

All but Small: miRNAs from Wharton’s Jelly-Mesenchymal Stromal Cell Small Extracellular Vesicles Rescue Premature White Matter Injury after Intranasal Administration
 , and
, and
Abstract
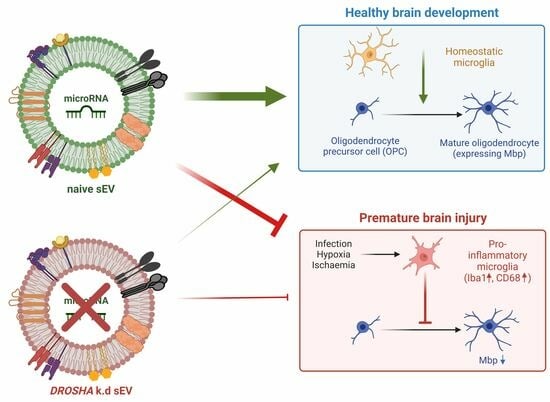
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Isolation of WJ-MSC-sEVs and DROSHA k.d sEVs
2.2. Preparation of DiR-Labeled WJ-MSC-sEVs to Visualize Their Biodistribution
2.3. Animal Model of Premature WMI
2.4. Analysis of WJ-MSC-sEV Biodistribution In Vivo and Ex Vivo
2.5. Immunohistochemistry
2.6. RNA and Protein Isolation
2.7. cDNA Synthesis and Real-Time Quantitative Reverse Transcription Polymerase Chain Reaction (qRT-PCR)
2.8. Western Blot Analysis
2.9. Detection of Neuronal Cell Death: TdT-Mediated dUTP-Biotin Nick End Labelling (TUNEL) Staining and Analysis of Cleaved Caspase 3 and Bad
2.10. Statistical Analysis
3. Results
3.1. Intranasally Administered WJ-MSC-sEVs Target the Brain
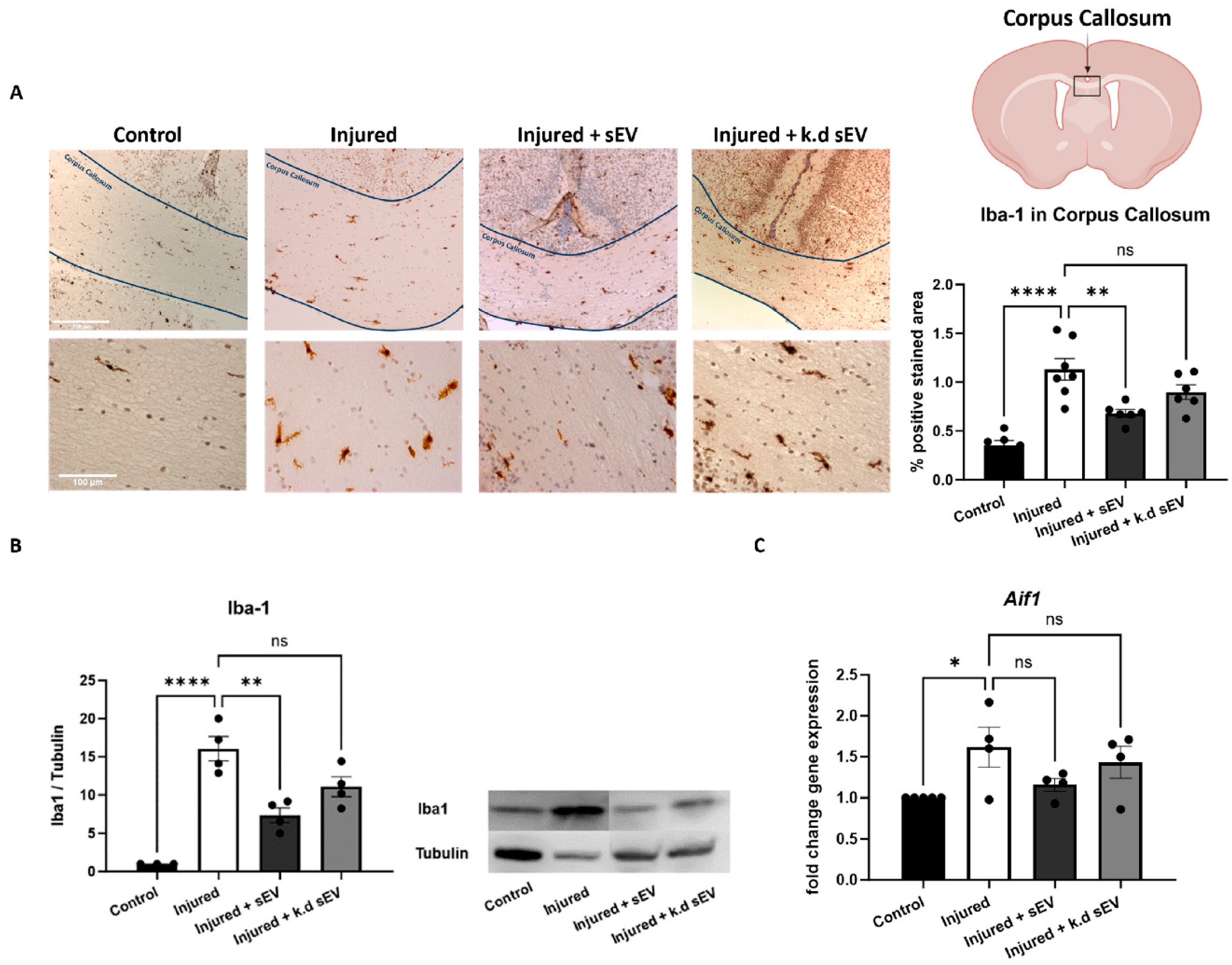
3.2. Intranasal Administration of WJ-MSC-sEVs Ameliorates the Inflammatory Response in Experimental Premature WMI
3.3. WJ-MSC-sEVs with Highly Abundant miRNA Cargo Increase OL Maturation
3.4. Hippocampal Cell Death Is Not Increased in Experimental Premature WMI
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Walani, S.R. Global burden of preterm birth. Int. J. Gynaecol. Obstet. 2020, 150, 31–33. [Google Scholar] [CrossRef]
- Khwaja, O.; Volpe, J.J. Pathogenesis of cerebral white matter injury of prematurity. Arch. Dis. Child. Fetal Neonatal Ed. 2008, 93, F153–F161. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.; Miller, S.P. Preterm brain Injury: White matter injury. Handb. Clin. Neurol. 2019, 162, 155–172. [Google Scholar] [CrossRef]
- Agut, T.; Alarcon, A.; Cabañas, F.; Bartocci, M.; Martinez-Biarge, M.; Horsch, S. Preterm white matter injury: Ultrasound diagnosis and classification. Pediatr. Res. 2020, 87, 37–49. [Google Scholar] [CrossRef]
- Arpino, C.; Compagnone, E.; Montanaro, M.L.; Cacciatore, D.; De Luca, A.; Cerulli, A.; Di Girolamo, S.; Curatolo, P. Preterm birth and neurodevelopmental outcome: A review. Child’s Nerv. Syst. 2010, 26, 1139–1149. [Google Scholar] [CrossRef]
- Hedderich, D.M.; Boeckh-Behrens, T.; Bäuml, J.G.; Menegaux, A.; Daamen, M.; Zimmer, C.; Bartmann, P.; Scheef, L.; Boecker, H.; Wolke, D.; et al. Sequelae of Premature Birth in Young Adults. Clin. Neuroradiol. 2021, 31, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Córcoles-Parada, M.; Giménez-Mateo, R.; Serrano-Del-Pueblo, V.; López, L.; Pérez-Hernández, E.; Mansilla, F.; Martínez, A.; Onsurbe, I.; San Roman, P.; Ubero-Martinez, M.; et al. Born Too Early and Too Small: Higher Order Cognitive Function and Brain at Risk at Ages 8–16. Front. Psychol. 2019, 10, 1942. [Google Scholar] [CrossRef]
- Nath, N.; Beltrano, W.; Haynes, L.; Dewey, D.; Bray, S. Long-Term Effects of Preterm Birth on Children’s Brain Structure: An Analysis of the Adolescent Brain Cognitive Development (ABCD) Study. eNeuro 2023, 10. [Google Scholar] [CrossRef] [PubMed]
- Cainelli, E.; Arrigoni, F.; Vedovelli, L. White matter injury and neurodevelopmental disabilities: A cross-disease (dis)connection. Prog. Neurobiol. 2020, 193, 101845. [Google Scholar] [CrossRef]
- Volpe, J.J. Brain injury in premature infants: A complex amalgam of destructive and developmental disturbances. Lancet Neurol. 2009, 8, 110–124. [Google Scholar] [CrossRef]
- Back, S.A.; Luo, N.L.; Borenstein, N.S.; Levine, J.M.; Volpe, J.J.; Kinney, H.C. Late oligodendrocyte progenitors coincide with the developmental window of vulnerability for human perinatal white matter injury. J. Neurosci. 2001, 21, 1302–1312. [Google Scholar] [CrossRef]
- Volpe, J.J.; Kinney, H.C.; Jensen, F.E.; Rosenberg, P.A. The developing oligodendrocyte: Key cellular target in brain injury in the premature infant. Int. J. Dev. Neurosci. 2011, 29, 423. [Google Scholar] [CrossRef]
- McNamara, N.B.; Miron, V.E. Microglia in developing white matter and perinatal brain injury. Neurosci. Lett. 2020, 714, 134539. [Google Scholar] [CrossRef] [PubMed]
- Back, S.A.; Han, B.H.; Luo, N.L.; Chricton, C.A.; Xanthoudakis, S.; Tam, J.; Arvin, K.L.; Holtzman, D.M. Selective Vulnerability of Late Oligodendrocyte Progenitors to Hypoxia–Ischemia. J. Neurosci. 2002, 22, 455. [Google Scholar] [CrossRef]
- Baburamani, A.A.; Supramaniam, V.G.; Hagberg, H.; Mallard, C. Microglia toxicity in preterm brain injury. Reprod. Toxicol. 2014, 48, 106–112. [Google Scholar] [CrossRef]
- Truttmann, A.C.; Ginet, V.; Puyal, J. Current Evidence on Cell Death in Preterm Brain Injury in Human and Preclinical Models. Front. Cell Dev. Biol. 2020, 8, 27. [Google Scholar] [CrossRef]
- Chavez-Valdez, R.; Emerson, P.; Goffigan-Holmes, J.; Kirkwood, A.; Martin, L.J.; Northington, F.J. Delayed injury of hippocampal interneurons after neonatal hypoxia-ischemia and therapeutic hypothermia in a murine model. Hippocampus 2018, 28, 617–630. [Google Scholar] [CrossRef]
- Back, S.A. Cerebral white and gray matter injury in newborns: New insights into pathophysiology and management. Clin. Perinatol. 2014, 41, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Cui, G.H.; Wu, J.; Mou, F.F.; Xie, W.H.; Wang, F.B.; Wang, Q.L.; Fang, J.; Xu, Y.W.; Dong, Y.R.; Liu, J.R.; et al. Exosomes derived from hypoxia-preconditioned mesenchymal stromal cells ameliorate cognitive decline by rescuing synaptic dysfunction and regulating inflammatory responses in APP/PS1 mice. FASEB J. 2018, 32, 654–668. [Google Scholar] [CrossRef] [PubMed]
- Harrell, C.R.; Volarevic, A.; Djonov, V.; Volarevic, V. Mesenchymal Stem Cell-Derived Exosomes as New Remedy for the Treatment of Neurocognitive Disorders. Int. J. Mol. Sci. 2021, 22, 1433. [Google Scholar] [CrossRef]
- Reza-Zaldivar, E.E.; Hernández-Sapiéns, M.A.; Minjarez, B.; Gutiérrez-Mercado, Y.K.; Márquez-Aguirre, A.L.; Canales-Aguirre, A.A. Potential Effects of MSC-Derived Exosomes in Neuroplasticity in Alzheimer’s Disease. Front. Cell. Neurosci. 2018, 12, 317. [Google Scholar] [CrossRef]
- Herman, S.; Fishel, I.; Offen, D. Intranasal delivery of mesenchymal stem cells-derived extracellular vesicles for the treatment of neurological diseases. Stem Cells 2021, 39, 1589–1600. [Google Scholar] [CrossRef]
- Turano, E.; Scambi, I.; Virla, F.; Bonetti, B.; Mariotti, R. Extracellular Vesicles from Mesenchymal Stem Cells: Towards Novel Therapeutic Strategies for Neurodegenerative Diseases. Int. J. Mol. Sci. 2023, 24, 2917. [Google Scholar] [CrossRef] [PubMed]
- Labusek, N.; Mouloud, Y.; Köster, C.; Diesterbeck, E.; Tertel, T.; Wiek, C.; Hanenberg, H.; Horn, P.A.; Felderhoff-Müser, U.; Bendix, I.; et al. Extracellular vesicles from immortalized mesenchymal stromal cells protect against neonatal hypoxic-ischemic brain injury. Inflamm. Regen. 2023, 43, 24. [Google Scholar] [CrossRef] [PubMed]
- Sisa, C.; Kholia, S.; Naylor, J.; Sanchez, M.B.H.; Bruno, S.; Deregibus, M.C.; Camussi, G.; Inal, J.M.; Lange, S.; Hristova, M. Mesenchymal Stromal Cell Derived Extracellular Vesicles Reduce Hypoxia-Ischaemia Induced Perinatal Brain Injury. Front. Physiol. 2019, 10, 442626. [Google Scholar] [CrossRef] [PubMed]
- Lawson, A.; Snyder, W.; Peeples, E.S. Intranasal Administration of Extracellular Vesicles Mitigates Apoptosis in a Mouse Model of Neonatal Hypoxic-Ischemic Brain Injury. Neonatology 2022, 119, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Kaminski, N.; Köster, C.; Mouloud, Y.; Börger, V.; Felderhoff-Müser, U.; Bendix, I.; Giebel, B.; Herz, J. Mesenchymal Stromal Cell-Derived Extracellular Vesicles Reduce Neuroinflammation, Promote Neural Cell Proliferation and Improve Oligodendrocyte Maturation in Neonatal Hypoxic-Ischemic Brain Injury. Front. Cell. Neurosci. 2020, 14, 601176. [Google Scholar] [CrossRef] [PubMed]
- Ophelders, D.R.M.G.; Wolfs, T.G.A.M.; Jellema, R.K.; Zwanenburg, A.; Andriessen, P.; Delhaas, T.; Ludwig, A.-K.; Radtke, S.; Peters, V.; Janssen, L.; et al. Mesenchymal Stromal Cell-Derived Extracellular Vesicles Protect the Fetal Brain after Hypoxia-Ischemia. Stem Cells Transl. Med. 2016, 5, 754–763. [Google Scholar] [CrossRef] [PubMed]
- Thomi, G.; Surbek, D.; Haesler, V.; Joerger-Messerli, M.; Schoeberlein, A. Exosomes derived from umbilical cord mesenchymal stem cells reduce microglia-mediated neuroinflammation in perinatal brain injury. Stem Cell Res. Ther. 2019, 10, 105. [Google Scholar] [CrossRef] [PubMed]
- Thomi, G.; Joerger-Messerli, M.; Haesler, V.; Muri, L.; Surbek, D.; Schoeberlein, A. Intranasally Administered Exosomes from Umbilical Cord Stem Cells Have Preventive Neuroprotective Effects and Contribute to Functional Recovery after Perinatal Brain Injury. Cells 2019, 8, 855. [Google Scholar] [CrossRef]
- Joerger-Messerli, M.S.; Oppliger, B.; Spinelli, M.; Thomi, G.; di Salvo, I.; Schneider, P.; Schoeberlein, A. Extracellular Vesicles Derived from Wharton’s Jelly Mesenchymal Stem Cells Prevent and Resolve Programmed Cell Death Mediated by Perinatal Hypoxia-Ischemia in Neuronal Cells. Cell Transplant. 2018, 27, 168–180. [Google Scholar] [CrossRef] [PubMed]
- Joerger-Messerli, M.S.; Thomi, G.; Haesler, V.; Keller, I.; Renz, P.; Surbek, D.V.; Schoeberlein, A. Human Wharton’s Jelly Mesenchymal Stromal Cell-Derived Small Extracellular Vesicles Drive Oligodendroglial Maturation by Restraining MAPK/ERK and Notch Signaling Pathways. Front. Cell Dev. Biol. 2021, 9, 622539. [Google Scholar] [CrossRef] [PubMed]
- Elsharkasy, O.M.; Nordin, J.Z.; Hagey, D.W.; de Jong, O.G.; Schiffelers, R.M.; Andaloussi, S.E.; Vader, P. Extracellular vesicles as drug delivery systems: Why and how? Adv. Drug Deliv. Rev. 2020, 159, 332–343. [Google Scholar] [CrossRef]
- Alzhrani, G.N.; Alanazi, S.T.; Alsharif, S.Y.; Albalawi, A.M.; Alsharif, A.A.; Abdel-Maksoud, M.S.; Elsherbiny, N. Exosomes: Isolation, characterization, and biomedical applications. Cell Biol Int 2021, 45, 1807–1831. [Google Scholar] [CrossRef]
- Lo Cicero, A.; Stahl, P.D.; Raposo, G. Extracellular vesicles shuffling intercellular messages: For good or for bad. Curr. Opin. Cell Biol. 2015, 35, 69–77. [Google Scholar] [CrossRef]
- Ranganathan, K.; Sivasankar, V. MicroRNAs—Biology and clinical applications. J. Oral Maxillofac. Pathol. 2014, 18, 229–234. [Google Scholar] [CrossRef]
- Han, J.; Lee, Y.; Yeom, K.H.; Kim, Y.K.; Jin, H.; Kim, V.N. The Drosha-DGCR8 complex in primary microRNA processing. Genes Dev. 2004, 18, 3016–3027. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.K.; Kim, B.; Kim, V.N. Re-evaluation of the roles of DROSHA, Exportin 5, and DICER in microRNA biogenesis. Proc. Natl. Acad. Sci. USA 2016, 113, E1881–E1889. [Google Scholar] [CrossRef]
- Tscherrig, V.; Cottagnoud, S.; Haesler, V.; Renz, P.; Surbek, D.; Schoeberlein, A.; Joerger-Messerli, M.S. MicroRNA Cargo in Wharton’s Jelly MSC Small Extracellular Vesicles: Key Functionality to In Vitro Prevention and Treatment of Premature White Matter Injury. Stem Cell Rev. Rep. 2023, 19, 2447–2464. [Google Scholar] [CrossRef]
- Collino, F.; Bruno, S.; Incarnato, D.; Dettori, D.; Neri, F.; Provero, P.; Pomatto, M.; Oliviero, S.; Tetta, C.; Quesenberry, P.J.; et al. AKI Recovery Induced by Mesenchymal Stromal Cell-Derived Extracellular Vesicles Carrying MicroRNAs. J. Am. Soc. Nephrol. 2015, 26, 2349–2360. [Google Scholar] [CrossRef]
- Luo, H.; Huang, F.; Huang, Z.; Huang, H.; Liu, C.; Feng, Y.; Qi, Z. microRNA-93 packaged in extracellular vesicles from mesenchymal stem cells reduce neonatal hypoxic-ischemic brain injury. Brain Res. 2022, 1794, 148042. [Google Scholar] [CrossRef]
- Gao, X.; Xiong, Y.; Li, Q.; Han, M.; Shan, D.; Yang, G.; Zhang, S.; Xin, D.; Zhao, R.; Wang, Z.; et al. Extracellular vesicle-mediated transfer of miR-21-5p from mesenchymal stromal cells to neurons alleviates early brain injury to improve cognitive function via the PTEN/Akt pathway after subarachnoid hemorrhage. Cell Death Dis. 2020, 11, 363. [Google Scholar] [CrossRef]
- Xin, D.Q.; Zhao, Y.J.; Li, T.T.; Ke, H.F.; Gai, C.C.; Guo, X.F.; Chen, W.Q.; Liu, D.X.; Wang, Z. The delivery of miR-21a-5p by extracellular vesicles induces microglial polarization via the STAT3 pathway following hypoxia-ischemia in neonatal mice. Neural. Regen. Res. 2022, 17, 2238–2246. [Google Scholar] [CrossRef]
- Xin, D.; Li, T.; Chu, X.; Ke, H.; Yu, Z.; Cao, L.; Bai, X.; Liu, D.; Wang, Z. Mesenchymal stromal cell-derived extracellular vesicles modulate microglia/macrophage polarization and protect the brain against hypoxia-ischemic injury in neonatal mice by targeting delivery of miR-21a-5p. Acta Biomater. 2020, 113, 597–613. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, J.; Su, M.; Wang, X.; Xie, C. Exosomal microRNA-22-3p alleviates cerebral ischemic injury by modulating KDM6B/BMP2/BMF axis. Stem Cell Res. Ther. 2021, 12, 111. [Google Scholar] [CrossRef]
- Garcia-Martin, R.; Wang, G.; Brandão, B.B.; Zanotto, T.M.; Shah, S.; Kumar Patel, S.; Schilling, B.; Kahn, C.R. MicroRNA sequence codes for small extracellular vesicle release and cellular retention. Nature 2022, 601, 446. [Google Scholar] [CrossRef]
- Messerli, M.; Wagner, A.; Sager, R.; Mueller, M.; Baumann, M.; Surbek, D.V.; Schoeberlein, A. Stem cells from umbilical cord Wharton’s jelly from preterm birth have neuroglial differentiation potential. Reprod. Sci. 2013, 20, 1455–1464. [Google Scholar] [CrossRef]
- Théry, C.; Amigorena, S.; Raposo, G.; Clayton, A. Isolation and Characterization of Exosomes from Cell Culture Supernatants and Biological Fluids. Curr. Protoc. Cell Biol. 2006, 30, 3–22. [Google Scholar] [CrossRef]
- Van Deun, J.; Mestdagh, P.; Agostinis, P.; Akay, Ö.; Anand, S.; Anckaert, J.; Martinez, Z.A.; Baetens, T.; Beghein, E.; Bertier, L.; et al. EV-TRACK: Transparent reporting and centralizing knowledge in extracellular vesicle research. Nat. Methods 2017, 14, 228–232. [Google Scholar] [CrossRef]
- Zhu, L.; Yu, Q.; Gao, P.; Liu, Q.; Luo, X.; Jiang, G.; Ji, R.; Yang, R.; Ma, X.; Xu, J.; et al. TAOK1 positively regulates TLR4-induced inflammatory responses by promoting ERK1/2 activation in macrophages. Mol. Immunol. 2020, 122, 124–131. [Google Scholar] [CrossRef]
- Fang, C.-Y.; Lai, T.-C.; Hsiao, M.; Chang, Y.-C. Molecular Sciences The Diverse Roles of TAO Kinases in Health and Diseases. Int. J. Mol. Sci. 2020, 21, 7463. [Google Scholar] [CrossRef]
- Zihni, C.; Mitsopoulos, C.; Tavares, I.A.; Baum, B.; Ridley, A.J.; Morris, J.D.H. Prostate-derived sterile 20-like kinase 1-alpha induces apoptosis. JNK- and caspase-dependent nuclear localization is a requirement for membrane blebbing. J. Biol. Chem. 2007, 282, 6484–6493. [Google Scholar] [CrossRef]
- Li, Y.; Wu, H.; Jiang, X.; Dong, Y.; Zheng, J.; Gao, J. New idea to promote the clinical applications of stem cells or their extracellular vesicles in central nervous system disorders: Combining with intranasal delivery. Acta Pharm. Sin. B 2022, 12, 3215–3232. [Google Scholar] [CrossRef]
- Chen, C.C.; Liu, L.; Ma, F.; Wong, C.W.; Guo, X.E.; Chacko, J.V.; Farhoodi, H.P.; Zhang, S.X.; Zimak, J.; Ségaliny, A.; et al. Elucidation of Exosome Migration across the Blood-Brain Barrier Model In Vitro HHS Public Access Author manuscript. Cell Mol. Bioeng. 2016, 9, 509–529. [Google Scholar] [CrossRef]
- Matsumoto, J.; Stewart, T.; Banks, W.A.; Zhang, J. The Transport Mechanism of Extracellular Vesicles at the Blood-Brain Barrier. Curr. Pharm. Des. 2018, 23, 6206–6214. [Google Scholar] [CrossRef]
- Attaluri, S.; Jaimes Gonzalez, J.; Kirmani, M.; Vogel, A.D.; Upadhya, R.; Kodali, M.; Madhu, L.N.; Rao, S.; Shuai, B.; Babu, R.S.; et al. Intranasally administered extracellular vesicles from human induced pluripotent stem cell-derived neural stem cells quickly incorporate into neurons and microglia in 5xFAD mice. Front. Aging Neurosci. 2023, 15, 1200445. [Google Scholar] [CrossRef]
- Betzer, O.; Shilo, M.; Opochinsky, R.; Barnoy, E.; Motiei, M.; Okun, E.; Yadid, G.; Popovtzer, R. The effect of nanoparticle size on the ability to cross the blood-brain barrier: An in vivo study. Nanomedicine 2017, 12, 1533–1546. [Google Scholar] [CrossRef]
- Liu, L.; Mao, Q.; Chu, S.; Mounayar, M.; Abdi, R.; Fodor, W.; Padbury, J.F.; De Paepe, M.E. Intranasal versus intraperitoneal delivery of human umbilical cord tissue-derived cultured mesenchymal stromal cells in a murine model of neonatal lung injury. Am. J. Pathol. 2014, 184, 3344–3358. [Google Scholar] [CrossRef]
- Freeman, S.C.; Karp, D.A.; Kahwaji, C.I. Physiology, Nasal. In StatPearls; StatPearls Publishing LLC.: Treasure Island, FL, USA, 2023. [Google Scholar]
- Drommelschmidt, K.; Serdar, M.; Bendix, I.; Herz, J.; Bertling, F.; Prager, S.; Keller, M.; Ludwig, A.K.; Duhan, V.; Radtke, S.; et al. Mesenchymal stem cell-derived extracellular vesicles ameliorate inflammation-induced preterm brain injury. Brain Behav. Immun. 2017, 60, 220–232. [Google Scholar] [CrossRef]
- Chu, X.; Liu, D.; Li, T.; Ke, H.; Xin, D.; Wang, S.; Cao, Y.; Xue, H.; Wang, Z. Hydrogen sulfide-modified extracellular vesicles from mesenchymal stem cells for treatment of hypoxic-ischemic brain injury. J. Control Release 2020, 328, 13–27. [Google Scholar] [CrossRef]
- Lier, J.; Streit, W.J.; Bechmann, I. Beyond Activation: Characterizing Microglial Functional Phenotypes. Cells 2021, 10, 2236. [Google Scholar] [CrossRef]
- Fu, R.; Shen, Q.; Xu, P.; Luo, J.J.; Tang, Y. Phagocytosis of microglia in the central nervous system diseases. Mol. Neurobiol. 2014, 49, 1422–1434. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.Y.; Cheng, A.C.; Wang, M.S.; Yin, Z.Q.; Jia, R.Y. Exosomes: Potential Therapies for Disease via Regulating TLRs. Mediat. Inflamm. 2020, 2020, 2319616. [Google Scholar] [CrossRef]
- Huang, S.; Liao, X.; Wu, J.; Zhang, X.; Li, Y.; Xiang, D.; Luo, S. The Microglial membrane receptor TREM2 mediates exosome secretion to promote phagocytosis of amyloid-β by microglia. FEBS Lett. 2022, 596, 1059–1071. [Google Scholar] [CrossRef]
- Zhang, Y.; Meng, J.; Zhang, L.; Ramkrishnan, S.; Roy, S. Extracellular Vesicles with Exosome-like Features Transfer TLRs between Dendritic Cells. Immunohorizons 2019, 3, 186–193. [Google Scholar] [CrossRef]
- Cieślik, M.; Bryniarski, K.; Nazimek, K. Biodelivery of therapeutic extracellular vesicles: Should mononuclear phagocytes always be feared? Front. Cell Dev. Biol. 2023, 11, 1211833. [Google Scholar] [CrossRef]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of microRNA biogenesis, mechanisms of actions, and circulation. Front. Endocrinol. 2018, 9, 388354. [Google Scholar] [CrossRef] [PubMed]
- Newville, J.; Jantzie, L.L.; Cunningham, L.A. Embracing oligodendrocyte diversity in the context of perinatal injury. Neural. Regen. Res. 2017, 12, 1575–1585. [Google Scholar] [CrossRef]
- Hou, H.; Wang, Y.; Yang, L.; Wang, Y. Exosomal miR-128-3p reversed fibrinogen-mediated inhibition of oligodendrocyte progenitor cell differentiation and remyelination after cerebral ischemia. CNS Neurosci. Ther. 2023, 29, 1405–1422. [Google Scholar] [CrossRef]
- Miller, C.J.; Turk, B.E. Homing in: Mechanisms of Substrate Targeting by Protein Kinases. Trends Biochem. Sci. 2018, 43, 380–394. [Google Scholar] [CrossRef]
- Kaidanovich-Beilin, O.; Eldar-Finkelman, H. Peptides targeting protein kinases: Strategies and implications. Physiology 2006, 21, 411–418. [Google Scholar] [CrossRef]
- Cho, K.H.T.; Xu, B.; Blenkiron, C.; Fraser, M. Emerging Roles of miRNAs in Brain Development and Perinatal Brain Injury. Front. Physiol. 2019, 10, 420089. [Google Scholar] [CrossRef]
- Qian, Y.; Li, Q.; Chen, L.; Sun, J.; Cao, K.; Mei, Z.; Lu, X. Mesenchymal Stem Cell-Derived Extracellular Vesicles Alleviate M1 Microglial Activation in Brain Injury of Mice with Subarachnoid Hemorrhage via microRNA-140-5p Delivery. Int. J. Neuropsychopharmacol. 2022, 25, 328–338. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, J.; Zhou, Y.; Zou, Z.; Xie, C.; Ma, L. miR-18a-5p shuttled by mesenchymal stem cell-derived extracellular vesicles alleviates early brain injury following subarachnoid hemorrhage through blockade of the ENC1/p62 axis. Cell Tissue Res. 2023, 392, 671–687. [Google Scholar] [CrossRef]
- Ligam, P.; Haynes, R.L.; Folkerth, R.D.; Liu, L.; Yang, M.; Volpe, J.J.; Kinney, H.C. Thalamic Damage in Periventricular Leukomalacia: Novel Pathologic Observations Relevant to Cognitive Deficits in Survivors of Prematurity. Pediatr. Res. 2009, 65, 524–529. [Google Scholar] [CrossRef] [PubMed]
- Pierson, C.R.; Folkerth, R.D.; Billiards, S.S.; Trachtenberg, F.L.; Drinkwater, M.E.; Volpe, J.J.; Kinney, H.C. Gray matter injury associated with periventricular leukomalacia in the premature infant. Acta Neuropathol. 2007, 114, 619–631. [Google Scholar] [CrossRef] [PubMed]
- Andiman, S.E.; Haynes, R.L.; Trachtenberg, F.L.; Billiards, S.S.; Folkerth, R.D.; Volpe, J.J.; Kinney, H.C. The Cerebral Cortex Overlying Periventricular Leukomalacia: Analysis of Pyramidal Neurons. Brain Pathol. 2010, 20, 803–814. [Google Scholar] [CrossRef] [PubMed]
- Kinney, H.C.; Haynes, R.L.; Xu, G.; Andiman, S.E.; Folkerth, R.D.; Sleeper, L.A.; Volpe, J.J. Neuron deficit in the white matter and subplate in periventricular leukomalacia. Ann. Neurol. 2012, 71, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Misumi, S.; Ueda, Y.; Nishigaki, R.; Suzuki, M.; Ishida, A.; Jung, C.-G.; Hida, H. Dysfunction in Motor Coordination in Neonatal White Matter Injury Model without Apparent Neuron Loss. Cell Transplant. 2016, 25, 1381–1393. [Google Scholar] [CrossRef]
- Back, S.A.; Miller, S.P. Brain injury in premature neonates: A primary cerebral dysmaturation disorder? Ann. Neurol. 2014, 75, 469–486. [Google Scholar] [CrossRef] [PubMed]
- Verney, C.; Pogledic, I.; Biran, V.; Adle-Biassette, H.; Fallet-Bianco, C.; Gressens, P. Microglial reaction in axonal crossroads is a hallmark of noncystic periventricular white matter injury in very preterm infants. J. Neuropathol. Exp. Neurol. 2012, 71, 251–264. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.M.; Li, Z.X.; Lin, R.H.; Shan, J.Q.; Yu, Q.W.; Wang, R.X.; Liao, L.S.; Yan, W.T.; Wang, Z.; Shang, L.; et al. Guidelines for Regulated Cell Death Assays: A Systematic Summary, A Categorical Comparison, A Prospective. Front. Cell Dev. Biol. 2021, 9, 634690. [Google Scholar] [CrossRef] [PubMed]
- Pong, S.K.; Gullerova, M. Noncanonical functions of microRNA pathway enzymes—Drosha, DGCR8, Dicer and Ago proteins. FEBS Lett. 2018, 592, 2973–2986. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tscherrig, V.; Steinfort, M.; Haesler, V.; Surbek, D.; Schoeberlein, A.; Joerger-Messerli, M.S. All but Small: miRNAs from Wharton’s Jelly-Mesenchymal Stromal Cell Small Extracellular Vesicles Rescue Premature White Matter Injury after Intranasal Administration. Cells 2024, 13, 543. https://doi.org/10.3390/cells13060543
Tscherrig V, Steinfort M, Haesler V, Surbek D, Schoeberlein A, Joerger-Messerli MS. All but Small: miRNAs from Wharton’s Jelly-Mesenchymal Stromal Cell Small Extracellular Vesicles Rescue Premature White Matter Injury after Intranasal Administration. Cells. 2024; 13(6):543. https://doi.org/10.3390/cells13060543
Chicago/Turabian StyleTscherrig, Vera, Marel Steinfort, Valérie Haesler, Daniel Surbek, Andreina Schoeberlein, and Marianne Simone Joerger-Messerli. 2024. "All but Small: miRNAs from Wharton’s Jelly-Mesenchymal Stromal Cell Small Extracellular Vesicles Rescue Premature White Matter Injury after Intranasal Administration" Cells 13, no. 6: 543. https://doi.org/10.3390/cells13060543