
Astrocytic GABAergic Regulation in Alcohol Use and Major Depressive Disorders


Abstract
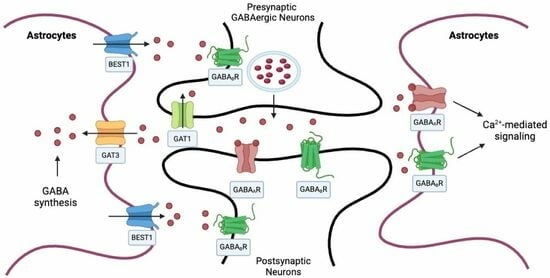
:
1. Introduction
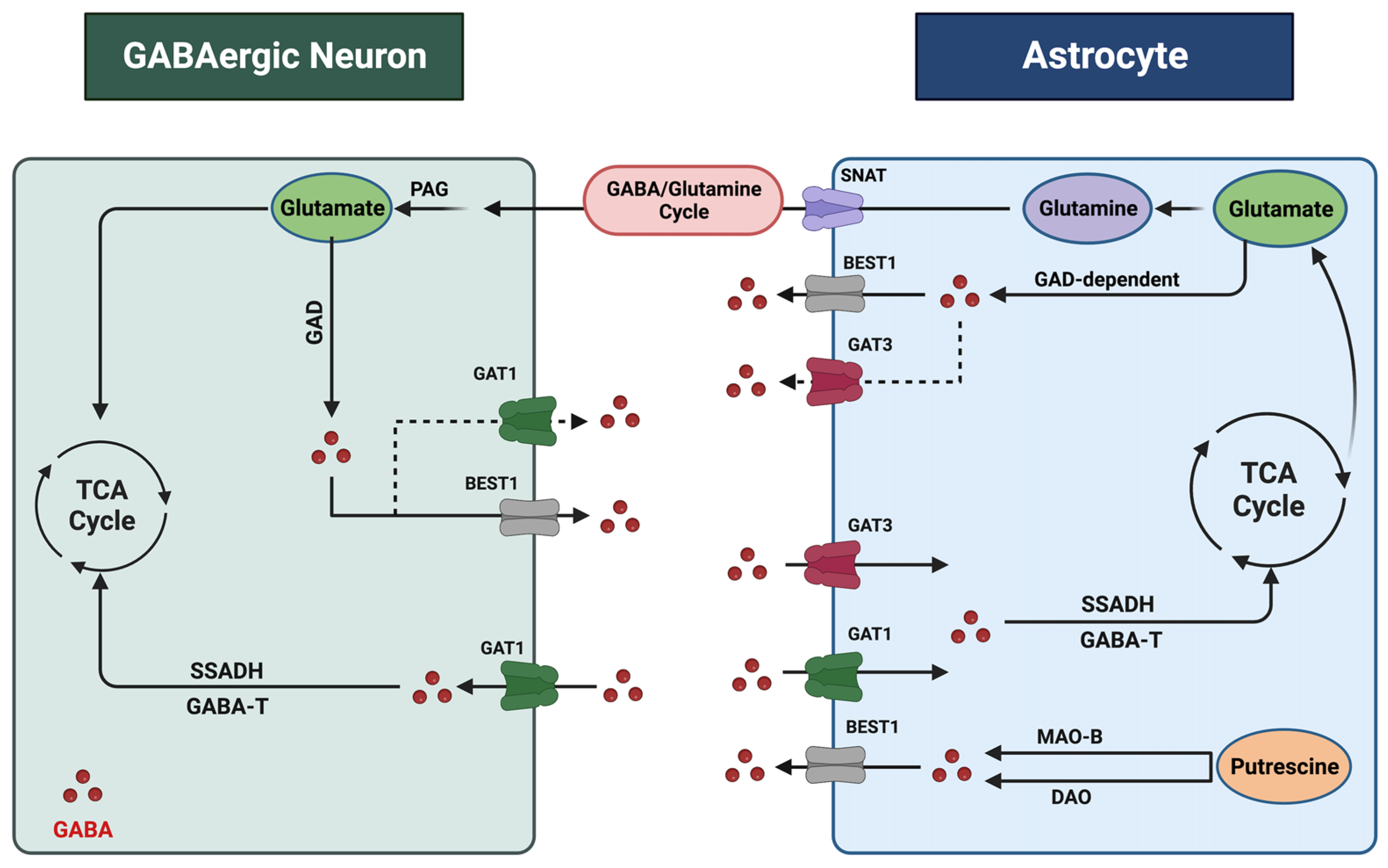
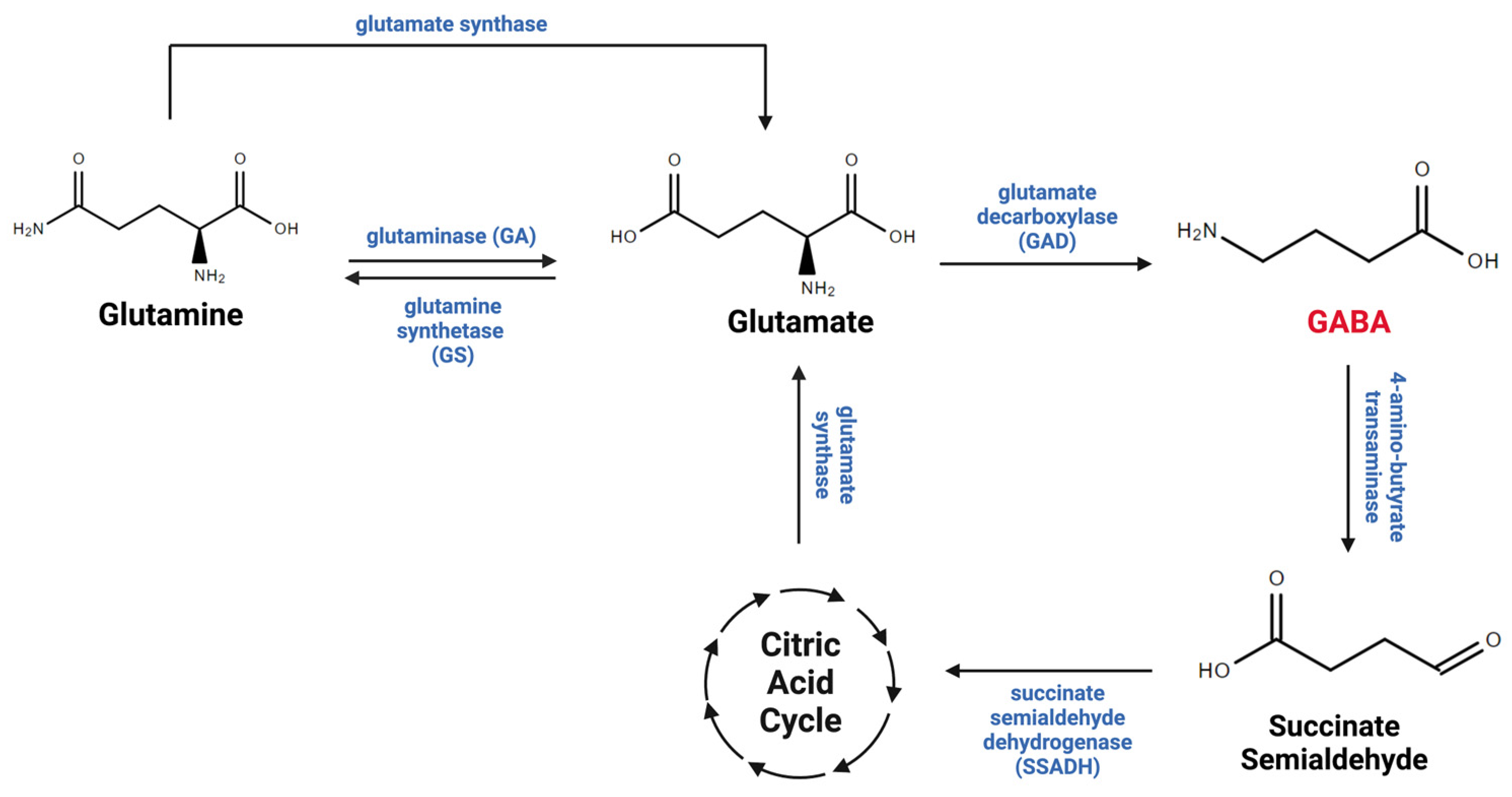
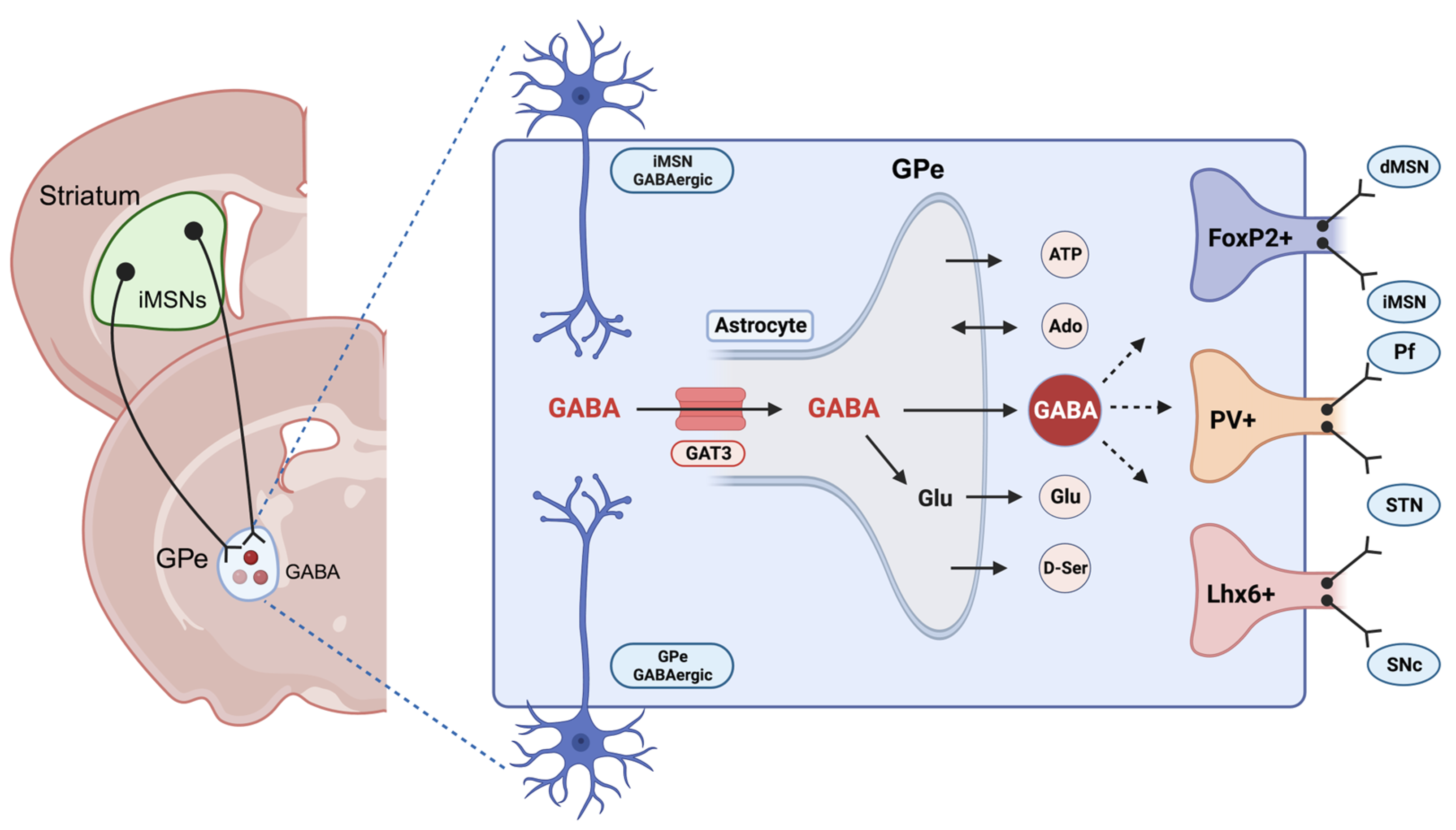
2. Astrocytic Regulation of GABA in the CNS
3. Astrocytes and GABA in Alcohol Use Disorder (AUD)
4. Astrocytes and GABA in Major Depressive Disorder (MDD)
5. Potential Therapeutic Targets in AUD and MDD
5.1. AUD

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Medication | Model | Target | Clinical Implications | References |
|---|---|---|---|---|
| Baclofen | Clinical Studies | GABAB receptor agonist | Reduces alcohol consumption and preference and decreases withdrawal symptoms in alcohol-dependent individuals; also supports the maintenance of abstinence from alcohol. | [131,132,134,135,138] |
| Benzodiazepines * | Rodents and Clinical Studies | GABAA receptor modulators | Can reduce the symptoms of alcohol withdrawal syndrome and reduce alcohol intake and alcohol seeking. | [143,144] |
| Allopregnanolone | Rodents and Clinical Studies | Neuroactive steroid | Serves as a safeguarding element in healthy control individuals, reducing the risk of developing AUD. | [145] |
| Vigabatrin | Rodents | GABA-transaminase inhibitor | Diminishes ethanol reinforcement and amplifies the discriminative stimulus effect of ethanol, leading to a significant decrease in ethanol consumption. | [146] |
| KK-92A | Rodents | Positive allosteric modulator of the GABAB receptor | Suppresses operant alcohol self-administration and reinstatement of alcohol seeking. | [147,148] |
| Semaglutide | Rodents and Clinical Studies | GLP-1 analogue increases GABA transmission in pyramidal neurons in layer 5 of the infralimbic cortex (ILC) and elevates dopamine levels in nucleus accumbens. | Decreases alcohol intake across different drinking models as it reduces alcohol intake and prevents relapse-like drinking. | [149] |
| Gabapentin | Clinical Studies and Rodents | Structural analog of GABA that binds to the alpha-2-delta type 1 subunit of voltage-gated calcium channels, reducing excitatory postsynaptic currents. | Most effective when implemented following the commencement of abstinence to maintain it, with its optimal performance likely observed in individuals with a track record of more intense alcohol withdrawal symptoms. | [139,140,150,151,152,153] |
| Muscimol | Rodents | GABAA receptor agonist | Intra-amygdala muscimol had a significant inhibitory effect on alcohol-seeking behavior in alcohol-dependent rats but had no impact on nondependent controls. In addition, it ameliorated the sleep–wake disruptions in alcohol-withdrawn rats by reducing the percentage of active wakefulness and increasing the percentage of REM sleep. | [110,154] |
| SR-95531 | Rodents | GABAA receptor antagonist | Decreased oral ethanol-seeking response in rats. | [87] |
| Tiagabine | Clinical Studies | Selective inhibitor of GABA reuptake by transporter subtype (GAT-1) | May reduce alcohol consumption and decrease alcohol dependence. | [155] |
5.2. MDD
| Medication | Model | Target | Clinical Implications | References |
|---|---|---|---|---|
| Tiagabine | Clinical Studies and Rodents | Selective inhibitor of GABA reuptake by transporter subtype (GAT1) | Demonstrated antidepressant-like properties in animal models. | [164,169] |
| * Brexanolone and zuranolone | Clinical Studies | Neuroactive steroids and positive allosteric modulators (PAM) of GABAA receptors | Zuranolone reduced depressive symptoms after 2 weeks of use. While brexanolone has been studied and approved for postpartum depression. | [165,166,167,170] |
| PRAX-114 | Clinical Studies and Rodents | Extrasynaptic GABAA receptor positive allosteric modulator | Attained antidepressant-like effects that varied based on the dosage. | [171,172] |
6. Discussion
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Whiteford, H.A.; Degenhardt, L.; Rehm, J.; Baxter, A.J.; Ferrari, A.J.; Erskine, H.E.; Charlson, F.J.; Norman, R.E.; Flaxman, A.D.; Johns, N. Global burden of disease attributable to mental and substance use disorders: Findings from the Global Burden of Disease Study 2010. Lancet 2013, 382, 1575–1586. [Google Scholar] [CrossRef]
- Rodgers, B.; Korten, A.E.; Jorm, A.F.; Jacomb, P.A.; Christensen, H.; Henderson, A.S. Non-linear relationships in associations of depression and anxiety with alcohol use. Psychol. Med. 2000, 30, 421–432. [Google Scholar] [CrossRef]
- Pedrelli, P.; Shapero, B.; Archibald, A.; Dale, C. Alcohol use and depression during adolescence and young adulthood: A summary and interpretation of mixed findings. Curr. Addict. Rep. 2016, 3, 91–97. [Google Scholar] [CrossRef]
- Miguel-Hidalgo, J.J.; Wilson, B.A.; Hussain, S.; Meshram, A.; Rajkowska, G.; Stockmeier, C.A. Reduced connexin 43 immunolabeling in the orbitofrontal cortex in alcohol dependence and depression. J. Psychiatr. Res. 2014, 55, 101–109. [Google Scholar] [CrossRef]
- Miguel-Hidalgo, J.J.; Waltzer, R.; Whittom, A.A.; Austin, M.C.; Rajkowska, G.; Stockmeier, C.A. Glial and glutamatergic markers in depression, alcoholism, and their comorbidity. J. Affect. Disord. 2010, 127, 230–240. [Google Scholar] [CrossRef]
- Brière, F.N.; Rohde, P.; Seeley, J.R.; Klein, D.; Lewinsohn, P.M. Comorbidity between major depression and alcohol use disorder from adolescence to adulthood. Compr. Psychiatry 2014, 55, 526–533. [Google Scholar] [CrossRef] [PubMed]
- Cornelius, J.R.; Bukstein, O.; Salloum, I.; Clark, D. Alcohol and psychiatric comorbidity. Recent. Dev. Alcohol. Res. Alcohol. Treat. 2002, 16, 361–374. [Google Scholar]
- Grant, B.F.; Harford, T.C. Comorbidity between DSM-IV alcohol use disorders and major depression: Results of a national survey. Drug Alcohol. Depend. 1995, 39, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Neupane, S.P. Neuroimmune interface in the comorbidity between alcohol use disorder and major depression. Front. Immunol. 2016, 7, 655. [Google Scholar] [CrossRef] [PubMed]
- Khalid, A.; Kunwar, A.R.; Rajbhandari, K.; Sharma, V.D.; Regmi, S.K. A study of prevalence and comorbidity of depression in alcohol dependence. Indian. J. Psychiatry 2000, 42, 434. [Google Scholar] [PubMed]
- Currie, S.R.; Patten, S.B.; Williams, J.V.; Wang, J.; Beck, C.A.; El-Guebaly, N.; Maxwell, C. Comorbidity of major depression with substance use disorders. Can. J. Psychiatry 2005, 50, 660–666. [Google Scholar] [CrossRef] [PubMed]
- Luscher, B.; Shen, Q.; Sahir, N. The GABAergic deficit hypothesis of major depressive disorder. Mol. Psychiatry 2011, 16, 383–406. [Google Scholar] [CrossRef]
- Koob, G.F. A role for GABA mechanisms in the motivational effects of alcohol. Biochem. Pharmacol. 2004, 68, 1515–1525. [Google Scholar] [CrossRef]
- Koob, G.F. A role for GABA in alcohol dependence1. Adv. Pharmacol. 2006, 54, 205–229. [Google Scholar] [PubMed]
- Jo, S.; Yarishkin, O.; Hwang, Y.J.; Chun, Y.E.; Park, M.; Woo, D.H.; Bae, J.Y.; Kim, T.; Lee, J.; Chun, H. GABA from reactive astrocytes impairs memory in mouse models of Alzheimer’s disease. Nat. Med. 2014, 20, 886–896. [Google Scholar] [CrossRef] [PubMed]
- Zurek, A.A.; Yu, J.; Wang, D.-S.; Haffey, S.C.; Bridgwater, E.M.; Penna, A.; Lecker, I.; Lei, G.; Chang, T.; Salter, E.W. Sustained increase in α5GABA A receptor function impairs memory after anesthesia. J. Clin. Investig. 2014, 124, 5437–5441. [Google Scholar] [CrossRef]
- Woo, J.; Min, J.O.; Kang, D.-S.; Kim, Y.S.; Jung, G.H.; Park, H.J.; Kim, S.; An, H.; Kwon, J.; Kim, J. Control of motor coordination by astrocytic tonic GABA release through modulation of excitation/inhibition balance in cerebellum. Proc. Natl. Acad. Sci. USA 2018, 115, 5004–5009. [Google Scholar] [CrossRef]
- Marowsky, A.; Rudolph, U.; Fritschy, J.-M.; Arand, M. Tonic inhibition in principal cells of the amygdala: A central role for α3 subunit-containing GABAA receptors. J. Neurosci. 2012, 32, 8611–8619. [Google Scholar] [CrossRef]
- Kwak, H.; Koh, W.; Kim, S.; Song, K.; Shin, J.-I.; Lee, J.M.; Lee, E.H.; Bae, J.Y.; Ha, G.E.; Oh, J.-E. Astrocytes control sensory acuity via tonic inhibition in the thalamus. Neuron 2020, 108, 691–706.e10. [Google Scholar] [CrossRef]
- Olsen, R.W.; Hanchar, H.J.; Meera, P.; Wallner, M. GABAA receptor subtypes: The “one glass of wine” receptors. Alcohol 2007, 41, 201–209. [Google Scholar] [CrossRef]
- Weyand, T.G.; Boudreaux, M.; Guido, W. Burst and tonic response modes in thalamic neurons during sleep and wakefulness. J. Neurophysiol. 2001, 85, 1107–1118. [Google Scholar] [CrossRef]
- Fogaça, M.V.; Duman, R.S. Cortical GABAergic Dysfunction in Stress and Depression: New Insights for Therapeutic Interventions. Front. Cell. Neurosci. 2019, 13, 87. [Google Scholar] [CrossRef]
- Roberto, M.; Kirson, D.; Khom, S. The Role of the Central Amygdala in Alcohol Dependence. Cold Spring Harb. Perspect. Med. 2021, 11, a039339. [Google Scholar] [CrossRef]
- Liljequist, S.; Engel, J. Effects of GABAergic agonists and antagonists on various ethanol-induced behavioral changes. Psychopharmacology 1982, 78, 71–75. [Google Scholar] [CrossRef]
- Vengeliene, V.; Bilbao, A.; Molander, A.; Spanagel, R. Neuropharmacology of alcohol addiction. Br. J. Pharmacol. 2008, 154, 299–315. [Google Scholar] [CrossRef]
- Cousins, M.S.; Roberts, D.C.; de Wit, H. GABA(B) receptor agonists for the treatment of drug addiction: A review of recent findings. Drug Alcohol. Depend. 2002, 65, 209–220. [Google Scholar] [CrossRef]
- Janak, P.H.; Michael Gill, T. Comparison of the effects of allopregnanolone with direct GABAergic agonists on ethanol self-administration with and without concurrently available sucrose. Alcohol 2003, 30, 1–7. [Google Scholar] [CrossRef]
- Grant, K.A.; Waters, C.A.; Green-Jordan, K.; Azarov, A.; Szeliga, K.T. Characterization of the discriminative stimulus effects of GABA(A) receptor ligands in Macaca fascicularis monkeys under different ethanol training conditions. Psychopharmacology 2000, 152, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Rassnick, S.; D’Amico, E.; Riley, E.; Koob, G.F. GABA antagonist and benzodiazepine partial inverse agonist reduce motivated responding for ethanol. Alcohol. Clin. Exp. Res. 1993, 17, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.V.; Schousboe, A.; Wellendorph, P. Astrocytes regulate inhibitory neurotransmission through GABA uptake, metabolism, and recycling. Essays Biochem. 2023, 67, 77–91. [Google Scholar] [CrossRef] [PubMed]
- Hasel, P.; Liddelow, S.A. Astrocytes. Curr. Biol. 2021, 31, R326–R327. [Google Scholar] [CrossRef] [PubMed]
- Chung, W.-S.; Allen, N.J.; Eroglu, C. Astrocytes control synapse formation, function, and elimination. Cold Spring Harb. Perspect. Biol. 2015, 7, a020370. [Google Scholar] [CrossRef] [PubMed]
- Faissner, A.; Pyka, M.; Geissler, M.; Sobik, T.; Frischknecht, R.; Gundelfinger, E.D.; Seidenbecher, C. Contributions of astrocytes to synapse formation and maturation—Potential functions of the perisynaptic extracellular matrix. Brain Res. Rev. 2010, 63, 26–38. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Benveniste, E.N. Immune function of astrocytes. Glia 2001, 36, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Swanson, R.A. Astrocytes and brain injury. J. Cereb. Blood Flow Metab. 2003, 23, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Deitmer, J.W.; Theparambil, S.M.; Ruminot, I.; Noor, S.I.; Becker, H.M. Energy dynamics in the brain: Contributions of astrocytes to metabolism and pH homeostasis. Front. Neurosci. 2019, 13, 1301. [Google Scholar] [CrossRef] [PubMed]
- Parpura, V.; Zorec, R. Gliotransmission: Exocytotic release from astrocytes. Brain Res. Rev. 2010, 63, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Angulo, M.C.; Le Meur, K.; Kozlov, A.S.; Charpak, S.; Audinat, E. GABA, a forgotten gliotransmitter. Prog. Neurobiol. 2008, 86, 297–303. [Google Scholar] [CrossRef]
- Yoon, B.-E.; Lee, C.J. GABA as a rising gliotransmitter. Front. Neural Circuits 2014, 8, 141. [Google Scholar] [CrossRef]
- Koh, W.; Kwak, H.; Cheong, E.; Lee, C.J. GABA tone regulation and its cognitive functions in the brain. Nat. Rev. Neurosci. 2023, 24, 523–539. [Google Scholar] [CrossRef]
- Adermark, L.; Bowers, M.S. Disentangling the role of astrocytes in alcohol use disorder. Alcohol. Clin. Exp. Res. 2016, 40, 1802–1816. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhu, X.; Ni, X.; Wen, Y.; Shang, D. Knowledge atlas of the involvement of glutamate and GABA in alcohol use disorder: A bibliometric and scientometric analysis. Front. Psychiatry 2022, 13, 965142. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.H.; Li, Z.L.; Liu, Y.S.; Chu, H.D.; Hu, N.Y.; Wu, D.Y.; Huang, L.; Li, S.J.; Li, X.W.; Yang, J.M.; et al. Astrocytic GABA(B) Receptors in Mouse Hippocampus Control Responses to Behavioral Challenges through Astrocytic BDNF. Neurosci. Bull. 2020, 36, 705–718. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, I.; Vazquez-Juarez, E.; Henning, L.; Gómez-Galán, M.; Lindskog, M. Blocking Astrocytic GABA Restores Synaptic Plasticity in Prefrontal Cortex of Rat Model of Depression. Cells 2020, 9, 1705. [Google Scholar] [CrossRef] [PubMed]
- Jazvinscak Jembrek, M.; Vlainic, J. GABA receptors: Pharmacological potential and pitfalls. Curr. Pharm. Des. 2015, 21, 4943–4959. [Google Scholar] [CrossRef] [PubMed]
- Sigel, E.; Steinmann, M.E. Structure, function, and modulation of GABAA receptors. J. Biol. Chem. 2012, 287, 40224–40231. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Maemura, K.; Kanbara, K.; Tamayama, T.; Hayasaki, H. GABA and GABA receptors in the central nervous system and other organs. Int. Rev. Cytol. 2002, 213, 1–47. [Google Scholar] [PubMed]
- Pinard, A.; Seddik, R.; Bettler, B. GABAB receptors: Physiological functions and mechanisms of diversity. Adv. Pharmacol. 2010, 58, 231–255. [Google Scholar]
- Lee, M.; Schwab, C.; Mcgeer, P.L. Astrocytes are GABAergic cells that modulate microglial activity. Glia 2011, 59, 152–165. [Google Scholar] [CrossRef]
- Liu, J.; Feng, X.; Wang, Y.; Xia, X.; Zheng, J.C. Astrocytes: GABAceptive and GABAergic cells in the brain. Front. Cell. Neurosci. 2022, 16, 892497. [Google Scholar] [CrossRef]
- Kilb, W.; Kirischuk, S. GABA release from astrocytes in health and disease. Int. J. Mol. Sci. 2022, 23, 15859. [Google Scholar] [CrossRef]
- Lee, M.; McGeer, E.G.; McGeer, P.L. Mechanisms of GABA release from human astrocytes. Glia 2011, 59, 1600–1611. [Google Scholar] [CrossRef]
- Schousboe, A.; Hertz, L.; Svenneby, G. Uptake and metabolism of GABA in astrocytes cultured from dissociated mouse brain hemispheres. Neurochem. Res. 1977, 2, 217–229. [Google Scholar] [CrossRef]
- Schousboe, A.; Waagepetersen, H.S. Role of astrocytes in homeostasis of glutamate and GABA during physiological and pathophysiological conditions. Adv. Mol. Cell Biol. 2003, 31, 461–474. [Google Scholar]
- Schousboe, A.; Westergaard, N.; Sonnewald, U.; Petersen, S.; Yu, A.; Hertz, L. Regulatory role of astrocytes for neuronal biosynthesis and homeostasis of glutamate and GABA. Prog. Brain Res. 1992, 94, 199–211. [Google Scholar]
- Ishibashi, M.; Egawa, K.; Fukuda, A. Diverse Actions of Astrocytes in GABAergic Signaling. Int. J. Mol. Sci. 2019, 20, 2964. [Google Scholar] [CrossRef]
- Roberts, E.; Frankel, S. γ-Aminobutyric acid in brain: Its formation from glutamic acid. J. Biol. Chem. 1950, 187, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Tao, R.; Davis, K.N.; Li, C.; Shin, J.H.; Gao, Y.; Jaffe, A.E.; Gondré-Lewis, M.C.; Weinberger, D.R.; Kleinman, J.E.; Hyde, T.M. GAD1 alternative transcripts and DNA methylation in human prefrontal cortex and hippocampus in brain development, schizophrenia. Mol. Psychiatry 2018, 23, 1496–1505. [Google Scholar] [CrossRef] [PubMed]
- Yoon, B.E.; Woo, J.; Chun, Y.E.; Chun, H.; Jo, S.; Bae, J.Y.; An, H.; Min, J.O.; Oh, S.J.; Han, K.S. Glial GABA, synthesized by monoamine oxidase B, mediates tonic inhibition. J. Physiol. 2014, 592, 4951–4968. [Google Scholar] [CrossRef] [PubMed]
- Losi, G.; Mariotti, L.; Carmignoto, G. GABAergic interneuron to astrocyte signalling: A neglected form of cell communication in the brain. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130609. [Google Scholar] [CrossRef] [PubMed]
- Scimemi, A. Structure, function, and plasticity of GABA transporters. Front. Cell. Neurosci. 2014, 8, 161. [Google Scholar] [CrossRef]
- Jin, X.T.; Galvan, A.; Wichmann, T.; Smith, Y. Localization and Function of GABA Transporters GAT-1 and GAT-3 in the Basal Ganglia. Front. Syst. Neurosci. 2011, 5, 63. [Google Scholar] [CrossRef]
- Fattorini, G.; Catalano, M.; Melone, M.; Serpe, C.; Bassi, S.; Limatola, C.; Conti, F. Microglial expression of GAT-1 in the cerebral cortex. Glia 2020, 68, 646–655. [Google Scholar] [CrossRef]
- Melone, M.; Ciappelloni, S.; Conti, F. A quantitative analysis of cellular and synaptic localization of GAT-1 and GAT-3 in rat neocortex. Brain Struct. Funct. 2015, 220, 885–897. [Google Scholar] [CrossRef]
- Minelli, A.; DeBiasi, S.; Brecha, N.C.; Zuccarello, L.V.; Conti, F. GAT-3, a high-affinity GABA plasma membrane transporter, is localized to astrocytic processes, and it is not confined to the vicinity of GABAergic synapses in the cerebral cortex. J. Neurosci. 1996, 16, 6255–6264. [Google Scholar] [CrossRef]
- Boddum, K.; Jensen, T.P.; Magloire, V.; Kristiansen, U.; Rusakov, D.A.; Pavlov, I.; Walker, M.C. Astrocytic GABA transporter activity modulates excitatory neurotransmission. Nat. Commun. 2016, 7, 13572. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.V.; Schousboe, A. Milestone Review: Metabolic dynamics of glutamate and GABA mediated neurotransmission—The essential roles of astrocytes. J. Neurochem. 2023, 166, 109–137. [Google Scholar] [CrossRef] [PubMed]
- Tritsch, N.X.; Granger, A.J.; Sabatini, B.L. Mechanisms and functions of GABA co-release. Nat. Rev. Neurosci. 2016, 17, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Murphy-Royal, C.; Ching, S.; Papouin, T. A conceptual framework for astrocyte function. Nat. Neurosci. 2023, 26, 1848–1856. [Google Scholar] [CrossRef] [PubMed]
- Waagepetersen, H.S.; Sonnewald, U.; Schousboe, A. Compartmentation of glutamine, glutamate, and GABA metabolism in neurons and astrocytes: Functional implications. Neurosci. 2003, 9, 398–403. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, A.F.; Heilig, M.; Perez, A.; Probst, C.; Rehm, J. Alcohol use disorders. Lancet 2019, 394, 781–792. [Google Scholar] [CrossRef]
- Lindberg, D.; Andres-Beck, L.; Jia, Y.F.; Kang, S.; Choi, D.S. Purinergic Signaling in Neuron-Astrocyte Interactions, Circadian Rhythms, and Alcohol Use Disorder. Front. Physiol. 2018, 9, 9. [Google Scholar] [CrossRef]
- Yang, W.; Singla, R.; Maheshwari, O.; Fontaine, C.J.; Gil-Mohapel, J. Alcohol Use Disorder: Neurobiology and Therapeutics. Biomedicines 2022, 10, 1192. [Google Scholar] [CrossRef]
- Griswold, M.G.; Fullman, N.; Hawley, C.; Arian, N.; Zimsen, S.R.; Tymeson, H.D.; Venkateswaran, V.; Tapp, A.D.; Forouzanfar, M.H.; Salama, J.S. Alcohol use and burden for 195 countries and territories, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet 2018, 392, 1015–1035. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Global Status Report on Alcohol and Health 2018; World Health Organization: Geneva, Switzerland, 2019. [Google Scholar]
- Collaborators, G.; Ärnlöv, J. Global burden of 87 risk factors in 204 countries and territories, 1990–2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet 2020, 396, 1223–1249. [Google Scholar]
- Koob, G.F.; Roberts, A.J.; Schulteis, G.; Parsons, L.H.; Heyser, C.J.; Hyytiä, P.; Merlo-Pich, E.; Weiss, F. Neurocircuitry targets in ethanol reward and dependence. Alcohol. Clin. Exp. Res. 1998, 22, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Augier, E.; Barbier, E.; Dulman, R.S.; Licheri, V.; Augier, G.; Domi, E.; Barchiesi, R.; Farris, S.; Nätt, D.; Mayfield, R.D.; et al. A molecular mechanism for choosing alcohol over an alternative reward. Science 2018, 360, 1321–1326. [Google Scholar] [CrossRef] [PubMed]
- Koob, G.F.; Volkow, N.D. Neurobiology of addiction: A neurocircuitry analysis. Lancet Psychiatry 2016, 3, 760–773. [Google Scholar] [CrossRef] [PubMed]
- Domi, E.; Domi, A.; Adermark, L.; Heilig, M.; Augier, E. Neurobiology of alcohol seeking behavior. J. Neurochem. 2021, 157, 1585–1614. [Google Scholar] [CrossRef] [PubMed]
- Abrahao, K.P.; Salinas, A.G.; Lovinger, D.M. Alcohol and the brain: Neuronal molecular targets, synapses, and circuits. Neuron 2017, 96, 1223–1238. [Google Scholar] [CrossRef] [PubMed]
- Koob, G.F.; Volkow, N.D. Neurocircuitry of addiction. Neuropsychopharmacology 2010, 35, 217–238. [Google Scholar] [CrossRef] [PubMed]
- Spanagel, R.; Zink, M.; Sommer, W.H. Neurobiology of Alcohol Addiction. In Neuroscience in the 21st Century: From Basic to Clinical; Pfaff, D.W., Ed.; Springer: New York, NY, USA, 2013; pp. 2745–2773. [Google Scholar] [CrossRef]
- Roberto, M.; Madamba, S.G.; Stouffer, D.G.; Parsons, L.H.; Siggins, G.R. Increased GABA release in the central amygdala of ethanol-dependent rats. J. Neurosci. 2004, 24, 10159–10166. [Google Scholar] [CrossRef]
- Colombo, G.; Serra, S.; Brunetti, G.; Vacca, G.; Carai, M.A.; Gessa, G.L. Suppression by baclofen of alcohol deprivation effect in Sardinian alcohol-preferring (sP) rats. Drug Alcohol. Depend. 2003, 70, 105–108. [Google Scholar] [CrossRef]
- GOLDSTEIN, D.B. Alcohol withdrawal reactions in mice: Effects of drugs that modify neurotransmission. J. Pharmacol. Exp. Ther. 1973, 186, 1–9. [Google Scholar]
- Hyytiä, P.; Koob, G.F. GABAA receptor antagonism in the extended amygdala decreases ethanol self-administration in rats. Eur. J. Pharmacol. 1995, 283, 151–159. [Google Scholar] [CrossRef]
- Foster, K.L.; McKay, P.F.; Seyoum, R.; Milbourne, D.; Yin, W.; Sarma, P.; Cook, J.M.; June, H.L. GABAA and opioid receptors of the central nucleus of the amygdala selectively regulate ethanol-maintained behaviors. Neuropsychopharmacology 2004, 29, 269–284. [Google Scholar] [CrossRef] [PubMed]
- Frye, G.D.; Mccown, T.J.; Breese, G.R. Characterization of susceptibility to audiogenic seizures in ethanol-dependent rats after microinjection of γ-aminobutyric acid (GABA) agonists into the inferior colliculus, substantia nigra or medial septum. J. Pharmacol. Exp. Ther. 1983, 227, 663. [Google Scholar] [PubMed]
- Cooper, B.; Viik, K.; Ferris, R.; White, H. Antagonism of the enhanced susceptibility to audiogenic seizures during alcohol withdrawal in the rat by gamma-aminobutyric acid (GABA) and” GABA-mimetic” agents. J. Pharmacol. Exp. Ther. 1979, 209, 396–403. [Google Scholar]
- Frye, G.D.; McCOWN, T.J.; Breese, G.R. Differential sensitivity of ethanol withdrawal signs in the rat to gamma-aminobutyric acid (GABA) mimetics: Blockade of audiogenic seizures but not forelimb tremors. J. Pharmacol. Exp. Ther. 1983, 226, 720–725. [Google Scholar]
- Valenzuela, C.F. Alcohol and neurotransmitter interactions. Alcohol. Health Res. World 1997, 21, 144–148. [Google Scholar]
- Mederos, S.; Perea, G. GABAergic-astrocyte signaling: A refinement of inhibitory brain networks. Glia 2019, 67, 1842–1851. [Google Scholar] [CrossRef]
- Corbit, L.H.; Nie, H.; Janak, P.H. Habitual alcohol seeking: Time course and the contribution of subregions of the dorsal striatum. Biol. Psychiatry 2012, 72, 389–395. [Google Scholar] [CrossRef]
- Barker, J.M.; Taylor, J.R. Habitual alcohol seeking: Modeling the transition from casual drinking to addiction. Neurosci. Biobehav. Rev. 2014, 47, 281–294. [Google Scholar] [CrossRef]
- Olsen, R.W.; Liang, J. Role of GABAA receptors in alcohol use disorders suggested by chronic intermittent ethanol (CIE) rodent model. Mol. Brain 2017, 10, 45. [Google Scholar] [CrossRef]
- Lovinger, D.M.; Kash, T.L. Mechanisms of neuroplasticity and ethanol’s effects on plasticity in the striatum and bed nucleus of the stria terminalis. Alcohol Res. Curr. Rev. 2015, 37, 109. [Google Scholar]
- Mederos, S.; Sánchez-Puelles, C.; Esparza, J.; Valero, M.; Ponomarenko, A.; Perea, G. GABAergic signaling to astrocytes in the prefrontal cortex sustains goal-directed behaviors. Nat. Neurosci. 2021, 24, 82–92. [Google Scholar] [CrossRef]
- Lipton, D.M.; Gonzales, B.J.; Citri, A. Dorsal Striatal Circuits for Habits, Compulsions and Addictions. Front. Syst. Neurosci. 2019, 13, 28. [Google Scholar] [CrossRef]
- Yin, H.H.; Knowlton, B.J. The role of the basal ganglia in habit formation. Nat. Rev. Neurosci. 2006, 7, 464–476. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Choi, D.S. Astrocyte adenosine signaling and neural mechanisms of goal-directed and habitual reward-seeking behaviors. Neuropsychopharmacology 2021, 46, 227–228. [Google Scholar] [CrossRef] [PubMed]
- Mallet, N.; Micklem, B.R.; Henny, P.; Brown, M.T.; Williams, C.; Bolam, J.P.; Nakamura, K.C.; Magill, P.J. Dichotomous organization of the external globus pallidus. Neuron 2012, 74, 1075–1086. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.-I.; Kang, S.; Song, M.; Yang, M.; Baker, M.; Kang, S.; Lee, J.; Lee, S.W.; Choi, D.-S. Astrocytes in the external globus pallidus coordinate flexibility of action strategy. Sci. Adv. 2021, 9, eadh9239. [Google Scholar]
- Hong, S.I.; Kang, S.; Baker, M.; Choi, D.S. Astrocyte-neuron interaction in the dorsal striatum-pallidal circuits and alcohol-seeking behaviors. Neuropharmacology 2021, 198, 108759. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Hong, S.I.; Kang, S.; Song, M.; Yang, M.A.; Essa, H.; Baker, M.; Lee, J.; Bruce, R.A.; Lee, S.W.; et al. Astrocyte activities in the external globus pallidus regulate action-selection strategies in reward-seeking behaviors. Sci. Adv. 2023, 9, eadh9239. [Google Scholar] [CrossRef] [PubMed]
- Chazalon, M.; Paredes-Rodriguez, E.; Morin, S.; Martinez, A.; Cristóvão-Ferreira, S.; Vaz, S.; Sebastiao, A.; Panatier, A.; Boué-Grabot, E.; Miguelez, C. GAT-3 dysfunction generates tonic inhibition in external globus pallidus neurons in parkinsonian rodents. Cell Rep. 2018, 23, 1678–1690. [Google Scholar] [CrossRef] [PubMed]
- Ribak, C.E.; Tong, W.M.; Brecha, N.C. GABA plasma membrane transporters, GAT-1 and GAT-3, display different distributions in the rat hippocampus. J. Comp. Neurol. 1996, 367, 595–606. [Google Scholar] [CrossRef]
- Morrow, A.L.; Suzdak, P.D.; Karanian, J.W.; Paul, S.M. Chronic ethanol administration alters gamma-aminobutyric acid, pentobarbital and ethanol-mediated 36Cl- uptake in cerebral cortical synaptoneurosomes. J. Pharmacol. Exp. Ther. 1988, 246, 158–164. [Google Scholar] [PubMed]
- Szmigielski, A.; Szmigielska, H.; Wejman, I. The effect of single and prolonged ethanol administration on the sensitivity of central GABA-A and benzodiazepine receptors in vivo. Pol. J. Pharmacol. Pharm. 1992, 44, 271–280. [Google Scholar]
- Roberts, A.J.; Cole, M.; Koob, G.F. Intra-amygdala muscimol decreases operant ethanol self-administration in dependent rats. Alcohol. Clin. Exp. Res. 1996, 20, 1289–1298. [Google Scholar] [CrossRef]
- Marti-Prats, L.; Belin-Rauscent, A.; Fouyssac, M.; Puaud, M.; Cocker, P.J.; Everitt, B.J.; Belin, D. Baclofen decreases compulsive alcohol drinking in rats characterized by reduced levels of GAT-3 in the central amygdala. Addict. Biol. 2021, 26, e13011. [Google Scholar] [CrossRef]
- Bachmann, S. Epidemiology of Suicide and the Psychiatric Perspective. Int. J. Environ. Res. Public. Health 2018, 15, 1425. [Google Scholar] [CrossRef]
- Chang, S.S.; Stuckler, D.; Yip, P.; Gunnell, D. Impact of 2008 global economic crisis on suicide: Time trend study in 54 countries. BMJ 2013, 347, f5239. [Google Scholar] [CrossRef] [PubMed]
- Hasin, D.S.; Goodwin, R.D.; Stinson, F.S.; Grant, B.F. Epidemiology of major depressive disorder: Results from the National Epidemiologic Survey on Alcoholism and Related Conditions. Arch. Gen. Psychiatry 2005, 62, 1097–1106. [Google Scholar] [CrossRef] [PubMed]
- Thompson, S.M.; Kallarackal, A.J.; Kvarta, M.D.; Van Dyke, A.M.; LeGates, T.A.; Cai, X. An excitatory synapse hypothesis of depression. Trends Neurosci. 2015, 38, 279–294. [Google Scholar] [CrossRef] [PubMed]
- Dean, J.; Keshavan, M. The neurobiology of depression: An integrated view. Asian J. Psychiatr. 2017, 27, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Kaltenboeck, A.; Harmer, C. The neuroscience of depressive disorders: A brief review of the past and some considerations about the future. Brain Neurosci. Adv. 2018, 2, 2398212818799269. [Google Scholar] [CrossRef] [PubMed]
- Marx, W.; Penninx, B.; Solmi, M.; Furukawa, T.A.; Firth, J.; Carvalho, A.F.; Berk, M. Major depressive disorder. Nat. Rev. Dis. Primers 2023, 9, 44. [Google Scholar] [CrossRef] [PubMed]
- Guglielmo, R.; de Filippis, R.; Ouanes, S.; Hasler, G. Editorial: The glutamate hypothesis of mood disorders: Neuroplasticity processes, clinical features, treatment perspectives. Front. Psychiatry 2022, 13, 1054887. [Google Scholar] [CrossRef] [PubMed]
- Duman, R.S.; Sanacora, G.; Krystal, J.H. Altered Connectivity in Depression: GABA and Glutamate Neurotransmitter Deficits and Reversal by Novel Treatments. Neuron 2019, 102, 75–90. [Google Scholar] [CrossRef]
- Godfrey, K.E.M.; Gardner, A.C.; Kwon, S.; Chea, W.; Muthukumaraswamy, S.D. Differences in excitatory and inhibitory neurotransmitter levels between depressed patients and healthy controls: A systematic review and meta-analysis. J. Psychiatr. Res. 2018, 105, 33–44. [Google Scholar] [CrossRef]
- Belleau, E.L.; Treadway, M.T.; Pizzagalli, D.A. The impact of stress and major depressive disorder on hippocampal and medial prefrontal cortex morphology. Biol. Psychiatry 2019, 85, 443–453. [Google Scholar] [CrossRef]
- Sanacora, G.; Mason, G.F.; Rothman, D.L.; Krystal, J.H. Increased occipital cortex GABA concentrations in depressed patients after therapy with selective serotonin reuptake inhibitors. Am. J. Psychiatry 2002, 159, 663–665. [Google Scholar] [CrossRef]
- Dubin, M.J.; Mao, X.; Banerjee, S.; Goodman, Z.; Lapidus, K.A.; Kang, G.; Liston, C.; Shungu, D.C. Elevated prefrontal cortex GABA in patients with major depressive disorder after TMS treatment measured with proton magnetic resonance spectroscopy. J. Psychiatry Neurosci. 2016, 41, E37–E45. [Google Scholar] [CrossRef]
- Kelly, T.J.; Bonniwell, E.M.; Mu, L.; Liu, X.; Hu, Y.; Friedman, V.; Yu, H.; Su, W.; McCorvy, J.D.; Liu, Q.S. Psilocybin analog 4-OH-DiPT enhances fear extinction and GABAergic inhibition of principal neurons in the basolateral amygdala. Neuropsychopharmacology 2023, E1–E9. [Google Scholar] [CrossRef]
- Lee, K.C.; Chen, J.J. Transdermal selegiline for the treatment of major depressive disorder. Neuropsychiatr. Dis. Treat. 2007, 3, 527–537. [Google Scholar]
- Zink, M.; Vollmayr, B.; Gebicke-Haerter, P.J.; Henn, F.A. Reduced Expression of GABA Transporter GAT3 in Helpless Rats, an Animal Model of Depression. Neurochem. Res. 2009, 34, 1584–1593. [Google Scholar] [CrossRef]
- Ma, K.; Zhang, H.; Wang, S.; Wang, H.; Wang, Y.; Liu, J.; Song, X.; Dong, Z.; Han, X.; Zhang, Y.; et al. The molecular mechanism underlying GABAergic dysfunction in nucleus accumbens of depression-like behaviours in mice. J. Cell Mol. Med. 2019, 23, 7021–7028. [Google Scholar] [CrossRef]
- Xu, L.; Nan, J.; Lan, Y. The Nucleus Accumbens: A Common Target in the Comorbidity of Depression and Addiction. Front. Neural Circuits 2020, 14, 37. [Google Scholar] [CrossRef]
- Lyu, S.; Guo, Y.; Zhang, L.; Wang, Y.; Tang, G.; Li, R.; Yang, J.; Gao, S.; Ma, B.; Liu, J. Blockade of GABA transporter-1 and GABA transporter-3 in the lateral habenula improves depressive-like behaviors in a rat model of Parkinson’s disease. Neuropharmacology 2020, 181, 108369. [Google Scholar] [CrossRef]
- Rose, A.K.; Jones, A. Baclofen: Its effectiveness in reducing harmful drinking, craving, and negative mood. A meta-analysis. Addiction 2018, 113, 1396–1406. [Google Scholar] [CrossRef]
- de Beaurepaire, R.; Rolland, B. Baclofen in alcohol use disorder: An analysis of the data provided by the French “Temporary Recommendation for Use” 2014–2017 cohort. Front. Psychiatry 2022, 13, 949750. [Google Scholar] [CrossRef]
- De Beaurepaire, R.; Sinclair, J.M.; Heydtmann, M.; Addolorato, G.; Aubin, H.-J.; Beraha, E.M.; Caputo, F.; Chick, J.D.; de La Selle, P.; Franchitto, N. The use of baclofen as a treatment for alcohol use disorder: A clinical practice perspective. Front. Psychiatry 2019, 9, 708. [Google Scholar] [CrossRef]
- Pierce, M.; Sutterland, A.; Beraha, E.M.; Morley, K.; van den Brink, W. Efficacy, tolerability, and safety of low-dose and high-dose baclofen in the treatment of alcohol dependence: A systematic review and meta-analysis. Eur. Neuropsychopharmacol. 2018, 28, 795–806. [Google Scholar] [CrossRef]
- Bschor, T.; Henssler, J.; Müller, M.; Baethge, C. Baclofen for alcohol use disorder—A systematic meta-analysis. Acta Psychiatr. Scand. 2018, 138, 232–242. [Google Scholar] [CrossRef]
- Krupitsky, E.; Burakov, A.; Ivanov, V.; Krandashova, G.; Lapin, I.; Grinenko, A.J.; Borodkin, Y.S. Baclofen administration for the treatment of affective disorders in alcoholic patients. Drug Alcohol Depend. 1993, 33, 157–163. [Google Scholar] [CrossRef]
- Morley, K.; Baillie, A.; Leung, S.; Addolorato, G.; Leggio, L.; Haber, P. Baclofen for the treatment of alcohol dependence and possible role of comorbid anxiety. Alcohol Alcohol. 2014, 49, 654–660. [Google Scholar] [CrossRef]
- Garbutt, J.C.; Kampov-Polevoy, A.B.; Pedersen, C.; Stansbury, M.; Jordan, R.; Willing, L.; Gallop, R.J. Efficacy and tolerability of baclofen in a U.S. community population with alcohol use disorder: A dose-response, randomized, controlled trial. Neuropsychopharmacology 2021, 46, 2250–2256. [Google Scholar] [CrossRef]
- Bates, R.E.; Leung, J.G.; Morgan, R.J.; Fischer, K.M.; Philbrick, K.L.; Kung, S. Retrospective Analysis of Gabapentin for Alcohol Withdrawal in the Hospital Setting: The Mayo Clinic Experience. Mayo Clin. Proc. Innov. Qual. Outcomes 2020, 4, 542–549. [Google Scholar] [CrossRef]
- Mason, B.J.; Quello, S.; Shadan, F. Gabapentin for the treatment of alcohol use disorder. Expert Opin. Investig. Drugs 2018, 27, 113–124. [Google Scholar] [CrossRef]
- Morley, K.C.; Perry, C.J.; Watt, J.; Hurzeler, T.; Leggio, L.; Lawrence, A.J.; Haber, P. New approved and emerging pharmacological approaches to alcohol use disorder: A review of clinical studies. Expert Opin. Pharmacother. 2021, 22, 1291–1303. [Google Scholar] [CrossRef]
- Kranzler, H.R.; Feinn, R.; Morris, P.; Hartwell, E.E. A meta-analysis of the efficacy of gabapentin for treating alcohol use disorder. Addiction 2019, 114, 1547–1555. [Google Scholar] [CrossRef]
- June, H.; Murphy, J.; Hewitt, R.; Greene, T.; Lin, M.; Mellorburke, J.; Lumeng, L.; Li, T. Benzodiazepine receptor ligands with different intrinsic efficacies alter ethanol intake in alcohol-nonpreferring (NP) rats. Neuropsychopharmacology 1996, 14, 55–66. [Google Scholar] [CrossRef]
- Sachdeva, A.; Choudhary, M.; Chandra, M. Alcohol Withdrawal Syndrome: Benzodiazepines and Beyond. J. Clin. Diagn. Res. 2015, 9, ve01–ve07. [Google Scholar] [CrossRef]
- Morrow, A.L.; Boero, G.; Porcu, P. A Rationale for Allopregnanolone Treatment of Alcohol Use Disorders: Basic and Clinical Studies. Alcohol. Clin. Exp. Res. 2020, 44, 320–339. [Google Scholar] [CrossRef]
- Griffin, W.C.; Nguyen, S.A.; Deleon, C.P.; Middaugh, L.D. Effects of vigabatrin, an irreversible GABA transaminase inhibitor, on ethanol reinforcement and ethanol discriminative stimuli in mice. Behav. Pharmacol. 2012, 23, 178–190. [Google Scholar] [CrossRef]
- Maccioni, P.; Kaczanowska, K.; Lawrence, H.; Yun, S.; Bratzu, J.; Gessa, G.L.; McDonald, P.; Colombo, G. The Novel Positive Allosteric Modulator of the GABAB Receptor, KK-92A, Suppresses Alcohol Self-Administration and Cue-Induced Reinstatement of Alcohol Seeking in Rats. Front. Cell Dev. Biol. 2021, 9, 727576. [Google Scholar] [CrossRef]
- Maccioni, P.; Kaczanowska, K.; Lobina, C.; Regonini Somenzi, L.; Bassareo, V.; Gessa, G.L.; Lawrence, H.R.; McDonald, P.; Colombo, G. Delving into the reducing effects of the GABAB positive allosteric modulator, KK-92A, on alcohol-related behaviors in rats. Alcohol 2023, 112, 61–70. [Google Scholar] [CrossRef]
- Chuong, V.; Farokhnia, M.; Khom, S.; Pince, C.L.; Elvig, S.K.; Vlkolinsky, R.; Marchette, R.C.N.; Koob, G.F.; Roberto, M.; Vendruscolo, L.F.; et al. The glucagon-like peptide-1 (GLP-1) analogue semaglutide reduces alcohol drinking and modulates central GABA neurotransmission. JCI Insight 2023, 8, e170671. [Google Scholar] [CrossRef]
- Anton, R.F.; Latham, P.; Voronin, K.; Book, S.; Hoffman, M.; Prisciandaro, J.; Bristol, E. Efficacy of Gabapentin for the Treatment of Alcohol Use Disorder in Patients With Alcohol Withdrawal Symptoms. JAMA Intern. Med. 2020, 180, 728. [Google Scholar] [CrossRef]
- Mariani, J.J.; Pavlicova, M.; Basaraba, C.; Mamczur-Fuller, A.; Brooks, D.J.; Bisaga, A.; Carpenter, K.M.; Nunes, E.V.; Levin, F.R. Pilot randomized placebo-controlled clinical trial of high-dose gabapentin for alcohol use disorder. Alcohol. Clin. Exp. Res. 2021, 45, 1639–1652. [Google Scholar] [CrossRef]
- Modesto-Lowe, V.; Barron, G.C.; Aronow, B.; Chaplin, M. Gabapentin for alcohol use disorder: A good option, or cause for concern? Clevel. Clin. J. Med. 2019, 86, 815–823. [Google Scholar] [CrossRef]
- Reus, V.I.; Fochtmann, L.J.; Bukstein, O.; Eyler, A.E.; Hilty, D.M.; Horvitz-Lennon, M.; Mahoney, J.; Pasic, J.; Weaver, M.; Wills, C.D.; et al. The American Psychiatric Association Practice Guideline for the Pharmacological Treatment of Patients With Alcohol Use Disorder. Am. J. Psychiatry 2018, 175, 86–90. [Google Scholar] [CrossRef]
- Rouhani, S.; Dall’Ava-Santucci, J.; Bajenaru, O.; Emmanouilidis, E.; Tran, G.; Manicom, R.; Dinh-Xuan, A.T.; Poenaru, S. Effects of muscimol or homotaurine on sleep-wake states in alcohol-dependent rats during withdrawal. Pharmacol. Biochem. Behav. 1998, 59, 955–960. [Google Scholar] [CrossRef]
- Paparrigopoulos, T.; Tzavellas, E.; Karaiskos, D.; Malitas, P.; Liappas, I. An open pilot study of tiagabine in alcohol dependence: Tolerability and clinical effects. J. Psychopharmacol. 2009, 24, 1375–1380. [Google Scholar] [CrossRef]
- Manthey, L.; Lohbeck, M.; Giltay, E.J.; van Veena, T.; Zitman, F.G.; Penninx, B.W. Correlates of benzodiazepine dependence in the N etherlands S tudy of D epression and A nxiety. Addiction 2012, 107, 2173–2182. [Google Scholar] [CrossRef]
- Twyman, R.E.; Rogers, C.J.; Macdonald, R.L. Differential regulation of γ-aminobutyric acid receptor channels by diazepam and phenobarbital. Ann. Neurol. 1989, 25, 213–220. [Google Scholar] [CrossRef]
- Bianchi, M.T.; Botzolakis, E.J.; Lagrange, A.H.; Macdonald, R.L. Benzodiazepine modulation of GABAA receptor opening frequency depends on activation context: A patch clamp and simulation study. Epilepsy Res. 2009, 85, 212–220. [Google Scholar] [CrossRef]
- Hanson, S.M.; Czajkowski, C. Structural mechanisms underlying benzodiazepine modulation of the GABAA receptor. J. Neurosci. 2008, 28, 3490–3499. [Google Scholar] [CrossRef]
- Petty, F.; Trivedi, M.H.; Fulton, M.; Rush, A.J. Benzodiazepines as antidepressants: Does GABA play a role in depression? Biol. Psychiatry 1995, 38, 578–591. [Google Scholar] [CrossRef]
- Lim, B.; Sproule, B.A.; Zahra, Z.; Sunderji, N.; Kennedy, S.H.; Rizvi, S.J. Understanding the effects of chronic benzodiazepine use in depression: A focus on neuropharmacology. Int. Clin. Psychopharmacol. 2020, 35, 243–253. [Google Scholar] [CrossRef]
- Lader, M. Benzodiazepines revisited—Will we ever learn? Addiction 2011, 106, 2086–2109. [Google Scholar] [CrossRef]
- Cutler, A.J.; Mattingly, G.W.; Maletic, V. Understanding the mechanism of action and clinical effects of neuroactive steroids and GABAergic compounds in major depressive disorder. Transl. Psychiatry 2023, 13, 228. [Google Scholar] [CrossRef]
- Carpenter, L.L.; Schecter, J.M.; Tyrka, A.R.; Mello, A.F.; Mello, M.F.; Haggarty, R.; Price, L.H. Open-label tiagabine monotherapy for major depressive disorder with anxiety. J. Clin. Psychiatry 2006, 67, 66–71. [Google Scholar] [CrossRef]
- Vecera, C.M.; Courtes, A.C.; Jones, G.; Soares, J.C.; Machado-Vieira, R. Pharmacotherapies Targeting GABA-Glutamate Neurotransmission for Treatment-Resistant Depression. Pharmaceuticals 2023, 16, 1572. [Google Scholar] [CrossRef]
- Gunduz-Bruce, H.; Silber, C.; Kaul, I.; Rothschild, A.J.; Riesenberg, R.; Sankoh, A.J.; Li, H.; Lasser, R.; Zorumski, C.F.; Rubinow, D.R.; et al. Trial of SAGE-217 in Patients with Major Depressive Disorder. New Engl. J. Med. 2019, 381, 903–911. [Google Scholar] [CrossRef]
- Epperson, C.N.; Rubinow, D.R.; Meltzer-Brody, S.; Deligiannidis, K.M.; Riesenberg, R.; Krystal, A.D.; Bankole, K.; Huang, M.-Y.; Li, H.; Brown, C.; et al. Effect of brexanolone on depressive symptoms, anxiety, and insomnia in women with postpartum depression: Pooled analyses from 3 double-blind, randomized, placebo-controlled clinical trials in the HUMMINGBIRD clinical program. J. Affect. Disord. 2023, 320, 353–359. [Google Scholar] [CrossRef]
- Evans, J.; Sun, Y.; McGregor, A.; Connor, B. Allopregnanolone regulates neurogenesis and depressive/anxiety-like behaviour in a social isolation rodent model of chronic stress. Neuropharmacology 2012, 63, 1315–1326. [Google Scholar] [CrossRef]
- Sałat, K.; Podkowa, A.; Kowalczyk, P.; Kulig, K.; Dziubina, A.; Filipek, B.; Librowski, T. Anticonvulsant active inhibitor of GABA transporter subtype 1, tiagabine, with activity in mouse models of anxiety, pain and depression. Pharmacol. Rep. 2015, 67, 465–472. [Google Scholar] [CrossRef]
- Kanes, S.J.; Colquhoun, H.; Doherty, J.; Raines, S.; Hoffmann, E.; Rubinow, D.R.; Meltzer-Brody, S. Open-label, proof-of-concept study of brexanolone in the treatment of severe postpartum depression. Hum. Psychopharmacol. 2017, 32, e2576. [Google Scholar] [CrossRef]
- Hughes, Z.; Scott, L.; Kahlig, K.; Wittmann, M. PRAX-114 is a Novel Extrasynaptic GABA-A Receptor Preferring Positive Allosteric Modulator With a Wide Separation Between a Translational Biomarker Signature Associated With Antidepressant-Like Activity, and Sedative Effects. Biol. Psychiatry 2021, 89, S204. [Google Scholar] [CrossRef]
- Scala, M.; Fanelli, G.; De Ronchi, D.; Serretti, A.; Fabbri, C. Clinical specificity profile for novel rapid acting antidepressant drugs. Int. Clin. Psychopharmacol. 2023, 38, 297–328. [Google Scholar] [CrossRef]
- Chowdhury, G.M.; Zhang, J.; Thomas, M.; Banasr, M.; Ma, X.; Pittman, B.; Bristow, L.; Schaeffer, E.; Duman, R.; Rothman, D. Transiently increased glutamate cycling in rat PFC is associated with rapid onset of antidepressant-like effects. Mol. Psychiatry 2017, 22, 120–126. [Google Scholar] [CrossRef]
- Evans, J.W.; Szczepanik, J.; Brutsché, N.; Park, L.T.; Nugent, A.C.; Zarate Jr, C.A. Default mode connectivity in major depressive disorder measured up to 10 days after ketamine administration. Biol. Psychiatry 2018, 84, 582–590. [Google Scholar] [CrossRef]
- Scheidegger, M.; Henning, A.; Walter, M.; Lehmann, M.; Kraehenmann, R.; Boeker, H.; Seifritz, E.; Grimm, S. Ketamine administration reduces amygdalo-hippocampal reactivity to emotional stimulation. Hum. Brain Mapp. 2016, 37, 1941–1952. [Google Scholar] [CrossRef]
- Scheidegger, M.; Walter, M.; Lehmann, M.; Metzger, C.; Grimm, S.; Boeker, H.; Boesiger, P.; Henning, A.; Seifritz, E. Ketamine decreases resting state functional network connectivity in healthy subjects: Implications for antidepressant drug action. PLoS ONE 2012, 7, e44799. [Google Scholar] [CrossRef]
- Milak, M.S.; Proper, C.J.; Mulhern, S.T.; Parter, A.L.; Kegeles, L.S.; Ogden, R.T.; Mao, X.; Rodriguez, C.I.; Oquendo, M.A.; Suckow, R.F.; et al. A pilot in vivo proton magnetic resonance spectroscopy study of amino acid neurotransmitter response to ketamine treatment of major depressive disorder. Mol. Psychiatry 2016, 21, 320–327. [Google Scholar] [CrossRef]
- Zarate, C.A.; Singh, J.B.; Carlson, P.J.; Brutsche, N.E.; Ameli, R.; Luckenbaugh, D.A.; Charney, D.S.; Manji, H.K. A Randomized Trial of an N-methyl-D-aspartate Antagonist in Treatment-Resistant Major Depression. Arch. General. Psychiatry 2006, 63, 856. [Google Scholar] [CrossRef]
- Zhang, K.; Hashimoto, K. An update on ketamine and its two enantiomers as rapid-acting antidepressants. Expert Rev. Neurother. 2019, 19, 83–92. [Google Scholar] [CrossRef]
- Hess, E.M.; Riggs, L.M.; Michaelides, M.; Gould, T.D. Mechanisms of ketamine and its metabolites as antidepressants. Biochem. Pharmacol. 2022, 197, 114892. [Google Scholar] [CrossRef]
- Hecking, J.; Davoudian, P.A.; Wilkinson, S.T. Emerging therapeutics based on the amino acid neurotransmitter system: An update on the pharmaceutical pipeline for mood disorders. Chronic Stress. 2021, 5, 24705470211020446. [Google Scholar] [CrossRef]
- Courtney, C.D.; Pamukcu, A.; Chan, C.S. Cell and circuit complexity of the external globus pallidus. Nat. Neurosci. 2023, 26, 1147–1159. [Google Scholar] [CrossRef]
- Lohoff, F.W. Targeting Unmet Clinical Needs in the Treatment of Alcohol Use Disorder. Front. Psychiatry 2022, 13, 767506. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ali, D.N.; Ali, H.M.; Lopez, M.R.; Kang, S.; Choi, D.-S. Astrocytic GABAergic Regulation in Alcohol Use and Major Depressive Disorders. Cells 2024, 13, 318. https://doi.org/10.3390/cells13040318
Ali DN, Ali HM, Lopez MR, Kang S, Choi D-S. Astrocytic GABAergic Regulation in Alcohol Use and Major Depressive Disorders. Cells. 2024; 13(4):318. https://doi.org/10.3390/cells13040318
Chicago/Turabian StyleAli, Dina N., Hossam M. Ali, Matthew R. Lopez, Shinwoo Kang, and Doo-Sup Choi. 2024. "Astrocytic GABAergic Regulation in Alcohol Use and Major Depressive Disorders" Cells 13, no. 4: 318. https://doi.org/10.3390/cells13040318