
miR-154-5p Is a Novel Endogenous Ligand for TLR7 Inducing Microglial Activation and Neuronal Injury
Abstract
:1. Introduction
2. Methods
2.1. Mice
2.2. Cell Lines
2.3. Primary Cell Culture
2.4. Tumor Necrosis Factor Enzyme-Linked Immunosorbent Assay (ELISA)
2.5. HEK-Blue TLR7/8 Activation Assay
2.6. Reverse Transcription Quantitative Polymerase Chain Reaction (RT-qPCR)
2.7. Synthetic Oligonucleotides and Cell Stimulation
2.8. Immunochemistry
2.9. Microscopy and Imaging
2.10. Intrathecal Injection
2.11. Statistics
3. Results
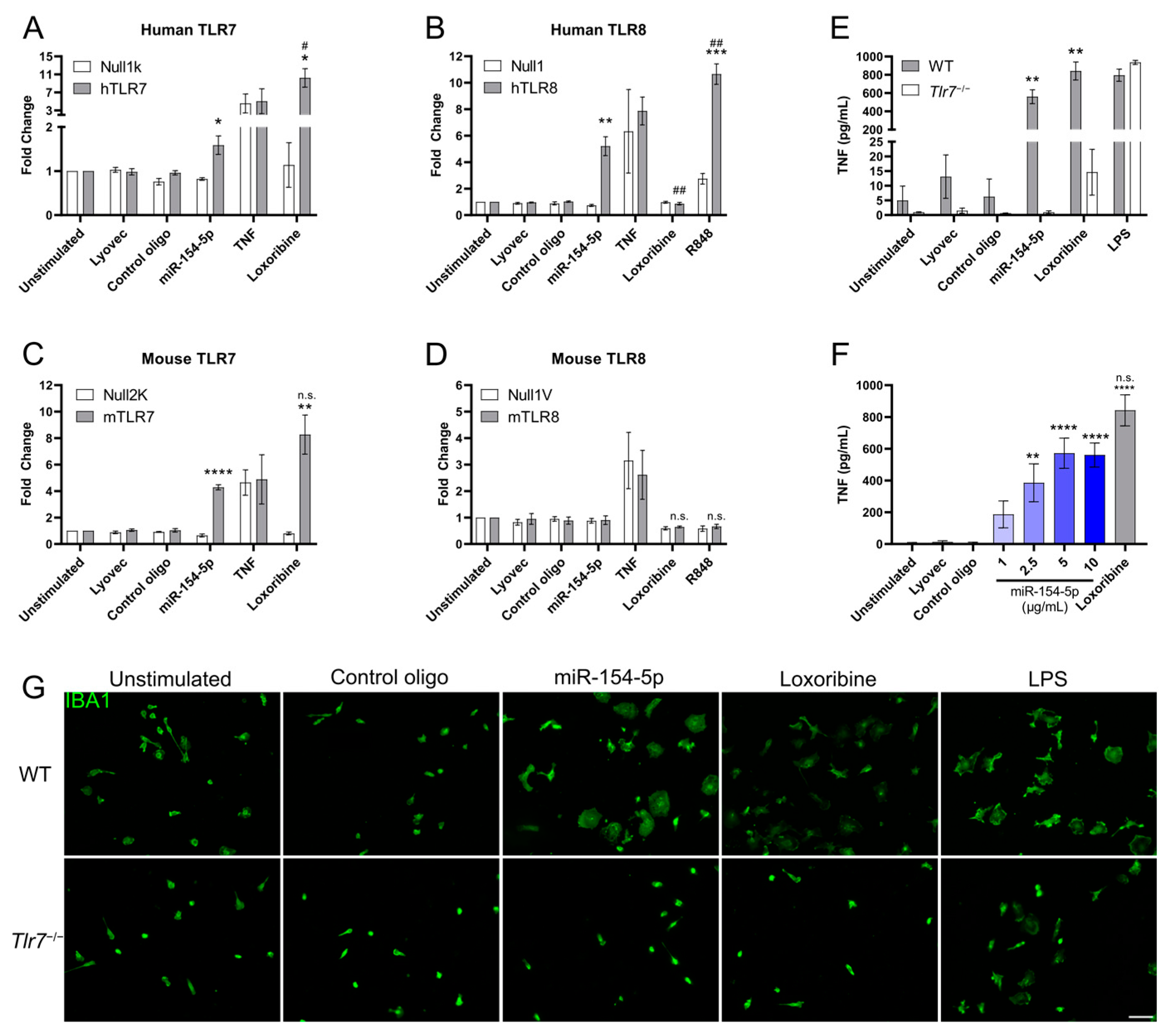
3.1. miR-154-5p Is an Endogenous Ligand for TLR7 and TLR8
3.2. miR-154-5p Alters the TLR7 Signalling Pathway in Microglia
3.3. Extracellular miR-154-5p Induces Neuronal Injury In Vitro
3.4. Intrathecal Injection of miR-154-5p into Mice Triggers Microglial Accumulation and Neuronal Injury via TLR7
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Khakpour, S.; Wilhelmsen, K.; Hellman, J. Vascular endothelial cell Toll-like receptor pathways in sepsis. Innate Immun. 2015, 21, 827–846. [Google Scholar] [CrossRef] [PubMed]
- Hanke, M.L.; Kielian, T. Toll-like receptors in health and disease in the brain: Mechanisms and therapeutic potential. Clin. Sci. Lond. Engl. 1979 2011, 121, 367–387. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Xiong, Y.; Li, Q.; Yang, H. Inhibition of toll-like receptor signaling as a promising therapy for inflammatory diseases: A journey from molecular to nano therapeutics. Front. Physiol. 2017, 8, 508. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Liu, S.; Han, J.; Li, S.; Gao, X.; Wang, M.; Zhu, J.; Jin, T. Role of Toll-Like Receptors in Neuroimmune Diseases: Therapeutic Targets and Problems. Front. Immunol. 2021, 12, 777606. [Google Scholar] [CrossRef] [PubMed]
- Mohammad Hosseini, A.; Majidi, J.; Baradaran, B.; Yousefi, M. Toll-Like Receptors in the Pathogenesis of Autoimmune Diseases. Adv. Pharm. Bull. 2015, 5, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, S.M.; Krüger, C.; Park, B.; Derkow, K.; Rosenberger, K.; Baumgart, J.; Trimbuch, T.; Eom, G.; Hinz, M.; Kaul, D.; et al. An unconventional role for miRNA: Let-7 activates Toll-like receptor 7 and causes neurodegeneration. Nat. Neurosci. 2012, 15, 827–835. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, S.M.; Rosenberger, K.; Krüger, C.; Habbel, P.; Derkow, K.; Kaul, D.; Rybak, A.; Brandt, C.; Schott, E.; Wulczyn, F.G.; et al. Extracellularly Delivered Single-Stranded Viral RNA Causes Neurodegeneration Dependent on TLR7. J. Immunol. 2012, 189, 1448–1458. [Google Scholar] [CrossRef]
- Luo, Z.; Su, R.; Wang, W.; Liang, Y.; Zeng, X.; Shereen, M.A.; Bashir, N.; Zhang, Q.; Zhao, L.; Wu, K.; et al. EV71 infection induces neurodegeneration via activating TLR7 signaling and IL-6 production. PLoS Pathog. 2019, 15, e1008142. [Google Scholar] [CrossRef]
- Deng, L.; Gao, R.; Chen, H.; Jiao, B.; Zhang, C.; Wei, L.; Yan, C.; Ye-Lehmann, S.; Zhu, T.; Chen, C. Let-7b-TLR7 Signaling Axis Contributes to the Anesthesia/Surgery-Induced Cognitive Impairment. Mol. Neurobiol. 2023. [Google Scholar] [CrossRef]
- Liu, H.Y.; Hung, Y.F.; Lin, H.R.; Yen, T.L.; Hsueh, Y.P. Tlr7 Deletion Selectively Ameliorates Spatial Learning but does not Influence beta Deposition and Inflammatory Response in an Alzheimers Disease Mouse Model. Neuropsychiatry 2017, 7, 509–521. [Google Scholar]
- Ha, M.; Kim, V.N. Regulation of microRNA biogenesis. Nat. Rev. Mol. Cell Biol. 2014, 15, 509–524. [Google Scholar] [CrossRef]
- Peng, Y.; Croce, C.M. The role of microRNAs in human cancer. Signal Transduct. Target. Ther. 2016, 1, 15004. [Google Scholar] [CrossRef]
- Peters, L.J.F.; Biessen, E.A.L.; Hohl, M.; Weber, C.; van der Vorst, E.P.C.; Santovito, D. Small Things Matter: Relevance of MicroRNAs in Cardiovascular Disease. Front. Physiol. 2020, 11, 793. [Google Scholar] [CrossRef]
- Pauley, K.M.; Cha, S.; Chan, E.K.L. MicroRNA in autoimmunity and autoimmune diseases. J. Autoimmun. 2009, 32, 189–194. [Google Scholar] [CrossRef]
- Kamal, M.A.; Mushtaq, G.; Greig, N.H. Current Update on Synopsis of miRNA Dysregulation in Neurological Disorders. CNS Neurol. Disord. Drug Targets 2015, 14, 492–501. [Google Scholar] [CrossRef]
- Zhang, Z.; Ohto, U.; Shibata, T.; Krayukhina, E.; Taoka, M.; Yamauchi, Y.; Tanji, H.; Isobe, T.; Uchiyama, S.; Miyake, K.; et al. Structural Analysis Reveals that Toll-like Receptor 7 Is a Dual Receptor for Guanosine and Single-Stranded RNA. Immunity 2016, 45, 737–748. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Kidder, K.; Liu, Y. Extracellular microRNAs initiate immunostimulation via activating toll-like receptor signaling pathways. ExRNA 2019, 1, 9. [Google Scholar] [CrossRef]
- Wu, N.; Morsey, B.M.; Emanuel, K.M.; Fox, H.S. Sequence-specific extracellular microRNAs activate TLR7 and induce cytokine secretion and leukocyte migration. Mol. Cell Biochem. 2021, 476, 4139–4151. [Google Scholar] [CrossRef] [PubMed]
- Wallach, T.; Mossmann, Z.J.; Szczepek, M.; Wetzel, M.; Machado, R.; Raden, M.; Miladi, M.; Kleinau, G.; Krüger, C.; Dembny, P.; et al. MicroRNA-100-5p and microRNA-298-5p released from apoptotic cortical neurons are endogenous Toll-like receptor 7/8 ligands that contribute to neurodegeneration. Mol. Neurodegener. 2021, 16, 80. [Google Scholar] [CrossRef] [PubMed]
- Wallach, T.; Wetzel, M.; Dembny, P.; Staszewski, O.; Krüger, C.; Buonfiglioli, A.; Prinz, M.; Lehnardt, S. Identification of CNS Injury-Related microRNAs as Novel Toll-Like Receptor 7/8 Signaling Activators by Small RNA Sequencing. Cells 2020, 9, 186. [Google Scholar] [CrossRef]
- Woodbury, M.E.; Freilich, R.W.; Cheng, C.J.; Asai, H.; Ikezu, S.; Boucher, J.D.; Slack, F.; Ikezu, T. miR-155 Is Essential for Inflammation-Induced Hippocampal Neurogenic Dysfunction. J. Neurosci. 2015, 35, 9764–9781. [Google Scholar] [CrossRef]
- Hamzei Taj, S.; Kho, W.; Aswendt, M.; Collmann, F.M.; Green, C.; Adamczak, J.; Tennstaedt, A.; Hoehn, M. Dynamic Modulation of Microglia/Macrophage Polarization by miR-124 after Focal Cerebral Ischemia. J. Neuroimmune Pharmacol. 2016, 11, 733–748. [Google Scholar] [CrossRef]
- Chen, S.; Wang, X.; Qian, Z.; Wang, M.; Zhang, F.; Zeng, T.; Li, L.; Gao, L. Exosomes from ADSCs ameliorate nerve damage in the hippocampus caused by post traumatic brain injury via the delivery of circ-Scmh1 promoting microglial M2 polarization. Injury 2023, 54, 110927. [Google Scholar] [CrossRef] [PubMed]
- Chi, J.-R.; Yu, Z.-H.; Liu, B.-W.; Zhang, D.; Ge, J.; Yu, Y.; Cao, X.-C. SNHG5 Promotes Breast Cancer Proliferation by Sponging the miR-154-5p/PCNA Axis. Mol. Ther.-Nucleic Acids 2019, 17, 138–149. [Google Scholar] [CrossRef]
- Xin, C.; Zhang, H.; Liu, Z. miR-154 suppresses colorectal cancer cell growth and motility by targeting TLR2. Mol. Cell Biochem. 2014, 387, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Wei, M.; Li, L.; Zhang, Y.; Zhang, Z.-J.; Liu, H.-L.; Bao, H.-G. LncRNA X inactive specific transcript contributes to neuropathic pain development by sponging miR-154-5p via inducing toll-like receptor 5 in CCI rat models. J. Cell Biochem. 2019, 120, 1271–1281. [Google Scholar] [CrossRef]
- Wang, X.; Sun, S.; Tong, X.; Ma, Q.; Di, H.; Fu, T.; Sun, Z.; Cai, Y.; Fan, W.; Wu, Q.; et al. MiRNA-154-5p inhibits cell proliferation and metastasis by targeting PIWIL1 in glioblastoma. Brain Res. 2017, 1676, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Guedes, J.R.; Santana, I.; Cunha, C.; Duro, D.; Almeida, M.R.; Cardoso, A.M.; de Lima, M.C.P.; Cardoso, A.L. MicroRNA deregulation and chemotaxis and phagocytosis impairment in Alzheimer’s disease. Alzheimers Dement. Diagn. Assess. Dis. Monit. 2016, 3, 7–17. [Google Scholar] [CrossRef]
- Ravanidis, S.; Bougea, A.; Papagiannakis, N.; Koros, C.; Simitsi, A.M.; Pachi, I.; Breza, M.; Stefanis, L.; Doxakis, E. Validation of differentially expressed brain-enriched microRNAs in the plasma of PD patients. Ann. Clin. Transl. Neurol. 2020, 7, 1594–1607. [Google Scholar] [CrossRef]
- Han, X.; Zhou, L.; Tu, Y.; Wei, J.; Zhang, J.; Jiang, G.; Shi, Q.; Ying, H. Circulating exo-miR-154-5p regulates vascular dementia through endothelial progenitor cell-mediated angiogenesis. Front. Cell Neurosci. 2022, 16, 881175. [Google Scholar] [CrossRef]
- Zhao, M.-W.; Qiu, W.-J.; Yang, P. SP1 activated-lncRNA SNHG1 mediates the development of epilepsy via miR-154-5p/TLR5 axis. Epilepsy Res. 2020, 168, 106476. [Google Scholar] [CrossRef]
- Chen, M.; Yang, Y.; Zhang, W.; Li, X.; Wu, J.; Zou, X.; Zeng, X. Long Noncoding RNA SNHG5 Knockdown Alleviates Neuropathic Pain by Targeting the miR-154-5p/CXCL13 Axis. Neurochem. Res. 2020, 45, 1566–1575. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, O.; Braun, J.S.; Becker, D.; Halle, A.; Freyer, D.; Dagand, E.; Lehnardt, S.; Weber, J.R. TLR2 Mediates Neuroinflammation and Neuronal Damage. J. Immunol. 2007, 178, 6476–6481. [Google Scholar] [CrossRef] [PubMed]
- Schön, M.P.; Schön, M. TLR7 and TLR8 as targets in cancer therapy. Oncogene 2008, 27, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Raden, M.; Wallach, T.; Miladi, M.; Zhai, Y.; Krüger, C.; Mossmann, Z.J.; Dembny, P.; Backofen, R.; Lehnardt, S. Structure-aware machine learning identifies microRNAs operating as Toll-like receptor 7/8 ligands. RNA Biol. 2021, 18, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Lax, N.; Fainstein, N.; Nishri, Y.; Ben-Zvi, A.; Ben-Hur, T. Systemic microbial TLR2 agonists induce neurodegeneration in Alzheimer’s disease mice. J. Neuroinflammation 2020, 17, 55. [Google Scholar] [CrossRef] [PubMed]
- Buchanan, M.M.; Hutchinson, M.; Watkins, L.R.; Yin, H. Toll-like receptor 4 in CNS pathologies. J. Neurochem. 2010, 114, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Ifuku, M.; Hinkelmann, L.; Kuhrt, L.D.; Efe, I.E.; Kumbol, V.; Buonfiglioli, A.; Krüger, C.; Jordan, P.; Fulde, M.; Noda, M.; et al. Activation of Toll-like receptor 5 in microglia modulates their function and triggers neuronal injury. Acta Neuropathol. Commun. 2020, 8, 1–19. [Google Scholar] [CrossRef]
- Derkow, K.; Krüger, C.; Dembny, P.; Lehnardt, S. Microglia Induce Neurotoxic IL-17+ γδ T Cells Dependent on TLR2, TLR4, and TLR9 Activation. PLoS ONE 2015, 10, e0135898. [Google Scholar] [CrossRef]
- Frederiksen, H.R.; Haukedal, H.; Freude, K. Cell Type Specific Expression of Toll-Like Receptors in Human Brains and Implications in Alzheimer’s Disease. BioMed Res. Int. 2019, 2019, e7420189. [Google Scholar] [CrossRef]
- Santovito, D.; Egea, V.; Bidzhekov, K.; Natarelli, L.; Mourão, A.; Blanchet, X.; Wichapong, K.; Aslani, M.; Brunßen, C.; Horckmans, M.; et al. Noncanonical inhibition of caspase-3 by a nuclear microRNA confers endothelial protection by autophagy in atherosclerosis. Sci. Transl. Med. 2020, 12, eaaz2294. [Google Scholar] [CrossRef]
- Yang, D.; Wan, X.; Dennis, A.T.; Bektik, E.; Wang, Z.; Costa, M.G.S.; Fagnen, C.; Xu, X.; Gratz, D.H.; Hund, T.J.; et al. MicroRNA Biophysically Modulates Cardiac Action Potential by Direct Binding to Ion Channel. Circulation 2021, 143, 1597–1613. [Google Scholar] [CrossRef]
- Fabbri, M.; Paone, A.; Calore, F.; Galli, R.; Gaudio, E.; Santhanam, R.; Lovat, F.; Fadda, P.; Mao, C.; Nuovo, G.J.; et al. MicroRNAs bind to Toll-like receptors to induce prometastatic inflammatory response. Proc. Natl. Acad. Sci. USA 2012, 109, E2110–E2116. [Google Scholar] [CrossRef] [PubMed]
- Heppner, F.L.; Ransohoff, R.M.; Becher, B. Immune attack: The role of inflammation in Alzheimer disease. Nat. Rev. Neurosci. 2015, 16, 358–372. [Google Scholar] [CrossRef]
- Tansey, M.G.; Wallings, R.L.; Houser, M.C.; Herrick, M.K.; Keating, C.E.; Joers, V. Inflammation and immune dysfunction in Parkinson disease. Nat. Rev. Immunol. 2022, 22, 657–673. [Google Scholar] [CrossRef]
- Sriram, U.; Xu, J.; Chain, R.W.; Varghese, L.; Chakhtoura, M.; Bennett, H.L.; Zoltick, P.W.; Gallucci, S. IL-4 Suppresses the Responses to TLR7 and TLR9 Stimulation and Increases the Permissiveness to Retroviral Infection of Murine Conventional Dendritic Cells. PLoS ONE 2014, 9, e87668. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Jo, H.; Kwon, Y.; Jeong, M.S.; Jung, H.S.; Kim, Y.; Jeoung, D. MiR-154-5p-MCP1 Axis Regulates Allergic Inflammation by Mediating Cellular Interactions. Front. Immunol. 2021, 12, 663726. [Google Scholar] [CrossRef]
- Jurk, M.; Heil, F.; Vollmer, J.; Schetter, C.; Krieg, A.M.; Wagner, H.; Lipford, G.; Bauer, S. Human TLR7 or TLR8 independently confer responsiveness to the antiviral compound R-848. Nat. Immunol. 2002, 3, 499. [Google Scholar] [CrossRef] [PubMed]
- Hemmi, H.; Kaisho, T.; Takeuchi, O.; Sato, S.; Sanjo, H.; Hoshino, K.; Horiuchi, T.; Tomizawa, H.; Takeda, K.; Akira, S. Small anti-viral compounds activate immune cells via the TLR7 MyD88–dependent signaling pathway. Nat. Immunol. 2002, 3, 196–200. [Google Scholar] [CrossRef]
- Liu, H.-Y.; Hong, Y.-F.; Huang, C.-M.; Chen, C.-Y.; Huang, T.-N.; Hsueh, Y.-P. TLR7 Negatively Regulates Dendrite Outgrowth through the Myd88–c-Fos–IL-6 Pathway. J. Neurosci. 2013, 33, 11479–11493. [Google Scholar] [CrossRef]
- Liu, J.; Ke, P.; Guo, H.; Gu, J.; Liu, Y.; Tian, X.; Wang, X.; Xiao, F. Activation of TLR7-mediated autophagy increases epileptic susceptibility via reduced KIF5A-dependent GABAA receptor transport in a murine model. Exp. Mol. Med. 2023, 55, 1159–1173. [Google Scholar] [CrossRef] [PubMed]
- Menassa, D.A.; Gomez-Nicola, D. Microglial dynamics during human brain development. Front. Immunol. 2018, 9, 1014. [Google Scholar] [CrossRef] [PubMed]
- Paolicelli, R.C.; Bolasco, G.; Pagani, F.; Maggi, L.; Scianni, M.; Panzanelli, P.; Giustetto, M.; Ferreira, T.A.; Guiducci, E.; Dumas, L.; et al. Synaptic pruning by microglia is necessary for normal brain development. Science 2011, 333, 1456–1458. [Google Scholar] [CrossRef] [PubMed]
- Cornell, J.; Salinas, S.; Huang, H.Y.; Zhou, M. Microglia regulation of synaptic plasticity and learning and memory. Neural Regen. Res. 2022, 17, 705–716. [Google Scholar]
- Arcuri, C.; Mecca, C.; Bianchi, R.; Giambanco, I.; Donato, R. The pathophysiological role of microglia in dynamic surveillance, phagocytosis and structural remodeling of the developing CNS. Front. Mol. Neurosci. 2017, 10, 191. [Google Scholar] [CrossRef]
- Custodero, C.; Ciavarella, A.; Panza, F.; Gnocchi, D.; Lenato, G.M.; Lee, J.; Mazzocca, A.; Sabbà, C.; Solfrizzi, V. Role of inflammatory markers in the diagnosis of vascular contributions to cognitive impairment and dementia: A systematic review and meta-analysis. GeroScience 2022, 44, 1373–1392. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Gene | Forward | Reverse |
|---|---|---|
| β-Actin | CCTGAACCCTAAGGCCAAC | GACAGCACAGCCTGGATGG |
| MyD88 | ACCTGTGTCTGGTCCATTGCCA | GCTGAGTGCAAACTTGGTCTGG |
| TICAM1 | ATCCATGCCAGGGCTGATGAAC | CGATGGCATCTTGGAGACAGTG |
| IRF7 | CCTCTGCTTTCTAGTGATGCCG | CGTAAACACGGTCTTGCTCCTG |
| p65 | TCCTGTTCGAGTCTCCATGCAG | GGTCTCATAGGTCCTTTTGCGC |
| TNF | GGTGCCTATGTCTCAGCCTCTT | GCCATAGAACTGATGAGAGGGAG |
| IFNβ | GCCTTTGCCATCCAAGAGATGC | ACACTGTCTGCTGGTGGAGTTC |
| IL6 | TACCACTTCACAAGTCGGAGGC | CTGCAAGTGCATCATCGTTGTTC |
| IRAK4 | CATACGCAACCTTAATGTGGGG | GGAACTGATTGTATCTGTCGTCG |
| SARM1 | TTCCTTGGCTCCAGAAATGCT | GACCCTGAGTTCCTCCGGTA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
McGurran, H.; Kumbol, V.; Krüger, C.; Wallach, T.; Lehnardt, S. miR-154-5p Is a Novel Endogenous Ligand for TLR7 Inducing Microglial Activation and Neuronal Injury. Cells 2024, 13, 407. https://doi.org/10.3390/cells13050407
McGurran H, Kumbol V, Krüger C, Wallach T, Lehnardt S. miR-154-5p Is a Novel Endogenous Ligand for TLR7 Inducing Microglial Activation and Neuronal Injury. Cells. 2024; 13(5):407. https://doi.org/10.3390/cells13050407
Chicago/Turabian StyleMcGurran, Hugo, Victor Kumbol, Christina Krüger, Thomas Wallach, and Seija Lehnardt. 2024. "miR-154-5p Is a Novel Endogenous Ligand for TLR7 Inducing Microglial Activation and Neuronal Injury" Cells 13, no. 5: 407. https://doi.org/10.3390/cells13050407