

Recent Research Trends in Neuroinflammatory and Neurodegenerative Disorders
 , ,
, ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
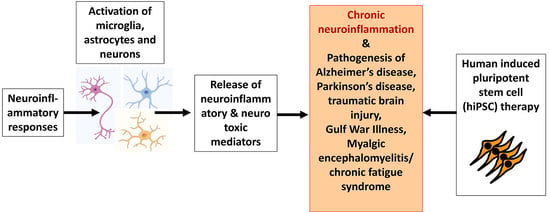
1. Introduction
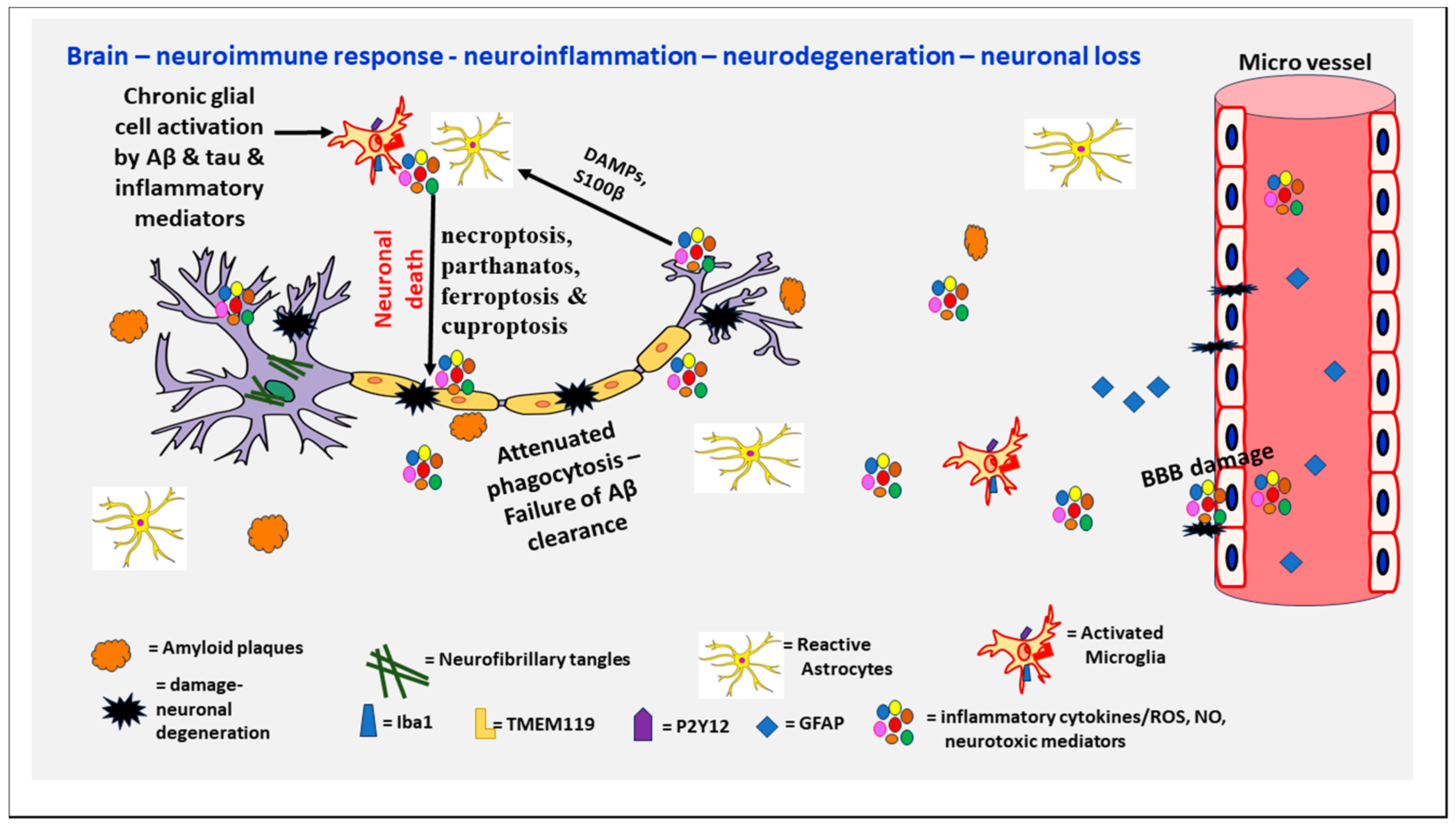
2. Alzheimer’s Disease (AD)
3. Parkinson’s Disease (PD)
4. Traumatic Brain Injury (TBI)
5. Gulf War Illness (GWI)
6. Myalgic Encephalomyelitis/Chronic Fatigue Syndrome
- ME/CFS is a complex, heterogeneous, and chronic neuroimmune disorder. The pathogenesis of ME/CFS is currently not known, but several factors including chemical exposures and infectious agents cause this condition [3]. ME/CFS and GWI show many symptoms such as fatigue, pain and cognitive disorders that are not controlled by rest [123]. Neurological symptoms of ME/CFS include muscle fatigue, mental fatigue, diminished cognition, psychomotor slowing, disrupted sleep, hypersensitivities to noise, light, and smells, headache, pain, paresthesia and extreme dysautonomia [124]. The unbearable physical and mental fatigue can remain in the ME/CFS patients for decades. About 836,000 to 2.5 million Americans have been reported to have ME/CFS, most common in people between 40 and 60 years old, where females appear to be more affected than men at a 4:1 ratio (Centers for Disease Control and Prevention (CDC) [125]. Both physiological and psychological stressors are implicated in the pathogenesis and severity of ME/CFS [3]. As the diagnosis of ME/CFS is difficult, another million may have this condition [3]. The “Three Pillars” hypothesis of ME/CFS pathogenesis [126] might be described as (a) dyshomeostasis of the CNS, (b) immune system, and (c) endocrine system. A disrupted BBB could potentially lead to chronic neuroinflammation observed in ME/CFS [127]. Neurological disruptions in ME/CSF include memory problems, headaches, sleep disturbances, unexplained joint and muscle pain, hypoactivation of the hypothalamic-pituitary-adrenal (HPA) axis and the basal ganglia, as well as unbalanced serotonin cycle, in addition to observed irregularities in the brain stem during brain imaging [128]. Presence of chronic neuroinflammation without apparent neurodegeneration and CNS pathology could occur in ME/CFS [3]. Neuroinflammation is present in the limbic system, midbrain and pons region of the brainstem [129]. Sleep disorders have been associated with damaged reticular formation, especially the reticular activating system (RAS) nuclei [130]. Hypocapnia and hyperventilation are also frequently associated with ME/CFS [124]. The post-exertional malaise (PEM) and orthostatic challenges in ME/CFS correlate with both diminished oxygen supply in muscles and low cerebral blood flow [131]. Furthermore, elevated levels of stress hormones, such as cortisol, may have immunosuppressive effects, acting on neutrophils and natural killer cells [132], contributing to the development of ME/CFS. Because the brain is unable to control stress in ME/CFS in the paraventricular nucleus (PVN) of the hypothalamus, the stress might be a constant stimulus of an immune response in the CNS, to trigger relapses and partial recovery cycles [133].
7. Amyotrophic Lateral Sclerosis (ALS)
8. The Use of iPSC-Derived Cells in Neuroinflammatory Disease Pathogenesis and Neurotherapeutics
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Lamptey, R.N.L.; Chaulagain, B.; Trivedi, R.; Gothwal, A.; Layek, B.; Singh, J. A Review of the Common Neurodegenerative Disorders: Current Therapeutic Approaches and the Potential Role of Nanotherapeutics. Int. J. Mol. Sci. 2022, 23, 1851. [Google Scholar] [CrossRef]
- Pathak, N.; Vimal, S.K.; Tandon, I.; Agrawal, L.; Hongyi, C.; Bhattacharyya, S. Neurodegenerative Disorders of Alzheimer, Parkinsonism, Amyotrophic Lateral Sclerosis and Multiple Sclerosis: An Early Diagnostic Approach for Precision Treatment. Metab. Brain Dis. 2022, 37, 67–104. [Google Scholar] [CrossRef]
- O’Callaghan, J.P.; Miller, D.B. Neuroinflammation disorders exacerbated by environmental stressors. Metabolism 2019, 100S, 153951. [Google Scholar] [CrossRef] [PubMed]
- Passeri, E.; Elkhoury, K.; Morsink, M.; Broersen, K.; Linder, M.; Tamayol, A.; Malaplate, C.; Yen, F.T.; Arab-Tehrany, E. Alzheimer’s Disease: Treatment Strategies and Their Limitations. Int. J. Mol. Sci. 2022, 23, 13954. [Google Scholar] [CrossRef] [PubMed]
- Brambilla, R. Neuroinflammation, the thread connecting neurological disease: Cluster: “Neuroinflammatory mechanisms in neurodegenerative disorders”. Acta Neuropathol. 2019, 137, 689–691. [Google Scholar] [CrossRef] [PubMed]
- Moyse, E.; Krantic, S.; Djellouli, N.; Roger, S.; Angoulvant, D.; Debacq, C.; Leroy, V.; Fougere, B.; Aidoud, A. Neuroinflammation: A Possible Link between Chronic Vascular Disorders and Neurodegenerative Diseases. Front. Aging Neurosci. 2022, 14, 827263. [Google Scholar] [CrossRef] [PubMed]
- Dhapola, R.; Hota, S.S.; Sarma, P.; Bhattacharyya, A.; Medhi, B.; Reddy, D.H. Recent advances in molecular pathways and therapeutic implications targeting neuroinflammation for Alzheimer’s disease. Inflammopharmacology 2021, 29, 1669–1681. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Liu, J.; Wang, B.; Sun, M.; Yang, H. Microglia in the Neuroinflammatory Pathogenesis of Alzheimer’s Disease and Related Therapeutic Targets. Front. Immunol. 2022, 13, 856376. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Sha, H.; Zhou, L.; Chen, Y.; Zhou, Q.; Dong, H.; Qian, Y. The Mast Cell Is an Early Activator of Lipopolysaccharide-Induced Neuroinflammation and Blood-Brain Barrier Dysfunction in the Hippocampus. Mediat. Inflamm. 2020, 2020, 8098439. [Google Scholar] [CrossRef]
- Thakur, S.; Dhapola, R.; Sarma, P.; Medhi, B.; Reddy, D.H. Neuroinflammation in Alzheimer’s Disease: Current Progress in Molecular Signaling and Therapeutics. Inflammation 2023, 46, 1–17. [Google Scholar] [CrossRef]
- Godinho-Silva, C.; Cardoso, F.; Veiga-Fernandes, H. Neuro-Immune Cell Units: A New Paradigm in Physiology. Annu. Rev. Immunol. 2019, 37, 19–46. [Google Scholar] [CrossRef]
- Zhang, W.; Xiao, D.; Mao, Q.; Xia, H. Role of neuroinflammation in neurodegeneration development. Signal Transduct. Target. Ther. 2023, 8, 267. [Google Scholar] [CrossRef]
- Kwon, H.S.; Koh, S.H. Neuroinflammation in neurodegenerative disorders: The roles of microglia and astrocytes. Transl. Neurodegener. 2020, 9, 42. [Google Scholar] [CrossRef]
- Zhou, R.; Ji, B.; Kong, Y.; Qin, L.; Ren, W.; Guan, Y.; Ni, R. PET Imaging of Neuroinflammation in Alzheimer’s Disease. Front. Immunol. 2021, 12, 739130. [Google Scholar] [CrossRef] [PubMed]
- Kempuraj, D.; Thangavel, R.; Selvakumar, G.P.; Zaheer, S.; Ahmed, M.E.; Raikwar, S.P.; Zahoor, H.; Saeed, D.; Natteru, P.A.; Iyer, S.; et al. Brain and Peripheral Atypical Inflammatory Mediators Potentiate Neuroinflammation and Neurodegeneration. Front. Cell. Neurosci. 2017, 11, 216. [Google Scholar] [CrossRef] [PubMed]
- Theoharides, T.C.; Twahir, A.; Kempuraj, D. Mast cells in the autonomic nervous system and potential role in disorders with dysautonomia and neuroinflammation. Ann. Allergy Asthma Immunol. 2023, S1081-1206(23)01397-2. [Google Scholar] [CrossRef]
- Carloni, S.; Rescigno, M. The gut-brain vascular axis in neuroinflammation. Semin. Immunol. 2023, 69, 101802. [Google Scholar] [CrossRef] [PubMed]
- Connolly, K.; Lehoux, M.; Assetta, B.; Huang, Y.A. Modeling Cellular Crosstalk of Neuroinflammation Axis by Tri-cultures of iPSC-Derived Human Microglia, Astrocytes, and Neurons. Methods Mol. Biol. 2023, 2683, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Karmirian, K.; Holubiec, M.; Goto-Silva, L.; Fernandez Bessone, I.; Vitoria, G.; Mello, B.; Alloatti, M.; Vanderborght, B.; Falzone, T.L.; Rehen, S. Modeling Alzheimer’s Disease Using Human Brain Organoids. Methods Mol. Biol. 2023, 2561, 135–158. [Google Scholar] [CrossRef]
- Barak, M.; Fedorova, V.; Pospisilova, V.; Raska, J.; Vochyanova, S.; Sedmik, J.; Hribkova, H.; Klimova, H.; Vanova, T.; Bohaciakova, D. Human iPSC-Derived Neural Models for Studying Alzheimer’s Disease: From Neural Stem Cells to Cerebral Organoids. Stem Cell Rev. Rep. 2022, 18, 792–820. [Google Scholar] [CrossRef]
- Akhtar, A.; Andleeb, A.; Waris, T.S.; Bazzar, M.; Moradi, A.R.; Awan, N.R.; Yar, M. Neurodegenerative diseases and effective drug delivery: A review of challenges and novel therapeutics. J. Control. Release 2021, 330, 1152–1167. [Google Scholar] [CrossRef] [PubMed]
- Achar, A.; Myers, R.; Ghosh, C. Drug Delivery Challenges in Brain Disorders across the Blood-Brain Barrier: Novel Methods and Future Considerations for Improved Therapy. Biomedicines 2021, 9, 1834. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s Association, Chicago, IL, 2023. Available online: https://www.alz.org/ (accessed on 20 December 2023).
- Leng, F.; Edison, P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat. Rev. Neurol. 2021, 17, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Zhong, R.J.; Cheng, C.; Li, S.; Le, W.D. New therapeutics beyond amyloid-beta and tau for the treatment of Alzheimer’s disease. Acta Pharmacol. Sin. 2021, 42, 1382–1389. [Google Scholar] [CrossRef] [PubMed]
- Mangalmurti, A.; Lukens, J.R. How neurons die in Alzheimer’s disease: Implications for neuroinflammation. Curr. Opin. Neurobiol. 2022, 75, 102575. [Google Scholar] [CrossRef] [PubMed]
- Golzari-Sorkheh, M.; Weaver, D.F.; Reed, M.A. COVID-19 as a Risk Factor for Alzheimer’s Disease. J. Alzheimers Dis. 2023, 91, 1–23. [Google Scholar] [CrossRef]
- Kempuraj, D.; Ahmed, M.E.; Selvakumar, G.P.; Thangavel, R.; Dhaliwal, A.S.; Dubova, I.; Mentor, S.; Premkumar, K.; Saeed, D.; Zahoor, H.; et al. Brain Injury-Mediated Neuroinflammatory Response and Alzheimer’s Disease. Neuroscientist 2020, 26, 134–155. [Google Scholar] [CrossRef]
- Soria Lopez, J.A.; Gonzalez, H.M.; Leger, G.C. Alzheimer’s disease. Handb. Clin. Neurol. 2019, 167, 231–255. [Google Scholar] [CrossRef]
- Moore, K.B.E.; Hung, T.J.; Fortin, J.S. Hyperphosphorylated tau (p-tau) and drug discovery in the context of Alzheimer’s disease and related tauopathies. Drug Discov. Today 2023, 28, 103487. [Google Scholar] [CrossRef]
- Long, J.M.; Holtzman, D.M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Halliwell, B. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat. Rev. Neurosci. 2019, 20, 148–160. [Google Scholar] [CrossRef]
- Goetzl, E.J.; Schwartz, J.B.; Abner, E.L.; Jicha, G.A.; Kapogiannis, D. High complement levels in astrocyte-derived exosomes of Alzheimer disease. Ann. Neurol. 2018, 83, 544–552. [Google Scholar] [CrossRef]
- Lananna, B.V.; McKee, C.A.; King, M.W.; Del-Aguila, J.L.; Dimitry, J.M.; Farias, F.H.G.; Nadarajah, C.J.; Xiong, D.D.; Guo, C.; Cammack, A.J.; et al. Chi3l1/YKL-40 is controlled by the astrocyte circadian clock and regulates neuroinflammation and Alzheimer’s disease pathogenesis. Sci. Transl. Med. 2020, 12, eaax3519. [Google Scholar] [CrossRef]
- Connolly, K.; Lehoux, M.; O’Rourke, R.; Assetta, B.; Erdemir, G.A.; Elias, J.A.; Lee, C.G.; Huang, Y.A. Potential role of chitinase-3-like protein 1 (CHI3L1/YKL-40) in neurodegeneration and Alzheimer’s disease. Alzheimers Dement. 2023, 19, 9–24. [Google Scholar] [CrossRef]
- Elahi, F.M.; Casaletto, K.B.; La Joie, R.; Walters, S.M.; Harvey, D.; Wolf, A.; Edwards, L.; Rivera-Contreras, W.; Karydas, A.; Cobigo, Y.; et al. Plasma biomarkers of astrocytic and neuronal dysfunction in early- and late-onset Alzheimer’s disease. Alzheimers Dement. 2020, 16, 681–695. [Google Scholar] [CrossRef]
- Pereira, J.B.; Janelidze, S.; Smith, R.; Mattsson-Carlgren, N.; Palmqvist, S.; Teunissen, C.E.; Zetterberg, H.; Stomrud, E.; Ashton, N.J.; Blennow, K.; et al. Plasma GFAP is an early marker of amyloid-beta but not tau pathology in Alzheimer’s disease. Brain 2021, 144, 3505–3516. [Google Scholar] [CrossRef]
- Horti, A.G.; Naik, R.; Foss, C.A.; Minn, I.; Misheneva, V.; Du, Y.; Wang, Y.; Mathews, W.B.; Wu, Y.; Hall, A.; et al. PET imaging of microglia by targeting macrophage colony-stimulating factor 1 receptor (CSF1R). Proc. Natl. Acad. Sci. USA 2019, 116, 1686–1691. [Google Scholar] [CrossRef] [PubMed]
- Maeda, J.; Minamihisamatsu, T.; Shimojo, M.; Zhou, X.; Ono, M.; Matsuba, Y.; Ji, B.; Ishii, H.; Ogawa, M.; Akatsu, H.; et al. Distinct microglial response against Alzheimer’s amyloid and tau pathologies characterized by P2Y12 receptor. Brain Commun. 2021, 3, fcab011. [Google Scholar] [CrossRef] [PubMed]
- Jackson, I.M.; Buccino, P.J.; Azevedo, E.C.; Carlson, M.L.; Luo, A.S.Z.; Deal, E.M.; Kalita, M.; Reyes, S.T.; Shao, X.; Beinat, C.; et al. Radiosynthesis and initial preclinical evaluation of [(11)C]AZD1283 as a potential P2Y12R PET radiotracer. Nucl. Med. Biol. 2022, 114–115, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Le Douce, J.; Maugard, M.; Veran, J.; Matos, M.; Jego, P.; Vigneron, P.A.; Faivre, E.; Toussay, X.; Vandenberghe, M.; Balbastre, Y.; et al. Impairment of Glycolysis-Derived l-Serine Production in Astrocytes Contributes to Cognitive Deficits in Alzheimer’s Disease. Cell Metab. 2020, 31, 503–517.e8. [Google Scholar] [CrossRef] [PubMed]
- Ramaswamy, S.B.; Bhagavan, S.M.; Kaur, H.; Giler, G.E.; Kempuraj, D.; Thangavel, R.; Ahmed, M.E.; Selvakumar, G.P.; Raikwar, S.P.; Zaheer, S.; et al. Glia Maturation Factor in the Pathogenesis of Alzheimer’s disease. Open Access J. Neurol. Neurosurg. 2019, 12, 79–82. [Google Scholar] [PubMed]
- Calsolaro, V.; Edison, P. Neuroinflammation in Alzheimer’s disease: Current evidence and future directions. Alzheimers Dement. 2016, 12, 719–732. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Barve, K.H.; Kumar, M.S. Recent Advancements in Pathogenesis, Diagnostics and Treatment of Alzheimer’s Disease. Curr. Neuropharmacol. 2020, 18, 1106–1125. [Google Scholar] [CrossRef] [PubMed]
- Weintraub, D.; Mamikonyan, E. The Neuropsychiatry of Parkinson Disease: A Perfect Storm. Am. J. Geriatr. Psychiatry 2019, 27, 998–1018. [Google Scholar] [CrossRef] [PubMed]
- Kempuraj, D.; Selvakumar, G.P.; Zaheer, S.; Thangavel, R.; Ahmed, M.E.; Raikwar, S.; Govindarajan, R.; Iyer, S.; Zaheer, A. Cross-Talk between Glia, Neurons and Mast Cells in Neuroinflammation Associated with Parkinson’s Disease. J. Neuroimmune Pharmacol. 2018, 13, 100–112. [Google Scholar] [CrossRef]
- Parkinsons Foundation, Miami, FL, 2023. Available online: https://www.parkinson.org/ (accessed on 20 December 2023).
- Pajares, M.; Rojo, A.I.; Manda, G.; Boscá, L.; Cuadrado, A. Inflammation in Parkinson’s Disease: Mechanisms and Therapeutic Implications. Cells 2020, 9, 1687. [Google Scholar] [CrossRef]
- Blommer, J.; Pitcher, T.; Mustapic, M.; Eren, E.; Yao, P.J.; Vreones, M.P.; Pucha, K.A.; Dalrymple-Alford, J.; Shoorangiz, R.; Meissner, W.G.; et al. Extracellular vesicle biomarkers for cognitive impairment in Parkinson’s disease. Brain 2023, 146, 195–208. [Google Scholar] [CrossRef] [PubMed]
- Dorsey, E.R.; Sherer, T.; Okun, M.S.; Bloem, B.R. The Emerging Evidence of the Parkinson Pandemic. J. Park. Dis. 2018, 8, S3–S8. [Google Scholar] [CrossRef]
- Costa, H.N.; Esteves, A.R.; Empadinhas, N.; Cardoso, S.M. Parkinson’s Disease: A Multisystem Disorder. Neurosci. Bull. 2023, 39, 113–124. [Google Scholar] [CrossRef]
- Ye, H.; Robak, L.A.; Yu, M.; Cykowski, M.; Shulman, J.M. Genetics and Pathogenesis of Parkinson’s Syndrome. Annu. Rev. Pathol. 2023, 18, 95–121. [Google Scholar] [CrossRef]
- Fernandes Gomes, B.; Farris, C.M.; Ma, Y.; Concha-Marambio, L.; Lebovitz, R.; Nellgard, B.; Dalla, K.; Constantinescu, J.; Constantinescu, R.; Gobom, J.; et al. alpha-Synuclein seed amplification assay as a diagnostic tool for parkinsonian disorders. Park. Relat. Disord. 2023, 117, 105807. [Google Scholar] [CrossRef]
- Kempuraj, D.; Thangavel, R.; Selvakumar, G.P.; Ahmed, M.E.; Zaheer, S.; Raikwar, S.P.; Zahoor, H.; Saeed, D.; Dubova, I.; Giler, G.; et al. Mast Cell Proteases Activate Astrocytes and Glia-Neurons and Release Interleukin-33 by Activating p38 and ERK1/2 MAPKs and NF-kappaB. Mol. Neurobiol. 2019, 56, 1681–1693. [Google Scholar] [CrossRef]
- Selvakumar, G.P.; Ahmed, M.E.; Thangavel, R.; Kempuraj, D.; Dubova, I.; Raikwar, S.P.; Zaheer, S.; Iyer, S.S.; Zaheer, A. A role for glia maturation factor dependent activation of mast cells and microglia in MPTP induced dopamine loss and behavioural deficits in mice. Brain Behav. Immun. 2020, 87, 429–443. [Google Scholar] [CrossRef]
- Limphaibool, N.; Iwanowski, P.; Holstad, M.J.V.; Kobylarek, D.; Kozubski, W. Infectious Etiologies of Parkinsonism: Pathomechanisms and Clinical Implications. Front. Neurol. 2019, 10, 652. [Google Scholar] [CrossRef]
- Seppi, K.; Ray Chaudhuri, K.; Coelho, M.; Fox, S.H.; Katzenschlager, R.; Perez Lloret, S.; Weintraub, D.; Sampaio, C.; the collaborators of the Parkinson’s Disease Update on Non-Motor Symptoms Study Group on behalf of the Movement Disorders Society Evidence-Based Medicine Committee. Update on treatments for nonmotor symptoms of Parkinson’s disease—An evidence-based medicine review. Mov. Disord. 2019, 34, 180–198. [Google Scholar] [CrossRef]
- Cheong, J.L.Y.; de Pablo-Fernandez, E.; Foltynie, T.; Noyce, A.J. The Association Between Type 2 Diabetes Mellitus and Parkinson’s Disease. J. Park. Dis. 2020, 10, 775–789. [Google Scholar] [CrossRef]
- Erekat, N.S. Autophagy and Its Association with Genetic Mutations in Parkinson Disease. Med. Sci. Monit. 2022, 28, e938519. [Google Scholar] [CrossRef] [PubMed]
- Blauwendraat, C.; Heilbron, K.; Vallerga, C.L.; Bandres-Ciga, S.; von Coelln, R.; Pihlstrom, L.; Simon-Sanchez, J.; Schulte, C.; Sharma, M.; Krohn, L.; et al. Parkinson’s disease age at onset genome-wide association study: Defining heritability, genetic loci, and alpha-synuclein mechanisms. Mov. Disord. 2019, 34, 866–875. [Google Scholar] [CrossRef] [PubMed]
- Brain Injury Association of America, Fairfax, VA. 2023. Available online: https://www.biausa.org/ (accessed on 10 December 2023).
- Ghaith, H.S.; Nawar, A.A.; Gabra, M.D.; Abdelrahman, M.E.; Nafady, M.H.; Bahbah, E.I.; Ebada, M.A.; Ashraf, G.M.; Negida, A.; Barreto, G.E. A Literature Review of Traumatic Brain Injury Biomarkers. Mol. Neurobiol. 2022, 59, 4141–4158. [Google Scholar] [CrossRef] [PubMed]
- Kempuraj, D.; Thangavel, R.; Natteru, P.A.; Selvakumar, G.P.; Saeed, D.; Zahoor, H.; Zaheer, S.; Iyer, S.S.; Zaheer, A. Neuroinflammation Induces Neurodegeneration. J. Neurol. Neurosurg. Spine 2016, 1, 1003. [Google Scholar]
- Thapa, K.; Khan, H.; Singh, T.G.; Kaur, A. Traumatic Brain Injury: Mechanistic Insight on Pathophysiology and Potential Therapeutic Targets. J. Mol. Neurosci. 2021, 71, 1725–1742. [Google Scholar] [CrossRef]
- Atif, H.; Hicks, S.D. A Review of MicroRNA Biomarkers in Traumatic Brain Injury. J. Exp. Neurosci. 2019, 13, 1179069519832286. [Google Scholar] [CrossRef]
- Dadas, A.; Washington, J.; Diaz-Arrastia, R.; Janigro, D. Biomarkers in traumatic brain injury (TBI): A review. Neuropsychiatr. Dis. Treat. 2018, 14, 2989–3000. [Google Scholar] [CrossRef]
- Brett, B.L.; Gardner, R.C.; Godbout, J.; Dams-O’Connor, K.; Keene, C.D. Traumatic Brain Injury and Risk of Neurodegenerative Disorder. Biol. Psychiatry 2022, 91, 498–507. [Google Scholar] [CrossRef]
- Hier, D.B.; Obafemi-Ajayi, T.; Thimgan, M.S.; Olbricht, G.R.; Azizi, S.; Allen, B.; Hadi, B.A.; Wunsch, D.C., 2nd. Blood biomarkers for mild traumatic brain injury: A selective review of unresolved issues. Biomark. Res. 2021, 9, 70. [Google Scholar] [CrossRef] [PubMed]
- Gutierre, M.U.; Telles, J.P.M.; Welling, L.C.; Rabelo, N.N.; Teixeira, M.J.; Figueiredo, E.G. Biomarkers for traumatic brain injury: A short review. Neurosurg. Rev. 2021, 44, 2091–2097. [Google Scholar] [CrossRef] [PubMed]
- Coppens, S.; Lehmann, S.; Hopley, C.; Hirtz, C. Neurofilament-Light, a Promising Biomarker: Analytical, Metrological and Clinical Challenges. Int. J. Mol. Sci. 2023, 24, 11624. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, M.L.; Tom, V.J. Neuroimmune System as a Driving Force for Plasticity Following CNS Injury. Front. Cell. Neurosci. 2020, 14, 187. [Google Scholar] [CrossRef]
- Ladak, A.A.; Enam, S.A.; Ibrahim, M.T. A Review of the Molecular Mechanisms of Traumatic Brain Injury. World Neurosurg. 2019, 131, 126–132. [Google Scholar] [CrossRef]
- Sivandzade, F.; Alqahtani, F.; Cucullo, L. Traumatic Brain Injury and Blood-Brain Barrier (BBB): Underlying Pathophysiological Mechanisms and the Influence of Cigarette Smoking as a Premorbid Condition. Int. J. Mol. Sci. 2020, 21, 2721. [Google Scholar] [CrossRef]
- Graham, N.S.; Sharp, D.J. Understanding neurodegeneration after traumatic brain injury: From mechanisms to clinical trials in dementia. J. Neurol. Neurosurg. Psychiatry 2019, 90, 1221–1233. [Google Scholar] [CrossRef]
- Cash, A.; Theus, M.H. Mechanisms of Blood-Brain Barrier Dysfunction in Traumatic Brain Injury. Int. J. Mol. Sci. 2020, 21, 3344. [Google Scholar] [CrossRef]
- Kempuraj, D.; Selvakumar, G.P.; Thangavel, R.; Ahmed, M.E.; Zaheer, S.; Raikwar, S.P.; Iyer, S.S.; Bhagavan, S.M.; Beladakere-Ramaswamy, S.; Zaheer, A. Mast Cell Activation in Brain Injury, Stress, and Post-traumatic Stress Disorder and Alzheimer’s Disease Pathogenesis. Front. Neurosci. 2017, 11, 703. [Google Scholar] [CrossRef]
- Eser Ocak, P.; Ocak, U.; Sherchan, P.; Zhang, J.H.; Tang, J. Insights into major facilitator superfamily domain-containing protein-2a (Mfsd2a) in physiology and pathophysiology. What do we know so far? J. Neurosci. Res. 2020, 98, 29–41. [Google Scholar] [CrossRef]
- Wang, K.K.; Yang, Z.; Zhu, T.; Shi, Y.; Rubenstein, R.; Tyndall, J.A.; Manley, G.T. An update on diagnostic and prognostic biomarkers for traumatic brain injury. Expert. Rev. Mol. Diagn. 2018, 18, 165–180. [Google Scholar] [CrossRef] [PubMed]
- Mozaffari, K.; Dejam, D.; Duong, C.; Ding, K.; French, A.; Ng, E.; Preet, K.; Franks, A.; Kwan, I.; Phillips, H.W.; et al. Systematic Review of Serum Biomarkers in Traumatic Brain Injury. Cureus 2021, 13, e17056. [Google Scholar] [CrossRef] [PubMed]
- Karantali, E.; Kazis, D.; McKenna, J.; Chatzikonstantinou, S.; Petridis, F.; Mavroudis, I. Neurofilament light chain in patients with a concussion or head impacts: A systematic review and meta-analysis. Eur. J. Trauma Emerg. Surg. 2022, 48, 1555–1567. [Google Scholar] [CrossRef] [PubMed]
- Barro, C.; Chitnis, T.; Weiner, H.L. Blood neurofilament light: A critical review of its application to neurologic disease. Ann. Clin. Transl. Neurol. 2020, 7, 2508–2523. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Zhang, Z.; Lv, X.; Wu, Q.; Yan, J.; Mao, G.; Xing, W. Neurofilament light chain level in traumatic brain injury: A system review and meta-analysis. Medicine 2020, 99, e22363. [Google Scholar] [CrossRef] [PubMed]
- Farragher, C.D.; Ku, Y.; Powers, J.E. The Potential Role of Neurofilament Light in Mild Traumatic Brain Injury Diagnosis: A Systematic Review. Cureus 2022, 14, e31301. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.K.; Munoz Pareja, J.C.; Mondello, S.; Diaz-Arrastia, R.; Wellington, C.; Kenney, K.; Puccio, A.M.; Hutchison, J.; McKinnon, N.; Okonkwo, D.O.; et al. Blood-based traumatic brain injury biomarkers–Clinical utilities and regulatory pathways in the United States, Europe and Canada. Expert. Rev. Mol. Diagn. 2021, 21, 1303–1321. [Google Scholar] [CrossRef]
- Papa, L.; Zonfrillo, M.R.; Welch, R.D.; Lewis, L.M.; Braga, C.F.; Tan, C.N.; Ameli, N.J.; Lopez, M.A.; Haeussler, C.A.; Mendez Giordano, D.; et al. Evaluating glial and neuronal blood biomarkers GFAP and UCH-L1 as gradients of brain injury in concussive, subconcussive and non-concussive trauma: A prospective cohort study. BMJ Paediatr. Open 2019, 3, e000473. [Google Scholar] [CrossRef]
- Lindblad, C.; Rostami, E.; Helmy, A. Interleukin-1 Receptor Antagonist as Therapy for Traumatic Brain Injury. Neurotherapeutics 2023, 20, 1508–1528. [Google Scholar] [CrossRef]
- Miskin, B.M.; Fox, L.A.; Abou-Al-Shaar, H.; Bin-Alamer, O.; Goertz, A.; Lipin, C.T.; Fertig, N.; Cox, N. Hyperbaric Oxygen Therapy for the Management of Mild and Moderate Traumatic Brain Injury: A Single-Center Experience. World Neurosurg. 2023, 176, e357–e370. [Google Scholar] [CrossRef]
- Bested, A.C.; Rana, A.M.; Hardigan, P.; Niyirora, J.; Cheema, A.; Antony, G.; Defina, P.A.; Machado, C. A case series of 39 United States Veterans with mild traumatic brain injury treated with hyperbaric oxygen therapy. Clin. Trans. Neurosci. 2022, 6, 21. [Google Scholar] [CrossRef]
- Saha, P.; Skidmore, P.T.; Holland, L.A.; Mondal, A.; Bose, D.; Seth, R.K.; Sullivan, K.; Janulewicz, P.A.; Horner, R.; Klimas, N.; et al. Andrographolide Attenuates Gut-Brain-Axis Associated Pathology in Gulf War Illness by Modulating Bacteriome-Virome Associated Inflammation and Microglia-Neuron Proinflammatory Crosstalk. Brain Sci. 2021, 11, 905. [Google Scholar] [CrossRef] [PubMed]
- Dickey, B.; Madhu, L.N.; Shetty, A.K. Gulf War Illness: Mechanisms Underlying Brain Dysfunction and Promising Therapeutic Strategies. Pharmacol. Ther. 2021, 220, 107716. [Google Scholar] [CrossRef] [PubMed]
- Jeffrey, M.G.; Krengel, M.; Kibler, J.L.; Zundel, C.; Klimas, N.G.; Sullivan, K.; Craddock, T.J.A. Neuropsychological Findings in Gulf War Illness: A Review. Front. Psychol. 2019, 10, 2088. [Google Scholar] [CrossRef] [PubMed]
- Reddy, D.S.; Wu, X.; Singh, T.; Neff, M. Experimental Models of Gulf War Illness, a Chronic Neuropsychiatric Disorder in Veterans. Curr. Protoc. 2023, 3, e707. [Google Scholar] [CrossRef] [PubMed]
- Georgopoulos, A.P.; James, L.M.; Carpenter, A.F.; Engdahl, B.E.; Leuthold, A.C.; Lewis, S.M. Gulf War illness (GWI) as a neuroimmune disease. Exp. Brain Res. 2017, 235, 3217–3225. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, S.; McCartan, R.; Browning, M.; Hahn-Townsend, C.; Gratkowski, A.; Morin, A.; Abdullah, L.; Ait-Ghezala, G.; Ojo, J.; Sullivan, K.; et al. Impact of gulf war toxic exposures after mild traumatic brain injury. Acta Neuropathol. Commun. 2022, 10, 147. [Google Scholar] [CrossRef] [PubMed]
- Jean-Pierre, M.; Michalovicz, L.T.; Kelly, K.A.; O’Callaghan, J.P.; Nathanson, L.; Klimas, N.; Craddock, T.J. A pilot reverse virtual screening study suggests toxic exposures caused long-term epigenetic changes in Gulf War Illness. Comput. Struct. Biotechnol. J. 2022, 20, 6206–6213. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, S.; Fried, D.E.; Grubisic, V.; McClain, J.L.; Gulbransen, B.D. Gastrointestinal neuroimmune disruption in a mouse model of Gulf War illness. FASEB J. 2019, 33, 6168–6184. [Google Scholar] [CrossRef]
- Elhaj, R.; Reynolds, J.M. Chemical exposures and suspected impact on Gulf War Veterans. Mil. Med. Res. 2023, 10, 11. [Google Scholar] [CrossRef]
- Nkiliza, A.; Joshi, U.; Evans, J.E.; Ait-Ghezala, G.; Parks, M.; Crawford, F.; Mullan, M.; Abdullah, L. Adaptive Immune Responses Associated with the Central Nervous System Pathology of Gulf War Illness. Neurosci. Insights 2021, 16, 26331055211018458. [Google Scholar] [CrossRef]
- Alshelh, Z.; Albrecht, D.S.; Bergan, C.; Akeju, O.; Clauw, D.J.; Conboy, L.; Edwards, R.R.; Kim, M.; Lee, Y.C.; Protsenko, E.; et al. In-vivo imaging of neuroinflammation in veterans with Gulf War illness. Brain Behav. Immun. 2020, 87, 498–507. [Google Scholar] [CrossRef]
- Madhu, L.N.; Attaluri, S.; Kodali, M.; Shuai, B.; Upadhya, R.; Gitai, D.; Shetty, A.K. Neuroinflammation in Gulf War Illness is linked with HMGB1 and complement activation, which can be discerned from brain-derived extracellular vesicles in the blood. Brain Behav. Immun. 2019, 81, 430–443. [Google Scholar] [CrossRef] [PubMed]
- Kodali, M.; Jankay, T.; Shetty, A.K.; Reddy, D.S. Pathophysiological basis and promise of experimental therapies for Gulf War Illness, a chronic neuropsychiatric syndrome in veterans. Psychopharmacology 2023, 240, 673–697. [Google Scholar] [CrossRef] [PubMed]
- Kimono, D.; Bose, D.; Seth, R.K.; Mondal, A.; Saha, P.; Janulewicz, P.; Sullivan, K.; Lasley, S.; Horner, R.; Klimas, N.; et al. Host Akkermansia muciniphila Abundance Correlates With Gulf War Illness Symptom Persistence via NLRP3-Mediated Neuroinflammation and Decreased Brain-Derived Neurotrophic Factor. Neurosci. Insights 2020, 15, 2633105520942480. [Google Scholar] [CrossRef] [PubMed]
- Seth, R.K.; Maqsood, R.; Mondal, A.; Bose, D.; Kimono, D.; Holland, L.A.; Janulewicz Lloyd, P.; Klimas, N.; Horner, R.D.; Sullivan, K.; et al. Gut DNA Virome Diversity and Its Association with Host Bacteria Regulate Inflammatory Phenotype and Neuronal Immunotoxicity in Experimental Gulf War Illness. Viruses 2019, 11, 968. [Google Scholar] [CrossRef] [PubMed]
- Bose, D.; Saha, P.; Mondal, A.; Fanelli, B.; Seth, R.K.; Janulewicz, P.; Sullivan, K.; Lasley, S.; Horner, R.; Colwell, R.R.; et al. Obesity Worsens Gulf War Illness Symptom Persistence Pathology by Linking Altered Gut Microbiome Species to Long-Term Gastrointestinal, Hepatic, and Neuronal Inflammation in a Mouse Model. Nutrients 2020, 12, 2764. [Google Scholar] [CrossRef] [PubMed]
- O’Callaghan, J.P.; Kelly, K.A.; Locker, A.R.; Miller, D.B.; Lasley, S.M. Corticosterone primes the neuroinflammatory response to DFP in mice: Potential animal model of Gulf War Illness. J. Neurochem. 2015, 133, 708–721. [Google Scholar] [CrossRef] [PubMed]
- Koo, B.B.; Michalovicz, L.T.; Calderazzo, S.; Kelly, K.A.; Sullivan, K.; Killiany, R.J.; O’Callaghan, J.P. Corticosterone potentiates DFP-induced neuroinflammation and affects high-order diffusion imaging in a rat model of Gulf War Illness. Brain Behav. Immun. 2018, 67, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Raju, R.P.; Terry, A.V. Dysregulation of cellular energetics in Gulf War Illness. Toxicology 2021, 461, 152894. [Google Scholar] [CrossRef] [PubMed]
- Gopinath, K.; Gandhi, P.; Goyal, A.; Jiang, L.; Fang, Y.; Ouyang, L.; Ganji, S.; Buhner, D.; Ringe, W.; Spence, J.; et al. FMRI reveals abnormal central processing of sensory and pain stimuli in ill Gulf War veterans. Neurotoxicology 2012, 33, 261–271. [Google Scholar] [CrossRef]
- Ashbrook, D.G.; Hing, B.; Michalovicz, L.T.; Kelly, K.A.; Miller, J.V.; de Vega, W.C.; Miller, D.B.; Broderick, G.; O’Callaghan, J.P.; McGowan, P.O. Epigenetic impacts of stress priming of the neuroinflammatory response to sarin surrogate in mice: A model of Gulf War illness. J. Neuroinflamm. 2018, 15, 86. [Google Scholar] [CrossRef]
- Kozlova, E.V.; Carabelli, B.; Bishay, A.E.; Liu, R.; Denys, M.E.; Macbeth, J.C.; Piamthai, V.; Crawford, M.S.; McCole, D.F.; Zur Nieden, N.I.; et al. Induction of distinct neuroinflammatory markers and gut dysbiosis by differential pyridostigmine bromide dosing in a chronic mouse model of GWI showing persistent exercise fatigue and cognitive impairment. Life Sci. 2022, 288, 120153. [Google Scholar] [CrossRef]
- Shetty, A.K.; Attaluri, S.; Kodali, M.; Shuai, B.; Shetty, G.A.; Upadhya, D.; Hattiangady, B.; Madhu, L.N.; Upadhya, R.; Bates, A.; et al. Monosodium luminol reinstates redox homeostasis, improves cognition, mood and neurogenesis, and alleviates neuro- and systemic inflammation in a model of Gulf War Illness. Redox Biol. 2020, 28, 101389. [Google Scholar] [CrossRef]
- Liu, L.; Wang, E.Q.; Du, C.; Chen, H.S.; Lv, Y. Minocycline alleviates Gulf War Illness rats via altering gut microbiome, attenuating neuroinflammation and enhancing hippocampal neurogenesis. Behav. Brain Res. 2021, 410, 113366. [Google Scholar] [CrossRef] [PubMed]
- Leibowitz, J.A.; Ormerod, B.K. The potential of treating Gulf War Illness with curcumin. Brain Behav. Immun. 2018, 70, 3–4. [Google Scholar] [CrossRef] [PubMed]
- Kodali, M.; Hattiangady, B.; Shetty, G.A.; Bates, A.; Shuai, B.; Shetty, A.K. Curcumin treatment leads to better cognitive and mood function in a model of Gulf War Illness with enhanced neurogenesis, and alleviation of inflammation and mitochondrial dysfunction in the hippocampus. Brain Behav. Immun. 2018, 69, 499–514. [Google Scholar] [CrossRef]
- Chester, J.E.; Rowneki, M.; Van Doren, W.; Helmer, D.A. Progression of intervention-focused research for Gulf War illness. Mil. Med. Res. 2019, 6, 31. [Google Scholar] [CrossRef]
- Cheema, A.K.; Wiener, L.E.; McNeil, R.B.; Abreu, M.M.; Craddock, T.; Fletcher, M.A.; Helmer, D.A.; Ashford, J.W.; Sullivan, K.; Klimas, N.G. A randomized phase II remote study to assess Bacopa for Gulf War Illness associated cognitive dysfunction: Design and methods of a national study. Life Sci. 2021, 282, 119819. [Google Scholar] [CrossRef] [PubMed]
- Helmer, D.A.; Van Doren, W.W.; Litke, D.R.; Tseng, C.L.; Ho, L.; Osinubi, O.; Pasinetti, G.M. Safety, Tolerability and Efficacy of Dietary Supplementation with Concord Grape Juice in Gulf War Veterans with Gulf War Illness: A Phase I/IIA, Randomized, Double-Blind, Placebo-Controlled Trial. Int. J. Environ. Res. Public Health 2020, 17, 3546. [Google Scholar] [CrossRef] [PubMed]
- McAndrew, L.M.; Quigley, K.S.; Lu, S.E.; Litke, D.; Rath, J.F.; Lange, G.; Santos, S.L.; Anastasides, N.; Petrakis, B.A.; Greenberg, L.; et al. Effect of Problem-solving Treatment on Self-reported Disability Among Veterans With Gulf War Illness: A Randomized Clinical Trial. JAMA Netw. Open 2022, 5, e2245272. [Google Scholar] [CrossRef]
- Niles, B.L.; Grossman, S.; McQuade, M.; Grossman, D.; Kaiser, A.P.; Muccio, B.; Warner, B.; Wang, C.; Mori, D.L. Study protocol for a revised randomized trial: Remotely delivered Tai Chi and wellness for Gulf War illness. Contemp. Clin. Trials 2023, 125, 107045. [Google Scholar] [CrossRef] [PubMed]
- Allende, S.; Mathersul, D.C.; Schulz-Heik, J.R.; Avery, T.J.; Mahoney, L.; Bayley, P.J. Yoga is effective for treating chronic pain in veterans with Gulf War Illness at long-term follow-up. BMC Complement. Med. Ther. 2023, 23, 319. [Google Scholar] [CrossRef]
- Chao, L.L.; Kanady, J.C.; Crocker, N.; Straus, L.D.; Hlavin, J.; Metzler, T.J.; Maguen, S.; Neylan, T.C. Cognitive behavioral therapy for insomnia in veterans with gulf war illness: Results from a randomized controlled trial. Life Sci. 2021, 279, 119147. [Google Scholar] [CrossRef]
- Donta, S.T.; Clauw, D.J.; Engel, C.C., Jr.; Guarino, P.; Peduzzi, P.; Williams, D.A.; Skinner, J.S.; Barkhuizen, A.; Taylor, T.; Kazis, L.E.; et al. Cognitive behavioral therapy and aerobic exercise for Gulf War veterans’ illnesses: A randomized controlled trial. JAMA 2003, 289, 1396–1404. [Google Scholar] [CrossRef]
- Baraniuk, J.N.; Kern, G.; Narayan, V.; Cheema, A. Exercise modifies glutamate and other metabolic biomarkers in cerebrospinal fluid from Gulf War Illness and Myalgic encephalomyelitis / Chronic Fatigue Syndrome. PLoS ONE 2021, 16, e0244116. [Google Scholar] [CrossRef]
- Wirth, K.J.; Scheibenbogen, C.; Paul, F. An attempt to explain the neurological symptoms of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. J. Transl. Med. 2021, 19, 471. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention (CDC). Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS); Centers for Disease Control and Prevention: Atlanta, GA, USA, 2023. [Google Scholar]
- Cortes Rivera, M.; Mastronardi, C.; Silva-Aldana, C.T.; Arcos-Burgos, M.; Lidbury, B.A. Myalgic Encephalomyelitis/Chronic Fatigue Syndrome: A Comprehensive Review. Diagnostics 2019, 9, 91. [Google Scholar] [CrossRef]
- Tate, W.; Walker, M.; Sweetman, E.; Helliwell, A.; Peppercorn, K.; Edgar, C.; Blair, A.; Chatterjee, A. Molecular Mechanisms of Neuroinflammation in ME/CFS and Long COVID to Sustain Disease and Promote Relapses. Front. Neurol. 2022, 13, 877772. [Google Scholar] [CrossRef]
- Kasimir, F.; Toomey, D.; Liu, Z.; Kaiping, A.C.; Ariza, M.E.; Prusty, B.K. Tissue specific signature of HHV-6 infection in ME/CFS. Front. Mol. Biosci. 2022, 9, 1044964. [Google Scholar] [CrossRef] [PubMed]
- Nakatomi, Y.; Mizuno, K.; Ishii, A.; Wada, Y.; Tanaka, M.; Tazawa, S.; Onoe, K.; Fukuda, S.; Kawabe, J.; Takahashi, K.; et al. Neuroinflammation in Patients with Chronic Fatigue Syndrome/Myalgic Encephalomyelitis: An (1)(1)C-(R)-PK11195 PET Study. J. Nucl. Med. 2014, 55, 945–950. [Google Scholar] [CrossRef]
- Nelson, T.; Zhang, L.X.; Guo, H.; Nacul, L.; Song, X. Brainstem Abnormalities in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome: A Scoping Review and Evaluation of Magnetic Resonance Imaging Findings. Front. Neurol. 2021, 12, 769511. [Google Scholar] [CrossRef]
- Marks, D.F. Converging Evidence of Similar Symptomatology of ME/CFS and PASC Indicating Multisystemic Dyshomeostasis. Biomedicines 2023, 11, 180. [Google Scholar] [CrossRef] [PubMed]
- Tonnesen, E.; Christensen, N.J.; Brinklov, M.M. Natural killer cell activity during cortisol and adrenaline infusion in healthy volunteers. Eur. J. Clin. Investig. 1987, 17, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Mackay, A. A Paradigm for Post-COVID-19 Fatigue Syndrome Analogous to ME/CFS. Front. Neurol. 2021, 12, 701419. [Google Scholar] [CrossRef]
- Sato, W.; Ono, H.; Matsutani, T.; Nakamura, M.; Shin, I.; Amano, K.; Suzuki, R.; Yamamura, T. Skewing of the B cell receptor repertoire in myalgic encephalomyelitis/chronic fatigue syndrome. Brain Behav. Immun. 2021, 95, 245–255. [Google Scholar] [CrossRef]
- Fluge, O.; Mella, O.; Bruland, O.; Risa, K.; Dyrstad, S.E.; Alme, K.; Rekeland, I.G.; Sapkota, D.; Rosland, G.V.; Fossa, A.; et al. Metabolic profiling indicates impaired pyruvate dehydrogenase function in myalgic encephalopathy/chronic fatigue syndrome. JCI Insight 2016, 1, e89376. [Google Scholar] [CrossRef]
- McCarthy, M.J. Circadian rhythm disruption in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome: Implications for the post-acute sequelae of COVID-19. Brain Behav. Immun. Health 2022, 20, 100412. [Google Scholar] [CrossRef]
- Komaroff, A.L.; Lipkin, W.I. Insights from myalgic encephalomyelitis/chronic fatigue syndrome may help unravel the pathogenesis of postacute COVID-19 syndrome. Trends Mol. Med. 2021, 27, 895–906. [Google Scholar] [CrossRef]
- Jason, L.A.; Porter, N.; Herrington, J.; Sorenson, M.; Kubow, S. Kindling and Oxidative Stress as Contributors to Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. J. Behav. Neurosci. Res. 2009, 7, 1–17. [Google Scholar]
- Marcocci, M.E.; Napoletani, G.; Protto, V.; Kolesova, O.; Piacentini, R.; Li Puma, D.D.; Lomonte, P.; Grassi, C.; Palamara, A.T.; De Chiara, G. Herpes Simplex Virus-1 in the Brain: The Dark Side of a Sneaky Infection. Trends Microbiol. 2020, 28, 808–820. [Google Scholar] [CrossRef]
- Domingues, T.D.; Malato, J.; Grabowska, A.D.; Lee, J.S.; Ameijeiras-Alonso, J.; Biecek, P.; Graca, L.; Mourino, H.; Scheibenbogen, C.; Westermeier, F.; et al. Association analysis between symptomology and herpesvirus IgG antibody concentrations in myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS) and multiple sclerosis. Heliyon 2023, 9, e18250. [Google Scholar] [CrossRef]
- Gravelsina, S.; Vilmane, A.; Svirskis, S.; Rasa-Dzelzkaleja, S.; Nora-Krukle, Z.; Vecvagare, K.; Krumina, A.; Leineman, I.; Shoenfeld, Y.; Murovska, M. Biomarkers in the diagnostic algorithm of myalgic encephalomyelitis/chronic fatigue syndrome. Front. Immunol. 2022, 13, 928945. [Google Scholar] [CrossRef]
- Sotzny, F.; Blanco, J.; Capelli, E.; Castro-Marrero, J.; Steiner, S.; Murovska, M.; Scheibenbogen, C.; on behalf of the European Network on ME/CFS (EUROMENE). Myalgic Encephalomyelitis/Chronic Fatigue Syndrome—Evidence for an autoimmune disease. Autoimmun. Rev. 2018, 17, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Loebel, M.; Eckey, M.; Sotzny, F.; Hahn, E.; Bauer, S.; Grabowski, P.; Zerweck, J.; Holenya, P.; Hanitsch, L.G.; Wittke, K.; et al. Serological profiling of the EBV immune response in Chronic Fatigue Syndrome using a peptide microarray. PLoS ONE 2017, 12, e0179124. [Google Scholar] [CrossRef] [PubMed]
- Kehnemouyi, Y.M.; Wilkins, K.B.; Anidi, C.M.; Anderson, R.W.; Afzal, M.F.; Bronte-Stewart, H.M. Modulation of beta bursts in subthalamic sensorimotor circuits predicts improvement in bradykinesia. Brain 2021, 144, 473–486. [Google Scholar] [CrossRef] [PubMed]
- Wirth, K.; Scheibenbogen, C. A Unifying Hypothesis of the Pathophysiology of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS): Recognitions from the finding of autoantibodies against ss2-adrenergic receptors. Autoimmun. Rev. 2020, 19, 102527. [Google Scholar] [CrossRef]
- Shukla, S.K.; Cook, D.; Meyer, J.; Vernon, S.D.; Le, T.; Clevidence, D.; Robertson, C.E.; Schrodi, S.J.; Yale, S.; Frank, D.N. Changes in Gut and Plasma Microbiome following Exercise Challenge in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS). PLoS ONE 2015, 10, e0145453. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.; Berk, M.; Galecki, P.; Maes, M. The emerging role of autoimmunity in myalgic encephalomyelitis/chronic fatigue syndrome (ME/cfs). Mol. Neurobiol. 2014, 49, 741–756. [Google Scholar] [CrossRef] [PubMed]
- Scheibenbogen, C.; Bellmann-Strobl, J.T.; Heindrich, C.; Wittke, K.; Stein, E.; Franke, C.; Pruss, H.; Pressler, H.; Machule, M.L.; Audebert, H.; et al. Fighting Post-COVID and ME/CFS—Development of curative therapies. Front. Med. 2023, 10, 1194754. [Google Scholar] [CrossRef]
- Gonzalez-Cebrian, A.; Almenar-Perez, E.; Xu, J.; Yu, T.; Huang, W.E.; Gimenez-Orenga, K.; Hutchinson, S.; Lodge, T.; Nathanson, L.; Morten, K.J.; et al. Diagnosis of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome With Partial Least Squares Discriminant Analysis: Relevance of Blood Extracellular Vesicles. Front. Med. 2022, 9, 842991. [Google Scholar] [CrossRef] [PubMed]
- Nepotchatykh, E.; Caraus, I.; Elremaly, W.; Leveau, C.; Elbakry, M.; Godbout, C.; Rostami-Afshari, B.; Petre, D.; Khatami, N.; Franco, A.; et al. Circulating microRNA expression signatures accurately discriminate myalgic encephalomyelitis from fibromyalgia and comorbid conditions. Sci. Rep. 2023, 13, 1896. [Google Scholar] [CrossRef]
- Helliwell, A.M.; Stockwell, P.A.; Edgar, C.D.; Chatterjee, A.; Tate, W.P. Dynamic Epigenetic Changes during a Relapse and Recovery Cycle in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. Int. J. Mol. Sci. 2022, 23, 11852. [Google Scholar] [CrossRef]
- Giloteaux, L.; Li, J.; Hornig, M.; Lipkin, W.I.; Ruppert, D.; Hanson, M.R. Proteomics and cytokine analyses distinguish myalgic encephalomyelitis/chronic fatigue syndrome cases from controls. J. Transl. Med. 2023, 21, 322. [Google Scholar] [CrossRef]
- Montoya, J.G.; Holmes, T.H.; Anderson, J.N.; Maecker, H.T.; Rosenberg-Hasson, Y.; Valencia, I.J.; Chu, L.; Younger, J.W.; Tato, C.M.; Davis, M.M. Cytokine signature associated with disease severity in chronic fatigue syndrome patients. Proc. Natl. Acad. Sci. USA 2017, 114, E7150–E7158. [Google Scholar] [CrossRef]
- Saury, J.M. The role of the hippocampus in the pathogenesis of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS). Med. Hypotheses 2016, 86, 30–38. [Google Scholar] [CrossRef]
- Vogl, T.; Kalka, I.N.; Klompus, S.; Leviatan, S.; Weinberger, A.; Segal, E. Systemic antibody responses against human microbiota flagellins are overrepresented in chronic fatigue syndrome patients. Sci. Adv. 2022, 8, eabq2422. [Google Scholar] [CrossRef] [PubMed]
- Cheema, A.K.; Sarria, L.; Bekheit, M.; Collado, F.; Almenar-Perez, E.; Martin-Martinez, E.; Alegre, J.; Castro-Marrero, J.; Fletcher, M.A.; Klimas, N.G.; et al. Unravelling myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS): Gender-specific changes in the microRNA expression profiling in ME/CFS. J. Cell. Mol. Med. 2020, 24, 5865–5877. [Google Scholar] [CrossRef] [PubMed]
- Tsilioni, I.; Natelson, B.; Theoharides, T.C. Exosome-Associated Mitochondrial DNA from Patients with ME/CFS Stimulates Human Cultured Microglia to Release IL-1beta. Eur. J. Neurosci. 2022, 56, 5784–5794. [Google Scholar] [CrossRef] [PubMed]
- Soffritti, I.; Gravelsina, S.; D’Accolti, M.; Bini, F.; Mazziga, E.; Vilmane, A.; Rasa-Dzelzkaleja, S.; Nora-Krukle, Z.; Krumina, A.; Murovska, M.; et al. Circulating miRNAs Expression in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. Int. J. Mol. Sci. 2023, 24, 10582. [Google Scholar] [CrossRef]
- Taccori, A.; Maksoud, R.; Eaton-Fitch, N.; Patel, M.; Marshall-Gradisnik, S. A systematic review and meta-analysis of urinary biomarkers in myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS). J. Transl. Med. 2023, 21, 440. [Google Scholar] [CrossRef]
- Castro-Marrero, J.; Saez-Francas, N.; Santillo, D.; Alegre, J. Treatment and management of chronic fatigue syndrome/myalgic encephalomyelitis: All roads lead to Rome. Br. J. Pharmacol. 2017, 174, 345–369. [Google Scholar] [CrossRef]
- Preßler, H.; Machule, M.L.; Ufer, F.; Bünger, I.; Li, L.Y.; Buchholz, E.; Werner, C.; Beraha, E.; Wagner, F.; Metz, M.; et al. IA-PACS-CFS: A double-blinded, randomized, sham-controlled, exploratory trial of immunoadsorption in patients with chronic fatigue syndrome (CFS) including patients with post-acute COVID-19 CFS (PACS-CFS). Trials 2024, 25, 172. [Google Scholar] [CrossRef] [PubMed]
- Crosby, L.D.; Kalanidhi, S.; Bonilla, A.; Subramanian, A.; Ballon, J.S.; Bonilla, H. Off label use of Aripiprazole shows promise as a treatment for Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS): A retrospective study of 101 patients treated with a low dose of Aripiprazole. J. Transl. Med. 2021, 19, 50. [Google Scholar] [CrossRef]
- Berger, A.; Locatelli, M.; Arcila-Londono, X.; Hayat, G.; Olney, N.; Wymer, J.; Gwathmey, K.; Lunetta, C.; Heiman-Patterson, T.; Ajroud-Driss, S.; et al. The natural history of ALS: Baseline characteristics from a multicenter clinical cohort. Amyotroph. Lateral Scler. Front. Degener. 2023, 24, 625–633. [Google Scholar] [CrossRef]
- Ilieva, H.; Vullaganti, M.; Kwan, J. Advances in molecular pathology, diagnosis, and treatment of amyotrophic lateral sclerosis. BMJ 2023, 383, e075037. [Google Scholar] [CrossRef]
- Zhu, L.; Li, S.; Li, X.J.; Yin, P. Pathological insights from amyotrophic lateral sclerosis animal models: Comparisons, limitations, and challenges. Transl. Neurodegener. 2023, 12, 46. [Google Scholar] [CrossRef] [PubMed]
- Donini, L.; Tanel, R.; Zuccarino, R.; Basso, M. Protein biomarkers for the diagnosis and prognosis of Amyotrophic Lateral Sclerosis. Neurosci. Res. 2023, 197, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Irwin, K.E.; Sheth, U.; Wong, P.C.; Gendron, T.F. Fluid biomarkers for amyotrophic lateral sclerosis: A review. Mol. Neurodegener. 2024, 19, 9. [Google Scholar] [CrossRef] [PubMed]
- Renton, A.E.; Chio, A.; Traynor, B.J. State of play in amyotrophic lateral sclerosis genetics. Nat. Neurosci. 2014, 17, 17–23. [Google Scholar] [CrossRef]
- Taylor, J.P.; Brown, R.H., Jr.; Cleveland, D.W. Decoding ALS: From genes to mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef]
- Bozzoni, V.; Pansarasa, O.; Diamanti, L.; Nosari, G.; Cereda, C.; Ceroni, M. Amyotrophic lateral sclerosis and environmental factors. Funct. Neurol. 2016, 31, 7–19. [Google Scholar] [CrossRef]
- Beers, D.R.; Appel, S.H. Immune dysregulation in amyotrophic lateral sclerosis: Mechanisms and emerging therapies. Lancet Neurol. 2019, 18, 211–220. [Google Scholar] [CrossRef]
- Wang, H.; Liu, Y.T.; Ren, Y.L.; Guo, X.Y.; Wang, Y. Association of peripheral immune activation with amyotrophic lateral sclerosis and Parkinson’s disease: A systematic review and meta-analysis. J. Neuroimmunol. 2024, 388, 578290. [Google Scholar] [CrossRef]
- Cao, W.; Fan, D. Neutrophils: A subgroup of neglected immune cells in ALS. Front. Immunol. 2023, 14, 1246768. [Google Scholar] [CrossRef]
- Nainu, F.; Mamada, S.S.; Harapan, H.; Emran, T.B. Inflammation-Mediated Responses in the Development of Neurodegenerative Diseases. Adv. Exp. Med. Biol. 2023, 1411, 39–70. [Google Scholar] [CrossRef] [PubMed]
- He, D.; Xu, Y.; Liu, M.; Cui, L. The Inflammatory Puzzle: Piecing together the Links between Neuroinflammation and Amyotrophic Lateral Sclerosis. Aging Dis. 2024, 15, 96–114. [Google Scholar] [CrossRef] [PubMed]
- Ravichandran, K.A.; Heneka, M.T. Inflammasomes in neurological disorders—Mechanisms and therapeutic potential. Nat. Rev. Neurol. 2024, 20, 67–83. [Google Scholar] [CrossRef]
- De Marchi, F.; Tondo, G.; Corrado, L.; Menegon, F.; Aprile, D.; Anselmi, M.; D’Alfonso, S.; Comi, C.; Mazzini, L. Neuroinflammatory Pathways in the ALS-FTD Continuum: A Focus on Genetic Variants. Genes 2023, 14, 1658. [Google Scholar] [CrossRef] [PubMed]
- Acosta-Galeana, I.; Hernandez-Martinez, R.; Reyes-Cruz, T.; Chiquete, E.; Aceves-Buendia, J.J. RNA-binding proteins as a common ground for neurodegeneration and inflammation in amyotrophic lateral sclerosis and multiple sclerosis. Front. Mol. Neurosci. 2023, 16, 1193636. [Google Scholar] [CrossRef] [PubMed]
- Carata, E.; Muci, M.; Di Giulio, S.; Mariano, S.; Panzarini, E. Looking to the Future of the Role of Macrophages and Extracellular Vesicles in Neuroinflammation in ALS. Int. J. Mol. Sci. 2023, 24, 11251. [Google Scholar] [CrossRef]
- Calafatti, M.; Cocozza, G.; Limatola, C.; Garofalo, S. Microglial crosstalk with astrocytes and immune cells in amyotrophic lateral sclerosis. Front. Immunol. 2023, 14, 1223096. [Google Scholar] [CrossRef]
- Yang, K.; Liu, Y.; Zhang, M. The Diverse Roles of Reactive Astrocytes in the Pathogenesis of Amyotrophic Lateral Sclerosis. Brain Sci. 2024, 14, 158. [Google Scholar] [CrossRef]
- Skaper, S.D.; Giusti, P.; Facci, L. Microglia and mast cells: Two tracks on the road to neuroinflammation. FASEB J. 2012, 26, 3103–3117. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, Y.; Dong, H.; Xu, Y.; Zhang, S. Induction of Microglial Activation by Mediators Released from Mast Cells. Cell. Physiol. Biochem. 2016, 38, 1520–1531. [Google Scholar] [CrossRef]
- Theoharides, T.C.; Perlman, A.I.; Twahir, A.; Kempuraj, D. Mast cell activation: Beyond histamine and tryptase. Expert. Rev. Clin. Immunol. 2023, 19, 639–654. [Google Scholar] [CrossRef]
- Mukai, K.; Tsai, M.; Saito, H.; Galli, S.J. Mast cells as sources of cytokines, chemokines, and growth factors. Immunol. Rev. 2018, 282, 121–150. [Google Scholar] [CrossRef]
- Trias, E.; King, P.H.; Si, Y.; Kwon, Y.; Varela, V.; Ibarburu, S.; Kovacs, M.; Moura, I.C.; Beckman, J.S.; Hermine, O.; et al. Mast cells and neutrophils mediate peripheral motor pathway degeneration in ALS. JCI Insight 2018, 3, e123249. [Google Scholar] [CrossRef]
- Granucci, E.J.; Griciuc, A.; Mueller, K.A.; Mills, A.N.; Le, H.; Dios, A.M.; McGinty, D.; Pereira, J.; Elmaleh, D.; Berry, J.D.; et al. Cromolyn sodium delays disease onset and is neuroprotective in the SOD1(G93A) Mouse Model of amyotrophic lateral sclerosis. Sci. Rep. 2019, 9, 17728. [Google Scholar] [CrossRef]
- Theoharides, T.C.; Tsilioni, I. Amyotrophic Lateral Sclerosis, Neuroinflammation, and Cromolyn. Clin. Ther. 2020, 42, 546–549. [Google Scholar] [CrossRef]
- Patel, A.B.; Tsilioni, I.; Leeman, S.E.; Theoharides, T.C. Neurotensin stimulates sortilin and mTOR in human microglia inhibitable by methoxyluteolin, a potential therapeutic target for autism. Proc. Natl. Acad. Sci. USA 2016, 113, E7049–E7058. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.B.; Theoharides, T.C. Methoxyluteolin Inhibits Neuropeptide-stimulated Proinflammatory Mediator Release via mTOR Activation from Human Mast Cells. J. Pharmacol. Exp. Ther. 2017, 361, 462–471. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.W.; Liu, X.; Yepes, M.; Shepherd, K.R.; Miller, G.W.; Liu, Y.; Wilson, W.D.; Xiao, G.; Blanchi, B.; Sun, Y.E.; et al. A selective TrkB agonist with potent neurotrophic activities by 7,8-dihydroxyflavone. Proc. Natl. Acad. Sci. USA 2010, 107, 2687–2692. [Google Scholar] [CrossRef] [PubMed]
- Stansberry, W.M.; Pierchala, B.A. Neurotrophic factors in the physiology of motor neurons and their role in the pathobiology and therapeutic approach to amyotrophic lateral sclerosis. Front. Mol. Neurosci. 2023, 16, 1238453. [Google Scholar] [CrossRef] [PubMed]
- Meanti, R.; Bresciani, E.; Rizzi, L.; Coco, S.; Zambelli, V.; Dimitroulas, A.; Molteni, L.; Omeljaniuk, R.J.; Locatelli, V.; Torsello, A. Potential Applications for Growth Hormone Secretagogues Treatment of Amyotrophic Lateral Sclerosis. Curr. Neuropharmacol. 2023, 21, 2376–2394. [Google Scholar] [CrossRef] [PubMed]
- Boylan, K. Familial Amyotrophic Lateral Sclerosis. Neurol. Clin. 2015, 33, 807–830. [Google Scholar] [CrossRef] [PubMed]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.X.; Hentati, A.; Tainer, J.A.; Iqbal, Z.; Cayabyab, A.; Hung, W.Y.; Getzoff, E.D.; Hu, P.; Herzfeldt, B.; Roos, R.P.; et al. Amyotrophic lateral sclerosis and structural defects in Cu,Zn superoxide dismutase. Science 1993, 261, 1047–1051. [Google Scholar] [CrossRef] [PubMed]
- Kaur, S.J.; McKeown, S.R.; Rashid, S. Mutant SOD1 mediated pathogenesis of Amyotrophic Lateral Sclerosis. Gene 2016, 577, 109–118. [Google Scholar] [CrossRef]
- Sreedharan, J.; Blair, I.P.; Tripathi, V.B.; Hu, X.; Vance, C.; Rogelj, B.; Ackerley, S.; Durnall, J.C.; Williams, K.L.; Buratti, E.; et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 2008, 319, 1668–1672. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowski, T.J., Jr.; Bosco, D.A.; Leclerc, A.L.; Tamrazian, E.; Vanderburg, C.R.; Russ, C.; Davis, A.; Gilchrist, J.; Kasarskis, E.J.; Munsat, T.; et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009, 323, 1205–1208. [Google Scholar] [CrossRef]
- Picher-Martel, V.; Valdmanis, P.N.; Gould, P.V.; Julien, J.P.; Dupre, N. From animal models to human disease: A genetic approach for personalized medicine in ALS. Acta Neuropathol. Commun. 2016, 4, 70. [Google Scholar] [CrossRef]
- Banci, L.; Bertini, I.; Boca, M.; Girotto, S.; Martinelli, M.; Valentine, J.S.; Vieru, M. SOD1 and amyotrophic lateral sclerosis: Mutations and oligomerization. PLoS ONE 2008, 3, e1677. [Google Scholar] [CrossRef]
- Mejzini, R.; Flynn, L.L.; Pitout, I.L.; Fletcher, S.; Wilton, S.D.; Akkari, P.A. ALS Genetics, Mechanisms, and Therapeutics: Where Are We Now? Front. Neurosci. 2019, 13, 1310. [Google Scholar] [CrossRef]
- Kodavati, M.; Wang, H.; Hegde, M.L. Altered Mitochondrial Dynamics in Motor Neuron Disease: An Emerging Perspective. Cells 2020, 9, 1065. [Google Scholar] [CrossRef]
- Wang, X.; Hu, Y.; Xu, R. The pathogenic mechanism of TAR DNA-binding protein 43 (TDP-43) in amyotrophic lateral sclerosis. Neural Regen. Res. 2024, 19, 800–806. [Google Scholar] [CrossRef]
- Prasad, A.; Bharathi, V.; Sivalingam, V.; Girdhar, A.; Patel, B.K. Molecular Mechanisms of TDP-43 Misfolding and Pathology in Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2019, 12, 25. [Google Scholar] [CrossRef]
- Cook, C.; Petrucelli, L. Genetic Convergence Brings Clarity to the Enigmatic Red Line in ALS. Neuron 2019, 101, 1057–1069. [Google Scholar] [CrossRef]
- Magrane, J.; Cortez, C.; Gan, W.B.; Manfredi, G. Abnormal mitochondrial transport and morphology are common pathological denominators in SOD1 and TDP43 ALS mouse models. Hum. Mol. Genet. 2014, 23, 1413–1424. [Google Scholar] [CrossRef]
- Madruga, E.; Maestro, I.; Martinez, A. Mitophagy Modulation, a New Player in the Race against ALS. Int. J. Mol. Sci. 2021, 22, 740. [Google Scholar] [CrossRef] [PubMed]
- Moller, A.; Bauer, C.S.; Cohen, R.N.; Webster, C.P.; De Vos, K.J. Amyotrophic lateral sclerosis-associated mutant SOD1 inhibits anterograde axonal transport of mitochondria by reducing Miro1 levels. Hum. Mol. Genet. 2017, 26, 4668–4679. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Zhang, J. Mitochondrial quality control and neurodegenerative diseases. Neuronal Signal 2018, 2, NS20180062. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Li, L.; Lin, W.L.; Dickson, D.W.; Petrucelli, L.; Zhang, T.; Wang, X. The ALS disease-associated mutant TDP-43 impairs mitochondrial dynamics and function in motor neurons. Hum. Mol. Genet. 2013, 22, 4706–4719. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Liu, H.; Itoh, K.; Oh, S.; Zhao, L.; Murata, D.; Sesaki, H.; Hartung, T.; Na, C.H.; Wang, J. C9orf72 regulates energy homeostasis by stabilizing mitochondrial complex I assembly. Cell Metab. 2021, 33, 531–546.e9. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Gonzalez, R.; Lu, Y.; Gendron, T.F.; Karydas, A.; Tran, H.; Yang, D.; Petrucelli, L.; Miller, B.L.; Almeida, S.; Gao, F.B. Poly(GR) in C9ORF72-Related ALS/FTD Compromises Mitochondrial Function and Increases Oxidative Stress and DNA Damage in iPSC-Derived Motor Neurons. Neuron 2016, 92, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Wang, W.; Perry, G.; Zhu, X.; Wang, X. Mitochondrial dynamic abnormalities in amyotrophic lateral sclerosis. Transl. Neurodegener. 2015, 4, 14. [Google Scholar] [CrossRef]
- Liu, J.; Wang, F. Role of Neuroinflammation in Amyotrophic Lateral Sclerosis: Cellular Mechanisms and Therapeutic Implications. Front. Immunol. 2017, 8, 1005. [Google Scholar] [CrossRef]
- Maragakis, N.J.; de Carvalho, M.; Weiss, M.D. Therapeutic targeting of ALS pathways: Refocusing an incomplete picture. Ann. Clin. Transl. Neurol. 2023, 10, 1948–1971. [Google Scholar] [CrossRef] [PubMed]
- Cunha-Oliveira, T.; Montezinho, L.; Simoes, R.F.; Carvalho, M.; Ferreiro, E.; Silva, F.S.G. Mitochondria: A Promising Convergent Target for the Treatment of Amyotrophic Lateral Sclerosis. Cells 2024, 13, 248. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.; Zhang, C.; Wu, W.; Lu, X.; Zhang, L. Modulation of the gut-brain axis via the gut microbiota: A new era in treatment of amyotrophic lateral sclerosis. Front. Neurol. 2023, 14, 1133546. [Google Scholar] [CrossRef] [PubMed]
- Van Daele, S.H.; Masrori, P.; Van Damme, P.; Van Den Bosch, L. The sense of antisense therapies in ALS. Trends Mol. Med. 2024, S1471-4914(23)00283-6. [Google Scholar] [CrossRef]
- Maharaj, D.; Kaur, K.; Saltese, A.; Gouvea, J. Personalized Precision Immunotherapy for Amyotrophic Lateral Sclerosis (ALS). Crit. Rev. Immunol. 2023, 43, 1–11. [Google Scholar] [CrossRef]
- Rizzuti, M.; Melzi, V.; Gagliardi, D.; Resnati, D.; Meneri, M.; Dioni, L.; Masrori, P.; Hersmus, N.; Poesen, K.; Locatelli, M.; et al. Insights into the identification of a molecular signature for amyotrophic lateral sclerosis exploiting integrated microRNA profiling of iPSC-derived motor neurons and exosomes. Cell. Mol. Life Sci. 2022, 79, 189. [Google Scholar] [CrossRef]
- Okano, H.; Morimoto, S.; Kato, C.; Nakahara, J.; Takahashi, S. Induced pluripotent stem cells-based disease modeling, drug screening, clinical trials, and reverse translational research for amyotrophic lateral sclerosis. J. Neurochem. 2023, 167, 603–614. [Google Scholar] [CrossRef]
- Liu, C.; Oikonomopoulos, A.; Sayed, N.; Wu, J.C. Modeling human diseases with induced pluripotent stem cells: From 2D to 3D and beyond. Development 2018, 145, dev156166. [Google Scholar] [CrossRef]
- Sharma, A.; Sances, S.; Workman, M.J.; Svendsen, C.N. Multi-lineage Human iPSC-Derived Platforms for Disease Modeling and Drug Discovery. Cell Stem Cell 2020, 26, 309–329. [Google Scholar] [CrossRef]
- Aboul-Soud, M.A.M.; Alzahrani, A.J.; Mahmoud, A. Induced Pluripotent Stem Cells (iPSCs)-Roles in Regenerative Therapies, Disease Modelling and Drug Screening. Cells 2021, 10, 2319. [Google Scholar] [CrossRef]
- Cecerska-Heryc, E.; Pekala, M.; Serwin, N.; Glizniewicz, M.; Grygorcewicz, B.; Michalczyk, A.; Heryc, R.; Budkowska, M.; Dolegowska, B. The Use of Stem Cells as a Potential Treatment Method for Selected Neurodegenerative Diseases: Review. Cell. Mol. Neurobiol. 2023, 43, 2643–2673. [Google Scholar] [CrossRef]
- Autar, K.; Guo, X.; Rumsey, J.W.; Long, C.J.; Akanda, N.; Jackson, M.; Narasimhan, N.S.; Caneus, J.; Morgan, D.; Hickman, J.J. A functional hiPSC-cortical neuron differentiation and maturation model and its application to neurological disorders. Stem Cell Rep. 2022, 17, 96–109. [Google Scholar] [CrossRef]
- Canfield, S.G.; Stebbins, M.J.; Faubion, M.G.; Gastfriend, B.D.; Palecek, S.P.; Shusta, E.V. An isogenic neurovascular unit model comprised of human induced pluripotent stem cell-derived brain microvascular endothelial cells, pericytes, astrocytes, and neurons. Fluids Barriers CNS 2019, 16, 25. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.Y.; Ting, H.C.; Liu, C.A.; Su, H.L.; Chiou, T.W.; Harn, H.J.; Lin, S.Z.; Ho, T.J. Differentiation of Human Pluripotent Stem Cells Into Specific Neural Lineages. Cell Transplant. 2021, 30, 9636897211017829. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Kuang, J.; Niu, Y.; Zhu, H.; Chen, X.; So, K.F.; Xu, A.; Shi, L. Multiple factors to assist human-derived induced pluripotent stem cells to efficiently differentiate into midbrain dopaminergic neurons. Neural Regen. Res. 2024, 19, 908–914. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Tang, Y. Transcription Factor-Mediated Differentiation of Motor Neurons from Human Pluripotent Stem Cells. Methods Mol. Biol. 2023, 2593, 245–258. [Google Scholar] [CrossRef] [PubMed]
- Marrone, L.; Bus, C.; Schondorf, D.; Fitzgerald, J.C.; Kubler, M.; Schmid, B.; Reinhardt, P.; Reinhardt, L.; Deleidi, M.; Levin, T.; et al. Generation of iPSCs carrying a common LRRK2 risk allele for in vitro modeling of idiopathic Parkinson’s disease. PLoS ONE 2018, 13, e0192497. [Google Scholar] [CrossRef] [PubMed]
- Ng, B.; Rowland, H.A.; Wei, T.; Arunasalam, K.; Hayes, E.M.; Koychev, I.; Hedegaard, A.; Ribe, E.M.; Chan, D.; Chessell, T.; et al. Neurons derived from individual early Alzheimer’s disease patients reflect their clinical vulnerability. Brain Commun. 2022, 4, fcac267. [Google Scholar] [CrossRef] [PubMed]
- Workman, M.J.; Lim, R.G.; Wu, J.; Frank, A.; Ornelas, L.; Panther, L.; Galvez, E.; Perez, D.; Meepe, I.; Lei, S.; et al. Large-scale differentiation of iPSC-derived motor neurons from ALS and control subjects. Neuron 2023, 111, 1191–1204.e5. [Google Scholar] [CrossRef]
- Alam, A.; Singh, T.; Kayhanian, S.; Tjerkaski, J.; Garcia, N.M.; Carpenter, K.L.H.; Patani, R.; Lindblad, C.; Thelin, E.P.; Syed, Y.A.; et al. Modeling the Inflammatory Response of Traumatic Brain Injury Using Human Induced Pluripotent Stem Cell Derived Microglia. J. Neurotrauma 2023, 40, 2164–2173. [Google Scholar] [CrossRef] [PubMed]
- Yates, P.L.; Patil, A.; Sun, X.; Niceforo, A.; Gill, R.; Callahan, P.; Beck, W.; Piermarini, E.; Terry, A.V.; Sullivan, K.A.; et al. A cellular approach to understanding and treating Gulf War Illness. Cell. Mol. Life Sci. 2021, 78, 6941–6961. [Google Scholar] [CrossRef] [PubMed]
- Monzon-Nomdedeu, M.B.; Morten, K.J.; Oltra, E. Induced pluripotent stem cells as suitable sensors for fibromyalgia and myalgic encephalomyelitis/chronic fatigue syndrome. World J. Stem Cells 2021, 13, 1134–1150. [Google Scholar] [CrossRef] [PubMed]
- Schweitzer, J.S.; Song, B.; Herrington, T.M.; Park, T.Y.; Lee, N.; Ko, S.; Jeon, J.; Cha, Y.; Kim, K.; Li, Q.; et al. Personalized iPSC-Derived Dopamine Progenitor Cells for Parkinson’s Disease. N. Engl. J. Med. 2020, 382, 1926–1932. [Google Scholar] [CrossRef] [PubMed]
- Cha, Y.; Park, T.Y.; Leblanc, P.; Kim, K.S. Current Status and Future Perspectives on Stem Cell-Based Therapies for Parkinson’s Disease. J. Mov. Disord. 2023, 16, 22–41. [Google Scholar] [CrossRef]
- Han, J.Y.; Lee, E.H.; Kim, S.M.; Park, C.H. Efficient Generation of Dopaminergic Neurons from Mouse Ventral Midbrain Astrocytes. Biomol. Ther. 2023, 31, 264–275. [Google Scholar] [CrossRef]
- Wang, Y.; Xia, Y.; Kou, L.; Yin, S.; Chi, X.; Li, J.; Sun, Y.; Wu, J.; Zhou, Q.; Zou, W.; et al. Astrocyte-to-neuron reprogramming and crosstalk in the treatment of Parkinson’s disease. Neurobiol. Dis. 2023, 184, 106224. [Google Scholar] [CrossRef]
- Leon, M.; Prieto, J.; Molina-Navarro, M.M.; Garcia-Garcia, F.; Barneo-Munoz, M.; Ponsoda, X.; Saez, R.; Palau, F.; Dopazo, J.; Izpisua Belmonte, J.C.; et al. Rapid degeneration of iPSC-derived motor neurons lacking Gdap1 engages a mitochondrial-sustained innate immune response. Cell Death Discov. 2023, 9, 217. [Google Scholar] [CrossRef]
- Ho, R.; Workman, M.J.; Mathkar, P.; Wu, K.; Kim, K.J.; O’Rourke, J.G.; Kellogg, M.; Montel, V.; Banuelos, M.G.; Arogundade, O.A.; et al. Cross-Comparison of Human iPSC Motor Neuron Models of Familial and Sporadic ALS Reveals Early and Convergent Transcriptomic Disease Signatures. Cell Syst. 2021, 12, 159–175.e9. [Google Scholar] [CrossRef]
- Yates, P.L.; Case, K.; Sun, X.; Sullivan, K.; Baas, P.W.; Qiang, L. Veteran-derived cerebral organoids display multifaceted pathological defects in studies on Gulf War Illness. Front. Cell. Neurosci. 2022, 16, 979652. [Google Scholar] [CrossRef]
- Kerkering, J.; Muinjonov, B.; Rosiewicz, K.S.; Diecke, S.; Biese, C.; Schiweck, J.; Chien, C.; Zocholl, D.; Conrad, T.; Paul, F.; et al. iPSC-derived reactive astrocytes from patients with multiple sclerosis protect cocultured neurons in inflammatory conditions. J. Clin. Investig. 2023, 133, e164637. [Google Scholar] [CrossRef]
- Delsing, L.; Herland, A.; Falk, A.; Hicks, R.; Synnergren, J.; Zetterberg, H. Models of the blood-brain barrier using iPSC-derived cells. Mol. Cell. Neurosci. 2020, 107, 103533. [Google Scholar] [CrossRef]
- Simoes Da Gama, C.; Morin-Brureau, M. Study of BBB Dysregulation in Neuropathogenicity Using Integrative Human Model of Blood-Brain Barrier. Front. Cell. Neurosci. 2022, 16, 863836. [Google Scholar] [CrossRef] [PubMed]
- Alhindi, A.; Boehm, I.; Chaytow, H. Small junction, big problems: Neuromuscular junction pathology in mouse models of amyotrophic lateral sclerosis (ALS). J. Anat. 2022, 241, 1089–1107. [Google Scholar] [CrossRef] [PubMed]
- Pereira, J.D.; DuBreuil, D.M.; Devlin, A.C.; Held, A.; Sapir, Y.; Berezovski, E.; Hawrot, J.; Dorfman, K.; Chander, V.; Wainger, B.J. Human sensorimotor organoids derived from healthy and amyotrophic lateral sclerosis stem cells form neuromuscular junctions. Nat. Commun. 2021, 12, 4744. [Google Scholar] [CrossRef] [PubMed]
- Massih, B.; Veh, A.; Schenke, M.; Mungwa, S.; Seeger, B.; Selvaraj, B.T.; Chandran, S.; Reinhardt, P.; Sterneckert, J.; Hermann, A.; et al. A 3D cell culture system for bioengineering human neuromuscular junctions to model ALS. Front. Cell. Dev. Biol. 2023, 11, 996952. [Google Scholar] [CrossRef]
- Kempuraj, D.; Mentor, S.; Thangavel, R.; Ahmed, M.E.; Selvakumar, G.P.; Raikwar, S.P.; Dubova, I.; Zaheer, S.; Iyer, S.S.; Zaheer, A. Mast Cells in Stress, Pain, Blood-Brain Barrier, Neuroinflammation and Alzheimer’s Disease. Front. Cell. Neurosci. 2019, 13, 54. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Nguyen, N.T.P.; Milanese, M.; Bonanno, G. Insights into Human-Induced Pluripotent Stem Cell-Derived Astrocytes in Neurodegenerative Disorders. Biomolecules 2022, 12, 344. [Google Scholar] [CrossRef] [PubMed]
- Yeap, Y.J.; Teddy, T.J.W.; Lee, M.J.; Goh, M.; Lim, K.L. From 2D to 3D: Development of Monolayer Dopaminergic Neuronal and Midbrain Organoid Cultures for Parkinson’s Disease Modeling and Regenerative Therapy. Int. J. Mol. Sci. 2023, 24, 2523. [Google Scholar] [CrossRef]
- Sozzi, E.; Nilsson, F.; Kajtez, J.; Parmar, M.; Fiorenzano, A. Generation of Human Ventral Midbrain Organoids Derived from Pluripotent Stem Cells. Curr. Protoc. 2022, 2, e555. [Google Scholar] [CrossRef]
- Bose, A.; Petsko, G.A.; Studer, L. Induced pluripotent stem cells: A tool for modeling Parkinson’s disease. Trends Neurosci. 2022, 45, 608–620. [Google Scholar] [CrossRef] [PubMed]
- Yefroyev, D.A.; Jin, S. Induced Pluripotent Stem Cells for Treatment of Alzheimer’s and Parkinson’s Diseases. Biomedicines 2022, 10, 208. [Google Scholar] [CrossRef] [PubMed]
- Jarazo, J.; Barmpa, K.; Modamio, J.; Saraiva, C.; Sabate-Soler, S.; Rosety, I.; Griesbeck, A.; Skwirblies, F.; Zaffaroni, G.; Smits, L.M.; et al. Parkinson’s Disease Phenotypes in Patient Neuronal Cultures and Brain Organoids Improved by 2-Hydroxypropyl-beta-Cyclodextrin Treatment. Mov. Disord. 2022, 37, 80–94. [Google Scholar] [CrossRef] [PubMed]
- Swingler, M.; Donadoni, M.; Bellizzi, A.; Cakir, S.; Sariyer, I.K. iPSC-derived three-dimensional brain organoid models and neurotropic viral infections. J. Neurovirol. 2023, 29, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Jacob, F.; Pather, S.R.; Huang, W.K.; Zhang, F.; Wong, S.Z.H.; Zhou, H.; Cubitt, B.; Fan, W.; Chen, C.Z.; Xu, M.; et al. Human Pluripotent Stem Cell-Derived Neural Cells and Brain Organoids Reveal SARS-CoV-2 Neurotropism Predominates in Choroid Plexus Epithelium. Cell Stem Cell 2020, 27, 937–950.e9. [Google Scholar] [CrossRef] [PubMed]
- Sahlgren Bendtsen, K.M.; Hall, V.J. The Breakthroughs and Caveats of Using Human Pluripotent Stem Cells in Modeling Alzheimer’s Disease. Cells 2023, 12, 420. [Google Scholar] [CrossRef]
- Pandey, S.; Jirasko, M.; Lochman, J.; Chvatal, A.; Chottova Dvorakova, M.; Kucera, R. iPSCs in Neurodegenerative Disorders: A Unique Platform for Clinical Research and Personalized Medicine. J. Pers. Med. 2022, 12, 1485. [Google Scholar] [CrossRef]
- Silva, M.C.; Haggarty, S.J. Human pluripotent stem cell-derived models and drug screening in CNS precision medicine. Ann. N. Y. Acad. Sci. 2020, 1471, 18–56. [Google Scholar] [CrossRef]
- Kim, S.H.; Chang, M.Y. Application of Human Brain Organoids-Opportunities and Challenges in Modeling Human Brain Development and Neurodevelopmental Diseases. Int. J. Mol. Sci. 2023, 24, 12528. [Google Scholar] [CrossRef]
- Parsaeimehr, A.; Ebirim, R.I.; Ozbay, G. CRISPR-Cas technology a new era in genomic engineering. Biotechnol. Rep. 2022, 34, e00731. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cohen, J.; Mathew, A.; Dourvetakis, K.D.; Sanchez-Guerrero, E.; Pangeni, R.P.; Gurusamy, N.; Aenlle, K.K.; Ravindran, G.; Twahir, A.; Isler, D.; et al. Recent Research Trends in Neuroinflammatory and Neurodegenerative Disorders. Cells 2024, 13, 511. https://doi.org/10.3390/cells13060511
Cohen J, Mathew A, Dourvetakis KD, Sanchez-Guerrero E, Pangeni RP, Gurusamy N, Aenlle KK, Ravindran G, Twahir A, Isler D, et al. Recent Research Trends in Neuroinflammatory and Neurodegenerative Disorders. Cells. 2024; 13(6):511. https://doi.org/10.3390/cells13060511
Chicago/Turabian StyleCohen, Jessica, Annette Mathew, Kirk D. Dourvetakis, Estella Sanchez-Guerrero, Rajendra P. Pangeni, Narasimman Gurusamy, Kristina K. Aenlle, Geeta Ravindran, Assma Twahir, Dylan Isler, and et al. 2024. "Recent Research Trends in Neuroinflammatory and Neurodegenerative Disorders" Cells 13, no. 6: 511. https://doi.org/10.3390/cells13060511