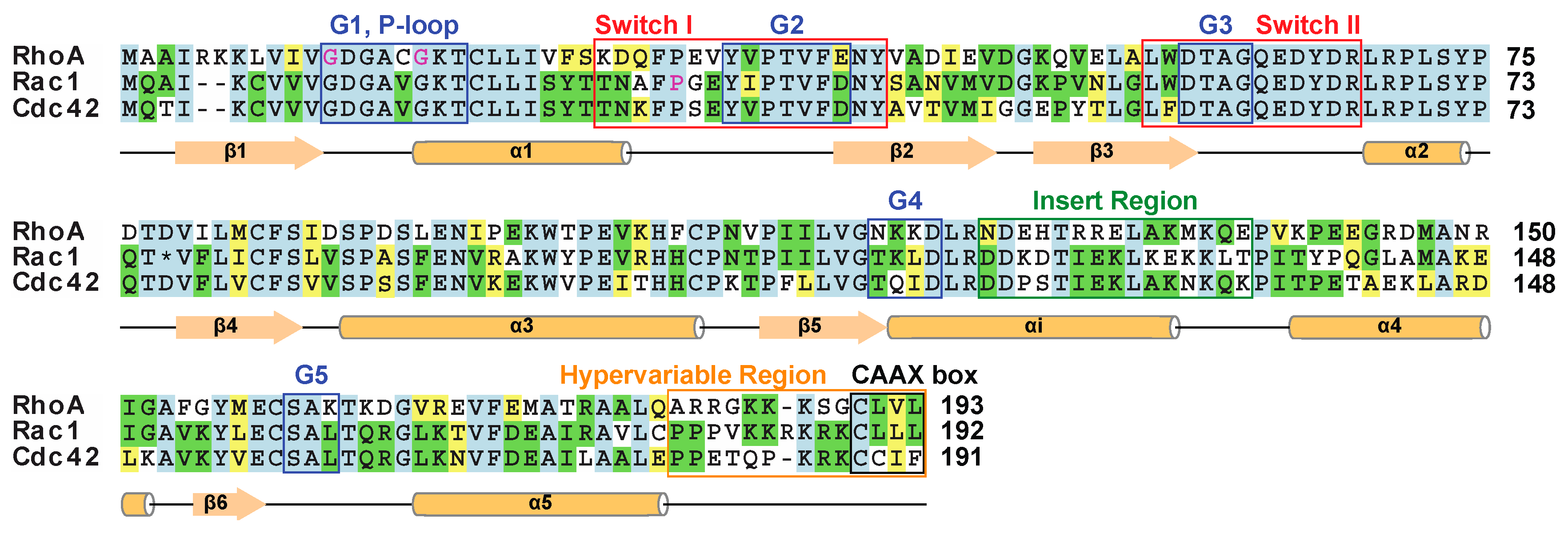
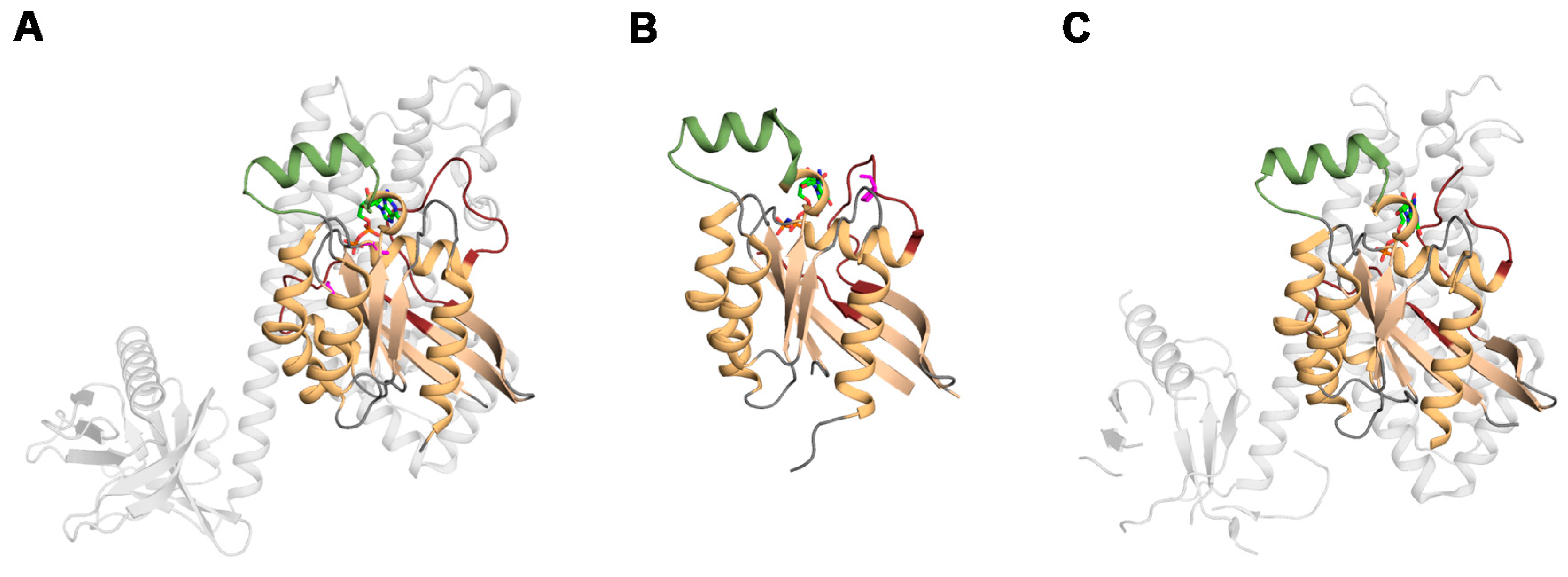
1.2. The Structure of Rho Family Members
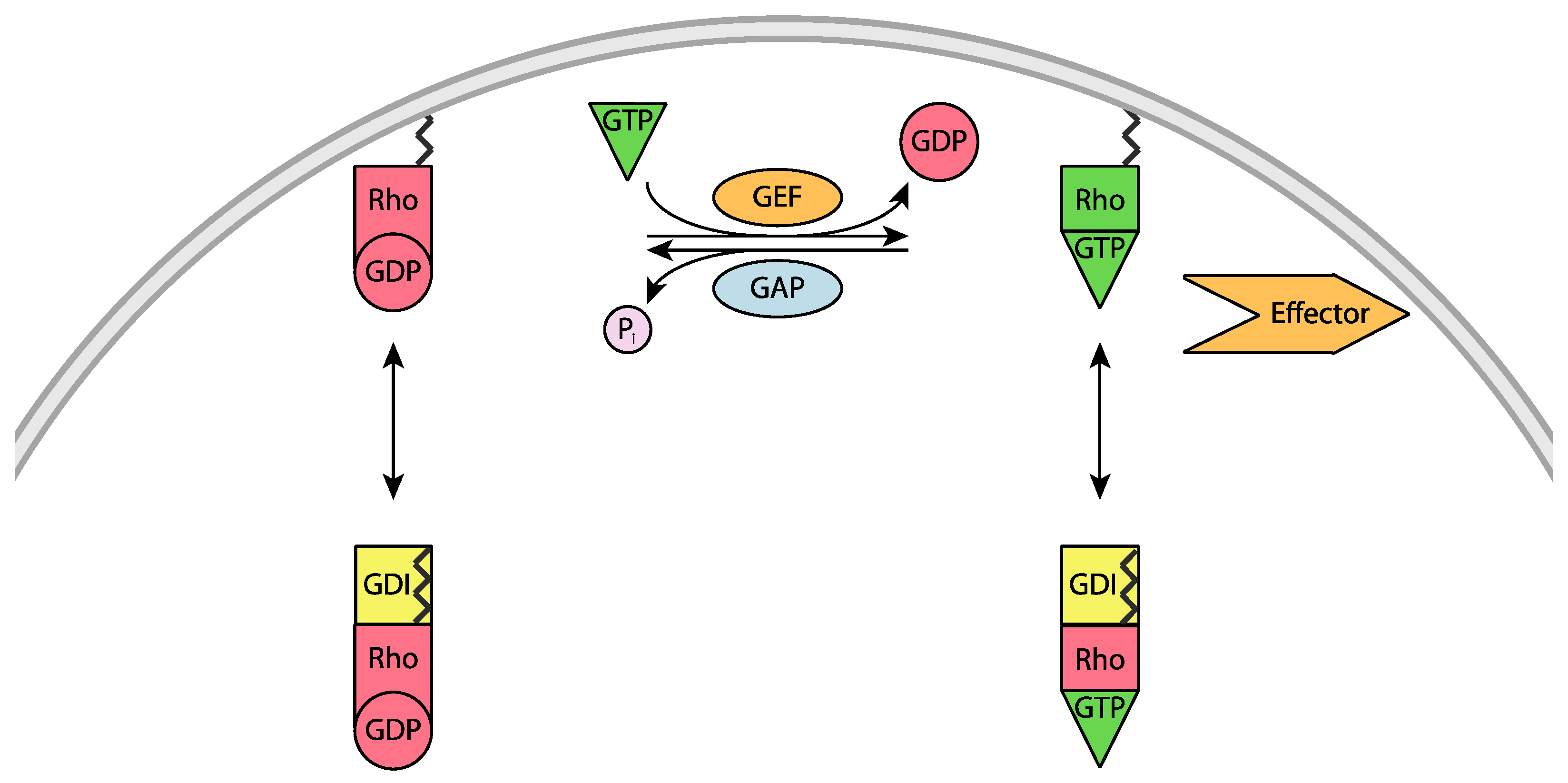
2. Regulation of Rho Mediated Cell Signaling
3. Role of GTPases in Cancer: RhoA, Rac1, and Cdc42
4. Targeting Rho GTPase Signaling for Therapeutic Intervention
5. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| GAP | GTPase Activating Protein |
| GEF | Guanine nucleotide Exchange Factor |
| GDI | GDO Dissociation Inhibitor |
| LARG | Leukaemia-Associated Rho GEF |
| AKAP | A-Kinase Associated Protein |
| mDia1 | Diadphanous-related formin-1 |
| eIF4E | Eukaryotic translation Initiation Factor 4E |
| 4E-BP1 | Eukaryotic translation initiation factor 4E Binding Proteim-1 |
| ROCK | Rho-associated protein Kinase |
| VEGF | Vascular Endothelial Growth Factor |
References
- Mott, H.R.; Owen, D. Structures of Ras superfamily effector complexes: What have we learnt in two decades? Crit. Rev. Biochem. Mol. Biol. 2015, 50, 85–133. [Google Scholar] [CrossRef] [PubMed]
- Wuichet, K.; Søgaard-Andersen, L. Evolution and diversity of the Ras superfamily of small GTPases in prokaryotes. Genome Biol. Evol. 2015, 7, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Rojas, A.M.; Fuentes, G.; Rausell, A.; Valencia, A. The Ras protein superfamily: Evolutionary tree and role of conserved amino acids. J. Cell Biol. 2012, 196, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Madaule, P.; Axel, R. A novel Ras-related gene family. Cell 1985, 41, 31–40. [Google Scholar] [CrossRef]
- Schmitt, H.D.; Wagner, P.; Pfaff, E.; Gallwitz, D. The Ras-related YPT1 gene product in yeast: A GTP-binding protein that might be involved in microtubule organization. Cell 1986, 47, 401–412. [Google Scholar] [CrossRef]
- Borda-d’Agua, B.; Infante, E.; Riou, P.; Tajadura, V.; Ridley, A.J. Atypical Rho family members. In Ras Superfamily Small G Proteins: Biology and Mechanisms 1; Wittinghofer, A., Ed.; Springer: Vienna, Austria, 2014; pp. 341–361. [Google Scholar]
- Buchsbaum, R.J. Rho activation at a glance. J. Cell Sci. 2007, 120, 1149–1152. [Google Scholar] [CrossRef] [PubMed]
- Hanna, S.; El-Sibai, M. Signaling networks of Rho GTPases in cell motility. Cell. Signal. 2013, 25, 1955–1961. [Google Scholar] [CrossRef] [PubMed]
- Sit, S.-T.; Manser, E. Rho GTPases and their role in organizing the actin cytoskeleton. J. Cell Sci. 2011, 124, 679–683. [Google Scholar] [CrossRef] [PubMed]
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Le Digabel, J.; Forcato, M.; Bicciato, S.; et al. Role of YAP/TAZ in mechanotransduction. Nature 2011, 474, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Miralles, F.; Posern, G.; Zaromytidou, A.-I.; Treisman, R. Actin dynamics control SRF activity by regulation of its coactivator MAL. Cell 2003, 113, 329–342. [Google Scholar] [CrossRef]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, S. The Biology Workbench—A seamless database and analysis environment for the biologist. Proteins 1998, 32, 1–2. [Google Scholar] [CrossRef]
- Wennerberg, K.; Rossman, K.L.; Der, C.J. The Ras superfamily at a glance. J. Cell Sci. 2005, 118, 843–846. [Google Scholar] [CrossRef] [PubMed]
- Parri, M.; Chiarugi, P. Rac and Rho GTPases in cancer cell motility control. Cell Commun. Signal. CCS 2010, 8, 23. [Google Scholar] [CrossRef] [PubMed]
- Pai, E.F.; Krengel, U.; Petsko, G.A.; Goody, R.S.; Kabsch, W.; Wittinghofer, A. Refined crystal structure of the triphosphate conformation of H-ras p21 at 1.35 A resolution: Implications for the mechanism of GTP hydrolysis. EMBO J. 1990, 9, 2351–2359. [Google Scholar] [PubMed]
- Cherfils, J.; Zeghouf, M. Chronicles of the GTPase switch. Nat. Chem. Biol. 2011, 7, 493–495. [Google Scholar] [CrossRef] [PubMed]
- Karnoub, A.E.; Symons, M.; Campbell, S.L.; Der, C.J. Molecular basis for Rho GTPase signaling specificity. Breast Cancer Res. Treat. 2004, 84, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Cherfils, J.; Zeghouf, M. Regulation of small GTPases by GEFs, GAPs, and GDIs. Physiol. Rev. 2013, 93, 269–309. [Google Scholar] [CrossRef] [PubMed]
- Wittinghofer, A.; Vetter, I.R. Structure-function relationships of the G domain, a canonical switch motif. Annu. Rev. Biochem. 2011, 80, 943–971. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, A.; Reinhard, N.R.; Hordijk, P.L. Toward understanding RhoGTPase specificity: Structure, function and local activation. Small GTPases 2014, 5, 6. [Google Scholar] [CrossRef] [PubMed]
- Hamel, B.; Monaghan-Benson, E.; Rojas, R.J.; Temple, B.R.S.; Marston, D.J.; Burridge, K.; Sondek, J. SmgGDS is a guanine nucleotide exchange factor that specifically activates RhoA and RhoC. J. Biol. Chem. 2011, 286, 12141–12148. [Google Scholar] [CrossRef] [PubMed]
- Ramos, S.; Khademi, F.; Somesh, B.P.; Rivero, F. Genomic organization and expression profile of the small GTPases of the RhoBTB family in human and mouse. Gene 2002, 298, 147–157. [Google Scholar] [CrossRef]
- Gao, J.; Liao, J.; Yang, G.-Y. CAAX-box protein, prenylation process and carcinogenesis. Am. J. Transl. Res. 2009, 1, 312–325. [Google Scholar] [PubMed]
- Winter-Vann, A.M.; Casey, P.J. Post-prenylation-processing enzymes as new targets in oncogenesis. Nat. Rev. Cancer 2005, 5, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Michaelson, D.; Silletti, J.; Murphy, G.; D’Eustachio, P.; Rush, M.; Philips, M.R. Differential localization of Rho GTPases in live cells regulation by hypervariable regions and Rhogdi binding. J. Cell Biol. 2001, 152, 111–126. [Google Scholar] [CrossRef] [PubMed]
- Roberts, P.J.; Mitin, N.; Keller, P.J.; Chenette, E.J.; Madigan, J.P.; Currin, R.O.; Cox, A.D.; Wilson, O.; Kirschmeier, P.; Der, C.J. Rho family GTPase modification and dependence on CAAX motif-signaled posttranslational modification. J. Biol. Chem. 2008, 283, 25150–25163. [Google Scholar] [CrossRef] [PubMed]
- Hakoshima, T. Structural basis of the Rho GTPase signaling. J. Biochem. (Tokyo) 2003, 134, 327–331. [Google Scholar] [CrossRef]
- Fiegen, D.; Haeusler, L.-C.; Blumenstein, L.; Herbrand, U.; Dvorsky, R.; Vetter, I.R.; Ahmadian, M.R. Alternative splicing of Rac1 generates Rac1b, a self-activating GTPase. J. Biol. Chem. 2004, 279, 4743–4749. [Google Scholar] [CrossRef] [PubMed]
- Stogios, P.J.; Downs, G.S.; Jauhal, J.J.; Nandra, S.K.; Privé, G.G. Sequence and structural analysis of BTB domain proteins. Genome Biol. 2005, 6, R82. [Google Scholar] [CrossRef] [PubMed]
- Perez-Torrado, R.; Yamada, D.; Defossez, P.-A. Born to bind: The BTB protein–protein interaction domain. BioEssays 2006, 28, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, A.; Ping, Q.; Carpenter, C.L. RhoBTB2 is a substrate of the mammalian Cul3 ubiquitin ligase complex. Genes Dev. 2004, 18, 856–861. [Google Scholar] [CrossRef] [PubMed]
- Abdul Azeez, K.R.; Knapp, S.; Fernandes, J.M.P.; Klussmann, E.; Elkins, J.M. The crystal structure of the RhoA–AKAP-Lbc DH–PH domain complex. Biochem. J. 2014, 464, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Kapp, G.T.; Liu, S.; Stein, A.; Wong, D.T.; Reményi, A.; Yeh, B.J.; Fraser, J.S.; Taunton, J.; Lim, W.A.; Kortemme, T. Control of protein signaling using a computationally designed GTPase/GEF orthogonal pair. Proc. Natl. Acad. Sci. USA 2012, 109, 5277–5282. [Google Scholar] [CrossRef] [PubMed]
- DerMardirossian, C.; Bokoch, G.M. GDIs: Central regulatory molecules in Rho GTPase activation. Trends Cell Biol. 2005, 15, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Bos, J.L.; Rehmann, H.; Wittinghofer, A. GEFs and GAPs: Critical elements in the control of small G proteins. Cell 2007, 129, 865–877. [Google Scholar] [CrossRef] [PubMed]
- Moon, S.Y.; Zheng, Y. Rho GTPase-activating proteins in cell regulation. Trends Cell Biol. 2003, 13, 13–22. [Google Scholar] [CrossRef]
- Lang, P.; Gesbert, F.; Thiberge, J.M.; Troalen, F.; Dutartre, H.; Chavrier, P.; Bertoglio, J. Characterization of a monoclonal antibody specific for the Ras-related GTP-binding protein Rho A. Biochem. Biophys. Res. Commun. 1993, 196, 1522–1528. [Google Scholar] [CrossRef] [PubMed]
- Olofsson, B. Rho Guanine dissociation inhibitors: Pivotal molecules in cellular signalling. Cell. Signal. 1999, 11, 545–554. [Google Scholar] [CrossRef]
- Shi, J.; Wei, L. Rho kinase in the regulation of cell death and survival. Arch. Immunol. Ther. Exp. (Warsz.) 2007, 55, 61–75. [Google Scholar] [CrossRef] [PubMed]
- Ishizaki, T.; Maekawa, M.; Fujisawa, K.; Okawa, K.; Iwamatsu, A.; Fujita, A.; Watanabe, N.; Saito, Y.; Kakizuka, A.; Morii, N.; et al. The small GTP-binding protein Rho binds to and activates a 160 kDa Ser/Thr protein kinase homologous to myotonic dystrophy kinase. EMBO J. 1996, 15, 1885–1893. [Google Scholar] [PubMed]
- Ongusaha, P.P.; Kim, H.-G.; Boswell, S.A.; Ridley, A.J.; Der, C.J.; Dotto, G.P.; Kim, Y.-B.; Aaronson, S.A.; Lee, S.W. RhoE is a pro-survival p53 target gene that inhibits ROCK I-mediated apoptosis in response to genotoxic stress. Curr. Biol. 2006, 16, 2466–2472. [Google Scholar] [CrossRef] [PubMed]
- Riento, K.; Totty, N.; Villalonga, P.; Garg, R.; Guasch, R.; Ridley, A.J. RhoE function is regulated by ROCK I-mediated phosphorylation. EMBO J. 2005, 24, 1170–1180. [Google Scholar] [CrossRef] [PubMed]
- Mitin, N.; Roberts, P.; Chenette, E.; Der, C. Posttranslational lipid modification of Rho family small GTPases. In Rho GTPases; Rivero, F., Ed.; Methods in Molecular Biology; Springer: New York, NY, USA, 2012; pp. 87–95. [Google Scholar]
- Dransart, E.; Olofsson, B.; Cherfils, J. RhoGDIs revisited: Novel roles in Rho regulation. Traffic 2005, 6, 957–966. [Google Scholar] [CrossRef] [PubMed]
- Aicart-Ramos, C.; Valero, R.A.; Rodriguez-Crespo, I. Protein palmitoylation and subcellular trafficking. Biochim. Biophys. Acta Biomembr. 2011, 1808, 2981–2994. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, A.; Linder, M.E. Identification of a novel prenyl and palmitoyl modification at the CaaX motif of Cdc42 that regulates RhoGDI binding. Mol. Cell. Biol. 2013, 33, 1417–1429. [Google Scholar] [CrossRef] [PubMed]
- Linder, M.E.; Deschenes, R.J. Palmitoylation: Policing protein stability and traffic. Nat. Rev. Mol. Cell Biol. 2007, 8, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Rocks, O.; Gerauer, M.; Vartak, N.; Koch, S.; Huang, Z.-P.; Pechlivanis, M.; Kuhlmann, J.; Brunsveld, L.; Chandra, A.; Ellinger, B.; et al. The Palmitoylation machinery is a spatially organizing system for peripheral membrane proteins. Cell 2010, 141, 458–471. [Google Scholar] [CrossRef] [PubMed]
- Hornbeck, P.V.; Zhang, B.; Murray, B.; Kornhauser, J.M.; Latham, V.; Skrzypek, E. PhosphoSitePlus, 2014: Mutations, PTMs and recalibrations. Nucleic Acids Res. 2015, 43, D512–D520. [Google Scholar] [CrossRef] [PubMed]
- Kwon, T.; Kwon, D.Y.; Chun, J.; Kim, J.H.; Kang, S.S. Akt protein kinase inhibits Rac1-GTP binding through phosphorylation at serine 71 of Rac1. J. Biol. Chem. 2000, 275, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Nusser, N.; Gosmanova, E.; Makarova, N.; Fujiwara, Y.; Yang, L.; Guo, F.; Luo, Y.; Zheng, Y.; Tigyi, G. Serine phosphorylation differentially affects RhoA binding to effectors: Implications to NGF-induced neurite outgrowth. Cell. Signal. 2006, 18, 704–714. [Google Scholar] [CrossRef] [PubMed]
- Forget, M.-A.; Desrosiers, R.R.; Gingras, D.; Béliveau, R. Phosphorylation states of Cdc42 and RhoA regulate their interactions with Rho GDP dissociation inhibitor and their extraction from biological membranes. Biochem. J. 2002, 361, 243. [Google Scholar] [CrossRef] [PubMed]
- Lemichez, E.; Aktories, K. Hijacking of Rho GTPases during bacterial infection. Exp. Cell Res. 2013, 319, 2329–2336. [Google Scholar] [CrossRef] [PubMed]
- Sehr, P.; Joseph, G.; Genth, H.; Just, I.; Pick, E.; Aktories, K. Glucosylation and ADP Ribosylation of Rho proteins: Effects on nucleotide binding, GTPase activity, and effector coupling. Biochemistry (Mosc.) 1998, 37, 5296–5304. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Peyrollier, K.; Kilic, G.; Brakebusch, C. Rho GTPases and cancer. BioFactors 2014, 40, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Fritz, G.; Just, I.; Kaina, B. Rho GTPases are over-expressed in human tumors. Int. J. Cancer 1999, 81, 682–687. [Google Scholar] [CrossRef]
- Pillé, J.-Y.; Denoyelle, C.; Varet, J.; Bertrand, J.-R.; Soria, J.; Opolon, P.; Lu, H.; Pritchard, L.-L.; Vannier, J.-P.; Malvy, C.; et al. Anti-RhoA and Anti-RhoC siRNAs inhibit the proliferation and invasiveness of MDA-MB-231 breast cancer cells in vitro and in vivo. Mol. Ther. 2005, 11, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Engers, R.; Ziegler, S.; Mueller, M.; Walter, A.; Willers, R.; Gabbert, H.E. Prognostic relevance of increased Rac GTPase expression in prostate carcinomas. Endocr. Relat. Cancer 2007, 14, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Zheng, Y. Approaches of targeting Rho GTPases in cancer drug discovery. Expert Opin. Drug Discov. 2015, 10, 991–1010. [Google Scholar] [CrossRef] [PubMed]
- Esufali, S.; Charames, G.S.; Pethe, V.V.; Buongiorno, P.; Bapat, B. Activation of tumor-specific splice variant Rac1b by dishevelled promotes canonical Wnt signaling and decreased adhesion of colorectal cancer cells. Cancer Res. 2007, 67, 2469–2479. [Google Scholar] [CrossRef] [PubMed]
- Matos, P.; Jordan, P. Increased Rac1b expression sustains colorectal tumor cell survival. Mol. Cancer Res. 2008, 6, 1178–1184. [Google Scholar] [CrossRef] [PubMed]
- Gómez del Pulgar, T.; Valdés-Mora, F.; Bandrés, E.; Pérez-Palacios, R.; Espina, C.; Cejas, P.; García-Cabezas, M.; Nistal, M.; Casado, E.; González-Barón, M.; et al. Cdc42 is highly expressed in colorectal adenocarcinoma and downregulates ID4 through an epigenetic mechanism. Int. J. Oncol. 2008, 33, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Song, N.; Ren, K.; Meng, S.; Xie, Y.; Long, Q.; Chen, X.; Zhao, X. Expression loss and revivification of RhoB gene in ovary carcinoma carcinogenesis and development. PLoS ONE 2013, 8, e78417. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Medarde, A.; Santos, E. Ras in cancer and developmental diseases. Genes Cancer 2011, 2, 344–358. [Google Scholar] [CrossRef] [PubMed]
- Forbes, S.A.; Beare, D.; Gunasekaran, P.; Leung, K.; Bindal, N.; Boutselakis, H.; Ding, M.; Bamford, S.; Cole, C.; Ward, S.; et al. COSMIC: Exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2015, 43, D805–D811. [Google Scholar] [CrossRef] [PubMed]
- Orgaz, J.L.; Herraiz, C.; Sanz-Moreno, V. Rho GTPases modulate malignant transformation of tumor cells. Small GTPases 2014, 5, e983867. [Google Scholar] [CrossRef] [PubMed]
- Hodis, E.; Watson, I.R.; Kryukov, G.V.; Arold, S.T.; Imielinski, M.; Theurillat, J.-P.; Nickerson, E.; Auclair, D.; Li, L.; Place, C.; et al. A landscape of driver mutations in melanoma. Cell 2012, 150, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Krauthammer, M.; Kong, Y.; Ha, B.H.; Evans, P.; Bacchiocchi, A.; McCusker, J.P.; Cheng, E.; Davis, M.J.; Goh, G.; Choi, M.; et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat. Genet. 2012, 44, 1006–1014. [Google Scholar] [CrossRef] [PubMed]
- Mar, V.J.; Wong, S.Q.; Logan, A.; Nguyen, T.; Cebon, J.; Kelly, J.W.; Wolfe, R.; Dobrovic, A.; McLean, C.; McArthur, G.A. Clinical and pathological associations of the activating RAC1 P29S mutation in primary cutaneous melanoma. Pigment Cell Melanoma Res. 2014, 27, 1117–1125. [Google Scholar] [CrossRef] [PubMed]
- Ridley, A.J.; Paterson, H.F.; Johnston, C.L.; Diekmann, D.; Hall, A. The small GTP-binding protein Rac regulates growth factor-induced membrane ruffling. Cell 1992, 70, 401–410. [Google Scholar] [CrossRef]
- Manso, R.; Sánchez-Beato, M.; Monsalvo, S.; Gómez, S.; Cereceda, L.; Llamas, P.; Rojo, F.; Mollejo, M.; Menárguez, J.; Alves, J.; et al. The RHOA G17V gene mutation occurs frequently in peripheral T-cell lymphoma and is associated with a characteristic molecular signature. Blood 2014, 123, 2893–2894. [Google Scholar] [CrossRef] [PubMed]
- Palomero, T.; Couronné, L.; Khiabanian, H.; Kim, M.-Y.; Ambesi-Impiombato, A.; Perez-Garcia, A.; Carpenter, Z.; Abate, F.; Allegretta, M.; Haydu, J.E.; et al. Recurrent mutations in epigenetic regulators, RHOA and FYN kinase in peripheral T cell lymphomas. Nat. Genet. 2014, 46, 166–170. [Google Scholar] [CrossRef] [PubMed]
- Sakata-Yanagimoto, M.; Enami, T.; Yoshida, K.; Shiraishi, Y.; Ishii, R.; Miyake, Y.; Muto, H.; Tsuyama, N.; Sato-Otsubo, A.; Okuno, Y.; et al. Somatic RHOA mutation in angioimmunoblastic T cell lymphoma. Nat. Genet. 2014, 46, 171–175. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.Y.; Sung, M.K.; Lee, S.H.; Kim, S.; Lee, H.; Park, S.; Kim, S.C.; Lee, B.; Rho, K.; Lee, J.-E.; et al. A recurrent inactivating mutation in RHOA GTPase in angioimmunoblastic T cell lymphoma. Nat. Genet. 2014, 46, 371–375. [Google Scholar] [CrossRef] [PubMed]
- Nagata, Y.; Kontani, K.; Enami, T.; Kataoka, K.; Ishii, R.; Totoki, Y.; Kataoka, T.R.; Hirata, M.; Aoki, K.; Nakano, K.; et al. Variegated RHOA mutations in adult T-cell leukemia/lymphoma. Blood 2016, 127, 596–604. [Google Scholar] [CrossRef] [PubMed]
- Mardis, E.R.; Ding, L.; Dooling, D.J.; Larson, D.E.; McLellan, M.D.; Chen, K.; Koboldt, D.C.; Fulton, R.S.; Delehaunty, K.D.; McGrath, S.D.; et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N. Engl. J. Med. 2009, 361, 1058–1066. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Zapico, M.E.; Gonzalez-Paz, N.C.; Weiss, E.; Savoy, D.N.; Molina, J.R.; Fonseca, R.; Smyrk, T.C.; Chari, S.T.; Urrutia, R.; Billadeau, D.D. Ectopic expression of VAV1 reveals an unexpected role in pancreatic cancer tumorigenesis. Cancer Cell 2005, 7, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Bartolomé, R.A.; Molina-Ortiz, I.; Samaniego, R.; Sánchez-Mateos, P.; Bustelo, X.R.; Teixidó, J. Activation of Vav/Rho GTPase signaling by CXCL12 Controls membrane-type matrix metalloproteinase-dependent melanoma cell invasion. Cancer Res. 2006, 66, 248–258. [Google Scholar] [CrossRef] [PubMed]
- Wirtenberger, M.; Tchatchou, S.; Hemminki, K.; Klaes, R.; Schmutzler, R.K.; Bermejo, J.L.; Chen, B.; Wappenschmidt, B.; Meindl, A.; Bartram, C.R.; et al. Association of genetic variants in the Rho guanine nucleotide exchange factor AKAP13 with familial breast cancer. Carcinogenesis 2006, 27, 593–598. [Google Scholar] [CrossRef] [PubMed]
- Toksoz, D.; Williams, D.A. Novel human oncogene LBC detected by transfection with distinct homology regions to signal transduction products. Oncogene 1994, 9, 621–628. [Google Scholar] [PubMed]
- Lenoir, M.; Sugawara, M.; Kaur, J.; Ball, L.J.; Overduin, M. Structural insights into the activation of the RhoA GTPase by the Lymphoid Blast Crisis (LBC) oncoprotein. J. Biol. Chem. 2014, 289, 23992–24004. [Google Scholar] [CrossRef] [PubMed]
- Olson, M.F.; Sterpetti, P.; Nagata, K.; Toksoz, D.; Hall, A. Distinct roles for DH and PH domains in the LBC oncogene. Oncogene 1998, 15, 2827–2831. [Google Scholar] [CrossRef] [PubMed]
- Kitzing, T.M.; Sahadevan, A.S.; Brandt, D.T.; Knieling, H.; Hannemann, S.; Fackler, O.T.; Großhans, J.; Grosse, R. Positive feedback between Dia1, LARG, and RhoA regulates cell morphology and invasion. Genes Dev. 2007, 21, 1478–1483. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Ma, R.; Wang, W.; Wang, N.; Sasaki, R.; Snyderman, D.; Wu, J.; Ruan, K. Automated NMR fragment based screening identified a novel interface blocker to the LARG/RhoA complex. PLoS ONE 2014, 9, e88098. [Google Scholar] [CrossRef] [PubMed]
- Flierman, D.; van der Heden van Noort, G.J.; Ekkebus, R.; Geurink, P.P.; Mevissen, T.E.T.; Hospenthal, M.K.; Komander, D.; Ovaa, H. Non-hydrolyzable Diubiquitin Probes Reveal Linkage-Specific Reactivity of Deubiquitylating Enzymes Mediated by S2 Pockets. Cell Chem. Biol. 2016, 23, 472–482. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, R.; Pedersen, E.D.; Wang, Z.; Brakebusch, C. Rho GTPase function in tumorigenesis. Biochim. Biophys. Acta Rev. Cancer 2009, 1796, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Hitomi, M.; Stacey, D.W. Variations in cyclin D1 levels through the cell cycle determine the proliferative fate of a cell. Cell Div. 2006, 1, 32. [Google Scholar] [CrossRef] [PubMed]
- Villalonga, P.; Ridley, A.J. Rho GTPases and cell cycle control. Growth Factors (Chur, Switz.) 2006, 24, 159–164. [Google Scholar] [CrossRef]
- Mettouchi, A.; Klein, S.; Guo, W.; Lopez-Lago, M.; Lemichez, E.; Westwick, J.K.; Giancotti, F.G. Integrin-specific activation of Rac controls progression through the G1 phase of the cell cycle. Mol. Cell 2001, 8, 115–127. [Google Scholar] [CrossRef]
- Qiu, R.G.; Abo, A.; McCormick, F.; Symons, M. Cdc42 regulates anchorage-independent growth and is necessary for Ras transformation. Mol. Cell. Biol. 1997, 17, 3449–3458. [Google Scholar] [CrossRef] [PubMed]
- Kyrkou, A.; Soufi, M.; Bahtz, R.; Ferguson, C.; Bai, M.; Parton, R.G.; Hoffmann, I.; Zerial, M.; Fotsis, T.; Murphy, C. RhoD participates in the regulation of cell-cycle progression and centrosome duplication. Oncogene 2013, 32, 1831–1842. [Google Scholar] [CrossRef] [PubMed]
- Villalonga, P.; de Mattos, S.F.; Ridley, A.J. RhoE inhibits 4E-BP1 phosphorylation and eIF4E function impairing cap-dependent translation. J. Biol. Chem. 2009, 284, 35287–35296. [Google Scholar] [CrossRef] [PubMed]
- Braga, V.M.M.; Machesky, L.M.; Hall, A.; Hotchin, N.A. The small GTPases Rho and Rac are required for the establishment of cadherin-dependent cell-cell contacts. J. Cell Biol. 1997, 137, 1421–1431. [Google Scholar] [CrossRef] [PubMed]
- Desai, R.A.; Gao, L.; Raghavan, S.; Liu, W.F.; Chen, C.S. Cell polarity triggered by cell-cell adhesion via E-cadherin. J. Cell Sci. 2009, 122, 905–911. [Google Scholar] [CrossRef] [PubMed]
- Elias, B.C.; Das, A.; Parekh, D.V.; Mernaugh, G.; Adams, R.; Yang, Z.; Brakebusch, C.; Pozzi, A.; Marciano, D.K.; Carroll, T.J.; et al. Cdc42 regulates epithelial cell polarity and cytoskeletal function during kidney tubule development. J. Cell Sci. 2015, 128, 4293–4305. [Google Scholar] [CrossRef] [PubMed]
- Royer, C.; Lu, X. Epithelial cell polarity: A major gatekeeper against cancer? Cell Death Differ. 2011, 18, 1470–1477. [Google Scholar] [CrossRef] [PubMed]
- Wheelock, M.J.; Shintani, Y.; Maeda, M.; Fukumoto, Y.; Johnson, K.R. Cadherin switching. J. Cell Sci. 2008, 121, 727–735. [Google Scholar] [CrossRef] [PubMed]
- Bendas, G.; Borsig, L. Cancer cell adhesion and metastasis: Selectins, integrins, and the inhibitory potential of heparins. Int. J. Cell Biol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Paňková, K.; Rösel, D.; Novotný, M.; Brábek, J. The molecular mechanisms of transition between mesenchymal and amoeboid invasiveness in tumor cells. Cell. Mol. Life Sci. 2010, 67, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Zegers, M.M.; Friedl, P. Rho GTPases in collective cell migration. Small GTPases 2014, 5, e983869. [Google Scholar] [CrossRef] [PubMed]
- Yumura, S.; Mori, H.; Fukui, Y. Localization of actin and myosin for the study of ameboid movement in Dictyostelium using improved immunofluorescence. J. Cell Biol. 1984, 99, 894–899. [Google Scholar] [CrossRef] [PubMed]
- Sahai, E.; Olson, M.F.; Marshall, C.J. Cross-talk between Ras and Rho signalling pathways in transformation favours proliferation and increased motility. EMBO J. 2001, 20, 755–766. [Google Scholar] [CrossRef] [PubMed]
- Friedl, P.; Borgmann, S.; Bröcker, E.-B. Amoeboid leukocyte crawling through extracellular matrix: Lessons from the Dictyostelium paradigm of cell movement. J. Leukoc. Biol. 2001, 70, 491–509. [Google Scholar] [PubMed]
- Vega, F.M.; Ridley, A.J. Rho GTPases in cancer cell biology. FEBS Lett. 2008, 582, 2093–2101. [Google Scholar] [CrossRef] [PubMed]
- Vega, F.M.; Fruhwirth, G.; Ng, T.; Ridley, A.J. RhoA and RhoC have distinct roles in migration and invasion by acting through different targets. J. Cell Biol. 2011, 193, 655–665. [Google Scholar] [CrossRef] [PubMed]
- Bryan, B.A.; D’Amore, P.A. What tangled webs they weave: Rho-GTPase control of angiogenesis. Cell. Mol. Life Sci. 2007, 64, 2053–2065. [Google Scholar] [CrossRef] [PubMed]
- Eichhorn, M.E.; Kleespies, A.; Angele, M.K.; Jauch, K.-W.; Bruns, C.J. Angiogenesis in cancer: Molecular mechanisms, clinical impact. Langenbecks Arch. Surg. 2007, 392, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Nishida, N.; Yano, H.; Nishida, T.; Kamura, T.; Kojiro, M. Angiogenesis in cancer. Vasc. Health Risk Manag. 2006, 2, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Holmgren, L.; O’Reilly, M.S.; Folkman, J. Dormancy of micrometastases: Balanced proliferation and apoptosis in the presence of angiogenesis suppression. Nat. Med. 1995, 1, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Parangi, S.; O’Reilly, M.; Christofori, G.; Holmgren, L.; Grosfeld, J.; Folkman, J.; Hanahan, D. Antiangiogenic therapy of transgenic mice impairs de novo tumor growth. Proc. Natl. Acad. Sci. USA 1996, 93, 2002–2007. [Google Scholar] [CrossRef] [PubMed]
- Jeeves, M.; Quill, L.; Overduin, M. Structure-based drug design using NMR. In NMR in Pharmaceutical Science; Everett, J.R., Harris, R.K., Lindon, J.C., Wilson, I.D., Eds.; John Wiley & Sons, Ltd.: Chichester, UK, 2015; pp. 317–330. [Google Scholar]
- Bouquier, N.; Vignal, E.; Charrasse, S.; Weill, M.; Schmidt, S.; Léonetti, J.-P.; Blangy, A.; Fort, P. A cell active chemical GEF inhibitor selectively targets the Trio/RhoG/Rac1 signaling pathway. Chem. Biol. 2009, 16, 657–666. [Google Scholar] [CrossRef] [PubMed]
- Shutes, A.; Onesto, C.; Picard, V.; Leblond, B.; Schweighoffer, F.; Der, C.J. Specificity and mechanism of action of EHT 1864, a novel small molecule inhibitor of Rac family small GTPases. J. Biol. Chem. 2007, 282, 35666–35678. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Dickerson, J.B.; Guo, F.; Zheng, J.; Zheng, Y. Rational design and characterization of a Rac GTPase-specific small molecule inhibitor. Proc. Natl. Acad. Sci. USA 2004, 101, 7618–7623. [Google Scholar] [CrossRef] [PubMed]
- Nassar, N.; Cancelas, J.; Zheng, J.; Williams, D.; Zheng, Y. Structure-function based design of small molecule inhibitors targeting Rho family GTPases. Curr. Top. Med. Chem. 2006, 6, 1109–1116. [Google Scholar] [CrossRef] [PubMed]
- Montalvo-Ortiz, B.L.; Castillo-Pichardo, L.; Hernández, E.; Humphries-Bickley, T.; Mota-Peynado, A.D.L.; Cubano, L.A.; Vlaar, C.P.; Dharmawardhane, S. Characterization of EHop-016, novel small molecule inhibitor of Rac GTPase. J. Biol. Chem. 2012, 287, 13228–13238. [Google Scholar] [CrossRef] [PubMed]
- Florian, M.C.; Dörr, K.; Niebel, A.; Daria, D.; Schrezenmeier, H.; Rojewski, M.; Filippi, M.-D.; Hasenberg, A.; Gunzer, M.; Scharffetter-Kochanek, K.; et al. Cdc42 activity regulates hematopoietic stem cell aging and rejuvenation. Cell Stem Cell 2012, 10, 520–530. [Google Scholar] [CrossRef] [PubMed]
- Hong, L.; Kenney, S.R.; Phillips, G.K.; Simpson, D.; Schroeder, C.E.; Nöth, J.; Romero, E.; Swanson, S.; Waller, A.; Strouse, J.J.; et al. Characterization of a Cdc42 protein inhibitor and its use as a molecular probe. J. Biol. Chem. 2013, 288, 8531–8543. [Google Scholar] [CrossRef] [PubMed]
- Friesland, A.; Zhao, Y.; Chen, Y.-H.; Wang, L.; Zhou, H.; Lu, Q. Small molecule targeting Cdc42-intersectin interaction disrupts Golgi organization and suppresses cell motility. Proc. Natl. Acad. Sci. USA 2013, 110, 1261–1266. [Google Scholar] [CrossRef] [PubMed]
- Shang, X.; Marchioni, F.; Sipes, N.; Evelyn, C.R.; Jerabek-Willemsen, M.; Duhr, S.; Seibel, W.; Wortman, M.; Zheng, Y. Rational design of small molecule inhibitors targeting RhoA subfamily Rho GTPases. Chem. Biol. 2012, 19, 699–710. [Google Scholar] [CrossRef] [PubMed]
- Evelyn, C.R.; Ferng, T.; Rojas, R.J.; Larsen, M.J.; Sondek, J.; Neubig, R.R. High-throughput screening for small molecule inhibitors of LARG-stimulated RhoA nucleotide binding via a novel fluorescence polarization assay. J. Biomol. Screen. 2009, 14, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Shang, X.; Marchioni, F.; Evelyn, C.R.; Sipes, N.; Zhou, X.; Seibel, W.; Wortman, M.; Zheng, Y. Small-molecule inhibitors targeting G-protein–coupled Rho guanine nucleotide exchange factors. Proc. Natl. Acad. Sci. USA 2013, 110, 3155–3160. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Feng, E.; Ma, S.; Zhang, Y.; Liu, X.; Li, H.; Huang, H.; Zhu, J.; Zhu, W.; Shen, X.; et al. Design and synthesis of small molecule RhoA inhibitors: A new promising therapy for cardiovascular diseases? J. Med. Chem. 2011, 54, 4508–4522. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Deng, J.; Li, B.; Li, X.; Yan, Z.; Zhu, J.; Chen, G.; Wang, Z.; Jiang, H.; Miao, L.; et al. Development of second-generation small-molecule RhoA inhibitors with enhanced water solubility, tissue potency, and significant in vivo efficacy. ChemMedChem 2015, 10, 193–206. [Google Scholar] [CrossRef] [PubMed]



© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smithers, C.C.; Overduin, M. Structural Mechanisms and Drug Discovery Prospects of Rho GTPases. Cells 2016, 5, 26. https://doi.org/10.3390/cells5020026
Smithers CC, Overduin M. Structural Mechanisms and Drug Discovery Prospects of Rho GTPases. Cells. 2016; 5(2):26. https://doi.org/10.3390/cells5020026
Chicago/Turabian StyleSmithers, Cameron C., and Michael Overduin. 2016. "Structural Mechanisms and Drug Discovery Prospects of Rho GTPases" Cells 5, no. 2: 26. https://doi.org/10.3390/cells5020026