

Protein Quality Control Systems and ER Stress as Key Players in SARS-CoV-2-Induced Neurodegeneration
 , , and
, , and
Abstract
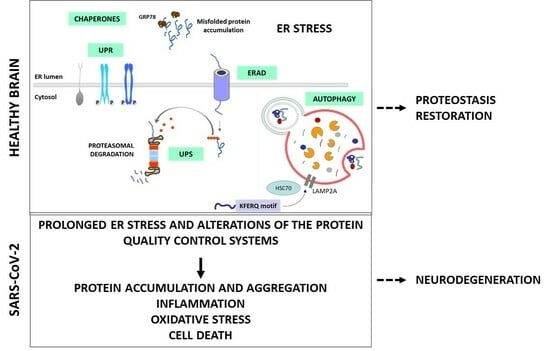
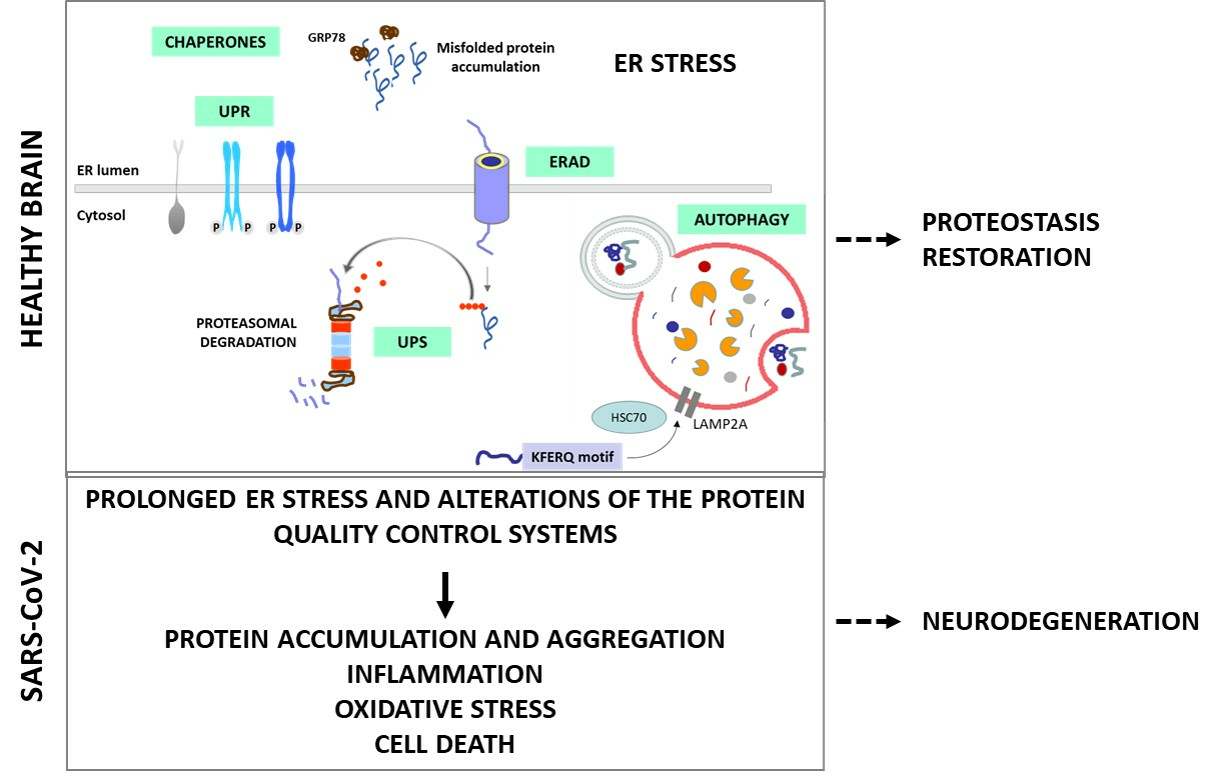
:
1. Introduction
- i)
- The critical role of protein quality control systems, such as the Unfolded Protein Response (UPR) induced by Endoplasmic Reticulum (ER) stress, the Endoplasmic Reticulum-Associated Degradation (ERAD) and the Ubiquitin-Proteasome System (UPS), the autophagic–lysosomal pathway and the molecular chaperones;
- ii)
- How these systems are manipulated during SARS-CoV-2 infection and potential therapeutic strategies targeting the viral manipulation of the protein quality control systems;
- iii)
- How ER stress and the manipulation of the protein quality control systems induced by SARS-CoV-2 in the central nervous system (CNS) could lead to neurodegeneration.
2. Role of Protein Quality Control Systems in SARS-CoV-2 Infection
2.1. ER Stress and UPR
- −
- IRE1 activation and splicing of XBP1 (X-box binding protein 1) mRNA, resulting in the production of sXBP1, an active transcription factor. sXBP1 regulates the expression of chaperones and ERAD components, reinforcing ER’s protein-folding and -degradation capacity. IRE1 also catalyzes the degradation of a large number of mRNAs and some pre-microRNAs (pre-miRNAs). This process is called regulated IRE1-dependent decay (RIDD) [23,24]. On the other hand, it is known that IRE1 is capable of forming high-order complexes in the ER membrane and interacting with a large number of proteins, among which the tumor necrosis factor (TNF) receptor-associated factor 2 (TRAF2) stands out. This interaction activates a cascade of signaling that leads to the activation of c-Jun N-terminal kinase (JNK), which, in turn, can inhibit some anti-apoptotic members of the BCL-2 family while activating pro-apoptotic proteins. Together, these two events lead to the oligomerization of BCL-2-like protein 4 (BAX) and BCL-2 antagonist/killer (BAK), initiating the apoptosis process [23,24,25];
- −
- PERK activation and the phosphorylation of the eukaryotic initiation factor 2α (eIF2α), a pivotal regulator of protein translation. Phosphorylated eIF2α reduces global protein synthesis, thereby alleviating the ER burden and allowing cells to cope with ER stress. However, this process can also induce apoptosis by upregulating the C/EBP homologous protein (CHOP). The prolonged upregulation of CHOP induces apoptosis through pathways involving the BCL2 binding component 3 (BBC3/PUMA) and the tribbles pseudokinase 3 (TRIB3).
- −
- ATF6 activation and translocation to the Golgi apparatus, where it is cleaved by site-1 (S1P) and site-2 (S2P) proteases, releasing a 50 kDa N-terminal fragment that translocates to the nucleus. This fragment acts as a transcription factor that subsequently upregulates genes encoding ER chaperones and other proteins involved in ER quality control.

2.1.1. ER Stress and UPR in Viral Infections

2.1.2. ER Stress and UPR in SARS-CoV-2 Infection
ER Stress/UPR-Related Antiviral Strategies against SARS-CoV-2

ER Stress/UPR-Related SARS-CoV-2 Strategies

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Key Components | SARS-CoV-2 Strategies | References | |
|---|---|---|---|
| ER Stress and UPR | ATF6 | Activated by SARS-CoV-2 ORF8 protein. | [36,40,41] |
| IRE1α | Contributes to viral replication. Activation in dDCNs. Activated by SARS-CoV-2 ORF8 protein. | [36,38,39,40,41] | |
| PERK | Activation plays a key role in SARS-CoV-2 infection in dDCNs. Reduced viral replication upon pharmacological inhibition. | [39,40,44] | |
| GRP78 PDIA4 CHOP | Upregulated by SARS-CoV-2 ORF8 protein. | [36,40,41] |
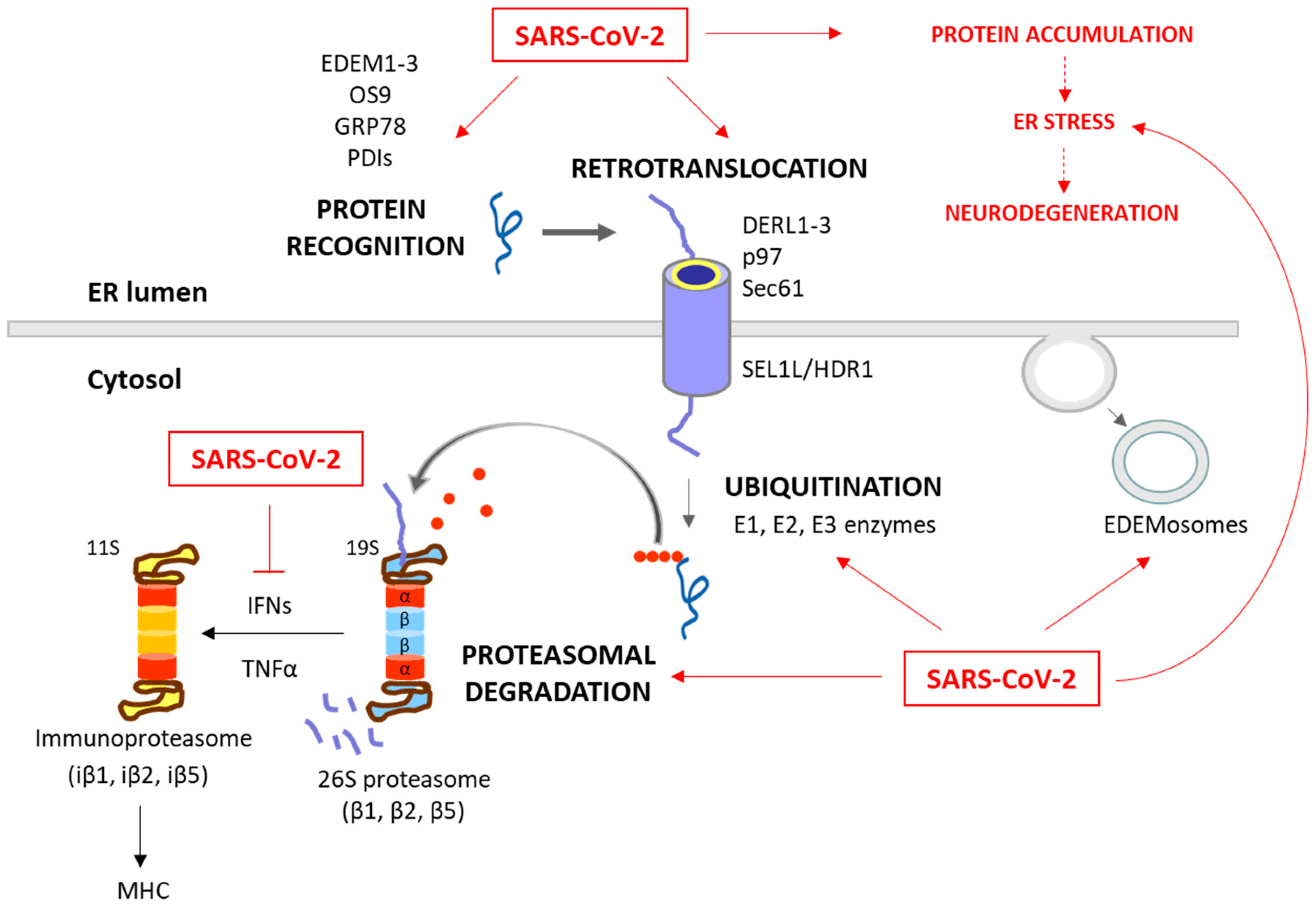
2.2. ERAD and UPS
2.2.1. ERAD and UPS in Viral Infections
- −
- −
- Proteasome activation. Modulating proteasome activity promotes the clearance of viral components within infected cells. Both 26S proteasome and immunoproteasome functions appear to be important for a variety of host responses to viral infection, degrading viral proteins and promoting the antigen presentation of viral particles [81,87];
- −
- −
- −
- Degradation of host proteins. Some viruses encode specific proteins that target host proteins for degradation. For example, in the case of Human cytomegalovirus (HCMV), the viral proteins US2 and US11 induce the degradation of major histocompatibility complex (MHC) class I molecules through ERAD [94]. By eliminating MHC class I molecules, which are crucial for immune recognition, infected cells can avoid detection and destruction by the immune system. Besides, the Human Immunodeficiency Virus (HIV), through the viral protein U (Vpu), targets the host’s cluster of differentiation 4 (CD4) protein for degradation [93]. By using the ERAD pathway, Vpu facilitates the degradation of CD4, the receptor for HIV entry, thereby reducing the number of available receptors on the cell surface. This downregulation of CD4 is beneficial for the virus, as it prevents superinfection (multiple viruses infecting the same cell);
- −
- Modulation of the expression or activity of ERAD/UPS-related proteins. Viruses can modulate the expression or activity of ERAD-related proteins to favor their replication and assembly processes. For instance, the ERAD pathway can reduce the amount of virus envelope proteins in order to control the level of virus particles, and thus facilitates chronic infections, as observed in Hepatitis B Virus (HBV) [28,95]. Zhou et al. (2022) recently published a comprehensive review on the topic of how viruses utilize the ERAD pathway to regulate their replication and propagation [92];
- −
- Manipulation of ER membrane dynamics. Viruses can also induce alterations in ER membrane dynamics, creating a specialized membrane structures that support viral replication. This is the case of the Japanese encephalitis virus (JEV), which modifies ERAD by confiscating EDEMosomes. These vesicles segregate ERAD factors such as EDEM1, OS9, and SEL1L from the ER lumen, which fall under the control of the virus [59];
- −
- Manipulation of ubiquitination. This viral strategy includes several possibilities—
- Ubiquitination of viral proteins as tools for assembly and entry. This post-translational modification serves as a molecular tag, facilitating the assembly and budding of new viral particles [82,96]. This ubiquitin-mediated process is crucial for the completion of the viral life cycle. Notably, this modification can also enhance virus–host interactions, promoting virus entry, replication, and pathogenesis [97,98,99];
2.2.2. SARS-CoV-2 and ERAD/UPS Interaction
ERAD/UPS-Related SARS-CoV-2 Strategies
| Key Components | SARS-CoV-2 Strategies | References | |
|---|---|---|---|
| ERAD and UPS | Ubiquitination | Facilitates viral replication of SARS-CoV-2 proteins. | [105] |
| E3 Ubiquitin Ligases and DUBs | Identified as potential antiviral targets. Utilized by the virus for invasion, replication, escape, and inflammation. | [68,105] | |
| Ubiquitin Variants (UbVs) | Block SARS-CoV-2 PLpro activity, offering insights for drug development. | [58,107] | |
| Heat Treatment | Promotes ubiquitin-mediated proteolysis of SARS-CoV-2 RNA polymerase (NSP12) through E3 ubiquitin ligase ZNF598. | [101] | |
| DUBs Inhibitors | Potential therapeutic targets against SARS-CoV-2 infection. | [107,109,110,111] | |
| Proteasome Inhibitors | MG132, epoxomycin, and bortezomib negatively impact SARS-CoV-2 replication. | [106,107] |
2.3. Autophagy
2.3.1. Autophagy in Viral Infections
- −
- The selective autophagic degradation of viral components. Autophagy can selectively target viral components, such as viral proteins and nucleic acids, for degradation. For example, selective autophagy has been observed to degrade the capsid protein of the Sindbis virus (SIN) in the CNS [133] or the p62 targeted Dengue virus (DENV) capsid protein [134]. Picornaviruses, such as poliovirus, are recognized by galectin 8, which limits viral infection by triggering the autophagic degradation of the viral RNA genome [135]. Activated antiviral autophagy in Drosophila melanogaster restricts ZIKV infection in the brain [136]. This targeted degradation hampers viral replication and restricts the spread of infection;
- −
- A role in antigen presentation and adaptive immunity. Autophagy plays a crucial role in antigen presentation. Viral antigens captured by autophagosomes can be processed and presented on MHC molecules, leading to the activation of virus-specific T cells [137,138]. This process enhances the adaptive immune response against viral infections.
- −
- A role in the innate immune response. There is an interplay between interferons and autophagy. Various antiviral signaling pathways, such as the IFN pathway, can induce autophagy in response to viral infections. This induction of autophagy is often linked to the restriction of viral replication [130,139]. At the same time, autophagy can promote interferon production, cooperating with pattern recognition receptor signaling [130,132].
- −
- Initiation of autophagy. Measles virus (MV) induces autophagy via CD46-Cyt-1, facilitating viral entry [141], and the HCV triggers autophagy through ER stress and reactive oxygen species (ROS), crucial for replication [142]. Other viruses can inhibit autophagy initiation, like HSV-1, which employs the infected cell protein 34.5 (ICP34.5) to interact with BECN1, impairing autophagy initiation [143]. Also, the HCMV utilizes BCL-2 and the viral proteins IRS1 and TRS1 to disrupt BECN1 function, hindering autophagosome formation [144]. Similarly, the Kaposi’s sarcoma-associated herpesvirus (KSHV) expresses a virally encoded G protein-coupled receptor (vGPCR), which activates the mammalian target of rapamycin (mTOR) pathway and downregulates ATG14L expression, leading to the inhibition of autophagy initiation [145];
- −
- −
- −
2.3.2. Autophagy and SARS-CoV-2
Proviral Role of Autophagy in SARS-CoV-2 Infection
| Key Components | SARS-CoV-2 Strategies | References | ||
|---|---|---|---|---|
| Autophagy | Early Autophagy Steps | NSP3, NSP6, NSP15, ORF3a, ORF8 | Block phagophore and autophagosome formation from the ER, but positive regulatory effect on autophagosome formation in general; smaller autophagosomes (related to DMVs). | [156,157,158,163,164,165,166,167,168] |
| Later Autophagy Steps | NSP3, NSP6, ORF3a, ORF7a, M, E | Block autophagosome–lysosome fusion and autolysosome acidification and degradation. | [123,156,157,169,170,171,172] | |
| Autophagy and Immune Evasion | ORF10, ORF8 | Impair mitophagy; downregulation of MHC-I molecules; inhibition of cGAS-STING-mediated autophagy flux. | [157,161,173,174,175,176,177] | |
| UPS and Autophagy Interaction | M protein | K63-ubiquitination of viral proteins for autophagic degradation. | [178,179] | |
| Autophagy Inhibition | ATG5, ATG7, BECN1, FIP200 | Suppression reduces SARS-CoV-2 replication. | [150] | |
2.4. Molecular Chaperones
2.4.1. Molecular Chaperones in Viral Infections
2.4.2. Molecular Chaperones and SARS-CoV-2 Infection
| Key Components | SARS-CoV-2 Strategies | References | |
|---|---|---|---|
| Molecular Chaperones | HSP90 | Essential for SARS-CoV-2 life cycle. HSP90 facilitates the correct folding and assembly of viral proteins, ensuring the proper formation of functional viral particles. | [193,224] |
| HSP90 inhibition | 17-AAG, 17-DMAG and Luminespib suppress SARS-CoV-2 replication. Reduction in pro-inflammatory cytokines modulating the host immune response during SARS-CoV-2 infection. | ||
| HSP70 | Crucial for SARS-CoV-2 gene expression, assists in capsid assembly. | [225] | |
| GRP78 | Upregulated during SARS-CoV-2 infection, acts as a pro-viral protein. | [45,227,228,229,230,231] |
3. Cell-Specific Effects of SARS-CoV-2 in the Brain
4. Link between SARS-CoV-2, ER Stress and Neurodegeneration
5. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Jackson, C.B.; Farzan, M.; Chen, B.; Choe, H. Mechanisms of SARS-CoV-2 entry into cells. Nat. Rev. Mol. Cell Biol. 2022, 23, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Kakavandi, S.; Zare, I.; VaezJalali, M.; Dadashi, M.; Azarian, M.; Akbari, A.; Ramezani Farani, M.; Zalpoor, H.; Hajikhani, B. Structural and non-structural proteins in SARS-CoV-2: Potential aspects to COVID-19 treatment or prevention of progression of related diseases. Cell Commun. Signal. 2023, 21, 110. [Google Scholar] [CrossRef] [PubMed]
- Benton, D.J.; Wrobel, A.G.; Xu, P.; Roustan, C.; Martin, S.R.; Rosenthal, P.B.; Skehel, J.J.; Gamblin, S.J. Receptor binding and priming of the spike protein of SARS-CoV-2 for membrane fusion. Nature 2020, 588, 327–330. [Google Scholar] [CrossRef]
- Barnes, C.O.; Jette, C.A.; Abernathy, M.E.; Dam, K.A.; Esswein, S.R.; Gristick, H.B.; Malyutin, A.G.; Sharaf, N.G.; Huey-Tubman, K.E.; Lee, Y.E.; et al. SARS-CoV-2 neutralizing antibody structures inform therapeutic strategies. Nature 2020, 588, 682–687. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Rao, Z. Structural biology of SARS-CoV-2 and implications for therapeutic development. Nat. Rev. Microbiol. 2021, 19, 685–700. [Google Scholar] [CrossRef] [PubMed]
- Harapan, B.N.; Yoo, H.J. Neurological symptoms, manifestations, and complications associated with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and coronavirus disease 19 (COVID-19). J. Neurol. 2021, 268, 3059–3071. [Google Scholar] [CrossRef] [PubMed]
- Maiese, A.; Manetti, A.C.; Bosetti, C.; Del Duca, F.; La Russa, R.; Frati, P.; Di Paolo, M.; Turillazzi, E.; Fineschi, V. SARS-CoV-2 and the brain: A review of the current knowledge on neuropathology in COVID-19. Brain Pathol. 2021, 31, e13013. [Google Scholar] [CrossRef]
- Rogers-Brown, J.S.; Wanga, V.; Okoro, C.; Brozowsky, D.; Evans, A.; Hopwood, D.; Cope, J.R.; Jackson, B.R.; Bushman, D.; Hernandez-Romieu, A.C.; et al. Outcomes Among Patients Referred to Outpatient Rehabilitation Clinics After COVID-19 diagnosis—United States, January 2020–March 2021. MMWR Morb. Mortal. Wkly. Rep. 2021, 70, 967–971. [Google Scholar] [CrossRef]
- Jha, N.K.; Ojha, S.; Jha, S.K.; Dureja, H.; Singh, S.K.; Shukla, S.D.; Chellappan, D.K.; Gupta, G.; Bhardwaj, S.; Kumar, N.; et al. Evidence of Coronavirus (CoV) Pathogenesis and Emerging Pathogen SARS-CoV-2 in the Nervous System: A Review on Neurological Impairments and Manifestations. J. Mol. Neurosci. 2021, 71, 2192–2209. [Google Scholar] [CrossRef]
- Villa, C.; Rivellini, E.; Lavitrano, M.; Combi, R. Can SARS-CoV-2 Infection Exacerbate Alzheimer’s Disease? An Overview of Shared Risk Factors and Pathogenetic Mechanisms. J. Pers. Med. 2022, 12, 29. [Google Scholar] [CrossRef] [PubMed]
- Boura, I.; Qamar, M.A.; Daddoveri, F.; Leta, V.; Poplawska-Domaszewicz, K.; Falup-Pecurariu, C.; Ray Chaudhuri, K. SARS-CoV-2 and Parkinson’s Disease: A Review of Where We Are Now. Biomedicines 2023, 11, 2524. [Google Scholar] [CrossRef]
- Al-Kuraishy, H.M.; Al-Gareeb, A.I.; Kaushik, A.; Kujawska, M.; Ahmed, E.A.; Batiha, G.E. SARS-CoV-2 infection and Parkinson’s disease: Possible links and perspectives. J. Neurosci. Res. 2023, 101, 952–975. [Google Scholar] [CrossRef] [PubMed]
- Ruano, D. Proteostasis Dysfunction in Aged Mammalian Cells. The Stressful Role of Inflammation. Front. Mol. Biosci. 2021, 8, 658742. [Google Scholar] [CrossRef]
- Ghemrawi, R.; Khair, M. Endoplasmic Reticulum Stress and Unfolded Protein Response in Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 6127. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Saxena, S. ER stress and the unfolded protein response in neurodegeneration. Nat. Rev. Neurol. 2017, 13, 477–491. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.S.; Yu, W.S.; Lim, L.W. Exploring ER stress response in cellular aging and neuroinflammation in Alzheimer’s disease. Ageing Res. Rev. 2021, 70, 101417. [Google Scholar] [CrossRef]
- Cirone, M. ER Stress, UPR Activation and the Inflammatory Response to Viral Infection. Viruses 2021, 13, 798. [Google Scholar] [CrossRef] [PubMed]
- Gavilan, M.P.; Pintado, C.; Gavilan, E.; Jimenez, S.; Rios, R.M.; Vitorica, J.; Castano, A.; Ruano, D. Dysfunction of the unfolded protein response increases neurodegeneration in aged rat hippocampus following proteasome inhibition. Aging Cell 2009, 8, 654–665. [Google Scholar] [CrossRef]
- Suaya, M.; Sanchez, G.M.; Vila, A.; Amante, A.; Cotarelo, M.; Garcia Carrillo, M.; Blaustein, M. Live and let die: Signaling AKTivation and UPRegulation dynamics in SARS-CoVs infection and cancer. Cell Death Dis. 2022, 13, 846. [Google Scholar] [CrossRef]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Gomora-Garcia, J.C.; Geronimo-Olvera, C.; Perez-Martinez, X.; Massieu, L. IRE1α RIDD activity induced under ER stress drives neuronal death by the degradation of 14-3-3 θ mRNA in cortical neurons during glucose deprivation. Cell Death Discov. 2021, 7, 131. [Google Scholar] [CrossRef] [PubMed]
- Tsuru, A.; Imai, Y.; Saito, M.; Kohno, K. Novel mechanism of enhancing IRE1α-XBP1 signalling via the PERK-ATF4 pathway. Sci. Rep. 2016, 6, 24217. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Bernasconi, P.; Fisher, J.; Lee, A.H.; Bassik, M.C.; Antonsson, B.; Brandt, G.S.; Iwakoshi, N.N.; Schinzel, A.; Glimcher, L.H.; et al. Proapoptotic BAX and BAK modulate the unfolded protein response by a direct interaction with IRE1α. Science 2006, 312, 572–576. [Google Scholar] [CrossRef]
- Inoue, T.; Tsai, B. How viruses use the endoplasmic reticulum for entry, replication, and assembly. Cold Spring Harb. Perspect. Biol. 2013, 5, a013250. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.A.; Song, C.H. Insights into the Role of Endoplasmic Reticulum Stress in Infectious Diseases. Front. Immunol. 2019, 10, 3147. [Google Scholar] [CrossRef]
- Li, S.; Kong, L.; Yu, X. The expanding roles of endoplasmic reticulum stress in virus replication and pathogenesis. Crit. Rev. Microbiol. 2015, 41, 150–164. [Google Scholar] [CrossRef]
- Prasad, V.; Greber, U.F. The endoplasmic reticulum unfolded protein response—Homeostasis, cell death and evolution in virus infections. FEMS Microbiol. Rev. 2021, 45, fuab016. [Google Scholar] [CrossRef]
- Santerre, M.; Arjona, S.P.; Allen, C.N.; Shcherbik, N.; Sawaya, B.E. Why do SARS-CoV-2 NSPs rush to the ER? J. Neurol. 2021, 268, 2013–2022. [Google Scholar] [CrossRef]
- Catanzaro, N.; Meng, X.J. Induction of the unfolded protein response (UPR) suppresses porcine reproductive and respiratory syndrome virus (PRRSV) replication. Virus Res. 2020, 276, 197820. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, A. Virus-induced ER stress and the unfolded protein response. Front. Plant Sci. 2012, 3, 293. [Google Scholar] [CrossRef]
- Rios-Ocampo, W.A.; Navas, M.C.; Buist-Homan, M.; Faber, K.N.; Daemen, T.; Moshage, H. Hepatitis C Virus Proteins Core and NS5A Are Highly Sensitive to Oxidative Stress-Induced Degradation after eIF2α/ATF4 Pathway Activation. Viruses 2020, 12, 425. [Google Scholar] [CrossRef] [PubMed]
- Kolpikova, E.P.; Tronco, A.R.; Hartigh, A.B.D.; Jackson, K.J.; Iwawaki, T.; Fink, S.L. IRE1α Promotes Zika Virus Infection via XBP1. Viruses 2020, 12, 278. [Google Scholar] [CrossRef]
- Johnston, B.P.; McCormick, C. Herpesviruses and the Unfolded Protein Response. Viruses 2019, 12, 17. [Google Scholar] [CrossRef] [PubMed]
- Fang, P.; Tian, L.; Zhang, H.; Xia, S.; Ding, T.; Zhu, X.; Zhang, J.; Ren, J.; Fang, L.; Xiao, S. Induction and modulation of the unfolded protein response during porcine deltacoronavirus infection. Vet. Microbiol. 2022, 271, 109494. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Czinn, S.J.; Reiter, R.J.; Blanchard, T.G. Crosstalk between endoplasmic reticulum stress and anti-viral activities: A novel therapeutic target for COVID-19. Life Sci. 2020, 255, 117842. [Google Scholar] [CrossRef] [PubMed]
- Yiang, G.T.; Wu, C.C.; Lu, C.L.; Hu, W.C.; Tsai, Y.J.; Huang, Y.M.; Su, W.L.; Lu, K.C. Endoplasmic Reticulum Stress in Elderly Patients with COVID-19: Potential of Melatonin Treatment. Viruses 2023, 15, 156. [Google Scholar] [CrossRef]
- Wang, X.; Wang, W.; Wang, T.; Wang, J.; Jiang, Y.; Wang, X.; Qiu, Z.; Feng, N.; Sun, W.; Li, C.; et al. SARS-CoV-2 ORF8 Protein Induces Endoplasmic Reticulum Stress-like Responses and Facilitates Virus Replication by Triggering Calnexin: An Unbiased Study. J. Virol. 2023, 97, e0001123. [Google Scholar] [CrossRef]
- Liu, X.; Wen, Y.Z.; Huang, Z.L.; Shen, X.; Wang, J.H.; Luo, Y.H.; Chen, W.X.; Lun, Z.R.; Li, H.B.; Qu, L.H.; et al. SARS-CoV-2 causes a significant stress response mediated by small RNAs in the blood of COVID-19 patients. Mol. Ther. Nucleic Acids 2022, 27, 751–762. [Google Scholar] [CrossRef]
- Bartolini, D.; Stabile, A.M.; Vacca, C.; Pistilli, A.; Rende, M.; Gioiello, A.; Cruciani, G.; Galli, F. Endoplasmic reticulum stress and NF-kB activation in SARS-CoV-2 infected cells and their response to antiviral therapy. IUBMB Life 2022, 74, 93–100. [Google Scholar] [CrossRef]
- Chaudhry, Z.L.; Gamal, M.; Ferhati, I.; Warda, M.; Ahmed, B.Y. ER Stress in COVID-19 and Parkinson’s Disease: In Vitro and In Silico Evidences. Brain Sci. 2022, 12, 507. [Google Scholar] [CrossRef]
- Echavarria-Consuegra, L.; Cook, G.M.; Busnadiego, I.; Lefevre, C.; Keep, S.; Brown, K.; Doyle, N.; Dowgier, G.; Franaszek, K.; Moore, N.A.; et al. Manipulation of the unfolded protein response: A pharmacological strategy against coronavirus infection. PLoS Pathog. 2021, 17, e1009644. [Google Scholar] [CrossRef] [PubMed]
- Rashid, F.; Dzakah, E.E.; Wang, H.; Tang, S. The ORF8 protein of SARS-CoV-2 induced endoplasmic reticulum stress and mediated immune evasion by antagonizing production of interferon beta. Virus Res. 2021, 296, 198350. [Google Scholar] [CrossRef]
- Su, W.Q.; Yu, X.J.; Zhou, C.M. SARS-CoV-2 ORF3a Induces Incomplete Autophagy via the Unfolded Protein Response. Viruses 2021, 13, 2467. [Google Scholar] [CrossRef] [PubMed]
- Xue, M.; Feng, L. The Role of Unfolded Protein Response in Coronavirus Infection and Its Implications for Drug Design. Front. Microbiol. 2021, 12, 808593. [Google Scholar] [CrossRef] [PubMed]
- Rozpedek-Kaminska, W.; Siwecka, N.; Wawrzynkiewicz, A.; Wojtczak, R.; Pytel, D.; Diehl, J.A.; Majsterek, I. The PERK-Dependent Molecular Mechanisms as a Novel Therapeutic Target for Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 2108. [Google Scholar] [CrossRef] [PubMed]
- Shaban, M.S.; Muller, C.; Mayr-Buro, C.; Weiser, H.; Meier-Soelch, J.; Albert, B.V.; Weber, A.; Linne, U.; Hain, T.; Babayev, I.; et al. Multi-level inhibition of coronavirus replication by chemical ER stress. Nat. Commun. 2021, 12, 5536. [Google Scholar] [CrossRef] [PubMed]
- Vembar, S.S.; Brodsky, J.L. One step at a time: Endoplasmic reticulum-associated degradation. Nat. Rev. Mol. Cell Biol. 2008, 9, 944–957. [Google Scholar] [CrossRef]
- Olivari, S.; Molinari, M. Glycoprotein folding and the role of EDEM1, EDEM2 and EDEM3 in degradation of folding-defective glycoproteins. FEBS Lett. 2007, 581, 3658–3664. [Google Scholar] [CrossRef]
- Morito, D.; Nagata, K. Pathogenic Hijacking of ER-Associated Degradation: Is ERAD Flexible? Mol. Cell 2015, 59, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.; Hong, E.H.; Ahn, J.H.; Cho, J.; Jeong, J.H.; Kim, C.W.; Yoon, B.I.; Koo, J.H.; Park, Y.Y.; Yang, Y.M.; et al. ERdj5 protects goblet cells from endoplasmic reticulum stress-mediated apoptosis under inflammatory conditions. Exp. Mol. Med. 2023, 55, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Lopata, A.; Kniss, A.; Lohr, F.; Rogov, V.V.; Dotsch, V. Ubiquitination in the ERAD Process. Int. J. Mol. Sci. 2020, 21, 5369. [Google Scholar] [CrossRef] [PubMed]
- Ohtake, F.; Saeki, Y.; Ishido, S.; Kanno, J.; Tanaka, K. The K48-K63 Branched Ubiquitin Chain Regulates NF-κB Signaling. Mol. Cell 2016, 64, 251–266. [Google Scholar] [CrossRef] [PubMed]
- Collins, G.A.; Goldberg, A.L. The Logic of the 26S Proteasome. Cell 2017, 169, 792–806. [Google Scholar] [CrossRef] [PubMed]
- Trulsson, F.; Akimov, V.; Robu, M.; van Overbeek, N.; Berrocal, D.A.P.; Shah, R.G.; Cox, J.; Shah, G.M.; Blagoev, B.; Vertegaal, A.C.O. Deubiquitinating enzymes and the proteasome regulate preferential sets of ubiquitin substrates. Nat. Commun. 2022, 13, 2736. [Google Scholar] [CrossRef] [PubMed]
- Paz Gavilan, M.; Vela, J.; Castano, A.; Ramos, B.; del Rio, J.C.; Vitorica, J.; Ruano, D. Cellular environment facilitates protein accumulation in aged rat hippocampus. Neurobiol. Aging 2006, 27, 973–982. [Google Scholar] [CrossRef] [PubMed]
- Movaqar, A.; Yaghoubi, A.; Rezaee, S.R.; Jamehdar, S.A.; Soleimanpour, S. Coronaviruses construct an interconnection way with ERAD and autophagy. Future Microbiol. 2021, 16, 1135–1151. [Google Scholar] [CrossRef]
- Noack, J.; Bernasconi, R.; Molinari, M. How viruses hijack the ERAD tuning machinery. J. Virol. 2014, 88, 10272–10275. [Google Scholar] [CrossRef]
- Rao, G.; Croft, B.; Teng, C.; Awasthi, V. Ubiquitin-Proteasome System in Neurodegenerative Disorders. J. Drug Metab. Toxicol. 2015, 6, 187. [Google Scholar] [CrossRef]
- Dantuma, N.P.; Bott, L.C. The ubiquitin-proteasome system in neurodegenerative diseases: Precipitating factor, yet part of the solution. Front. Mol. Neurosci. 2014, 7, 70. [Google Scholar] [CrossRef]
- Schmidt, M.F.; Gan, Z.Y.; Komander, D.; Dewson, G. Ubiquitin signalling in neurodegeneration: Mechanisms and therapeutic opportunities. Cell Death Differ. 2021, 28, 570–590. [Google Scholar] [CrossRef]
- van Vliet, V.J.E.; Huynh, N.; Pala, J.; Patel, A.; Singer, A.; Slater, C.; Chung, J.; van Huizen, M.; Teyra, J.; Miersch, S.; et al. Ubiquitin variants potently inhibit SARS-CoV-2 PLpro and viral replication via a novel site distal to the protease active site. PLoS Pathog. 2022, 18, e1011065. [Google Scholar] [CrossRef] [PubMed]
- Camborde, L.; Planchais, S.; Tournier, V.; Jakubiec, A.; Drugeon, G.; Lacassagne, E.; Pflieger, S.; Chenon, M.; Jupin, I. The ubiquitin-proteasome system regulates the accumulation of Turnip yellow mosaic virus RNA-dependent RNA polymerase during viral infection. Plant Cell 2010, 22, 3142–3152. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Mar, K.B.; Sari, L.; Gaszek, I.K.; Cheng, Q.; Evers, B.M.; Shelton, J.M.; Wight-Carter, M.; Siegwart, D.J.; Lin, M.M.; et al. TRIM7 inhibits enterovirus replication and promotes emergence of a viral variant with increased pathogenicity. Cell 2021, 184, 3410–3425.e3417. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Wu, P.; Chen, H.; Li, G. Pleiotropic roles of the ubiquitin-proteasome system during viral propagation. Life Sci. 2018, 207, 350–354. [Google Scholar] [CrossRef] [PubMed]
- Kong, F.; You, H.; Kong, D.; Zheng, K.; Tang, R. The interaction of hepatitis B virus with the ubiquitin proteasome system in viral replication and associated pathogenesis. Virol. J. 2019, 16, 73. [Google Scholar] [CrossRef] [PubMed]
- Choi, A.G.; Wong, J.; Marchant, D.; Luo, H. The ubiquitin-proteasome system in positive-strand RNA virus infection. Rev. Med. Virol. 2013, 23, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Hao, P.; Zhao, Z.; Gao, W.; Huan, C.; Li, L.; Chen, X.; Wang, H.; Jin, N.; Luo, Z.Q.; et al. The E3 ligase RNF5 restricts SARS-CoV-2 replication by targeting its envelope protein for degradation. Signal Transduct. Target. Ther. 2023, 8, 53. [Google Scholar] [CrossRef]
- Chaudhary, P.; Proulx, J.; Park, I.W. Ubiquitin-protein ligase E3A (UBE3A) mediation of viral infection and human diseases. Virus Res. 2023, 335, 199191. [Google Scholar] [CrossRef]
- Rojas, V.K.; Park, I.W. Role of the Ubiquitin Proteasome System (UPS) in the HIV-1 Life Cycle. Int. J. Mol. Sci. 2019, 20, 2984. [Google Scholar] [CrossRef] [PubMed]
- Voss, M.; Braun, V.; Bredow, C.; Kloetzel, P.M.; Beling, A. Coxsackievirus B3 Exploits the Ubiquitin-Proteasome System to Facilitate Viral Replication. Viruses 2021, 13, 1360. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Zhang, M.; Yang, Z.; Zhou, Z.; Huang, J.; Zhao, B. Role of E3 ubiquitin ligases and deubiquitinating enzymes in SARS-CoV-2 infection. Front. Cell. Infect. Microbiol. 2023, 13, 1217383. [Google Scholar] [CrossRef] [PubMed]
- Zu, S.; Li, C.; Li, L.; Deng, Y.Q.; Chen, X.; Luo, D.; Ye, Q.; Huang, Y.J.; Li, X.F.; Zhang, R.R.; et al. TRIM22 suppresses Zika virus replication by targeting NS1 and NS3 for proteasomal degradation. Cell Biosci. 2022, 12, 139. [Google Scholar] [CrossRef] [PubMed]
- Le-Trilling, V.T.K.; Trilling, M. Ub to no good: How cytomegaloviruses exploit the ubiquitin proteasome system. Virus Res. 2020, 281, 197938. [Google Scholar] [CrossRef] [PubMed]
- Han, K.; Zhao, D.; Liu, Y.; Liu, Q.; Huang, X.; Yang, J.; Zhang, L.; Li, Y. The ubiquitin-proteasome system is necessary for the replication of duck Tembusu virus. Microb. Pathog. 2019, 132, 362–368. [Google Scholar] [CrossRef] [PubMed]
- Pang, Y.; Li, M.; Zhou, Y.; Liu, W.; Tao, R.; Zhang, H.; Xiao, S.; Fang, L. The ubiquitin proteasome system is necessary for efficient proliferation of porcine reproductive and respiratory syndrome virus. Vet. Microbiol. 2021, 253, 108947. [Google Scholar] [CrossRef]
- Seyoum Tola, F. The Role of Ubiquitin-Proteasome System in the Pathogenesis of Severe Acute Respiratory Syndrome Coronavirus-2 Disease. Int. J. Inflam. 2023, 2023, 6698069. [Google Scholar] [CrossRef]
- Gao, W.; Wang, L.; Ju, X.; Zhao, S.; Li, Z.; Su, M.; Xu, J.; Wang, P.; Ding, Q.; Lv, G.; et al. The Deubiquitinase USP29 Promotes SARS-CoV-2 Virulence by Preventing Proteasome Degradation of ORF9b. mBio 2022, 13, e0130022. [Google Scholar] [CrossRef]
- Ming, S.L.; Zhang, S.; Wang, Q.; Zeng, L.; Zhou, L.Y.; Wang, M.D.; Ma, Y.X.; Han, L.Q.; Zhong, K.; Zhu, H.S.; et al. Inhibition of USP14 influences alphaherpesvirus proliferation by degrading viral VP16 protein via ER stress-triggered selective autophagy. Autophagy 2022, 18, 1801–1821. [Google Scholar] [CrossRef]
- Schneider, S.M.; Lee, B.H.; Nicola, A.V. Viral entry and the ubiquitin-proteasome system. Cell Microbiol. 2021, 23, e13276. [Google Scholar] [CrossRef]
- Luo, H. Interplay between the virus and the ubiquitin-proteasome system: Molecular mechanism of viral pathogenesis. Curr. Opin. Virol. 2016, 17, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Dubiella, U.; Serrano, I. The Ubiquitin Proteasome System as a Double Agent in Plant-Virus Interactions. Plants 2021, 10, 928. [Google Scholar] [CrossRef] [PubMed]
- Alcaide-Loridan, C.; Jupin, I. Ubiquitin and plant viruses, let’s play together! Plant Physiol. 2012, 160, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.; Luo, H. The ubiquitin-proteasome pathway in viral infections. Can. J. Physiol. Pharmacol. 2006, 84, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Isaacson, M.K.; Ploegh, H.L. Ubiquitination, ubiquitin-like modifiers, and deubiquitination in viral infection. Cell Host Microbe 2009, 5, 559–570. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, M.K.; Weinberg, J.B. The immunoproteasome and viral infection: A complex regulator of inflammation. Front. Microbiol. 2015, 6, 21. [Google Scholar] [CrossRef] [PubMed]
- Cetin, G.; Klafack, S.; Studencka-Turski, M.; Kruger, E.; Ebstein, F. The Ubiquitin-Proteasome System in Immune Cells. Biomolecules 2021, 11, 60. [Google Scholar] [CrossRef] [PubMed]
- Bhat, S.A.; Vasi, Z.; Adhikari, R.; Gudur, A.; Ali, A.; Jiang, L.; Ferguson, R.; Liang, D.; Kuchay, S. Ubiquitin proteasome system in immune regulation and therapeutics. Curr. Opin. Pharmacol. 2022, 67, 102310. [Google Scholar] [CrossRef]
- Katze, M.G.; He, Y.; Gale, M., Jr. Viruses and interferon: A fight for supremacy. Nat. Rev. Immunol. 2002, 2, 675–687. [Google Scholar] [CrossRef]
- Dalskov, L.; Gad, H.H.; Hartmann, R. Viral recognition and the antiviral interferon response. EMBO J. 2023, 42, e112907. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.; Wang, X.; Zhao, F.; Wu, K.; Li, X.; Li, Z.; Li, Y.; Chen, W.; Zeng, S.; Liu, X.; et al. Viruses Hijack ERAD to Regulate Their Replication and Propagation. Int. J. Mol. Sci. 2022, 23, 9398. [Google Scholar] [CrossRef] [PubMed]
- Magadan, J.G.; Perez-Victoria, F.J.; Sougrat, R.; Ye, Y.; Strebel, K.; Bonifacino, J.S. Multilayered mechanism of CD4 downregulation by HIV-1 Vpu involving distinct ER retention and ERAD targeting steps. PLoS Pathog. 2010, 6, e1000869. [Google Scholar] [CrossRef] [PubMed]
- van den Boomen, D.J.; Lehner, P.J. Identifying the ERAD ubiquitin E3 ligases for viral and cellular targeting of MHC class I. Mol. Immunol. 2015, 68, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Lazar, C.; Macovei, A.; Petrescu, S.; Branza-Nichita, N. Activation of ERAD pathway by human hepatitis B virus modulates viral and subviral particle production. PLoS ONE 2012, 7, e34169. [Google Scholar] [CrossRef] [PubMed]
- Park, E.S.; Dezhbord, M.; Lee, A.R.; Kim, K.H. The Roles of Ubiquitination in Pathogenesis of Influenza Virus Infection. Int. J. Mol. Sci. 2022, 23, 4593. [Google Scholar] [CrossRef] [PubMed]
- Cohen, F.S. How Viruses Invade Cells. Biophys. J. 2016, 110, 1028–1032. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, C.; Dybas, J.M.; Liddle, J.C.; Price, A.M.; Hayer, K.E.; Lauman, R.; Purman, C.E.; Charman, M.; Kim, E.T.; Garcia, B.A.; et al. Adenovirus-mediated ubiquitination alters protein-RNA binding and aids viral RNA processing. Nat. Microbiol. 2020, 5, 1217–1231. [Google Scholar] [CrossRef] [PubMed]
- Cai, D.; Liu, L.; Tian, B.; Fu, X.; Yang, Q.; Chen, J.; Zhang, Y.; Fang, J.; Shen, L.; Wang, Y.; et al. Dual-Role Ubiquitination Regulation Shuttling the Entire Life Cycle of the Flaviviridae. Front. Microbiol. 2022, 13, 835344. [Google Scholar] [CrossRef]
- Valerdi, K.M.; Hage, A.; van Tol, S.; Rajsbaum, R.; Giraldo, M.I. The Role of the Host Ubiquitin System in Promoting Replication of Emergent Viruses. Viruses 2021, 13, 369. [Google Scholar] [CrossRef]
- Imbert, F.; Langford, D. Viruses, SUMO, and immunity: The interplay between viruses and the host SUMOylation system. J. Neurovirol. 2021, 27, 531–541. [Google Scholar] [CrossRef]
- Zhang, Q.; Jia, Q.; Gao, W.; Zhang, W. The Role of Deubiquitinases in Virus Replication and Host Innate Immune Response. Front. Microbiol. 2022, 13, 839624. [Google Scholar] [CrossRef]
- Bailey-Elkin, B.A.; Knaap, R.C.M.; Kikkert, M.; Mark, B.L. Structure and Function of Viral Deubiquitinating Enzymes. J. Mol. Biol. 2017, 429, 3441–3470. [Google Scholar] [CrossRef] [PubMed]
- Raaben, M.; Posthuma, C.C.; Verheije, M.H.; te Lintelo, E.G.; Kikkert, M.; Drijfhout, J.W.; Snijder, E.J.; Rottier, P.J.; de Haan, C.A. The ubiquitin-proteasome system plays an important role during various stages of the coronavirus infection cycle. J. Virol. 2010, 84, 7869–7879. [Google Scholar] [CrossRef] [PubMed]
- Che, Y.; Jiang, D.; Zhang, Y.; Zhang, J.; Xu, T.; Sun, Y.; Fan, J.; Wang, J.; Chang, N.; Wu, Y.; et al. Elevated ubiquitination contributes to protective immunity against severe SARS-CoV-2 infection. Clin. Transl. Med. 2022, 12, e1103. [Google Scholar] [CrossRef]
- Maimaitiyiming, Y.; Yang, T.; Wang, Q.Q.; Feng, Y.; Chen, Z.; Bjorklund, M.; Wang, F.; Hu, C.; Hsu, C.H.; Naranmandura, H. Heat Treatment Promotes Ubiquitin-Mediated Proteolysis of SARS-CoV-2 RNA Polymerase and Decreases Viral Load. Research 2022, 2022, 9802969. [Google Scholar] [CrossRef] [PubMed]
- Baskol, G.; Ozel, M.; Saracoglu, H.; Ulger, B.; Kalin Unuvar, G.; Onuk, S.; Bayram, A.; Karayol Akin, A.; Muhtaroglu, S.; Sagiroglu, P.; et al. New Avenues to Explore in SARS-CoV-2 Infection: Both TRIM25 and TRIM56 Positively Correlate with VEGF, GAS6, and sAXL in COVID-19 Patients. Viral Immunol. 2022, 35, 690–699. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Afolabi, L.O.; Wan, X.; Shim, J.S.; Chen, L. TRIM family proteins: Roles in proteostasis and neurodegenerative diseases. Open Biol. 2022, 12, 220098. [Google Scholar] [CrossRef] [PubMed]
- Vanderboom, P.M.; Mun, D.G.; Madugundu, A.K.; Mangalaparthi, K.K.; Saraswat, M.; Garapati, K.; Chakraborty, R.; Ebihara, H.; Sun, J.; Pandey, A. Proteomic Signature of Host Response to SARS-CoV-2 Infection in the Nasopharynx. Mol. Cell. Proteom. 2021, 20, 100134. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Wu, Y.; Xiao, T.; Qi, F.; Fan, L.; Zhang, S.; Zhou, J.; He, Y.; Gao, X.; Zeng, H.; et al. Multiomics approach reveals the ubiquitination-specific processes hijacked by SARS-CoV-2. Signal Transduct. Target. Ther. 2022, 7, 312. [Google Scholar] [CrossRef]
- Longhitano, L.; Tibullo, D.; Giallongo, C.; Lazzarino, G.; Tartaglia, N.; Galimberti, S.; Li Volti, G.; Palumbo, G.A.; Liso, A. Proteasome Inhibitors as a Possible Therapy for SARS-CoV-2. Int. J. Mol. Sci. 2020, 21, 3622. [Google Scholar] [CrossRef] [PubMed]
- Grosse, M.; Setz, C.; Rauch, P.; Auth, J.; Morokutti-Kurz, M.; Temchura, V.; Schubert, U. Inhibitors of Deubiquitinating Enzymes Interfere with the SARS-CoV-2 Papain-like Protease and Block Virus Replication In Vitro. Viruses 2022, 14, 1404. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Zhao, Y.G.; Zhang, H. Endomembrane remodeling in SARS-CoV-2 infection. Cell Insight 2022, 1, 100031. [Google Scholar] [CrossRef] [PubMed]
- Clemente, V.; D’Arcy, P.; Bazzaro, M. Deubiquitinating Enzymes in Coronaviruses and Possible Therapeutic Opportunities for COVID-19. Int. J. Mol. Sci. 2020, 21, 3492. [Google Scholar] [CrossRef] [PubMed]
- Freitas, B.T.; Durie, I.A.; Murray, J.; Longo, J.E.; Miller, H.C.; Crich, D.; Hogan, R.J.; Tripp, R.A.; Pegan, S.D. Characterization and Noncovalent Inhibition of the Deubiquitinase and deISGylase Activity of SARS-CoV-2 Papain-Like Protease. ACS Infect. Dis. 2020, 6, 2099–2109. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, A.; Narwal, M.; Majowicz, S.A.; Varricchio, C.; Toner, S.A.; Ballatore, C.; Brancale, A.; Murakami, K.S.; Jose, J. Identification of SARS-CoV-2 inhibitors targeting Mpro and PLpro using in-cell-protease assay. Commun. Biol. 2022, 5, 169. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Levine, B. Autophagy in mammalian development and differentiation. Nat. Cell Biol. 2010, 12, 823–830. [Google Scholar] [CrossRef] [PubMed]
- Parzych, K.R.; Klionsky, D.J. An overview of autophagy: Morphology, mechanism, and regulation. Antioxid. Redox Signal. 2014, 20, 460–473. [Google Scholar] [CrossRef]
- Levine, B.; Kroemer, G. SnapShot: Macroautophagy. Cell 2008, 132, 162.e1–162.e3. [Google Scholar] [CrossRef]
- Li, W.W.; Li, J.; Bao, J.K. Microautophagy: Lesser-known self-eating. Cell. Mol. Life Sci. 2012, 69, 1125–1136. [Google Scholar] [CrossRef]
- Bejarano, E.; Cuervo, A.M. Chaperone-mediated autophagy. Proc. Am. Thorac. Soc. 2010, 7, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; He, D.; Yao, Z.; Klionsky, D.J. The machinery of macroautophagy. Cell Res. 2014, 24, 24–41. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Klionsky, D.J.; Shen, H.M. The emerging mechanisms and functions of microautophagy. Nat. Rev. Mol. Cell Biol. 2023, 24, 186–203. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Cuervo, A.M. The coming of age of chaperone-mediated autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 365–381. [Google Scholar] [CrossRef] [PubMed]
- Gavilan, E.; Pintado, C.; Gavilan, M.P.; Daza, P.; Sanchez-Aguayo, I.; Castano, A.; Ruano, D. Age-related dysfunctions of the autophagy lysosomal pathway in hippocampal pyramidal neurons under proteasome stress. Neurobiol. Aging 2015, 36, 1953–1963. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Klionsky, D.J. Autophagy and neurodegeneration. ACS Chem. Biol. 2006, 1, 211–213. [Google Scholar] [CrossRef] [PubMed]
- Aman, Y.; Schmauck-Medina, T.; Hansen, M.; Morimoto, R.I.; Simon, A.K.; Bjedov, I.; Palikaras, K.; Simonsen, A.; Johansen, T.; Tavernarakis, N.; et al. Autophagy in healthy aging and disease. Nat. Aging 2021, 1, 634–650. [Google Scholar] [CrossRef]
- Chen, T.; Tu, S.; Ding, L.; Jin, M.; Chen, H.; Zhou, H. The role of autophagy in viral infections. J. Biomed. Sci. 2023, 30, 5. [Google Scholar] [CrossRef]
- Mao, J.; Lin, E.; He, L.; Yu, J.; Tan, P.; Zhou, Y. Autophagy and Viral Infection. Adv. Exp. Med. Biol. 2019, 1209, 55–78. [Google Scholar] [CrossRef]
- Choi, Y.; Bowman, J.W.; Jung, J.U. Autophagy during viral infection—A double-edged sword. Nat. Rev. Microbiol. 2018, 16, 341–354. [Google Scholar] [CrossRef]
- Rubinsztein, D.C.; Codogno, P.; Levine, B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat. Rev. Drug Discov. 2012, 11, 709–730. [Google Scholar] [CrossRef]
- Liang, S.; Wu, Y.S.; Li, D.Y.; Tang, J.X.; Liu, H.F. Autophagy in Viral Infection and Pathogenesis. Front. Cell Dev. Biol. 2021, 9, 766142. [Google Scholar] [CrossRef]
- Orvedahl, A.; MacPherson, S.; Sumpter, R., Jr.; Talloczy, Z.; Zou, Z.; Levine, B. Autophagy protects against Sindbis virus infection of the central nervous system. Cell Host Microbe 2010, 7, 115–127. [Google Scholar] [CrossRef]
- Wu, Y.; Zhou, T.; Hu, J.; Liu, Y.; Jin, S.; Wu, J.; Guan, X.; Cui, J. Autophagy Activation Induces p62-Dependent Autophagic Degradation of Dengue Virus Capsid Protein During Infection. Front. Microbiol. 2022, 13, 889693. [Google Scholar] [CrossRef] [PubMed]
- Staring, J.; von Castelmur, E.; Blomen, V.A.; van den Hengel, L.G.; Brockmann, M.; Baggen, J.; Thibaut, H.J.; Nieuwenhuis, J.; Janssen, H.; van Kuppeveld, F.J.; et al. PLA2G16 represents a switch between entry and clearance of Picornaviridae. Nature 2017, 541, 412–416. [Google Scholar] [CrossRef] [PubMed]
- Delorme-Axford, E.; Klionsky, D.J. Inflammatory-dependent Sting activation induces antiviral autophagy to limit zika virus in the Drosophila brain. Autophagy 2019, 15, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Oynebraten, I. Involvement of autophagy in MHC class I antigen presentation. Scand. J. Immunol. 2020, 92, e12978. [Google Scholar] [CrossRef] [PubMed]
- Munz, C. Autophagy Beyond Intracellular MHC Class II Antigen Presentation. Trends Immunol. 2016, 37, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Wang, M.L.; Zhao, J. Crosstalk between Autophagy and Type I Interferon Responses in Innate Antiviral Immunity. Viruses 2019, 11, 132. [Google Scholar] [CrossRef] [PubMed]
- Chawla, K.; Subramanian, G.; Rahman, T.; Fan, S.; Chakravarty, S.; Gujja, S.; Demchak, H.; Chakravarti, R.; Chattopadhyay, S. Autophagy in Virus Infection: A Race between Host Immune Response and Viral Antagonism. Immuno 2022, 2, 153–169. [Google Scholar] [CrossRef] [PubMed]
- Joubert, P.E.; Meiffren, G.; Gregoire, I.P.; Pontini, G.; Richetta, C.; Flacher, M.; Azocar, O.; Vidalain, P.O.; Vidal, M.; Lotteau, V.; et al. Autophagy induction by the pathogen receptor CD46. Cell Host Microbe 2009, 6, 354–366. [Google Scholar] [CrossRef] [PubMed]
- Gregoire, I.P.; Richetta, C.; Meyniel-Schicklin, L.; Borel, S.; Pradezynski, F.; Diaz, O.; Deloire, A.; Azocar, O.; Baguet, J.; Le Breton, M.; et al. IRGM is a common target of RNA viruses that subvert the autophagy network. PLoS Pathog. 2011, 7, e1002422. [Google Scholar] [CrossRef] [PubMed]
- Orvedahl, A.; Alexander, D.; Talloczy, Z.; Sun, Q.; Wei, Y.; Zhang, W.; Burns, D.; Leib, D.A.; Levine, B. HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe 2007, 1, 23–35. [Google Scholar] [CrossRef]
- Mouna, L.; Hernandez, E.; Bonte, D.; Brost, R.; Amazit, L.; Delgui, L.R.; Brune, W.; Geballe, A.P.; Beau, I.; Esclatine, A. Analysis of the role of autophagy inhibition by two complementary human cytomegalovirus BECN1/Beclin 1-binding proteins. Autophagy 2016, 12, 327–342. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, A.P.; Damania, B. AKTivation of PI3K/AKT/mTOR signaling pathway by KSHV. Front. Immunol. 2012, 3, 401. [Google Scholar] [CrossRef] [PubMed]
- Prentice, E.; Jerome, W.G.; Yoshimori, T.; Mizushima, N.; Denison, M.R. Coronavirus replication complex formation utilizes components of cellular autophagy. J. Biol. Chem. 2004, 279, 10136–10141. [Google Scholar] [CrossRef]
- Paul, D.; Madan, V.; Ramirez, O.; Bencun, M.; Stoeck, I.K.; Jirasko, V.; Bartenschlager, R. Glycine Zipper Motifs in Hepatitis C Virus Nonstructural Protein 4B Are Required for the Establishment of Viral Replication Organelles. J. Virol. 2018, 92, 10–1128. [Google Scholar] [CrossRef]
- Gannage, M.; Dormann, D.; Albrecht, R.; Dengjel, J.; Torossi, T.; Ramer, P.C.; Lee, M.; Strowig, T.; Arrey, F.; Conenello, G.; et al. Matrix protein 2 of influenza A virus blocks autophagosome fusion with lysosomes. Cell Host Microbe 2009, 6, 367–380. [Google Scholar] [CrossRef]
- Ding, B.; Zhang, G.; Yang, X.; Zhang, S.; Chen, L.; Yan, Q.; Xu, M.; Banerjee, A.K.; Chen, M. Phosphoprotein of human parainfluenza virus type 3 blocks autophagosome-lysosome fusion to increase virus production. Cell Host Microbe 2014, 15, 564–577. [Google Scholar] [CrossRef]
- Wu, Y.W.; Mettling, C.; Wu, S.R.; Yu, C.Y.; Perng, G.C.; Lin, Y.S.; Lin, Y.L. Autophagy-associated dengue vesicles promote viral transmission avoiding antibody neutralization. Sci. Rep. 2016, 6, 32243. [Google Scholar] [CrossRef]
- Chen, Y.H.; Du, W.; Hagemeijer, M.C.; Takvorian, P.M.; Pau, C.; Cali, A.; Brantner, C.A.; Stempinski, E.S.; Connelly, P.S.; Ma, H.C.; et al. Phosphatidylserine vesicles enable efficient en bloc transmission of enteroviruses. Cell 2015, 160, 619–630. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; He, X.; Ma, L.; Zhuang, Z.; Wang, Y.; Lin, M.; Cai, S.; Wei, L.; Wang, Z.; Zhao, Z.; et al. Suppression of ACE2 SUMOylation protects against SARS-CoV-2 infection through TOLLIP-mediated selective autophagy. Nat. Commun. 2022, 13, 5204. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Li, J.; Wang, P.H.; Yang, N.; Huang, J.; Ou, J.; Xu, T.; Zhao, X.; Liu, T.; Huang, X.; et al. SARS-CoV-2 spike promotes inflammation and apoptosis through autophagy by ROS-suppressed PI3K/AKT/mTOR signaling. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166260. [Google Scholar] [CrossRef] [PubMed]
- Silva, R.; Travassos, L.H. Autophagy: A self-guard against SARS-CoV-2. Med. Hypotheses 2022, 169, 110984. [Google Scholar] [CrossRef] [PubMed]
- Gassen, N.C.; Papies, J.; Bajaj, T.; Emanuel, J.; Dethloff, F.; Chua, R.L.; Trimpert, J.; Heinemann, N.; Niemeyer, C.; Weege, F.; et al. SARS-CoV-2-mediated dysregulation of metabolism and autophagy uncovers host-targeting antivirals. Nat. Commun. 2021, 12, 3818. [Google Scholar] [CrossRef]
- Rayner, J.O.; Roberts, R.A.; Kim, J.; Poklepovic, A.; Roberts, J.L.; Booth, L.; Dent, P. AR12 (OSU-03012) suppresses GRP78 expression and inhibits SARS-CoV-2 replication. Biochem. Pharmacol. 2020, 182, 114227. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Ferretti, M.; Ying, B.; Descamps, H.; Lee, E.; Dittmar, M.; Lee, J.S.; Whig, K.; Kamalia, B.; Dohnalova, L.; et al. Pharmacological activation of STING blocks SARS-CoV-2 infection. Sci. Immunol. 2021, 6, eabi9007. [Google Scholar] [CrossRef]
- Tomic, S.; Dokic, J.; Stevanovic, D.; Ilic, N.; Gruden-Movsesijan, A.; Dinic, M.; Radojevic, D.; Bekic, M.; Mitrovic, N.; Tomasevic, R.; et al. Reduced Expression of Autophagy Markers and Expansion of Myeloid-Derived Suppressor Cells Correlate with Poor T Cell Response in Severe COVID-19 Patients. Front. Immunol. 2021, 12, 614599. [Google Scholar] [CrossRef]
- Shang, C.; Zhuang, X.; Zhang, H.; Li, Y.; Zhu, Y.; Lu, J.; Ge, C.; Cong, J.; Li, T.; Li, N.; et al. Inhibition of Autophagy Suppresses SARS-CoV-2 Replication and Ameliorates Pneumonia in hACE2 Transgenic Mice and Xenografted Human Lung Tissues. J. Virol. 2021, 95, e0153721. [Google Scholar] [CrossRef]
- Verma, R.; Saha, S.; Kumar, S.; Mani, S.; Maiti, T.K.; Surjit, M. RNA-Protein Interaction Analysis of SARS-CoV-2 5’ and 3’ Untranslated Regions Reveals a Role of Lysosome-Associated Membrane Protein-2a during Viral Infection. mSystems 2021, 6, e0064321. [Google Scholar] [CrossRef]
- Zhou, H.; Hu, Z.; Castro-Gonzalez, S. Bidirectional interplay between SARS-CoV-2 and autophagy. mBio 2023, 14, e0102023. [Google Scholar] [CrossRef]
- Ivanova, T.; Mariienko, Y.; Mehterov, N.; Kazakova, M.; Sbirkov, Y.; Todorova, K.; Hayrabedyan, S.; Sarafian, V. Autophagy and SARS-CoV-2-Old Players in New Games. Int. J. Mol. Sci. 2023, 24, 7734. [Google Scholar] [CrossRef]
- Tan, X.; Cai, K.; Li, J.; Yuan, Z.; Chen, R.; Xiao, H.; Xu, C.; Hu, B.; Qin, Y.; Ding, B. Coronavirus subverts ER-phagy by hijacking FAM134B and ATL3 into p62 condensates to facilitate viral replication. Cell Rep. 2023, 42, 112286. [Google Scholar] [CrossRef] [PubMed]
- Samimi, N.; Farjam, M.; Klionsky, D.J.; Rezaei, N. The role of autophagy in the pathogenesis of SARS-CoV-2 infection in different cell types. Autophagy 2022, 18, 1728–1731. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sun, H.; Pei, R.; Mao, B.; Zhao, Z.; Li, H.; Lin, Y.; Lu, K. The SARS-CoV-2 protein ORF3a inhibits fusion of autophagosomes with lysosomes. Cell Discov. 2021, 7, 31. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, Y.; Li, Y.; Huang, F.; Luo, B.; Yuan, Y.; Xia, B.; Ma, X.; Yang, T.; Yu, F.; et al. The ORF8 protein of SARS-CoV-2 mediates immune evasion through down-regulating MHC-Iota. Proc. Natl. Acad. Sci. USA 2021, 118, e2024202118. [Google Scholar] [CrossRef]
- Sargazi, S.; Sheervalilou, R.; Rokni, M.; Shirvaliloo, M.; Shahraki, O.; Rezaei, N. The role of autophagy in controlling SARS-CoV-2 infection: An overview on virophagy-mediated molecular drug targets. Cell Biol. Int. 2021, 45, 1599–1612. [Google Scholar] [CrossRef] [PubMed]
- Hayn, M.; Hirschenberger, M.; Koepke, L.; Nchioua, R.; Straub, J.H.; Klute, S.; Hunszinger, V.; Zech, F.; Prelli Bozzo, C.; Aftab, W.; et al. Systematic functional analysis of SARS-CoV-2 proteins uncovers viral innate immune antagonists and remaining vulnerabilities. Cell Rep. 2021, 35, 109126. [Google Scholar] [CrossRef]
- Zou, D.; Xu, J.; Duan, X.; Xu, X.; Li, P.; Cheng, L.; Zheng, L.; Li, X.; Zhang, Y.; Wang, X.; et al. Porcine epidemic diarrhea virus ORF3 protein causes endoplasmic reticulum stress to facilitate autophagy. Vet. Microbiol. 2019, 235, 209–219. [Google Scholar] [CrossRef]
- Mohamud, Y.; Xue, Y.C.; Liu, H.; Ng, C.S.; Bahreyni, A.; Jan, E.; Luo, H. The papain-like protease of coronaviruses cleaves ULK1 to disrupt host autophagy. Biochem. Biophys. Res. Commun. 2021, 540, 75–82. [Google Scholar] [CrossRef]
- Benvenuto, D.; Angeletti, S.; Giovanetti, M.; Bianchi, M.; Pascarella, S.; Cauda, R.; Ciccozzi, M.; Cassone, A. Evolutionary analysis of SARS-CoV-2: How mutation of Non-Structural Protein 6 (NSP6) could affect viral autophagy. J. Infect. 2020, 81, e24–e27. [Google Scholar] [CrossRef]
- Yang, N.; Shen, H.M. Targeting the Endocytic Pathway and Autophagy Process as a Novel Therapeutic Strategy in COVID-19. Int. J. Biol. Sci. 2020, 16, 1724–1731. [Google Scholar] [CrossRef]
- Liang, H.; Luo, D.; Liao, H.; Li, S. Coronavirus Usurps the Autophagy-Lysosome Pathway and Induces Membranes Rearrangement for Infection and Pathogenesis. Front. Microbiol. 2022, 13, 846543. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.; Wang, X.; Zhu, Y.; Wang, W.; Wang, Y.; Hu, G.; Liu, C.; Li, J.; Ren, S.; Xiao, M.Z.X.; et al. ORF3a-Mediated Incomplete Autophagy Facilitates Severe Acute Respiratory Syndrome Coronavirus-2 Replication. Front. Cell Dev. Biol. 2021, 9, 716208. [Google Scholar] [CrossRef] [PubMed]
- Koepke, L.; Hirschenberger, M.; Hayn, M.; Kirchhoff, F.; Sparrer, K.M. Manipulation of autophagy by SARS-CoV-2 proteins. Autophagy 2021, 17, 2659–2661. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Luo, X.; Ren, M. Clearance or Hijack: Universal Interplay Mechanisms Between Viruses and Host Autophagy from Plants to Animals. Front. Cell. Infect. Microbiol. 2021, 11, 786348. [Google Scholar] [CrossRef] [PubMed]
- Hou, P.; Wang, X.; Wang, H.; Wang, T.; Yu, Z.; Xu, C.; Zhao, Y.; Wang, W.; Zhao, Y.; Chu, F.; et al. The ORF7a protein of SARS-CoV-2 initiates autophagy and limits autophagosome-lysosome fusion via degradation of SNAP29 to promote virus replication. Autophagy 2023, 19, 551–569. [Google Scholar] [CrossRef]
- Li, X.; Hou, P.; Ma, W.; Wang, X.; Wang, H.; Yu, Z.; Chang, H.; Wang, T.; Jin, S.; Wang, X.; et al. SARS-CoV-2 ORF10 suppresses the antiviral innate immune response by degrading MAVS through mitophagy. Cell. Mol. Immunol. 2022, 19, 67–78. [Google Scholar] [CrossRef]
- Ouyang, L.; Gong, J. Mitochondrial-targeted ubiquinone: A potential treatment for COVID-19. Med. Hypotheses 2020, 144, 110161. [Google Scholar] [CrossRef] [PubMed]
- Domizio, J.D.; Gulen, M.F.; Saidoune, F.; Thacker, V.V.; Yatim, A.; Sharma, K.; Nass, T.; Guenova, E.; Schaller, M.; Conrad, C.; et al. The cGAS-STING pathway drives type I IFN immunopathology in COVID-19. Nature 2022, 603, 145–151. [Google Scholar] [CrossRef]
- Su, J.; Shen, S.; Hu, Y.; Chen, S.; Cheng, L.; Cai, Y.; Wei, W.; Wang, Y.; Rui, Y.; Yu, X.F. SARS-CoV-2 ORF3a inhibits cGAS-STING-mediated autophagy flux and antiviral function. J. Med. Virol. 2023, 95, e28175. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Zheng, Y.; Deng, J.; Nan, M.L.; Xiao, Y.; Zhuang, M.W.; Zhang, J.; Wang, W.; Gao, C.; Wang, P.H. SARS-CoV-2 ORF10 antagonizes STING-dependent interferon activation and autophagy. J. Med. Virol. 2022, 94, 5174–5188. [Google Scholar] [CrossRef] [PubMed]
- Madiraju, C.; Novack, J.P.; Reed, J.C.; Matsuzawa, S.I. K63 ubiquitination in immune signaling. Trends Immunol. 2022, 43, 148–162. [Google Scholar] [CrossRef] [PubMed]
- Rui, Y.; Su, J.; Shen, S.; Hu, Y.; Huang, D.; Zheng, W.; Lou, M.; Shi, Y.; Wang, M.; Chen, S.; et al. Unique and complementary suppression of cGAS-STING and RNA sensing- triggered innate immune responses by SARS-CoV-2 proteins. Signal Transduct. Target. Ther. 2021, 6, 123. [Google Scholar] [CrossRef] [PubMed]
- Hartl, F.U.; Bracher, A.; Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature 2011, 475, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Adriaenssens, E.; Asselbergh, B.; Rivera-Mejias, P.; Bervoets, S.; Vendredy, L.; De Winter, V.; Spaas, K.; de Rycke, R.; van Isterdael, G.; Impens, F.; et al. Small heat shock proteins operate as molecular chaperones in the mitochondrial intermembrane space. Nat. Cell Biol. 2023, 25, 467–480. [Google Scholar] [CrossRef] [PubMed]
- Bobori, C.; Theocharopoulou, G.; Vlamos, P. Molecular Chaperones in Neurodegenerative Diseases: A Short Review. Adv. Exp. Med. Biol. 2017, 987, 219–231. [Google Scholar] [CrossRef]
- Morimoto, R.I. Proteotoxic stress and inducible chaperone networks in neurodegenerative disease and aging. Genes. Dev. 2008, 22, 1427–1438. [Google Scholar] [CrossRef]
- Witt, S.N. Molecular chaperones, alpha-synuclein, and neurodegeneration. Mol. Neurobiol. 2013, 47, 552–560. [Google Scholar] [CrossRef]
- Franklin, T.B.; Krueger-Naug, A.M.; Clarke, D.B.; Arrigo, A.P.; Currie, R.W. The role of heat shock proteins Hsp70 and Hsp27 in cellular protection of the central nervous system. Int. J. Hyperth. 2005, 21, 379–392. [Google Scholar] [CrossRef]
- Bohush, A.; Bieganowski, P.; Filipek, A. Hsp90 and Its Co-Chaperones in Neurodegenerative Diseases. Int. J. Mol. Sci. 2019, 20, 4976. [Google Scholar] [CrossRef]
- Hooper, P.L.; Hightower, L.E.; Hooper, P.L. Loss of stress response as a consequence of viral infection: Implications for disease and therapy. Cell Stress Chaperones 2012, 17, 647–655. [Google Scholar] [CrossRef] [PubMed]
- Paladino, L.; Vitale, A.M.; Caruso Bavisotto, C.; Conway de Macario, E.; Cappello, F.; Macario, A.J.L.; Gammazza, A.M. The Role of Molecular Chaperones in Virus Infection and Implications for Understanding and Treating COVID-19. J. Clin. Med. 2020, 9, 3518. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Belshan, M.; Ratner, L. Hsp40 facilitates nuclear import of the human immunodeficiency virus type 2 Vpx-mediated preintegration complex. J. Virol. 2008, 82, 1229–1237. [Google Scholar] [CrossRef] [PubMed]
- Oglesbee, M.J.; Pratt, M.; Carsillo, T. Role for heat shock proteins in the immune response to measles virus infection. Viral Immunol. 2002, 15, 399–416. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Kanai, F.; Kawakami, T.; Tateishi, K.; Ijichi, H.; Kawabe, T.; Arakawa, Y.; Kawakami, T.; Nishimura, T.; Shirakata, Y.; et al. Interaction of the hepatitis B virus X protein (HBx) with heat shock protein 60 enhances HBx-mediated apoptosis. Biochem. Biophys. Res. Commun. 2004, 318, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.M.; Kim, S.J.; Kim, J.H.; Lee, W.; Kim, G.W.; Lee, K.H.; Choi, K.Y.; Oh, J.W. Interaction of hepatitis C virus core protein with Hsp60 triggers the production of reactive oxygen species and enhances TNF-α-mediated apoptosis. Cancer Lett. 2009, 279, 230–237. [Google Scholar] [CrossRef]
- Zhang, X.; Yu, W. Heat shock proteins and viral infection. Front. Immunol. 2022, 13, 947789. [Google Scholar] [CrossRef]
- Wang, Z.; Li, Y.; Yang, X.; Zhao, J.; Cheng, Y.; Wang, J. Mechanism and Complex Roles of HSC70 in Viral Infections. Front. Microbiol. 2020, 11, 1577. [Google Scholar] [CrossRef]
- Hauser, H.; Shen, L.; Gu, Q.L.; Krueger, S.; Chen, S.Y. Secretory heat-shock protein as a dendritic cell-targeting molecule: A new strategy to enhance the potency of genetic vaccines. Gene Ther. 2004, 11, 924–932. [Google Scholar] [CrossRef]
- Takashima, K.; Oshiumi, H.; Matsumoto, M.; Seya, T. DNAJB1/HSP40 Suppresses Melanoma Differentiation-Associated Gene 5-Mitochondrial Antiviral Signaling Protein Function in Conjunction with HSP70. J. Innate Immun. 2018, 10, 44–55. [Google Scholar] [CrossRef]
- Sun, M.; Yu, Z.; Ma, J.; Pan, Z.; Lu, C.; Yao, H. Down-regulating heat shock protein 27 is involved in porcine epidemic diarrhea virus escaping from host antiviral mechanism. Vet. Microbiol. 2017, 205, 6–13. [Google Scholar] [CrossRef] [PubMed]
- Le, Y.; Jia, P.; Jin, Y.; Liu, W.; Jia, K.; Yi, M. The antiviral role of heat shock protein 27 against red spotted grouper nervous necrosis virus infection in sea perch. Fish. Shellfish. Immunol. 2017, 70, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Rawat, P.; Khan, S.Z.; Dhamija, N.; Chaudhary, P.; Ravi, D.S.; Mitra, D. Reciprocal regulation of human immunodeficiency virus-1 gene expression and replication by heat shock proteins 40 and 70. J. Mol. Biol. 2011, 410, 944–958. [Google Scholar] [CrossRef] [PubMed]
- Batra, J.; Tripathi, S.; Kumar, A.; Katz, J.M.; Cox, N.J.; Lal, R.B.; Sambhara, S.; Lal, S.K. Human Heat shock protein 40 (Hsp40/DnaJB1) promotes influenza A virus replication by assisting nuclear import of viral ribonucleoproteins. Sci. Rep. 2016, 6, 19063. [Google Scholar] [CrossRef] [PubMed]
- Park, S.G.; Lee, S.M.; Jung, G. Antisense oligodeoxynucleotides targeted against molecular chaperonin Hsp60 block human hepatitis B virus replication. J. Biol. Chem. 2003, 278, 39851–39857. [Google Scholar] [CrossRef]
- Wan, Q.; Song, D.; Li, H.; He, M.L. Stress proteins: The biological functions in virus infection, present and challenges for target-based antiviral drug development. Signal Transduct. Target. Ther. 2020, 5, 125. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Del Valle, J.; Chavez-Salinas, S.; Medina, F.; Del Angel, R.M. Heat shock protein 90 and heat shock protein 70 are components of dengue virus receptor complex in human cells. J. Virol. 2005, 79, 4557–4567. [Google Scholar] [CrossRef] [PubMed]
- Thongtan, T.; Wikan, N.; Wintachai, P.; Rattanarungsan, C.; Srisomsap, C.; Cheepsunthorn, P.; Smith, D.R. Characterization of putative Japanese encephalitis virus receptor molecules on microglial cells. J. Med. Virol. 2012, 84, 615–623. [Google Scholar] [CrossRef]
- Lubkowska, A.; Pluta, W.; Stronska, A.; Lalko, A. Role of Heat Shock Proteins (HSP70 and HSP90) in Viral Infection. Int. J. Mol. Sci. 2021, 22, 9366. [Google Scholar] [CrossRef]
- Okamoto, T.; Nishimura, Y.; Ichimura, T.; Suzuki, K.; Miyamura, T.; Suzuki, T.; Moriishi, K.; Matsuura, Y. Hepatitis C virus RNA replication is regulated by FKBP8 and Hsp90. EMBO J. 2006, 25, 5015–5025. [Google Scholar] [CrossRef] [PubMed]
- Chase, G.; Deng, T.; Fodor, E.; Leung, B.W.; Mayer, D.; Schwemmle, M.; Brownlee, G. Hsp90 inhibitors reduce influenza virus replication in cell culture. Virology 2008, 377, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Aviner, R.; Frydman, J. Proteostasis in Viral Infection: Unfolding the Complex Virus-Chaperone Interplay. Cold Spring Harb. Perspect. Biol. 2020, 12, a034090. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Mitra, D. Heat shock protein 40 is necessary for human immunodeficiency virus-1 Nef-mediated enhancement of viral gene expression and replication. J. Biol. Chem. 2005, 280, 40041–40050. [Google Scholar] [CrossRef] [PubMed]
- Shelton, M.N.; Huang, M.B.; Ali, S.A.; Powell, M.D.; Bond, V.C. Secretion modification region-derived peptide disrupts HIV-1 Nef’s interaction with mortalin and blocks virus and Nef exosome release. J. Virol. 2012, 86, 406–419. [Google Scholar] [CrossRef] [PubMed]
- Geiger, R.; Andritschke, D.; Friebe, S.; Herzog, F.; Luisoni, S.; Heger, T.; Helenius, A. BAP31 and BiP are essential for dislocation of SV40 from the endoplasmic reticulum to the cytosol. Nat. Cell Biol. 2011, 13, 1305–1314. [Google Scholar] [CrossRef] [PubMed]
- Khongwichit, S.; Sornjai, W.; Jitobaom, K.; Greenwood, M.; Greenwood, M.P.; Hitakarun, A.; Wikan, N.; Murphy, D.; Smith, D.R. A functional interaction between GRP78 and Zika virus E protein. Sci. Rep. 2021, 11, 393. [Google Scholar] [CrossRef]
- Chu, H.; Chan, C.M.; Zhang, X.; Wang, Y.; Yuan, S.; Zhou, J.; Au-Yeung, R.K.; Sze, K.H.; Yang, D.; Shuai, H.; et al. Middle East respiratory syndrome coronavirus and bat coronavirus HKU9 both can utilize GRP78 for attachment onto host cells. J. Biol. Chem. 2018, 293, 11709–11726. [Google Scholar] [CrossRef]
- Elfiky, A.A.; Amr, A.; Mosaad, A.; Mubarak, A.K.; Sayed, M.A.; El-Halwany, K.K. Cs-GRP78 recognition site on dengue virus envelope protein: In silico perspective. Future Virol. 2023, 18, 411–420. [Google Scholar] [CrossRef]
- Buchkovich, N.J.; Maguire, T.G.; Yu, Y.; Paton, A.W.; Paton, J.C.; Alwine, J.C. Human cytomegalovirus specifically controls the levels of the endoplasmic reticulum chaperone BiP/GRP78, which is required for virion assembly. J. Virol. 2008, 82, 31–39. [Google Scholar] [CrossRef]
- Maruri-Avidal, L.; Lopez, S.; Arias, C.F. Endoplasmic reticulum chaperones are involved in the morphogenesis of rotavirus infectious particles. J. Virol. 2008, 82, 5368–5380. [Google Scholar] [CrossRef]
- Fislova, T.; Thomas, B.; Graef, K.M.; Fodor, E. Association of the influenza virus RNA polymerase subunit PB2 with the host chaperonin CCT. J. Virol. 2010, 84, 8691–8699. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ye, C.; Ruan, X.; Zan, J.; Xu, Y.; Liao, M.; Zhou, J. The chaperonin CCTα is required for efficient transcription and replication of rabies virus. Microbiol. Immunol. 2014, 58, 590–599. [Google Scholar] [CrossRef] [PubMed]
- Knowlton, J.J.; Fernandez de Castro, I.; Ashbrook, A.W.; Gestaut, D.R.; Zamora, P.F.; Bauer, J.A.; Forrest, J.C.; Frydman, J.; Risco, C.; Dermody, T.S. The TRiC chaperonin controls reovirus replication through outer-capsid folding. Nat. Microbiol. 2018, 3, 481–493. [Google Scholar] [CrossRef] [PubMed]
- Tahmaz, I.; Shahmoradi Ghahe, S.; Topf, U. Prefoldin Function in Cellular Protein Homeostasis and Human Diseases. Front. Cell Dev. Biol. 2022, 9, 816214. [Google Scholar] [CrossRef]
- Knowlton, J.J.; Gestaut, D.; Ma, B.; Taylor, G.; Seven, A.B.; Leitner, A.; Wilson, G.J.; Shanker, S.; Yates, N.A.; Prasad, B.V.V.; et al. Structural and functional dissection of reovirus capsid folding and assembly by the prefoldin-TRiC/CCT chaperone network. Proc. Natl. Acad. Sci. USA 2021, 118, e2018127118. [Google Scholar] [CrossRef] [PubMed]
- Tsao, M.L.; Chao, C.H.; Yeh, C.T. Interaction of hepatitis C virus F protein with prefoldin 2 perturbs tubulin cytoskeleton organization. Biochem. Biophys. Res. Commun. 2006, 348, 271–277. [Google Scholar] [CrossRef]
- Rain, J.C.; Cribier, A.; Gerard, A.; Emiliani, S.; Benarous, R. Yeast two-hybrid detection of integrase-host factor interactions. Methods 2009, 47, 291–297. [Google Scholar] [CrossRef]
- Li, C.; Chu, H.; Liu, X.; Chiu, M.C.; Zhao, X.; Wang, D.; Wei, Y.; Hou, Y.; Shuai, H.; Cai, J.; et al. Human coronavirus dependency on host heat shock protein 90 reveals an antiviral target. Emerg. Microbes Infect. 2020, 9, 2663–2672. [Google Scholar] [CrossRef]
- Zhao, Z.; Xu, L.D.; Zhang, F.; Liang, Q.Z.; Jiao, Y.; Shi, F.S.; He, B.; Xu, P.; Huang, Y.W. Heat shock protein 90 facilitates SARS-CoV-2 structural protein-mediated virion assembly and promotes virus-induced pyroptosis. J. Biol. Chem. 2023, 299, 104668. [Google Scholar] [CrossRef]
- Wyler, E.; Mosbauer, K.; Franke, V.; Diag, A.; Gottula, L.T.; Arsie, R.; Klironomos, F.; Koppstein, D.; Honzke, K.; Ayoub, S.; et al. Transcriptomic profiling of SARS-CoV-2 infected human cell lines identifies HSP90 as target for COVID-19 therapy. iScience 2021, 24, 102151. [Google Scholar] [CrossRef]
- Sabirli, R.; Koseler, A.; Goren, T.; Turkcuer, I.; Kurt, O. High GRP78 levels in COVID-19 infection: A case-control study. Life Sci. 2021, 265, 118781. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.; Toyoda, S.; Fukuhara, A.; Shimomura, I. GRP78, a Novel Host Factor for SARS-CoV-2: The Emerging Roles in COVID-19 Related to Metabolic Risk Factors. Biomedicines 2022, 10, 1995. [Google Scholar] [CrossRef] [PubMed]
- Carlos, A.J.; Ha, D.P.; Yeh, D.W.; Van Krieken, R.; Tseng, C.C.; Zhang, P.; Gill, P.; Machida, K.; Lee, A.S. The chaperone GRP78 is a host auxiliary factor for SARS-CoV-2 and GRP78 depleting antibody blocks viral entry and infection. J. Biol. Chem. 2021, 296, 100759. [Google Scholar] [CrossRef] [PubMed]
- Shin, W.J.; Ha, D.P.; Machida, K.; Lee, A.S. The stress-inducible ER chaperone GRP78/BiP is upregulated during SARS-CoV-2 infection and acts as a pro-viral protein. Nat. Commun. 2022, 13, 6551. [Google Scholar] [CrossRef] [PubMed]
- Ha, D.P.; Shin, W.J.; Hernandez, J.C.; Neamati, N.; Dubeau, L.; Machida, K.; Lee, A.S. GRP78 Inhibitor YUM70 Suppresses SARS-CoV-2 Viral Entry, Spike Protein Production and Ameliorates Lung Damage. Viruses 2023, 15, 1118. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhou, L.; Bao, L.; Liu, J.; Zhu, H.; Lv, Q.; Liu, R.; Chen, W.; Tong, W.; Wei, Q.; et al. SARS-CoV-2 crosses the blood-brain barrier accompanied with basement membrane disruption without tight junctions alteration. Signal Transduct. Target. Ther. 2021, 6, 337. [Google Scholar] [CrossRef] [PubMed]
- Butowt, R.; Meunier, N.; Bryche, B.; von Bartheld, C.S. The olfactory nerve is not a likely route to brain infection in COVID-19: A critical review of data from humans and animal models. Acta Neuropathol. 2021, 141, 809–822. [Google Scholar] [CrossRef]
- Villadiego, J.; Garcia-Arriaza, J.; Ramirez-Lorca, R.; Garcia-Swinburn, R.; Cabello-Rivera, D.; Rosales-Nieves, A.E.; Alvarez-Vergara, M.I.; Cala-Fernandez, F.; Garcia-Roldan, E.; Lopez-Ogayar, J.L.; et al. Full protection from SARS-CoV-2 brain infection and damage in susceptible transgenic mice conferred by MVA-CoV2-S vaccine candidate. Nat. Neurosci. 2023, 26, 226–238. [Google Scholar] [CrossRef]
- Tavassoly, O.; Safavi, F.; Tavassoly, I. Seeding Brain Protein Aggregation by SARS-CoV-2 as a Possible Long-Term Complication of COVID-19 Infection. ACS Chem. Neurosci. 2020, 11, 3704–3706. [Google Scholar] [CrossRef]
- Butowt, R.; von Bartheld, C.S. The route of SARS-CoV-2 to brain infection: Have we been barking up the wrong tree? Mol. Neurodegener. 2022, 17, 20. [Google Scholar] [CrossRef]
- Sollmann, N.; Beer, A.J.; Kirchhoff, F. SARS-CoV-2 infection and the brain: Direct evidence for brain changes in milder cases. Signal Transduct. Target. Ther. 2022, 7, 230. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.; Michalak, M.; Agellon, L.B. Endoplasmic Reticulum Malfunction in the Nervous System. Front. Neurosci. 2017, 11, 220. [Google Scholar] [CrossRef] [PubMed]
- Choi, I.; Zhang, Y.; Seegobin, S.P.; Pruvost, M.; Wang, Q.; Purtell, K.; Zhang, B.; Yue, Z. Microglia clear neuron-released α-synuclein via selective autophagy and prevent neurodegeneration. Nat. Commun. 2020, 11, 1386. [Google Scholar] [CrossRef] [PubMed]
- Srimat Kandadai, K.; Kotur, M.B.; Dokalis, N.; Amrein, I.; Keller, C.W.; Munz, C.; Wolfer, D.; Prinz, M.; Lunemann, J.D. ATG5 in microglia does not contribute vitally to autoimmune neuroinflammation in mice. Autophagy 2021, 17, 3566–3576. [Google Scholar] [CrossRef]
- Tydlacka, S.; Wang, C.E.; Wang, X.; Li, S.; Li, X.J. Differential activities of the ubiquitin-proteasome system in neurons versus glia may account for the preferential accumulation of misfolded proteins in neurons. J. Neurosci. 2008, 28, 13285–13295. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, S. Selective Neuronal Death in Neurodegenerative Diseases: The Ongoing Mystery. Yale J. Biol. Med. 2019, 92, 695–705. [Google Scholar] [PubMed]
- Bedford, L.; Hay, D.; Devoy, A.; Paine, S.; Powe, D.G.; Seth, R.; Gray, T.; Topham, I.; Fone, K.; Rezvani, N.; et al. Depletion of 26S proteasomes in mouse brain neurons causes neurodegeneration and Lewy-like inclusions resembling human pale bodies. J. Neurosci. 2008, 28, 8189–8198. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Kominami, E.; Tanaka, K. Autophagy and neurodegeneration. Autophagy 2006, 2, 315–317. [Google Scholar] [CrossRef]
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 441, 885–889. [Google Scholar] [CrossRef]
- Bjornevik, K.; Cortese, M.; Healy, B.C.; Kuhle, J.; Mina, M.J.; Leng, Y.; Elledge, S.J.; Niebuhr, D.W.; Scher, A.I.; Munger, K.L.; et al. Longitudinal analysis reveals high prevalence of Epstein-Barr virus associated with multiple sclerosis. Science 2022, 375, 296–301. [Google Scholar] [CrossRef]
- Fulop, T.; Witkowski, J.M.; Larbi, A.; Khalil, A.; Herbein, G.; Frost, E.H. Does HIV infection contribute to increased beta-amyloid synthesis and plaque formation leading to neurodegeneration and Alzheimer’s disease? J. Neurovirol 2019, 25, 634–647. [Google Scholar] [CrossRef] [PubMed]
- De Chiara, G.; Marcocci, M.E.; Sgarbanti, R.; Civitelli, L.; Ripoli, C.; Piacentini, R.; Garaci, E.; Grassi, C.; Palamara, A.T. Infectious agents and neurodegeneration. Mol. Neurobiol. 2012, 46, 614–638. [Google Scholar] [CrossRef] [PubMed]
- Kristen, H.; Santana, S.; Sastre, I.; Recuero, M.; Bullido, M.J.; Aldudo, J. Herpes simplex virus type 2 infection induces AD-like neurodegeneration markers in human neuroblastoma cells. Neurobiol. Aging 2015, 36, 2737–2747. [Google Scholar] [CrossRef] [PubMed]
- Bortolotti, D.; Gentili, V.; Rotola, A.; Caselli, E.; Rizzo, R. HHV-6A infection induces amyloid-beta expression and activation of microglial cells. Alzheimers Res. Ther. 2019, 11, 104. [Google Scholar] [CrossRef] [PubMed]
- Levine, K.S.; Leonard, H.L.; Blauwendraat, C.; Iwaki, H.; Johnson, N.; Bandres-Ciga, S.; Ferrucci, L.; Faghri, F.; Singleton, A.B.; Nalls, M.A. Virus exposure and neurodegenerative disease risk across national biobanks. Neuron 2023, 111, 1086–1093.e2. [Google Scholar] [CrossRef] [PubMed]
- Leblanc, P.; Vorberg, I.M. Viruses in neurodegenerative diseases: More than just suspects in crimes. PLoS Pathog. 2022, 18, e1010670. [Google Scholar] [CrossRef] [PubMed]
- Romeo, M.A.; Gilardini Montani, M.S.; Gaeta, A.; D’Orazi, G.; Faggioni, A.; Cirone, M. HHV-6A infection dysregulates autophagy/UPR interplay increasing beta amyloid production and tau phosphorylation in astrocytoma cells as well as in primary neurons, possible molecular mechanisms linking viral infection to Alzheimer’s disease. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165647. [Google Scholar] [CrossRef]
- Marreiros, R.; Muller-Schiffmann, A.; Trossbach, S.V.; Prikulis, I.; Hansch, S.; Weidtkamp-Peters, S.; Moreira, A.R.; Sahu, S.; Soloviev, I.; Selvarajah, S.; et al. Disruption of cellular proteostasis by H1N1 influenza A virus causes α-synuclein aggregation. Proc. Natl. Acad. Sci. USA 2020, 117, 6741–6751. [Google Scholar] [CrossRef]
- Rieder, A.S.; Deniz, B.F.; Netto, C.A.; Wyse, A.T.S. A Review of In Silico Research, SARS-CoV-2, and Neurodegeneration: Focus on Papain-Like Protease. Neurotox. Res. 2022, 40, 1553–1569. [Google Scholar] [CrossRef]
- Karvandi, M.S.; Sheikhzadeh Hesari, F.; Aref, A.R.; Mahdavi, M. The neuroprotective effects of targeting key factors of neuronal cell death in neurodegenerative diseases: The role of ER stress, oxidative stress, and neuroinflammation. Front. Cell. Neurosci. 2023, 17, 1105247. [Google Scholar] [CrossRef]
- Rifino, N.; Censori, B.; Agazzi, E.; Alimonti, D.; Bonito, V.; Camera, G.; Conti, M.Z.; Foresti, C.; Frigeni, B.; Gerevini, S.; et al. Neurologic manifestations in 1760 COVID-19 patients admitted to Papa Giovanni XXIII Hospital, Bergamo, Italy. J Neurol 2021, 268, 2331–2338. [Google Scholar] [CrossRef]
- Natoli, S.; Oliveira, V.; Calabresi, P.; Maia, L.F.; Pisani, A. Does SARS-CoV-2 invade the brain? Translational lessons from animal models. Eur. J. Neurol. 2020, 27, 1764–1773. [Google Scholar] [CrossRef]
- Baazaoui, N.; Iqbal, K. COVID-19 and Neurodegenerative Diseases: Prion-Like Spread and Long-Term Consequences. J. Alzheimers Dis. 2022, 88, 399–416. [Google Scholar] [CrossRef]
- Krey, L.; Huber, M.K.; Hoglinger, G.U.; Wegner, F. Can SARS-CoV-2 Infection Lead to Neurodegeneration and Parkinson’s Disease? Brain Sci. 2021, 11, 1654. [Google Scholar] [CrossRef] [PubMed]
- Idrees, D.; Kumar, V. SARS-CoV-2 spike protein interactions with amyloidogenic proteins: Potential clues to neurodegeneration. Biochem. Biophys. Res. Commun. 2021, 554, 94–98. [Google Scholar] [CrossRef]
- Bowen, D.R.; Pathak, S.; Nadar, R.M.; Parise, R.D.; Ramesh, S.; Govindarajulu, M.; Moore, A.; Ren, J.; Moore, T.; Dhanasekaran, M. Oxidative stress and COVID-19-associated neuronal dysfunction: Mechanisms and therapeutic implications. Acta Biochim. Biophys. Sin. 2023, 55, 1153–1167. [Google Scholar] [CrossRef] [PubMed]
- Lingor, P.; Demleitner, A.F.; Wolff, A.W.; Feneberg, E. SARS-CoV-2 and neurodegenerative diseases: What we know and what we don’t. J. Neural Transm. 2022, 129, 1155–1167. [Google Scholar] [CrossRef] [PubMed]
- Tonges, L.; Klebe, S. SARS-CoV-2, COVID-19 and Neurodegeneration. Brain Sci. 2022, 12, 897. [Google Scholar] [CrossRef]
- Strong, M.J. SARS-CoV-2, aging, and Post-COVID-19 neurodegeneration. J. Neurochem. 2023, 165, 115–130. [Google Scholar] [CrossRef] [PubMed]
- Salari, M.; Etemadifar, M. Can COVID-19 accelerate neurodegeneration? Clin. Case Rep. 2021, 9, e04433. [Google Scholar] [CrossRef] [PubMed]
- Ramani, A.; Muller, L.; Ostermann, P.N.; Gabriel, E.; Abida-Islam, P.; Muller-Schiffmann, A.; Mariappan, A.; Goureau, O.; Gruell, H.; Walker, A.; et al. SARS-CoV-2 targets neurons of 3D human brain organoids. EMBO J. 2020, 39, e106230. [Google Scholar] [CrossRef] [PubMed]
- Prasad, K.; AlOmar, S.Y.; Alqahtani, S.A.M.; Malik, M.Z.; Kumar, V. Brain Disease Network Analysis to Elucidate the Neurological Manifestations of COVID-19. Mol. Neurobiol. 2021, 58, 1875–1893. [Google Scholar] [CrossRef] [PubMed]
- Paniz-Mondolfi, A.; Bryce, C.; Grimes, Z.; Gordon, R.E.; Reidy, J.; Lednicky, J.; Sordillo, E.M.; Fowkes, M. Central nervous system involvement by severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2). J. Med. Virol. 2020, 92, 699–702. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.M.; Wang, F.; Qi, D.; Liu, W.W.; Gu, C.; Mao, C.J.; Yang, Y.P.; Zhao, Z.; Hu, L.F.; Liu, C.F. A Critical Role of Autophagy in Regulating Microglia Polarization in Neurodegeneration. Front. Aging Neurosci. 2018, 10, 378. [Google Scholar] [CrossRef] [PubMed]
- Oslowski, C.M.; Urano, F. Measuring ER stress and the unfolded protein response using mammalian tissue culture system. Methods Enzymol. 2011, 490, 71–92. [Google Scholar] [CrossRef] [PubMed]
- Macauslane, K.L.; Pegg, C.L.; Short, K.R.; Schulz, B.L. Modulation of endoplasmic reticulum stress response pathways by respiratory viruses. Crit. Rev. Microbiol. 2023, 7, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Thakur, A.; Sharma, V.; Averbek, S.; Liang, L.; Pandya, N.; Kumar, G.; Cili, A.; Zhang, K. Immune landscape and redox imbalance during neurological disorders in COVID-19. Cell Death Dis. 2023, 14, 593. [Google Scholar] [CrossRef]
- Saleh, J.; Peyssonnaux, C.; Singh, K.K.; Edeas, M. Mitochondria and microbiota dysfunction in COVID-19 pathogenesis. Mitochondrion 2020, 54, 1–7. [Google Scholar] [CrossRef]
- Matias-Perez, D.; Hernandez-Bautista, E.; Antonio Garcia-Montalvo, I. Oxidative Stress Derived from COVID-19 and Its Possible Association with the Development of Neurodegenerative Diseases. Arch. Neurosci. 2022, 9, e123302. [Google Scholar] [CrossRef]
- Hohn, A.; Tramutola, A.; Cascella, R. Proteostasis Failure in Neurodegenerative Diseases: Focus on Oxidative Stress. Oxid. Med. Cell. Longev. 2020, 2020, 5497046. [Google Scholar] [CrossRef]
- Holubiec, M.I.; Gellert, M.; Hanschmann, E.M. Redox signaling and metabolism in Alzheimer’s disease. Front. Aging Neurosci. 2022, 14, 1003721. [Google Scholar] [CrossRef]
- Passos, F.R.S.; Heimfarth, L.; Monteiro, B.S.; Correa, C.B.; Moura, T.R.; Araujo, A.A.S.; Martins-Filho, P.R.; Quintans-Junior, L.J.; Quintans, J.S.S. Oxidative stress and inflammatory markers in patients with COVID-19: Potential role of RAGE, HMGB1, GFAP and COX-2 in disease severity. Int. Immunopharmacol. 2022, 104, 108502. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gavilán, E.; Medina-Guzman, R.; Bahatyrevich-Kharitonik, B.; Ruano, D. Protein Quality Control Systems and ER Stress as Key Players in SARS-CoV-2-Induced Neurodegeneration. Cells 2024, 13, 123. https://doi.org/10.3390/cells13020123
Gavilán E, Medina-Guzman R, Bahatyrevich-Kharitonik B, Ruano D. Protein Quality Control Systems and ER Stress as Key Players in SARS-CoV-2-Induced Neurodegeneration. Cells. 2024; 13(2):123. https://doi.org/10.3390/cells13020123
Chicago/Turabian StyleGavilán, Elena, Rafael Medina-Guzman, Bazhena Bahatyrevich-Kharitonik, and Diego Ruano. 2024. "Protein Quality Control Systems and ER Stress as Key Players in SARS-CoV-2-Induced Neurodegeneration" Cells 13, no. 2: 123. https://doi.org/10.3390/cells13020123