
Neuronal Autophagy: Regulations and Implications in Health and Disease
Abstract
:1. Introduction
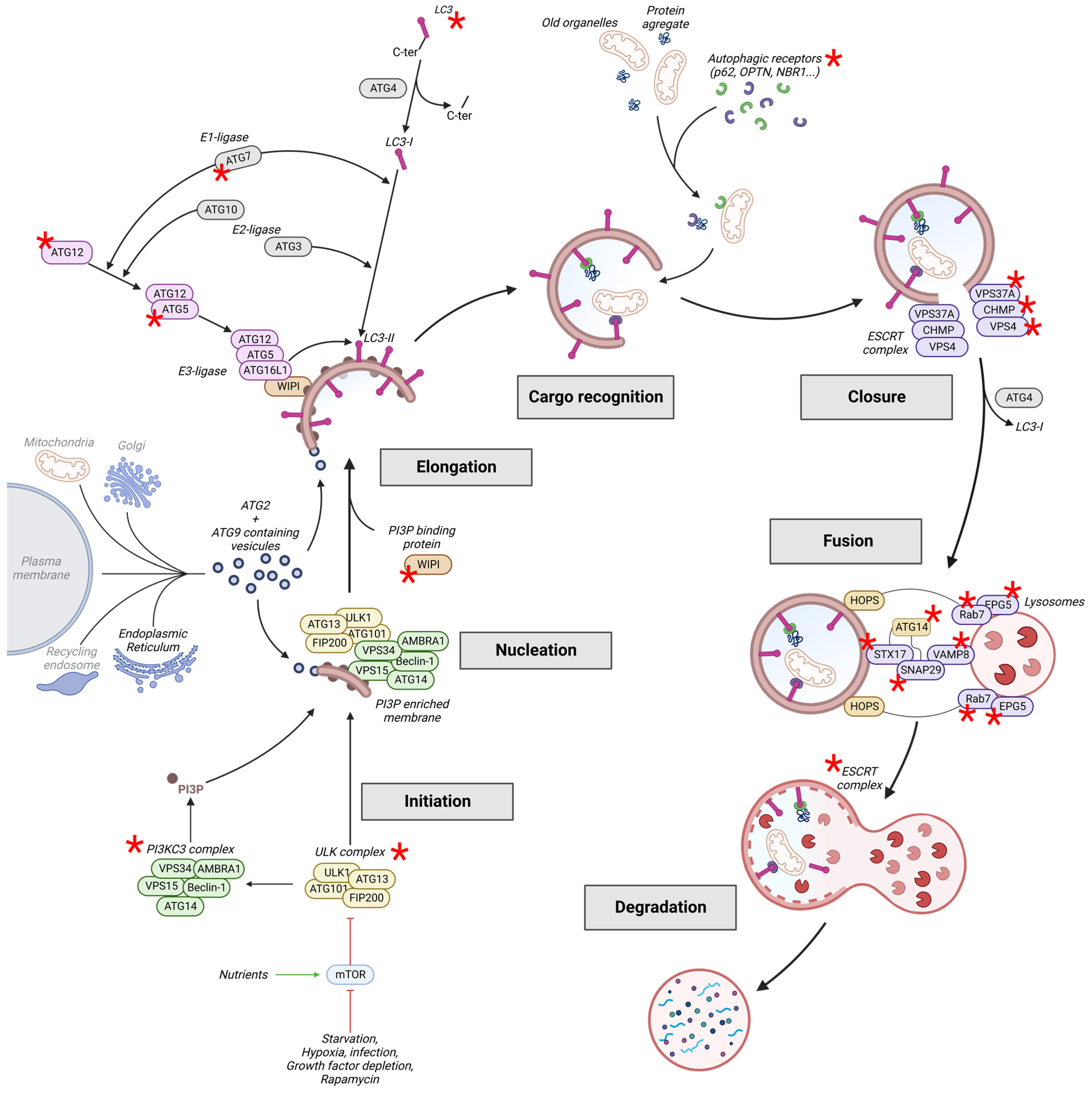
2. The Autophagy Pathway
2.1. Induction: Initiation and Nucleation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Autophagic Step | Protein Mutated | Pathology | Refs. |
|---|---|---|---|
| Induction and nucleation | FIP200 | Psychiatric disorders | [26,27,28,29] |
| AMBRA1 | NDD with encephalocele, spina bifida and NTD | [30] | |
| VPS15 | Cortical atrophy, Epilepsy | [31] | |
| VPS35 | PD | [32,33,34] | |
| Elongation | ATG7 | NDD with cerebellar hypoplasia and ataxia, PD | [35,36] |
| ATG5 | PD, SCA25, Cerebral Palsy | [37,38,39] | |
| ATG12 | PD | [40] | |
| WIPI2 WIPI4 | NDD with mental retardation, BPAN | [41,42,43] | |
| LC3 | PD (risk factor), ASD | [44,45] | |
| Cargo recognition | ALFY | Microcephaly with ID | [46] |
| p62/SQSTM1 | NADGP, ALS, FTD, AD (risk factor) | [47,48,49,50,51,52,53,54] | |
| NBR1 | PD, DLB, AD | [55] | |
| Optineurin | ALS | [56,57,58] | |
| Ubiquilin-2 | ALS | [59,60] | |
| Maturation: Closure and Fusion | VPS37A | HSP | [61] |
| VPS4A | DD and ID | [62] | |
| CHMP1A CHMP2B | pontocerebellar hypoplasia with microcephaly, ALS, FTD | [63,64,65,66,67,68] | |
| Rab7 | CMT type 2B | [69,70] | |
| EPG5 | Vici syndrome | [71] | |
| WIPI3/4 | NDD, BPAN | [41,42,43,72] | |
| SNX14 | SCAR17/20 | [73,74,75] |
2.2. Elongation
2.2.1. Membrane Expansion
2.2.2. Cargo Recognition
2.3. Closure and Fusion
2.3.1. Closure
2.3.2. Fusion
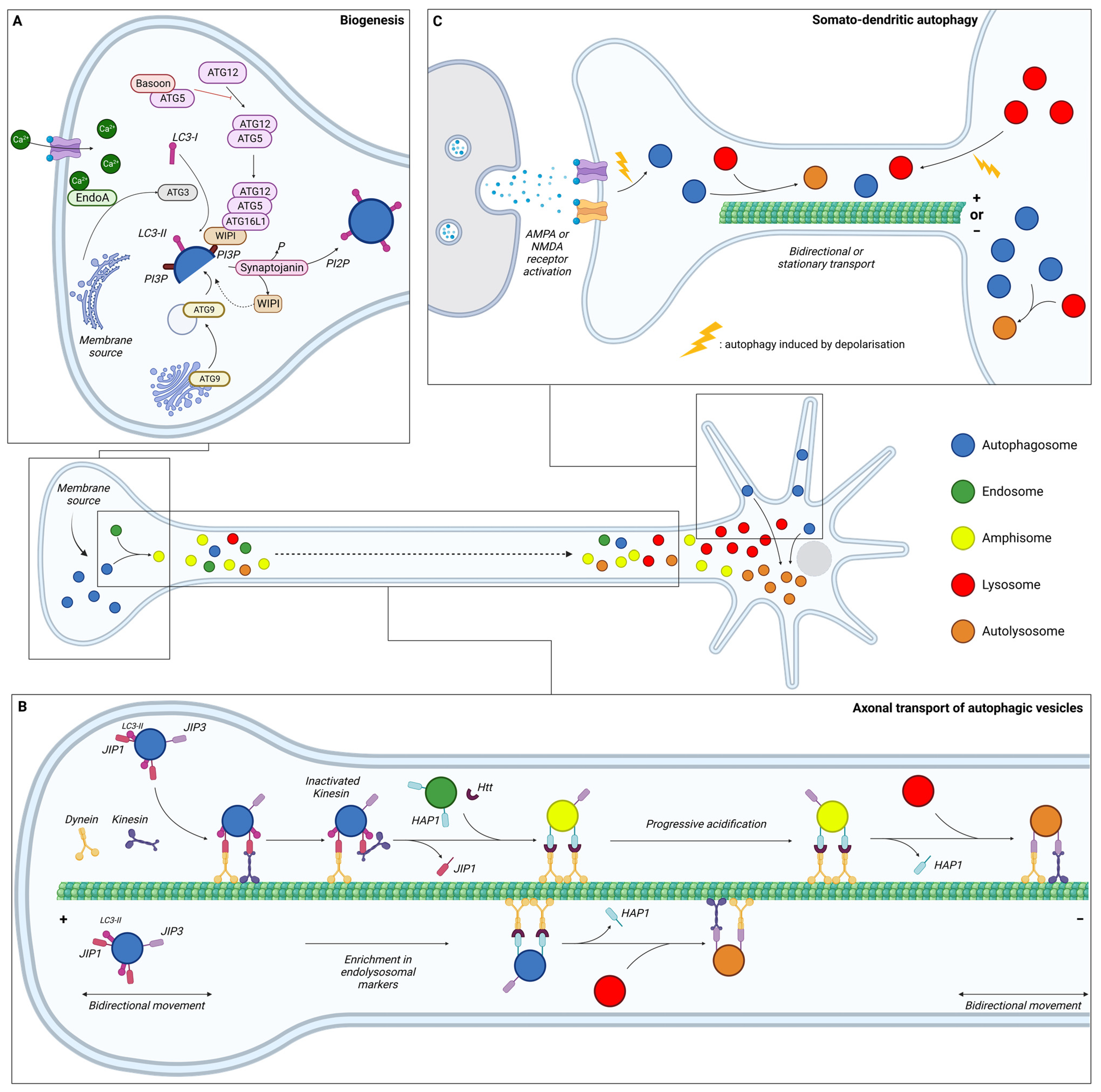
3. Compartmentalisation of Neuronal Autophagy
3.1. Autophagy in the Axon
3.1.1. Biogenesis
3.1.2. Maturation during Transport
3.1.3. Degradation
3.2. Autophagy in the Soma
3.3. Autophagy in Dendrites
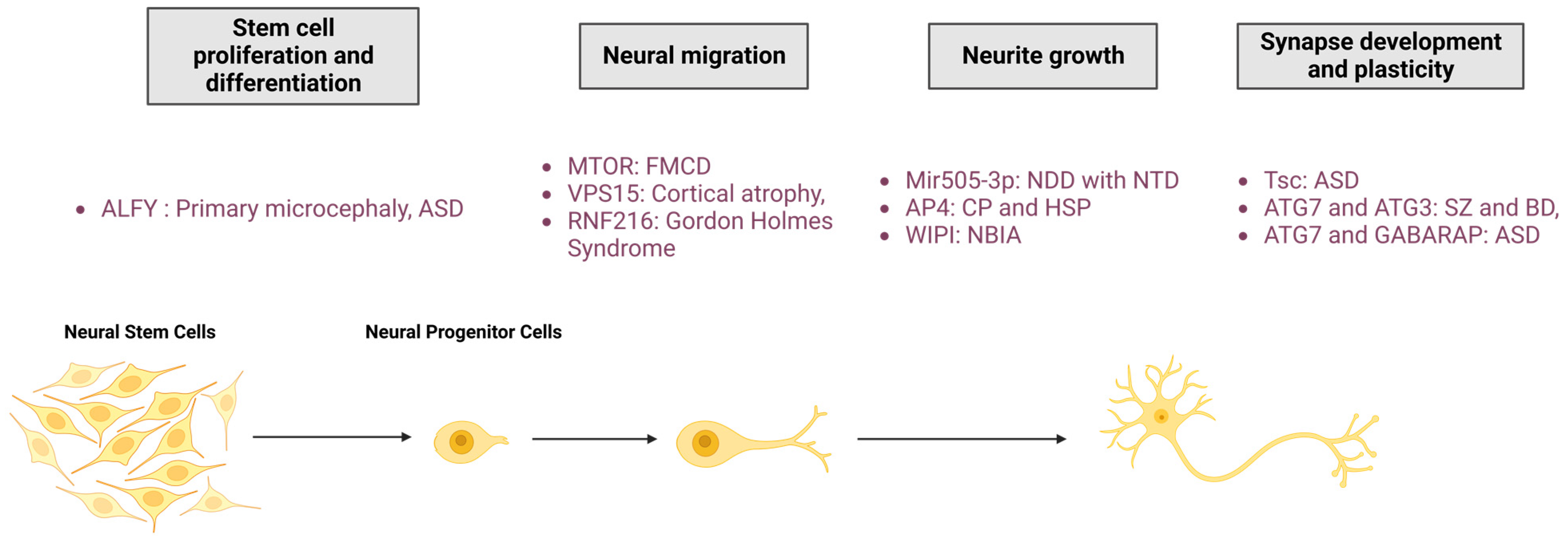
4. Autophagy in Neurological Physiopathology
4.1. Neural Proliferation and Differentiation
4.2. Neural Migration
4.3. Neurite Outgrowth
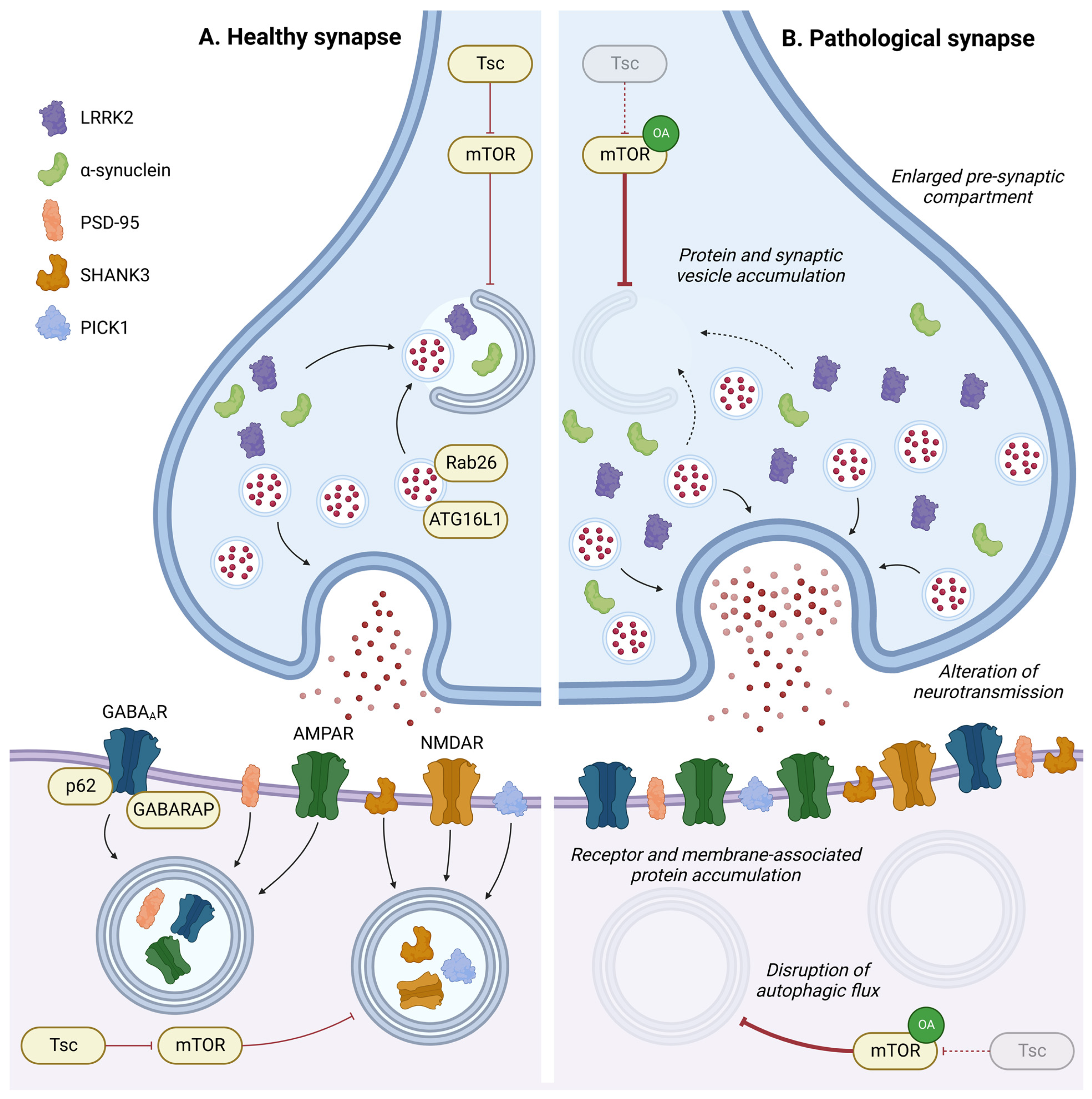
4.4. Synapse Development and Plasticity
5. Concluding Remarks and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| ALFY | Autophagy-linked FYVE protein |
| ALS | Amyotrophic lateral sclerosis |
| AMBRA1 | Activating molecule in BECN1-regulated autophagy protein 1 |
| AMPA (R) | α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (receptor) |
| AP | Adaptor protein |
| ASD | Autism spectrum disorder |
| ATG | Autophagy-related proteins |
| AV | Autophagic vesicle |
| BPAN | β-propeller protein-associated neurodegeneration |
| BD | Bipolar disorders |
| CHMP | Charged multivesicular body protein |
| CMT | Charcot–Marie–Tooth disease |
| DD | Developmental delay |
| DQ-BSA | dye quenched-bovine serum albumin |
| EndoA | Endophilin A |
| EPG5 | Ectopic P granules protein 5 homolog |
| Eva1a | Epithelial v-like antigen 1 |
| FIP200 | Focal adhesion kinase family interacting protein of 200 kDa |
| FTD | Fronto-temporal dementia |
| FYVE | Fab1, YOTB, Vac1 and EEA1 domain |
| GABA | Gamma-aminobutyric acid |
| GABARAP | Gamma-aminobutyric acid receptor-associated protein |
| GAN | Giant axonal neuropathy |
| HD | Huntington’s Disease |
| HOPS | Homotypic fusion and protein sorting |
| HAP1 | Huntingtin-associated protein 1 |
| HSP | Hereditary Spastic Paraplegia |
| HSPB1 | Heat shock protein beta-1 |
| HTT | Huntingtin |
| ID | Intellectual disability |
| JIP | JNK-interacting protein |
| LAMP | Lysosomal membrane protein |
| LC3 | Microtubule-associated protein light chain 3 |
| LRRK2 | Leucine-rich repeat serine/threonine-protein kinase 2 |
| MT | Microtubules |
| mTOR | mammalian target of rapamycin |
| NBIA | Neurodegeneration with brain iron accumulation |
| NBR1 | Next to BRCA1 gene 1 protein |
| NDD | Neurodevelopmental disorders |
| NDP52 | Calcium-binding and coiled-coil domain-containing protein 2 (Calcoco2) |
| NMDA (R) | N-methyl-D-aspartate (receptor) |
| NSCs | Neural stem cells |
| NPCs | Neural progenitor cells |
| OPTN | Optineurin |
| PD | Parkinson’s disease |
| PI3KC3 | class III phosphatidylinositol 3-kinase |
| PI3P | phosphatidylinositol-3-phosphate |
| PICK1 | Protein interacting with C kinase 1 |
| PSD-95 | Postsynaptic density protein 95 |
| RNF | Ring Finger Protein |
| SCA | Spinocerebellar ataxia |
| SHANK3 | SH3 and multiple ankyrin repeat domains protein 3 |
| SNAP | Synaptosomal-Associated Protein |
| SNARE | SNAp Receptor |
| SZ | Schizophrenia |
| Tsc (1) | Tuberous sclerosis protein (1) |
| ULK | Unc-51-like kinase |
| VAMP | Vesicle-associated membrane protein |
| VPS | Vacuolar protein sorting |
| WIPI | WD repeat domain phosphoinositide-interacting protein |
References
- Parzych, K.R.; Klionsky, D.J. An Overview of Autophagy: Morphology, Mechanism, and Regulation. Antioxid. Redox Signal. 2014, 20, 460–473. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Petroni, G.; Amaravadi, R.K.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cadwell, K.; Cecconi, F.; Choi, A.M.K.; et al. Autophagy in Major Human Diseases. EMBO J. 2021, 40, e108863. [Google Scholar] [CrossRef] [PubMed]
- Ichimiya, T.; Yamakawa, T.; Hirano, T.; Yokoyama, Y.; Hayashi, Y.; Hirayama, D.; Wagatsuma, K.; Itoi, T.; Nakase, H. Autophagy and Autophagy-Related Diseases: A Review. Int. J. Mol. Sci. 2020, 21, 8974. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Kroemer, G. Biological Functions of Autophagy Genes: A Disease Perspective. Cell 2019, 176, 11–42. [Google Scholar] [CrossRef]
- Mizushima, N.; Yamamoto, A.; Matsui, M.; Yoshimori, T.; Ohsumi, Y. In Vivo Analysis of Autophagy in Response to Nutrient Starvation Using Transgenic Mice Expressing a Fluorescent Autophagosome Marker. Mol. Biol. Cell 2004, 15, 1101–1111. [Google Scholar] [CrossRef]
- Deng, Z.; Zhou, X.; Lu, J.-H.; Yue, Z. Autophagy Deficiency in Neurodevelopmental Disorders. Cell Biosci. 2021, 11, 214. [Google Scholar] [CrossRef]
- Fleming, A.; Bourdenx, M.; Fujimaki, M.; Karabiyik, C.; Krause, G.J.; Lopez, A.; Martín-Segura, A.; Puri, C.; Scrivo, A.; Skidmore, J.; et al. The Different Autophagy Degradation Pathways and Neurodegeneration. Neuron 2022, 110, 935–966. [Google Scholar] [CrossRef]
- Zatyka, M.; Sarkar, S.; Barrett, T. Autophagy in Rare (NonLysosomal) Neurodegenerative Diseases. J. Mol. Biol. 2020, 432, 2735–2753. [Google Scholar] [CrossRef]
- Ashford, T.P.; Porter, K.R. Cytoplasmic components in hepatic cell lysosomes. J. Cell Biol. 1962, 12, 198–202. [Google Scholar] [CrossRef]
- Deter, R.L.; De Duve, C. Influence of glucagon, an inducer of cellular autophagy, on some physical properties of rat liver lysosomes. J. Cell Biol. 1967, 33, 437–449. [Google Scholar] [CrossRef]
- Deter, R.L.; Baudhuin, P.; de Duve, C. Participation of lysosomes in cellular autophagy induced in rat liver by glucagon. J. Cell Biol. 1967, 35, C11–C16. [Google Scholar] [CrossRef] [PubMed]
- Mortimore, G.E.; Schworer, C.M. Induction of Autophagy by Amino-Acid Deprivation in Perfused Rat Liver. Nature 1977, 270, 174–176. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; He, P.; Huang, Y.; Li, Y.-F.; Lu, J.; Li, M.; Kurihara, H.; Luo, Z.; Meng, T.; Onishi, M.; et al. Selective Autophagy of Intracellular Organelles: Recent Research Advances. Theranostics 2021, 11, 222–256. [Google Scholar] [CrossRef] [PubMed]
- Egan, D.; Kim, J.; Shaw, R.J.; Guan, K.-L. The Autophagy Initiating Kinase ULK1 Is Regulated via Opposing Phosphorylation by AMPK and mTOR. Autophagy 2011, 7, 643–644. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.-L. AMPK and mTOR Regulate Autophagy through Direct Phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef]
- Noda, N.N.; Fujioka, Y. Atg1 Family Kinases in Autophagy Initiation. Cell. Mol. Life Sci. 2015, 72, 3083–3096. [Google Scholar] [CrossRef]
- Kim, B.-W.; Jin, Y.; Kim, J.; Kim, J.H.; Jung, J.; Kang, S.; Kim, I.Y.; Kim, J.; Cheong, H.; Song, H.K. The C-Terminal Region of ATG101 Bridges ULK1 and PtdIns3K Complex in Autophagy Initiation. Autophagy 2018, 14, 2104–2116. [Google Scholar] [CrossRef]
- Petiot, A.; Ogier-Denis, E.; Blommaart, E.F.; Meijer, A.J.; Codogno, P. Distinct Classes of Phosphatidylinositol 3′-Kinases Are Involved in Signaling Pathways That Control Macroautophagy in HT-29 Cells. J. Biol. Chem. 2000, 275, 992–998. [Google Scholar] [CrossRef]
- Polson, H.E.J.; de Lartigue, J.; Rigden, D.J.; Reedijk, M.; Urbé, S.; Clague, M.J.; Tooze, S.A. Mammalian Atg18 (WIPI2) Localizes to Omegasome-Anchored Phagophores and Positively Regulates LC3 Lipidation. Autophagy 2010, 6, 506–522. [Google Scholar] [CrossRef]
- Kishi-Itakura, C.; Koyama-Honda, I.; Itakura, E.; Mizushima, N. Ultrastructural Analysis of Autophagosome Organization Using Mammalian Autophagy-Deficient Cells. J. Cell Sci. 2014, 127, 4984. [Google Scholar] [CrossRef]
- Dooley, H.C.; Razi, M.; Polson, H.E.J.; Girardin, S.E.; Wilson, M.I.; Tooze, S.A. WIPI2 Links LC3 Conjugation with PI3P, Autophagosome Formation, and Pathogen Clearance by Recruiting Atg12–5-16L1. Mol. Cell 2014, 55, 238–252. [Google Scholar] [CrossRef] [PubMed]
- Bakula, D.; Müller, A.J.; Zuleger, T.; Takacs, Z.; Franz-Wachtel, M.; Thost, A.-K.; Brigger, D.; Tschan, M.P.; Frickey, T.; Robenek, H.; et al. WIPI3 and WIPI4 β-Propellers Are Scaffolds for LKB1-AMPK-TSC Signalling Circuits in the Control of Autophagy. Nat. Commun. 2017, 8, 15637. [Google Scholar] [CrossRef] [PubMed]
- Maeda, S.; Otomo, C.; Otomo, T. The Autophagic Membrane Tether ATG2A Transfers Lipids between Membranes. eLife 2019, 8, e45777. [Google Scholar] [CrossRef] [PubMed]
- Valverde, D.P.; Yu, S.; Boggavarapu, V.; Kumar, N.; Lees, J.A.; Walz, T.; Reinisch, K.M.; Melia, T.J. ATG2 Transports Lipids to Promote Autophagosome Biogenesis. J. Cell Biol. 2019, 218, 1787–1798. [Google Scholar] [CrossRef] [PubMed]
- Young, A.R.J.; Chan, E.Y.W.; Hu, X.W.; Köchl, R.; Crawshaw, S.G.; High, S.; Hailey, D.W.; Lippincott-Schwartz, J.; Tooze, S.A. Starvation and ULK1-Dependent Cycling of Mammalian Atg9 between the TGN and Endosomes. J. Cell Sci. 2006, 119, 3888–3900. [Google Scholar] [CrossRef] [PubMed]
- Cooper, G.M.; Coe, B.P.; Girirajan, S.; Rosenfeld, J.A.; Vu, T.H.; Baker, C.; Williams, C.; Stalker, H.; Hamid, R.; Hannig, V.; et al. A Copy Number Variation Morbidity Map of Developmental Delay. Nat. Genet. 2011, 43, 838–846. [Google Scholar] [CrossRef] [PubMed]
- Degenhardt, F.; Priebe, L.; Meier, S.; Lennertz, L.; Streit, F.; Witt, S.H.; Hofmann, A.; Becker, T.; Mössner, R.; Maier, W.; et al. Duplications in RB1CC1 Are Associated with Schizophrenia; Identification in Large European Sample Sets. Transl. Psychiatry 2013, 3, e326. [Google Scholar] [CrossRef]
- Marshall, C.R.; Noor, A.; Vincent, J.B.; Lionel, A.C.; Feuk, L.; Skaug, J.; Shago, M.; Moessner, R.; Pinto, D.; Ren, Y.; et al. Structural Variation of Chromosomes in Autism Spectrum Disorder. Am. J. Hum. Genet. 2008, 82, 477–488. [Google Scholar] [CrossRef]
- Xu, B.; Roos, J.L.; Dexheimer, P.; Boone, B.; Plummer, B.; Levy, S.; Gogos, J.A.; Karayiorgou, M. Exome Sequencing Supports a de Novo Mutational Paradigm for Schizophrenia. Nat. Genet. 2011, 43, 864–868. [Google Scholar] [CrossRef]
- Ye, J.; Tong, Y.; Lv, J.; Peng, R.; Chen, S.; Kuang, L.; Su, K.; Zheng, Y.; Zhang, T.; Zhang, F.; et al. Rare Mutations in the Autophagy-Regulating Gene AMBRA1 Contribute to Human Neural Tube Defects. Hum. Mutat. 2020, 41, 1383–1393. [Google Scholar] [CrossRef]
- Gstrein, T.; Edwards, A.; Přistoupilová, A.; Leca, I.; Breuss, M.; Pilat-Carotta, S.; Hansen, A.H.; Tripathy, R.; Traunbauer, A.K.; Hochstoeger, T.; et al. Mutations in Vps15 Perturb Neuronal Migration in Mice and Are Associated with Neurodevelopmental Disease in Humans. Nat. Neurosci. 2018, 21, 207–217. [Google Scholar] [CrossRef]
- Vilariño-Güell, C.; Wider, C.; Ross, O.A.; Dachsel, J.C.; Kachergus, J.M.; Lincoln, S.J.; Soto-Ortolaza, A.I.; Cobb, S.A.; Wilhoite, G.J.; Bacon, J.A.; et al. VPS35 Mutations in Parkinson Disease. Am. J. Hum. Genet. 2011, 89, 162–167. [Google Scholar] [CrossRef]
- Zimprich, A.; Benet-Pagès, A.; Struhal, W.; Graf, E.; Eck, S.H.; Offman, M.N.; Haubenberger, D.; Spielberger, S.; Schulte, E.C.; Lichtner, P.; et al. A Mutation in VPS35, Encoding a Subunit of the Retromer Complex, Causes Late-Onset Parkinson Disease. Am. J. Hum. Genet. 2011, 89, 168–175. [Google Scholar] [CrossRef]
- Zavodszky, E.; Seaman, M.N.J.; Moreau, K.; Jimenez-Sanchez, M.; Breusegem, S.Y.; Harbour, M.E.; Rubinsztein, D.C. Mutation in VPS35 Associated with Parkinson’s Disease Impairs WASH Complex Association and Inhibits Autophagy. Nat. Commun. 2014, 5, 3828. [Google Scholar] [CrossRef]
- Collier, J.J.; Guissart, C.; Oláhová, M.; Sasorith, S.; Piron-Prunier, F.; Suomi, F.; Zhang, D.; Martinez-Lopez, N.; Leboucq, N.; Bahr, A.; et al. Developmental Consequences of Defective ATG7-Mediated Autophagy in Humans. N. Engl. J. Med. 2021, 384, 2406–2417. [Google Scholar] [CrossRef]
- Chen, D.; Pang, S.; Feng, X.; Huang, W.; Hawley, R.G.; Yan, B. Genetic Analysis of the ATG7 Gene Promoter in Sporadic Parkinson’s Disease. Neurosci. Lett. 2013, 534, 193–198. [Google Scholar] [CrossRef]
- Chen, D.; Zhu, C.; Wang, X.; Feng, X.; Pang, S.; Huang, W.; Hawley, R.G.; Yan, B. A Novel and Functional Variant within the ATG5 Gene Promoter in Sporadic Parkinson’s Disease. Neurosci. Lett. 2013, 538, 49–53. [Google Scholar] [CrossRef]
- Kim, M.; Sandford, E.; Gatica, D.; Qiu, Y.; Liu, X.; Zheng, Y.; Schulman, B.A.; Xu, J.; Semple, I.; Ro, S.-H.; et al. Mutation in ATG5 Reduces Autophagy and Leads to Ataxia with Developmental Delay. eLife 2016, 5, e12245. [Google Scholar] [CrossRef]
- Xu, J.; Xia, L.; Shang, Q.; Du, J.; Zhu, D.; Wang, Y.; Bi, D.; Song, J.; Ma, C.; Gao, C.; et al. A Variant of the Autophagy-Related 5 Gene Is Associated with Child Cerebral Palsy. Front. Cell. Neurosci. 2017, 11, 407. [Google Scholar] [CrossRef]
- Li, Y.; Huang, J.; Pang, S.; Wang, H.; Zhang, A.; Hawley, R.G.; Yan, B. Novel and Functional ATG12 Gene Variants in Sporadic Parkinson’s Disease. Neurosci. Lett. 2017, 643, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Saitsu, H.; Nishimura, T.; Muramatsu, K.; Kodera, H.; Kumada, S.; Sugai, K.; Kasai-Yoshida, E.; Sawaura, N.; Nishida, H.; Hoshino, A.; et al. De Novo Mutations in the Autophagy Gene WDR45 Cause Static Encephalopathy of Childhood with Neurodegeneration in Adulthood. Nat. Genet. 2013, 45, 445–449. [Google Scholar] [CrossRef]
- Jelani, M.; Dooley, H.C.; Gubas, A.; Mohamoud, H.S.A.; Khan, M.T.M.; Ali, Z.; Kang, C.; Rahim, F.; Jan, A.; Vadgama, N.; et al. A Mutation in the Major Autophagy Gene, WIPI2, Associated with Global Developmental Abnormalities. Brain 2019, 142, 1242–1254. [Google Scholar] [CrossRef] [PubMed]
- Maroofian, R.; Gubas, A.; Kaiyrzhanov, R.; Scala, M.; Hundallah, K.; Severino, M.; Abdel-Hamid, M.S.; Rosenfeld, J.A.; Ebrahimi-Fakhari, D.; Ali, Z.; et al. Homozygous Missense WIPI2 Variants Cause a Congenital Disorder of Autophagy with Neurodevelopmental Impairments of Variable Clinical Severity and Disease Course. Brain Commun. 2021, 3, fcab183. [Google Scholar] [CrossRef]
- Xu, J.; Yang, Y.; Pang, S.; Huang, W.; Qin, X.; Hawley, R.G.; Yan, B. Identification of a Novel 21bp-Insertion Variant within the LC3B Gene Promoter in Sporadic Parkinson’s Disease. Transl. Res. 2013, 161, 441–443. [Google Scholar] [CrossRef] [PubMed]
- Poultney, C.S.; Goldberg, A.P.; Drapeau, E.; Kou, Y.; Harony-Nicolas, H.; Kajiwara, Y.; De Rubeis, S.; Durand, S.; Stevens, C.; Rehnström, K.; et al. Identification of Small Exonic CNV from Whole-Exome Sequence Data and Application to Autism Spectrum Disorder. Am. J. Hum. Genet. 2013, 93, 607–619. [Google Scholar] [CrossRef] [PubMed]
- Kadir, R.; Harel, T.; Markus, B.; Perez, Y.; Bakhrat, A.; Cohen, I.; Volodarsky, M.; Feintsein-Linial, M.; Chervinski, E.; Zlotogora, J.; et al. ALFY-Controlled DVL3 Autophagy Regulates Wnt Signaling, Determining Human Brain Size. PLoS Genet. 2016, 12, e1005919. [Google Scholar] [CrossRef]
- Haack, T.B.; Ignatius, E.; Calvo-Garrido, J.; Iuso, A.; Isohanni, P.; Maffezzini, C.; Lönnqvist, T.; Suomalainen, A.; Gorza, M.; Kremer, L.S.; et al. Absence of the Autophagy Adaptor SQSTM1/P62 Causes Childhood-Onset Neurodegeneration with Ataxia, Dystonia, and Gaze Palsy. Am. J. Hum. Genet. 2016, 99, 735–743. [Google Scholar] [CrossRef]
- Fecto, F. SQSTM1 Mutations in Familial and Sporadic Amyotrophic Lateral Sclerosis. Arch. Neurol. 2011, 68, 1440. [Google Scholar] [CrossRef]
- Rubino, E.; Rainero, I.; Chio, A.; Rogaeva, E.; Galimberti, D.; Fenoglio, P.; Grinberg, Y.; Isaia, G.; Calvo, A.; Gentile, S.; et al. SQSTM1 Mutations in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Neurology 2012, 79, 1556–1562. [Google Scholar] [CrossRef]
- Le Ber, I. SQSTM1 Mutations in French Patients with Frontotemporal Dementia or Frontotemporal Dementia with Amyotrophic Lateral Sclerosis. JAMA Neurol. 2013, 70, 1403–1410. [Google Scholar] [CrossRef]
- Boutoleau-Bretonnière, C.; Camuzat, A.; Le Ber, I.; Bouya-Ahmed, K.; Guerreiro, R.; Deruet, A.-L.; Evrard, C.; Bras, J.; Lamy, E.; Auffray-Calvier, E.; et al. A Phenotype of Atypical Apraxia of Speech in a Family Carrying SQSTM1 Mutation. J. Alzheimers Dis. 2014, 43, 625–630. [Google Scholar] [CrossRef] [PubMed]
- Goode, A.; Butler, K.; Long, J.; Cavey, J.; Scott, D.; Shaw, B.; Sollenberger, J.; Gell, C.; Johansen, T.; Oldham, N.J.; et al. Defective Recognition of LC3B by Mutant SQSTM1/P62 Implicates Impairment of Autophagy as a Pathogenic Mechanism in ALS-FTLD. Autophagy 2016, 12, 1094–1104. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Lim, J.; Wang, Q.; Purtell, K.; Wu, S.; Palomo, G.M.; Tan, H.; Manfredi, G.; Zhao, Y.; Peng, J.; et al. ALS-FTLD-Linked Mutations of SQSTM1/P62 Disrupt Selective Autophagy and NFE2L2/NRF2 Anti-Oxidative Stress Pathway. Autophagy 2020, 16, 917–931. [Google Scholar] [CrossRef]
- Cuyvers, E.; van der Zee, J.; Bettens, K.; Engelborghs, S.; Vandenbulcke, M.; Robberecht, C.; Dillen, L.; Merlin, C.; Geerts, N.; Graff, C.; et al. Genetic Variability in SQSTM1 and Risk of Early-Onset Alzheimer Dementia: A European Early-Onset Dementia Consortium Study. Neurobiol. Aging 2015, 36, 2005.e15–2005.e22. [Google Scholar] [CrossRef]
- Odagiri, S.; Tanji, K.; Mori, F.; Kakita, A.; Takahashi, H.; Wakabayashi, K. Autophagic Adapter Protein NBR1 Is Localized in Lewy Bodies and Glial Cytoplasmic Inclusions and Is Involved in Aggregate Formation in α-Synucleinopathy. Acta Neuropathol. 2012, 124, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.-X. Differential Involvement of Optineurin in Amyotrophic Lateral Sclerosis with or without SOD1 Mutations. Arch. Neurol. 2011, 68, 1057. [Google Scholar] [CrossRef]
- Maruyama, H.; Morino, H.; Ito, H.; Izumi, Y.; Kato, H.; Watanabe, Y.; Kinoshita, Y.; Kamada, M.; Nodera, H.; Suzuki, H.; et al. Mutations of Optineurin in Amyotrophic Lateral Sclerosis. Nature 2010, 465, 223–226. [Google Scholar] [CrossRef]
- Shen, W.-C.; Li, H.-Y.; Chen, G.-C.; Chern, Y.; Tu, P. Mutations in the Ubiquitin-Binding Domain of OPTN/Optineurin Interfere with Autophagy-Mediated Degradation of Misfolded Proteins by a Dominant-Negative Mechanism. Autophagy 2015, 11, 685–700. [Google Scholar] [CrossRef]
- Synofzik, M.; Maetzler, W.; Grehl, T.; Prudlo, J.; vom Hagen, J.M.; Haack, T.; Rebassoo, P.; Munz, M.; Schöls, L.; Biskup, S. Screening in ALS and FTD Patients Reveals 3 Novel UBQLN2 Mutations Outside the PXX Domain and a Pure FTD Phenotype. Neurobiol. Aging 2012, 33, 2949.e13–2949.e17. [Google Scholar] [CrossRef]
- Deng, H.-X.; Chen, W.; Hong, S.-T.; Boycott, K.M.; Gorrie, G.H.; Siddique, N.; Yang, Y.; Fecto, F.; Shi, Y.; Zhai, H.; et al. Mutations in UBQLN2 Cause Dominant X-Linked Juvenile and Adult-Onset ALS and ALS/Dementia. Nature 2011, 477, 211–215. [Google Scholar] [CrossRef]
- Zivony-Elboum, Y.; Westbroek, W.; Kfir, N.; Savitzki, D.; Shoval, Y.; Bloom, A.; Rod, R.; Khayat, M.; Gross, B.; Samri, W.; et al. A Founder Mutation in Vps37A Causes Autosomal Recessive Complex Hereditary Spastic Paraparesis. J. Med. Genet. 2012, 49, 462–472. [Google Scholar] [CrossRef] [PubMed]
- Rodger, C.; Flex, E.; Allison, R.J.; Sanchis-Juan, A.; Hasenahuer, M.A.; Cecchetti, S.; French, C.E.; Edgar, J.R.; Carpentieri, G.; Ciolfi, A.; et al. De Novo VPS4A Mutations Cause Multisystem Disease with Abnormal Neurodevelopment. Am. J. Hum. Genet. 2020, 107, 1129–1148. [Google Scholar] [CrossRef] [PubMed]
- Mochida, G.H.; Ganesh, V.S.; de Michelena, M.I.; Dias, H.; Atabay, K.D.; Kathrein, K.L.; Huang, H.-T.; Hill, R.S.; Felie, J.M.; Rakiec, D.; et al. CHMP1A Encodes an Essential Regulator of BMI1-INK4A in Cerebellar Development. Nat. Genet. 2012, 44, 1260–1264. [Google Scholar] [CrossRef]
- Parkinson, N.; Ince, P.G.; Smith, M.O.; Highley, R.; Skibinski, G.; Andersen, P.M.; Morrison, K.E.; Pall, H.S.; Hardiman, O.; Collinge, J.; et al. ALS Phenotypes with Mutations in CHMP2B (Charged Multivesicular Body Protein 2B). Neurology 2006, 67, 1074–1077. [Google Scholar] [CrossRef] [PubMed]
- Skibinski, G.; Parkinson, N.J.; Brown, J.M.; Chakrabarti, L.; Lloyd, S.L.; Hummerich, H.; Nielsen, J.E.; Hodges, J.R.; Spillantini, M.G.; Thusgaard, T.; et al. Mutations in the Endosomal ESCRTIII-Complex Subunit CHMP2B in Frontotemporal Dementia. Nat. Genet. 2005, 37, 806–808. [Google Scholar] [CrossRef]
- Filimonenko, M.; Stuffers, S.; Raiborg, C.; Yamamoto, A.; Malerød, L.; Fisher, E.M.C.; Isaacs, A.; Brech, A.; Stenmark, H.; Simonsen, A. Functional Multivesicular Bodies Are Required for Autophagic Clearance of Protein Aggregates Associated with Neurodegenerative Disease. J. Cell Biol. 2007, 179, 485–500. [Google Scholar] [CrossRef]
- Lee, J.-A.; Beigneux, A.; Ahmad, S.T.; Young, S.G.; Gao, F.-B. ESCRT-III Dysfunction Causes Autophagosome Accumulation and Neurodegeneration. Curr. Biol. 2007, 17, 1561–1567. [Google Scholar] [CrossRef]
- Lee, J.-A.; Gao, F.-B. Inhibition of Autophagy Induction Delays Neuronal Cell Loss Caused by Dysfunctional ESCRT-III in Frontotemporal Dementia. J. Neurosci. 2009, 29, 8506–8511. [Google Scholar] [CrossRef]
- Verhoeven, K.; De Jonghe, P.; Coen, K.; Verpoorten, N.; Auer-Grumbach, M.; Kwon, J.M.; FitzPatrick, D.; Schmedding, E.; De Vriendt, E.; Jacobs, A.; et al. Mutations in the Small GTP-Ase Late Endosomal Protein RAB7 Cause Charcot-Marie-Tooth Type 2B Neuropathy. Am. J. Hum. Genet. 2003, 72, 722–727. [Google Scholar] [CrossRef]
- Romano, R.; Del Fiore, V.S.; Saveri, P.; Palamà, I.E.; Pisciotta, C.; Pareyson, D.; Bucci, C.; Guerra, F. Autophagy and Lysosomal Functionality in CMT2B Fibroblasts Carrying the RAB7K126R Mutation. Cells 2022, 11, 496. [Google Scholar] [CrossRef]
- Cullup, T.; Kho, A.L.; Dionisi-Vici, C.; Brandmeier, B.; Smith, F.; Urry, Z.; Simpson, M.A.; Yau, S.; Bertini, E.; McClelland, V.; et al. Recessive Mutations in EPG5 Cause Vici Syndrome, a Multisystem Disorder with Defective Autophagy. Nat. Genet. 2013, 45, 83–87. [Google Scholar] [CrossRef]
- Ji, C.; Zhao, H.; Chen, D.; Zhang, H.; Zhao, Y.G. β-Propeller Proteins WDR45 and WDR45B Regulate Autophagosome Maturation into Autolysosomes in Neural Cells. Curr. Biol. 2021, 31, 1666–1677.e6. [Google Scholar] [CrossRef]
- Akizu, N.; Cantagrel, V.; Zaki, M.S.; Al-Gazali, L.; Wang, X.; Rosti, R.O.; Dikoglu, E.; Gelot, A.B.; Rosti, B.; Vaux, K.K.; et al. Biallelic Mutations in SNX14 Cause a Syndromic Form of Cerebellar Atrophy and Lysosome-Autophagosome Dysfunction. Nat. Genet. 2015, 47, 528–534. [Google Scholar] [CrossRef]
- Bryant, D.; Liu, Y.; Datta, S.; Hariri, H.; Seda, M.; Anderson, G.; Peskett, E.; Demetriou, C.; Sousa, S.; Jenkins, D.; et al. SNX14 Mutations Affect Endoplasmic Reticulum-Associated Neutral Lipid Metabolism in Autosomal Recessive Spinocerebellar Ataxia 20. Hum. Mol. Genet. 2018, 27, 1927–1940. [Google Scholar] [CrossRef]
- Thomas, A.C.; Williams, H.; Setó-Salvia, N.; Bacchelli, C.; Jenkins, D.; O’Sullivan, M.; Mengrelis, K.; Ishida, M.; Ocaka, L.; Chanudet, E.; et al. Mutations in SNX14 Cause a Distinctive Autosomal-Recessive Cerebellar Ataxia and Intellectual Disability Syndrome. Am. J. Hum. Genet. 2014, 95, 611–621. [Google Scholar] [CrossRef]
- Liang, C.-C.; Wang, C.; Peng, X.; Gan, B.; Guan, J.-L. Neural-Specific Deletion of FIP200 Leads to Cerebellar Degeneration Caused by Increased Neuronal Death and Axon Degeneration. J. Biol. Chem. 2010, 285, 3499–3509. [Google Scholar] [CrossRef]
- Wen, J.; Zellner, A.; Braun, N.C.; Bajaj, T.; Gassen, N.C.; Peitz, M.; Brüstle, O. Loss of Function of FIP200 in Human Pluripotent Stem Cell-Derived Neurons Leads to Axonal Pathology and Hyperactivity. Transl. Psychiatry 2023, 13, 143. [Google Scholar] [CrossRef]
- Maria Fimia, G.; Stoykova, A.; Romagnoli, A.; Giunta, L.; Di Bartolomeo, S.; Nardacci, R.; Corazzari, M.; Fuoco, C.; Ucar, A.; Schwartz, P.; et al. Ambra1 Regulates Autophagy and Development of the Nervous System. Nature 2007, 447, 1121–1125. [Google Scholar] [CrossRef]
- Mattera, R.; Park, S.Y.; De Pace, R.; Guardia, C.M.; Bonifacino, J.S. AP-4 Mediates Export of ATG9A from the Trans -Golgi Network to Promote Autophagosome Formation. Proc. Natl. Acad. Sci. USA 2017, 114, E10697–E10706. [Google Scholar] [CrossRef]
- De Pace, R.; Skirzewski, M.; Damme, M.; Mattera, R.; Mercurio, J.; Foster, A.M.; Cuitino, L.; Jarnik, M.; Hoffmann, V.; Morris, H.D.; et al. Altered Distribution of ATG9A and Accumulation of Axonal Aggregates in Neurons from a Mouse Model of AP-4 Deficiency Syndrome. PLoS Genet. 2018, 14, e1007363. [Google Scholar] [CrossRef]
- Behne, R.; Teinert, J.; Wimmer, M.; D’Amore, A.; Davies, A.K.; Scarrott, J.M.; Eberhardt, K.; Brechmann, B.; Chen, I.P.-F.; Buttermore, E.D.; et al. Adaptor Protein Complex 4 Deficiency: A Paradigm of Childhood-Onset Hereditary Spastic Paraplegia Caused by Defective Protein Trafficking. Hum. Mol. Genet. 2020, 29, 320–334. [Google Scholar] [CrossRef]
- Mizushima, N.; Noda, T.; Yoshimori, T.; Tanaka, Y.; Ishii, T.; George, M.D.; Klionsky, D.J.; Ohsumi, M.; Ohsumi, Y. A Protein Conjugation System Essential for Autophagy. Nature 1998, 395, 395–398. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Dalton, V.M.; Eggerton, K.P.; Scott, S.V.; Klionsky, D.J. Apg7p/Cvt2p Is Required for the Cytoplasm-to-Vacuole Targeting, Macroautophagy, and Peroxisome Degradation Pathways. Mol. Biol. Cell 1999, 10, 1337–1351. [Google Scholar] [CrossRef] [PubMed]
- Shintani, T.; Mizushima, N.; Ogawa, Y.; Matsuura, A.; Noda, T.; Ohsumi, Y. Apg10p, a Novel Protein-Conjugating Enzyme Essential for Autophagy in Yeast. EMBO J. 1999, 18, 5234–5241. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, T.; Kaizuka, T.; Cadwell, K.; Sahani, M.H.; Saitoh, T.; Akira, S.; Virgin, H.W.; Mizushima, N. FIP200 Regulates Targeting of Atg16L1 to the Isolation Membrane. EMBO Rep. 2013, 14, 284–291. [Google Scholar] [CrossRef] [PubMed]
- Kuma, A.; Mizushima, N.; Ishihara, N.; Ohsumi, Y. Formation of the Approximately 350-kDa Apg12-Apg5.Apg16 Multimeric Complex, Mediated by Apg16 Oligomerization, Is Essential for Autophagy in Yeast. J. Biol. Chem. 2002, 277, 18619–18625. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Kuma, A.; Kobayashi, Y.; Yamamoto, A.; Matsubae, M.; Takao, T.; Natsume, T.; Ohsumi, Y.; Yoshimori, T. Mouse Apg16L, a Novel WD-Repeat Protein, Targets to the Autophagic Isolation Membrane with the Apg12-Apg5 Conjugate. J. Cell Sci. 2003, 116, 1679–1688. [Google Scholar] [CrossRef]
- Kirisako, T.; Ichimura, Y.; Okada, H.; Kabeya, Y.; Mizushima, N.; Yoshimori, T.; Ohsumi, M.; Takao, T.; Noda, T.; Ohsumi, Y. The Reversible Modification Regulates the Membrane-Binding State of Apg8/Aut7 Essential for Autophagy and the Cytoplasm to Vacuole Targeting Pathway. J. Cell Biol. 2000, 151, 263–276. [Google Scholar] [CrossRef]
- Ichimura, Y.; Kirisako, T.; Takao, T.; Satomi, Y.; Shimonishi, Y.; Ishihara, N.; Mizushima, N.; Tanida, I.; Kominami, E.; Ohsumi, M.; et al. A Ubiquitin-like System Mediates Protein Lipidation. Nature 2000, 408, 488–492. [Google Scholar] [CrossRef]
- Hanada, T.; Noda, N.N.; Satomi, Y.; Ichimura, Y.; Fujioka, Y.; Takao, T.; Inagaki, F.; Ohsumi, Y. The Atg12-Atg5 Conjugate Has a Novel E3-like Activity for Protein Lipidation in Autophagy. J. Biol. Chem. 2007, 282, 37298–37302. [Google Scholar] [CrossRef]
- Hanada, T.; Satomi, Y.; Takao, T.; Ohsumi, Y. The Amino-Terminal Region of Atg3 Is Essential for Association with Phosphatidylethanolamine in Atg8 Lipidation. FEBS Lett. 2009, 583, 1078–1083. [Google Scholar] [CrossRef] [PubMed]
- Fujita, N.; Itoh, T.; Omori, H.; Fukuda, M.; Noda, T.; Yoshimori, T. The Atg16L Complex Specifies the Site of LC3 Lipidation for Membrane Biogenesis in Autophagy. Mol. Biol. Cell 2008, 19, 2092–2100. [Google Scholar] [CrossRef]
- Komatsu, M.; Wang, Q.J.; Holstein, G.R.; Friedrich, V.L.; Iwata, J.; Kominami, E.; Chait, B.T.; Tanaka, K.; Yue, Z. Essential Role for Autophagy Protein Atg7 in the Maintenance of Axonal Homeostasis and the Prevention of Axonal Degeneration. Proc. Natl. Acad. Sci. USA 2007, 104, 14489–14494. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of Basal Autophagy in Neural Cells Causes Neurodegenerative Disease in Mice. Nature 2006, 441, 885–889. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.G.; Sun, L.; Miao, G.; Ji, C.; Zhao, H.; Sun, H.; Miao, L.; Yoshii, S.R.; Mizushima, N.; Wang, X.; et al. The Autophagy Gene Wdr45/Wipi4 Regulates Learning and Memory Function and Axonal Homeostasis. Autophagy 2015, 11, 881–890. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Wang, X.; Feng, X.; Zhang, A.; Li, J.; Gu, K.; Huang, J.; Pang, S.; Dong, H.; Gao, H.; et al. Altered Expression of Autophagic Genes in the Peripheral Leukocytes of Patients with Sporadic Parkinson’s Disease. Brain Res. 2011, 1394, 105–111. [Google Scholar] [CrossRef]
- N’Diaye, E.-N.; Debnath, J.; Brown, E.J. Ubiquilins Accelerate Autophagosome Maturation and Promote Cell Survival during Nutrient Starvation. Autophagy 2009, 5, 573–575. [Google Scholar] [CrossRef]
- Rothenberg, C.; Srinivasan, D.; Mah, L.; Kaushik, S.; Peterhoff, C.M.; Ugolino, J.; Fang, S.; Cuervo, A.M.; Nixon, R.A.; Monteiro, M.J. Ubiquilin Functions in Autophagy and Is Degraded by Chaperone-Mediated Autophagy. Hum. Mol. Genet. 2010, 19, 3219–3232. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Monteiro, M.J. Ubiquilin Interacts and Enhances the Degradation of Expanded-Polyglutamine Proteins. Biochem. Biophys. Res. Commun. 2007, 360, 423–427. [Google Scholar] [CrossRef]
- Rutherford, N.J.; Lewis, J.; Clippinger, A.K.; Thomas, M.A.; Adamson, J.; Cruz, P.E.; Cannon, A.; Xu, G.; Golde, T.E.; Shaw, G.; et al. Unbiased Screen Reveals Ubiquilin-1 and -2 Highly Associated with Huntingtin Inclusions. Brain Res. 2013, 1524, 62–73. [Google Scholar] [CrossRef]
- Khaminets, A.; Behl, C.; Dikic, I. Ubiquitin-Dependent And Independent Signals In Selective Autophagy. Trends Cell Biol. 2016, 26, 6–16. [Google Scholar] [CrossRef] [PubMed]
- Adriaenssens, E.; Ferrari, L.; Martens, S. Orchestration of Selective Autophagy by Cargo Receptors. Curr. Biol. 2022, 32, R1357–R1371. [Google Scholar] [CrossRef] [PubMed]
- Bjørkøy, G.; Lamark, T.; Brech, A.; Outzen, H.; Perander, M.; Øvervatn, A.; Stenmark, H.; Johansen, T. P62/SQSTM1 Forms Protein Aggregates Degraded by Autophagy and Has a Protective Effect on Huntingtin-Induced Cell Death. J. Cell Biol. 2005, 171, 603–614. [Google Scholar] [CrossRef]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.-A.; Outzen, H.; Øvervatn, A.; Bjørkøy, G.; Johansen, T. P62/SQSTM1 Binds Directly to Atg8/LC3 to Facilitate Degradation of Ubiquitinated Protein Aggregates by Autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef] [PubMed]
- Lamark, T.; Svenning, S.; Johansen, T. Regulation of Selective Autophagy: The P62/SQSTM1 Paradigm. Essays Biochem. 2017, 61, 609–624. [Google Scholar] [CrossRef] [PubMed]
- Simonsen, A.; Birkeland, H.C.G.; Gillooly, D.J.; Mizushima, N.; Kuma, A.; Yoshimori, T.; Slagsvold, T.; Brech, A.; Stenmark, H. Alfy, a Novel FYVE-Domain-Containing Protein Associated with Protein Granules and Autophagic Membranes. J. Cell Sci. 2004, 117, 4239–4251. [Google Scholar] [CrossRef] [PubMed]
- Clausen, T.H.; Lamark, T.; Isakson, P.; Finley, K.D.; Larsen, K.B.; Brech, A.; Øvervatn, A.; Stenmark, H.; Bjørkøy, G.; Simonsen, A.; et al. P62/SQSTM1 and ALFY Interact to Facilitate the Formation of P62 Bodies/ALIS and Their Degradation by Autophagy. Autophagy 2010, 6, 330–344. [Google Scholar] [CrossRef]
- Filimonenko, M.; Isakson, P.; Finley, K.D.; Anderson, M.; Jeong, H.; Melia, T.J.; Bartlett, B.J.; Myers, K.M.; Birkeland, H.C.G.; Lamark, T.; et al. The Selective Macroautophagic Degradation of Aggregated Proteins Requires the PI3P-Binding Protein Alfy. Mol. Cell 2010, 38, 265–279. [Google Scholar] [CrossRef]
- Deng, Z.; Purtell, K.; Lachance, V.; Wold, M.S.; Chen, S.; Yue, Z. Autophagy Receptors and Neurodegenerative Diseases. Trends Cell Biol. 2017, 27, 491–504. [Google Scholar] [CrossRef]
- Rué, L.; López-Soop, G.; Gelpi, E.; Martínez-Vicente, M.; Alberch, J.; Pérez-Navarro, E. Brain Region- and Age-Dependent Dysregulation of P62 and NBR1 in a Mouse Model of Huntington’s Disease. Neurobiol. Dis. 2013, 52, 219–228. [Google Scholar] [CrossRef]
- Wu, J.J.; Cai, A.; Greenslade, J.E.; Higgins, N.R.; Fan, C.; Le, N.T.T.; Tatman, M.; Whiteley, A.M.; Prado, M.A.; Dieriks, B.V.; et al. ALS/FTD Mutations in UBQLN2 Impede Autophagy by Reducing Autophagosome Acidification through Loss of Function. Proc. Natl. Acad. Sci. USA 2020, 117, 15230–15241. [Google Scholar] [CrossRef]
- Le, N.T.T.; Chang, L.; Kovlyagina, I.; Georgiou, P.; Safren, N.; Braunstein, K.E.; Kvarta, M.D.; Van Dyke, A.M.; LeGates, T.A.; Philips, T.; et al. Motor Neuron Disease, TDP-43 Pathology, and Memory Deficits in Mice Expressing ALS–FTD-Linked UBQLN2 Mutations. Proc. Natl. Acad. Sci. USA 2016, 113, E7580–E7589. [Google Scholar] [CrossRef]
- Jiang, W.; Chen, X.; Ji, C.; Zhang, W.; Song, J.; Li, J.; Wang, J. Key Regulators of Autophagosome Closure. Cells 2021, 10, 2814. [Google Scholar] [CrossRef]
- Sou, Y.; Waguri, S.; Iwata, J.; Ueno, T.; Fujimura, T.; Hara, T.; Sawada, N.; Yamada, A.; Mizushima, N.; Uchiyama, Y.; et al. The Atg8 Conjugation System Is Indispensable for Proper Development of Autophagic Isolation Membranes in Mice. Mol. Biol. Cell 2008, 19, 4762–4775. [Google Scholar] [CrossRef]
- Velikkakath, A.K.G.; Nishimura, T.; Oita, E.; Ishihara, N.; Mizushima, N. Mammalian Atg2 Proteins Are Essential for Autophagosome Formation and Important for Regulation of Size and Distribution of Lipid Droplets. Mol. Biol. Cell 2012, 23, 896–909. [Google Scholar] [CrossRef]
- Bozic, M.; van den Bekerom, L.; Milne, B.A.; Goodman, N.; Roberston, L.; Prescott, A.R.; Macartney, T.J.; Dawe, N.; McEwan, D.G. A Conserved ATG2-GABARAP Family Interaction Is Critical for Phagophore Formation. EMBO Rep. 2020, 21, e48412. [Google Scholar] [CrossRef]
- Takahashi, Y.; Liang, X.; Hattori, T.; Tang, Z.; He, H.; Chen, H.; Liu, X.; Abraham, T.; Imamura-Kawasawa, Y.; Buchkovich, N.J.; et al. VPS37A Directs ESCRT Recruitment for Phagophore Closure. J. Cell Biol. 2019, 218, 3336–3354. [Google Scholar] [CrossRef]
- Takahashi, Y.; He, H.; Tang, Z.; Hattori, T.; Liu, Y.; Young, M.M.; Serfass, J.M.; Chen, L.; Gebru, M.; Chen, C.; et al. An Autophagy Assay Reveals the ESCRT-III Component CHMP2A as a Regulator of Phagophore Closure. Nat. Commun. 2018, 9, 2855. [Google Scholar] [CrossRef]
- Zhou, F.; Wu, Z.; Zhao, M.; Murtazina, R.; Cai, J.; Zhang, A.; Li, R.; Sun, D.; Li, W.; Zhao, L.; et al. Rab5-Dependent Autophagosome Closure by ESCRT. J. Cell Biol. 2019, 218, 1908–1927. [Google Scholar] [CrossRef]
- Itakura, E.; Kishi-Itakura, C.; Mizushima, N. The Hairpin-Type Tail-Anchored SNARE Syntaxin 17 Targets to Autophagosomes for Fusion with Endosomes/Lysosomes. Cell 2012, 151, 1256–1269. [Google Scholar] [CrossRef]
- Furuta, N.; Fujita, N.; Noda, T.; Yoshimori, T.; Amano, A. Combinational Soluble N-Ethylmaleimide-Sensitive Factor Attachment Protein Receptor Proteins VAMP8 and Vti1b Mediate Fusion of Antimicrobial and Canonical Autophagosomes with Lysosomes. Mol. Biol. Cell 2010, 21, 1001–1010. [Google Scholar] [CrossRef]
- Diao, J.; Liu, R.; Rong, Y.; Zhao, M.; Zhang, J.; Lai, Y.; Zhou, Q.; Wilz, L.M.; Li, J.; Vivona, S.; et al. ATG14 Promotes Membrane Tethering and Fusion of Autophagosomes to Endolysosomes. Nature 2015, 520, 563–566. [Google Scholar] [CrossRef]
- Liu, R.; Zhi, X.; Zhong, Q. ATG14 Controls SNARE-Mediated Autophagosome Fusion with a Lysosome. Autophagy 2015, 11, 847–849. [Google Scholar] [CrossRef]
- Wang, Z.; Miao, G.; Xue, X.; Guo, X.; Yuan, C.; Wang, Z.; Zhang, G.; Chen, Y.; Feng, D.; Hu, J.; et al. The Vici Syndrome Protein EPG5 Is a Rab7 Effector That Determines the Fusion Specificity of Autophagosomes with Late Endosomes/Lysosomes. Mol. Cell 2016, 63, 781–795. [Google Scholar] [CrossRef]
- Tian, Y.; Li, Z.; Hu, W.; Ren, H.; Tian, E.; Zhao, Y.; Lu, Q.; Huang, X.; Yang, P.; Li, X.; et al. C. elegans Screen Identifies Autophagy Genes Specific to Multicellular Organisms. Cell 2010, 141, 1042–1055. [Google Scholar] [CrossRef]
- Zhao, H.; Zhao, Y.G.; Wang, X.; Xu, L.; Miao, L.; Feng, D.; Chen, Q.; Kovács, A.L.; Fan, D.; Zhang, H. Mice Deficient in Epg5 Exhibit Selective Neuronal Vulnerability to Degeneration. J. Cell Biol. 2013, 200, 731–741. [Google Scholar] [CrossRef]
- Metcalf, D.; Isaacs, A.M. The Role of ESCRT Proteins in Fusion Events Involving Lysosomes, Endosomes and Autophagosomes. Biochem. Soc. Trans. 2010, 38, 1469–1473. [Google Scholar] [CrossRef]
- Colecchia, D.; Stasi, M.; Leonardi, M.; Manganelli, F.; Nolano, M.; Veneziani, B.M.; Santoro, L.; Eskelinen, E.-L.; Chiariello, M.; Bucci, C. Alterations of Autophagy in the Peripheral Neuropathy Charcot-Marie-Tooth Type 2B. Autophagy 2018, 14, 930–941. [Google Scholar] [CrossRef]
- Dionisi Vici, C.; Sabetta, G.; Gambarara, M.; Vigevano, F.; Bertini, E.; Boldrini, R.; Parisi, S.G.; Quinti, I.; Aiuti, F.; Fiorilli, M. Agenesis of the Corpus Callosum, Combined Immunodeficiency, Bilateral Cataract, and Hypopigmentation in Two Brothers. Am. J. Med. Genet. 1988, 29, 1–8. [Google Scholar] [CrossRef]
- Maday, S.; Holzbaur, E.L.F. Compartment-Specific Regulation of Autophagy in Primary Neurons. J. Neurosci. 2016, 36, 5933–5945. [Google Scholar] [CrossRef]
- Bunge, M.B. Fine structure of nerve fibers and growth cones of isolated sympathetic neurons in culture. J. Cell Biol. 1973, 56, 713–735. [Google Scholar] [CrossRef] [PubMed]
- Hollenbeck, P.J. Products of Endocytosis and Autophagy Are Retrieved from Axons by Regulated Retrograde Organelle Transport. J. Cell Biol. 1993, 121, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Maday, S.; Holzbaur, E.L.F. Autophagosome Assembly and Cargo Capture in the Distal Axon. Autophagy 2012, 8, 858–860. [Google Scholar] [CrossRef] [PubMed]
- Matoba, K.; Kotani, T.; Tsutsumi, A.; Tsuji, T.; Mori, T.; Noshiro, D.; Sugita, Y.; Nomura, N.; Iwata, S.; Ohsumi, Y.; et al. Atg9 Is a Lipid Scramblase That Mediates Autophagosomal Membrane Expansion. Nat. Struct. Mol. Biol. 2020, 27, 1185–1193. [Google Scholar] [CrossRef] [PubMed]
- Ivankovic, D.; Drew, J.; Lesept, F.; White, I.J.; López Doménech, G.; Tooze, S.A.; Kittler, J.T. Axonal Autophagosome Maturation Defect through Failure of ATG9A Sorting Underpins Pathology in AP-4 Deficiency Syndrome. Autophagy 2020, 16, 391–407. [Google Scholar] [CrossRef]
- Yang, S.; Park, D.; Manning, L.; Hill, S.E.; Cao, M.; Xuan, Z.; Gonzalez, I.; Dong, Y.; Clark, B.; Shao, L.; et al. Presynaptic Autophagy Is Coupled to the Synaptic Vesicle Cycle via ATG-9. Neuron 2022, 110, 824–840.e10. [Google Scholar] [CrossRef]
- Stavoe, A.K.H.; Hill, S.E.; Hall, D.H.; Colón-Ramos, D.A. KIF1A/UNC-104 Transports ATG-9 to Regulate Neurodevelopment and Autophagy at Synapses. Dev. Cell 2016, 38, 171–185. [Google Scholar] [CrossRef]
- Xuan, Z.; Yang, S.; Clark, B.; Hill, S.E.; Manning, L.; Colón-Ramos, D.A. The Active Zone Protein Clarinet Regulates Synaptic Sorting of ATG-9 and Presynaptic Autophagy. PLoS Biol. 2023, 21, e3002030. [Google Scholar] [CrossRef]
- Maday, S.; Holzbaur, E.L.F. Autophagosome Biogenesis in Primary Neurons Follows an Ordered and Spatially Regulated Pathway. Dev. Cell 2014, 30, 71–85. [Google Scholar] [CrossRef]
- Okerlund, N.D.; Schneider, K.; Leal-Ortiz, S.; Montenegro-Venegas, C.; Kim, S.A.; Garner, L.C.; Waites, C.L.; Gundelfinger, E.D.; Reimer, R.J.; Garner, C.C. Bassoon Controls Presynaptic Autophagy through Atg5. Neuron 2017, 93, 897–913.e7. [Google Scholar] [CrossRef]
- Stavoe, A.K.; Gopal, P.P.; Gubas, A.; Tooze, S.A.; Holzbaur, E.L. Expression of WIPI2B Counteracts Age-Related Decline in Autophagosome Biogenesis in Neurons. eLife 2019, 8, e44219. [Google Scholar] [CrossRef]
- Soukup, S.-F.; Kuenen, S.; Vanhauwaert, R.; Manetsberger, J.; Hernández-Díaz, S.; Swerts, J.; Schoovaerts, N.; Vilain, S.; Gounko, N.V.; Vints, K.; et al. A LRRK2-Dependent EndophilinA Phosphoswitch Is Critical for Macroautophagy at Presynaptic Terminals. Neuron 2016, 92, 829–844. [Google Scholar] [CrossRef] [PubMed]
- Murdoch, J.D.; Rostosky, C.M.; Gowrisankaran, S.; Arora, A.S.; Soukup, S.-F.; Vidal, R.; Capece, V.; Freytag, S.; Fischer, A.; Verstreken, P.; et al. Endophilin-A Deficiency Induces the Foxo3a-Fbxo32 Network in the Brain and Causes Dysregulation of Autophagy and the Ubiquitin-Proteasome System. Cell Rep. 2016, 17, 1071–1086. [Google Scholar] [CrossRef] [PubMed]
- Vanhauwaert, R.; Kuenen, S.; Masius, R.; Bademosi, A.; Manetsberger, J.; Schoovaerts, N.; Bounti, L.; Gontcharenko, S.; Swerts, J.; Vilain, S.; et al. The SAC1 Domain in Synaptojanin Is Required for Autophagosome Maturation at Presynaptic Terminals. EMBO J. 2017, 36, 1392–1411. [Google Scholar] [CrossRef] [PubMed]
- Zimprich, A.; Biskup, S.; Leitner, P.; Lichtner, P.; Farrer, M.; Lincoln, S.; Kachergus, J.; Hulihan, M.; Uitti, R.J.; Calne, D.B.; et al. Mutations in LRRK2 Cause Autosomal-Dominant Parkinsonism with Pleomorphic Pathology. Neuron 2004, 44, 601–607. [Google Scholar] [CrossRef] [PubMed]
- Matta, S.; Van Kolen, K.; da Cunha, R.; van den Bogaart, G.; Mandemakers, W.; Miskiewicz, K.; De Bock, P.-J.; Morais, V.A.; Vilain, S.; Haddad, D.; et al. LRRK2 Controls an EndoA Phosphorylation Cycle in Synaptic Endocytosis. Neuron 2012, 75, 1008–1021. [Google Scholar] [CrossRef] [PubMed]
- Bademosi, A.T.; Decet, M.; Kuenen, S.; Calatayud, C.; Swerts, J.; Gallego, S.F.; Schoovaerts, N.; Karamanou, S.; Louros, N.; Martin, E.; et al. EndophilinA-Dependent Coupling between Activity-Induced Calcium Influx and Synaptic Autophagy Is Disrupted by a Parkinson-Risk Mutation. Neuron 2023, 111, 1402–1422.e13. [Google Scholar] [CrossRef]
- Decet, M.; Soukup, S.-F. Endophilin-A/SH3GL2 Calcium Switch for Synaptic Autophagy Induction Is Impaired by a Parkinson’s Risk Variant. Autophagy, 2023; 1–3, online ahead of print. [Google Scholar] [CrossRef]
- Maday, S.; Wallace, K.E.; Holzbaur, E.L.F. Autophagosomes Initiate Distally and Mature during Transport toward the Cell Soma in Primary Neurons. J. Cell Biol. 2012, 196, 407–417. [Google Scholar] [CrossRef]
- Lee, S.; Sato, Y.; Nixon, R.A. Lysosomal Proteolysis Inhibition Selectively Disrupts Axonal Transport of Degradative Organelles and Causes an Alzheimer’s-Like Axonal Dystrophy. J. Neurosci. 2011, 31, 7817–7830. [Google Scholar] [CrossRef]
- Cheng, X.-T.; Zhou, B.; Lin, M.-Y.; Cai, Q.; Sheng, Z.-H. Axonal Autophagosomes Recruit Dynein for Retrograde Transport through Fusion with Late Endosomes. J. Cell Biol. 2015, 209, 377–386. [Google Scholar] [CrossRef]
- Calderilla-Barbosa, L.; Seibenhener, M.L.; Du, Y.; Diaz-Meco, M.-T.; Moscat, J.; Yan, J.; Wooten, M.W.; Wooten, M.C. Interaction of SQSTM1 with the Motor Protein Dynein: SQSTM1 Is Required for Normal Dynein Function and Trafficking. J. Cell Sci. 2014, 127, 4052–4063. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Fang, Y.; Cheng, X.; Lian, Y.; Xu, H. Interaction between TRPML1 and P62 in Regulating Autophagosome-Lysosome Fusion and Impeding Neuroaxonal Dystrophy in Alzheimer’s Disease. Oxid. Med. Cell. Longev. 2022, 2022, 8096009. [Google Scholar] [CrossRef] [PubMed]
- Overly, C.C.; Lee, K.D.; Berthiaume, E.; Hollenbeck, P.J. Quantitative Measurement of Intraorganelle pH in the Endosomal-Lysosomal Pathway in Neurons by Using Ratiometric Imaging with Pyranine. Proc. Natl. Acad. Sci. USA 1995, 92, 3156–3160. [Google Scholar] [CrossRef] [PubMed]
- Aplin, A.; Jasionowski, T.; Tuttle, D.L.; Lenk, S.E.; Dunn, W.A. Cytoskeletal Elements Are Required for the Formation and Maturation of Autophagic Vacuoles. J. Cell. Physiol. 1992, 152, 458–466. [Google Scholar] [CrossRef] [PubMed]
- Köchl, R.; Hu, X.W.; Chan, E.Y.W.; Tooze, S.A. Microtubules Facilitate Autophagosome Formation and Fusion of Autophagosomes with Endosomes. Traffic 2006, 7, 129–145. [Google Scholar] [CrossRef]
- Fu, M.; Nirschl, J.J.; Holzbaur, E.L.F. LC3 Binding to the Scaffolding Protein JIP1 Regulates Processive Dynein-Driven Transport of Autophagosomes. Dev. Cell 2014, 29, 577–590. [Google Scholar] [CrossRef]
- Wong, Y.C.; Holzbaur, E.L.F. The Regulation of Autophagosome Dynamics by Huntingtin and HAP1 Is Disrupted by Expression of Mutant Huntingtin, Leading to Defective Cargo Degradation. J. Neurosci. 2014, 34, 1293–1305. [Google Scholar] [CrossRef]
- Cason, S.E.; Carman, P.J.; Van Duyne, C.; Goldsmith, J.; Dominguez, R.; Holzbaur, E.L.F. Sequential Dynein Effectors Regulate Axonal Autophagosome Motility in a Maturation-Dependent Pathway. J. Cell Biol. 2021, 220, e202010179. [Google Scholar] [CrossRef]
- Nieto-Torres, J.L.; Shanahan, S.-L.; Chassefeyre, R.; Chaiamarit, T.; Zaretski, S.; Landeras-Bueno, S.; Verhelle, A.; Encalada, S.E.; Hansen, M. LC3B Phosphorylation Regulates FYCO1 Binding and Directional Transport of Autophagosomes. Curr. Biol. CB 2021, 31, 3440–3449.e7. [Google Scholar] [CrossRef]
- Pankiv, S.; Alemu, E.A.; Brech, A.; Bruun, J.-A.; Lamark, T.; Øvervatn, A.; Bjørkøy, G.; Johansen, T. FYCO1 Is a Rab7 Effector That Binds to LC3 and PI3P to Mediate Microtubule plus End–Directed Vesicle Transport. J. Cell Biol. 2010, 188, 253–269. [Google Scholar] [CrossRef]
- Jordens, I.; Fernandez-Borja, M.; Marsman, M.; Dusseljee, S.; Janssen, L.; Calafat, J.; Janssen, H.; Wubbolts, R.; Neefjes, J. The Rab7 Effector Protein RILP Controls Lysosomal Transport by Inducing the Recruitment of Dynein-Dynactin Motors. Curr. Biol. 2001, 11, 1680–1685. [Google Scholar] [CrossRef]
- Wijdeven, R.H.; Janssen, H.; Nahidiazar, L.; Janssen, L.; Jalink, K.; Berlin, I.; Neefjes, J. Cholesterol and ORP1L-Mediated ER Contact Sites Control Autophagosome Transport and Fusion with the Endocytic Pathway. Nat. Commun. 2016, 7, 11808. [Google Scholar] [CrossRef] [PubMed]
- Fu, M.; Holzbaur, E.L.F. JIP1 Regulates the Directionality of APP Axonal Transport by Coordinating Kinesin and Dynein Motors. J. Cell Biol. 2013, 202, 495–508. [Google Scholar] [CrossRef] [PubMed]
- Fu, M.; Holzbaur, E.L.F. MAPK8IP1/JIP1 Regulates the Trafficking of Autophagosomes in Neurons. Autophagy 2014, 10, 2079–2081. [Google Scholar] [CrossRef] [PubMed]
- Brown, H.M.; Van Epps, H.A.; Goncharov, A.; Grant, B.D.; Jin, Y. The JIP3 Scaffold Protein UNC-16 Regulates RAB-5 Dependent Membrane Trafficking at C. elegans Synapses. Dev. Neurobiol. 2009, 69, 174–190. [Google Scholar] [CrossRef]
- Drerup, C.M.; Nechiporuk, A.V. JNK-Interacting Protein 3 Mediates the Retrograde Transport of Activated c-Jun N-Terminal Kinase and Lysosomes. PLoS Genet. 2013, 9, e1003303. [Google Scholar] [CrossRef] [PubMed]
- Gowrishankar, S.; Wu, Y.; Ferguson, S.M. Impaired JIP3-Dependent Axonal Lysosome Transport Promotes Amyloid Plaque Pathology. J. Cell Biol. 2017, 216, 3291–3305. [Google Scholar] [CrossRef]
- Hill, S.E.; Kauffman, K.J.; Krout, M.; Richmond, J.E.; Melia, T.J.; Colón-Ramos, D.A. Maturation and Clearance of Autophagosomes in Neurons Depends on a Specific Cysteine Protease Isoform, ATG-4.2. Dev. Cell 2019, 49, 251–266.e8. [Google Scholar] [CrossRef]
- Li, T.; Lu, D.; Yao, C.; Li, T.; Dong, H.; Li, Z.; Xu, G.; Chen, J.; Zhang, H.; Yi, X.; et al. Kansl1 Haploinsufficiency Impairs Autophagosome-Lysosome Fusion and Links Autophagic Dysfunction with Koolen-de Vries Syndrome in Mice. Nat. Commun. 2022, 13, 931. [Google Scholar] [CrossRef]
- Linda, K.; Lewerissa, E.I.; Verboven, A.H.A.; Gabriele, M.; Frega, M.; Klein Gunnewiek, T.M.; Devilee, L.; Ulferts, E.; Hommersom, M.; Oudakker, A.; et al. Imbalanced Autophagy Causes Synaptic Deficits in a Human Model for Neurodevelopmental Disorders. Autophagy 2022, 18, 423–442. [Google Scholar] [CrossRef]
- Nixon, R.A.; Wegiel, J.; Kumar, A.; Yu, W.H.; Peterhoff, C.; Cataldo, A.; Cuervo, A.M. Extensive Involvement of Autophagy in Alzheimer Disease: An Immuno-Electron Microscopy Study. J. Neuropathol. Exp. Neurol. 2005, 64, 10. [Google Scholar] [CrossRef] [PubMed]
- Tammineni, P.; Ye, X.; Feng, T.; Aikal, D.; Cai, Q. Impaired Retrograde Transport of Axonal Autophagosomes Contributes to Autophagic Stress in Alzheimer’s Disease Neurons. eLife 2017, 6, e21776. [Google Scholar] [CrossRef] [PubMed]
- Hafezparast, M.; Klocke, R.; Ruhrberg, C.; Marquardt, A.; Ahmad-Annuar, A.; Bowen, S.; Lalli, G.; Witherden, A.S.; Hummerich, H.; Nicholson, S.; et al. Mutations in Dynein Link Motor Neuron Degeneration to Defects in Retrograde Transport. Science 2003, 300, 808–812. [Google Scholar] [CrossRef]
- Ravikumar, B.; Acevedo-Arozena, A.; Imarisio, S.; Berger, Z.; Vacher, C.; O’Kane, C.J.; Brown, S.D.M.; Rubinsztein, D.C. Dynein Mutations Impair Autophagic Clearance of Aggregate-Prone Proteins. Nat. Genet. 2005, 37, 771–776. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Shi, J.-J.; Mao, C.-J.; Liu, S.; Wang, J.-D.; Chen, J.; Wang, F.; Yang, Y.-P.; Hu, W.-D.; Hu, L.-F.; et al. Alteration of Dynein Function Affects α-Synuclein Degradation via the Autophagosome-Lysosome Pathway. Int. J. Mol. Sci. 2013, 14, 24242–24254. [Google Scholar] [CrossRef] [PubMed]
- Poirier, K.; Lebrun, N.; Broix, L.; Tian, G.; Saillour, Y.; Boscheron, C.; Parrini, E.; Valence, S.; SaintPierre, B.; Oger, M.; et al. Mutations in TUBG1, DYNC1H1, KIF5C and KIF2A Cause Malformations of Cortical Development and Microcephaly. Nat. Genet. 2013, 45, 639–647. [Google Scholar] [CrossRef] [PubMed]
- Harms, M.B.; Ori-McKenney, K.M.; Scoto, M.; Tuck, E.P.; Bell, S.; Ma, D.; Masi, S.; Allred, P.; Al-Lozi, M.; Reilly, M.M.; et al. Mutations in the Tail Domain of DYNC1H1 Cause Dominant Spinal Muscular Atrophy. Neurology 2012, 78, 1714–1720. [Google Scholar] [CrossRef]
- Weedon, M.N.; Hastings, R.; Caswell, R.; Xie, W.; Paszkiewicz, K.; Antoniadi, T.; Williams, M.; King, C.; Greenhalgh, L.; Newbury-Ecob, R.; et al. Exome Sequencing Identifies a DYNC1H1 Mutation in a Large Pedigree with Dominant Axonal Charcot-Marie-Tooth Disease. Am. J. Hum. Genet. 2011, 89, 308–312. [Google Scholar] [CrossRef]
- Boecker, C.A.; Goldsmith, J.; Dou, D.; Cajka, G.G.; Holzbaur, E.L.F. Increased LRRK2 Kinase Activity Alters Neuronal Autophagy by Disrupting the Axonal Transport of Autophagosomes. Curr. Biol. 2021, 31, 2140–2154.e6. [Google Scholar] [CrossRef]
- Dou, D.; Smith, E.M.; Evans, C.S.; Boecker, C.A.; Holzbaur, E.L.F. Regulatory Imbalance between LRRK2 Kinase, PPM1H Phosphatase, and ARF6 GTPase Disrupts the Axonal Transport of Autophagosomes. Cell Rep. 2023, 42, 112448. [Google Scholar] [CrossRef]
- Sleigh, J.N.; Rossor, A.M.; Fellows, A.D.; Tosolini, A.P.; Schiavo, G. Axonal Transport and Neurological Disease. Nat. Rev. Neurol. 2019, 15, 691–703. [Google Scholar] [CrossRef] [PubMed]
- Perlson, E.; Maday, S.; Fu, M.; Moughamian, A.J.; Holzbaur, E.L.F. Retrograde Axonal Transport: Pathways To Cell Death? Trends Neurosci. 2010, 33, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Almeida-Souza, L.; Asselbergh, B.; d’Ydewalle, C.; Moonens, K.; Goethals, S.; de Winter, V.; Azmi, A.; Irobi, J.; Timmermans, J.-P.; Gevaert, K.; et al. Small Heat-Shock Protein HSPB1 Mutants Stabilize Microtubules in Charcot-Marie-Tooth Neuropathy. J. Neurosci. Off. J. Soc. Neurosci. 2011, 31, 15320–15328. [Google Scholar] [CrossRef]
- Zhang, X.; Qiao, Y.; Han, R.; Gao, Y.; Yang, X.; Zhang, Y.; Wan, Y.; Yu, W.; Pan, X.; Xing, J. A Charcot-Marie-Tooth-Causing Mutation in HSPB1 Decreases Cell Adaptation to Repeated Stress by Disrupting Autophagic Clearance of Misfolded Proteins. Cells 2022, 11, 2886. [Google Scholar] [CrossRef] [PubMed]
- Haidar, M.; Asselbergh, B.; Adriaenssens, E.; De Winter, V.; Timmermans, J.-P.; Auer-Grumbach, M.; Juneja, M.; Timmerman, V. Neuropathy-Causing Mutations in HSPB1 Impair Autophagy by Disturbing the Formation of SQSTM1/P62 Bodies. Autophagy 2019, 15, 1051–1068. [Google Scholar] [CrossRef]
- Gowrishankar, S.; Yuan, P.; Wu, Y.; Schrag, M.; Paradise, S.; Grutzendler, J.; De Camilli, P.; Ferguson, S.M. Massive Accumulation of Luminal Protease-Deficient Axonal Lysosomes at Alzheimer’s Disease Amyloid Plaques. Proc. Natl. Acad. Sci. USA 2015, 112, E3699–E3708. [Google Scholar] [CrossRef]
- Farfel-Becker, T.; Roney, J.C.; Cheng, X.-T.; Li, S.; Cuddy, S.R.; Sheng, Z.-H. Neuronal Soma-Derived Degradative Lysosomes Are Continuously Delivered to Distal Axons to Maintain Local Degradation Capacity. Cell Rep. 2019, 28, 51–64.e4. [Google Scholar] [CrossRef]
- Özkan, N.; Koppers, M.; van Soest, I.; van Harten, A.; Jurriens, D.; Liv, N.; Klumperman, J.; Kapitein, L.C.; Hoogenraad, C.C.; Farías, G.G. ER—Lysosome Contacts at a Pre-Axonal Region Regulate Axonal Lysosome Availability. Nat. Commun. 2021, 12, 4493. [Google Scholar] [CrossRef]
- Cheng, X.-T.; Xie, Y.-X.; Zhou, B.; Huang, N.; Farfel-Becker, T.; Sheng, Z.-H. Characterization of LAMP1-Labeled Nondegradative Lysosomal and Endocytic Compartments in Neurons. J. Cell Biol. 2018, 217, 3127–3139. [Google Scholar] [CrossRef]
- Bomont, P.; Cavalier, L.; Blondeau, F.; Hamida, C.B.; Belal, S.; Tazir, M.; Demir, E.; Topaloglu, H.; Korinthenberg, R.; Tüysüz, B.; et al. The Gene Encoding Gigaxonin, a New Member of the Cytoskeletal BTB/Kelch Repeat Family, Is Mutated in Giant Axonal Neuropathy. Nat. Genet. 2000, 26, 370–374. [Google Scholar] [CrossRef]
- Scrivo, A.; Codogno, P.; Bomont, P. Gigaxonin E3 Ligase Governs ATG16L1 Turnover to Control Autophagosome Production. Nat. Commun. 2019, 10, 780. [Google Scholar] [CrossRef]
- Cai, Q.; Zakaria, H.M.; Simone, A.; Sheng, Z.-H. Spatial Parkin Translocation and Degradation of Damaged Mitochondria via Mitophagy in Live Cortical Neurons. Curr. Biol. 2012, 22, 545–552. [Google Scholar] [CrossRef]
- Zheng, Y.; Zhang, X.; Wu, X.; Jiang, L.; Ahsan, A.; Ma, S.; Xiao, Z.; Han, F.; Qin, Z.-H.; Hu, W.; et al. Somatic Autophagy of Axonal Mitochondria in Ischemic Neurons. J. Cell Biol. 2019, 218, 1891–1907. [Google Scholar] [CrossRef]
- Shehata, M.; Matsumura, H.; Okubo-Suzuki, R.; Ohkawa, N.; Inokuchi, K. Neuronal Stimulation Induces Autophagy in Hippocampal Neurons That Is Involved in AMPA Receptor Degradation after Chemical Long-Term Depression. J. Neurosci. 2012, 32, 10413–10422. [Google Scholar] [CrossRef] [PubMed]
- Kallergi, E.; Daskalaki, A.-D.; Kolaxi, A.; Camus, C.; Ioannou, E.; Mercaldo, V.; Haberkant, P.; Stein, F.; Sidiropoulou, K.; Dalezios, Y.; et al. Dendritic Autophagy Degrades Postsynaptic Proteins and Is Required for Long-Term Synaptic Depression in Mice. Nat. Commun. 2022, 13, 680. [Google Scholar] [CrossRef] [PubMed]
- Baas, P.W.; Black, M.M.; Banker, G.A. Changes in Microtubule Polarity Orientation during the Development of Hippocampal Neurons in Culture. J. Cell Biol. 1989, 109, 3085–3094. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, V.V.; Anand, A.; Herr, J.B.; Miranda, C.; Vogel, M.C.; Maday, S. Synaptic Activity Controls Autophagic Vacuole Motility and Function in Dendrites. J. Cell Biol. 2021, 220, e202002084. [Google Scholar] [CrossRef] [PubMed]
- Goo, M.S.; Sancho, L.; Slepak, N.; Boassa, D.; Deerinck, T.J.; Ellisman, M.H.; Bloodgood, B.L.; Patrick, G.N. Activity-Dependent Trafficking of Lysosomes in Dendrites and Dendritic Spines. J. Cell Biol. 2017, 216, 2499–2513. [Google Scholar] [CrossRef]
- Yap, C.C.; Digilio, L.; McMahon, L.P.; Garcia, A.D.R.; Winckler, B. Degradation of Dendritic Cargos Requires Rab7-Dependent Transport to Somatic Lysosomes. J. Cell Biol. 2018, 217, 3141–3159. [Google Scholar] [CrossRef]
- Giehl, K.M. Neuronal Development. Prog. Exp. Tumor Res. 2007, 39, 1–29. [Google Scholar] [CrossRef]
- Molnár, Z.; Clowry, G.J.; Šestan, N.; Alzu’bi, A.; Bakken, T.; Hevner, R.F.; Hüppi, P.S.; Kostović, I.; Rakic, P.; Anton, E.S.; et al. New Insights into the Development of the Human Cerebral Cortex. J. Anat. 2019, 235, 432–451. [Google Scholar] [CrossRef]
- Fleming, A.; Rubinsztein, D.C. Autophagy in Neuronal Development and Plasticity. Trends Neurosci. 2020, 43, 767–779. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Fleming, A.; Ricketts, T.; Pavel, M.; Virgin, H.; Menzies, F.M.; Rubinsztein, D.C. Autophagy Regulates Notch Degradation and Modulates Stem Cell Development and Neurogenesis. Nat. Commun. 2016, 7, 10533. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Lu, G.; Hu, J.; Shen, X.; Ju, J.; Gao, Y.; Qu, L.; Xia, Y.; Chen, Y.; Bai, Y. EVA1A/TMEM166 Regulates Embryonic Neurogenesis by Autophagy. Stem Cell Rep. 2016, 6, 396–410. [Google Scholar] [CrossRef] [PubMed]
- Le Duc, D.; Giulivi, C.; Hiatt, S.M.; Napoli, E.; Panoutsopoulos, A.; Harlan De Crescenzo, A.; Kotzaeridou, U.; Syrbe, S.; Anagnostou, E.; Azage, M.; et al. Pathogenic WDFY3 Variants Cause Neurodevelopmental Disorders and Opposing Effects on Brain Size. Brain 2019, 142, 2617–2630. [Google Scholar] [CrossRef] [PubMed]
- Orosco, L.A.; Ross, A.P.; Cates, S.L.; Scott, S.E.; Wu, D.; Sohn, J.; Pleasure, D.; Pleasure, S.J.; Adamopoulos, I.E.; Zarbalis, K.S. Loss of Wdfy3 in Mice Alters Cerebral Cortical Neurogenesis Reflecting Aspects of the Autism Pathology. Nat. Commun. 2014, 5, 4692. [Google Scholar] [CrossRef]
- Jacquemont, M.; Sanlaville, D.; Redon, R.; Raoul, O.; Cormier-Daire, V.; Lyonnet, S.; Amiel, J.; Merrer, M.L.; Heron, D.; de Blois, M.; et al. Array-based Comparative Genomic Hybridisation Identifies High Frequency of Cryptic Chromosomal Rearrangements in Patients with Syndromic Autism Spectrum Disorders. J. Med. Genet. 2006, 43, 843–849. [Google Scholar] [CrossRef] [PubMed]
- Iossifov, I.; Ronemus, M.; Levy, D.; Wang, Z.; Hakker, I.; Rosenbaum, J.; Yamrom, B.; Lee, Y.; Narzisi, G.; Leotta, A.; et al. De Novo Gene Disruptions in Children on the Autistic Spectrum. Neuron 2012, 74, 285–299. [Google Scholar] [CrossRef]
- Vázquez, P.; Arroba, A.I.; Cecconi, F.; De La Rosa, E.J.; Boya, P.; De Pablo, F. Atg5 and Ambra1 Differentially Modulate Neurogenesis in Neural Stem Cells. Autophagy 2012, 8, 187–199. [Google Scholar] [CrossRef]
- Yazdankhah, M.; Farioli-Vecchioli, S.; Tonchev, A.B.; Stoykova, A.; Cecconi, F. The Autophagy Regulators Ambra1 and Beclin 1 Are Required for Adult Neurogenesis in the Brain Subventricular Zone. Cell Death Dis. 2014, 5, e1403. [Google Scholar] [CrossRef]
- Wang, C.; Liang, C.-C.; Bian, Z.C.; Zhu, Y.; Guan, J.-L. FIP200 Is Required for Maintenance and Differentiation of Postnatal Neural Stem Cells. Nat. Neurosci. 2013, 16, 532–542. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Yeo, S.; Haas, M.A.; Guan, J.-L. Autophagy Gene FIP200 in Neural Progenitors Non-Cell Autonomously Controls Differentiation by Regulating Microglia. J. Cell Biol. 2017, 216, 2581–2596. [Google Scholar] [CrossRef] [PubMed]
- Lv, X.; Jiang, H.; Li, B.; Liang, Q.; Wang, S.; Zhao, Q.; Jiao, J. The Crucial Role of Atg5 in Cortical Neurogenesis During Early Brain Development. Sci. Rep. 2014, 4, 6010. [Google Scholar] [CrossRef] [PubMed]
- Kenific, C.M.; Stehbens, S.J.; Goldsmith, J.; Leidal, A.M.; Faure, N.; Ye, J.; Wittmann, T.; Debnath, J. NBR1 Enables Autophagy-Dependent Focal Adhesion Turnover. J. Cell Biol. 2016, 212, 577–590. [Google Scholar] [CrossRef] [PubMed]
- Sharifi, M.N.; Mowers, E.E.; Drake, L.E.; Collier, C.; Chen, H.; Zamora, M.; Mui, S.; Macleod, K.F. Autophagy Promotes Focal Adhesion Disassembly and Cell Motility of Metastatic Tumor Cells through the Direct Interaction of Paxillin with LC3. Cell Rep. 2016, 15, 1660–1672. [Google Scholar] [CrossRef] [PubMed]
- Bressan, C.; Pecora, A.; Gagnon, D.; Snapyan, M.; Labrecque, S.; De Koninck, P.; Parent, M.; Saghatelyan, A. The Dynamic Interplay between ATP/ADP Levels and Autophagy Sustain Neuronal Migration in Vivo. eLife 2020, 9, e56006. [Google Scholar] [CrossRef] [PubMed]
- Molnár, M.; Sőth, Á.; Simon-Vecsei, Z. Pathways of Integrins in the Endo-Lysosomal System. Biol. Futura 2022, 73, 171–185. [Google Scholar] [CrossRef]
- Peng, C.; Ye, J.; Yan, S.; Kong, S.; Shen, Y.; Li, C.; Li, Q.; Zheng, Y.; Deng, K.; Xu, T.; et al. Ablation of Vacuole Protein Sorting 18 (Vps18) Gene Leads to Neurodegeneration and Impaired Neuronal Migration by Disrupting Multiple Vesicle Transport Pathways to Lysosomes. J. Biol. Chem. 2012, 287, 32861–32873. [Google Scholar] [CrossRef]
- Petri, R.; Pircs, K.; Jönsson, M.E.; Åkerblom, M.; Brattås, P.L.; Klussendorf, T.; Jakobsson, J. Let-7 Regulates Radial Migration of New-born Neurons through Positive Regulation of Autophagy. EMBO J. 2017, 36, 1379–1391. [Google Scholar] [CrossRef]
- Li, F.; Li, D.; Liu, H.; Cao, B.-B.; Jiang, F.; Chen, D.-N.; Li, J.-D. RNF216 Regulates the Migration of Immortalized GnRH Neurons by Suppressing Beclin1-Mediated Autophagy. Front. Endocrinol. 2019, 10, 12. [Google Scholar] [CrossRef]
- Baybis, M.; Yu, J.; Lee, A.; Golden, J.A.; Weiner, H.; McKhann, G.; Aronica, E.; Crino, P.B. mTOR Cascade Activation Distinguishes Tubers from Focal Cortical Dysplasia. Ann. Neurol. 2004, 56, 478–487. [Google Scholar] [CrossRef] [PubMed]
- Ljungberg, M.C.; Bhattacharjee, M.B.; Lu, Y.; Armstrong, D.L.; Yoshor, D.; Swann, J.W.; Sheldon, M.; D’Arcangelo, G. Activation of Mammalian Target of Rapamycin in Cytomegalic Neurons of Human Cortical Dysplasia. Ann. Neurol. 2006, 60, 420–429. [Google Scholar] [CrossRef]
- Lee, J.H.; Huynh, M.; Silhavy, J.L.; Kim, S.; Dixon-Salazar, T.; Heiberg, A.; Scott, E.; Bafna, V.; Hill, K.J.; Collazo, A.; et al. De Novo Somatic Mutations in Components of the PI3K-AKT3-mTOR Pathway Cause Hemimegalencephaly. Nat. Genet. 2012, 44, 941–945. [Google Scholar] [CrossRef]
- Leventer, R.J.; Scerri, T.; Marsh, A.P.L.; Pope, K.; Gillies, G.; Maixner, W.; MacGregor, D.; Harvey, A.S.; Delatycki, M.B.; Amor, D.J.; et al. Hemispheric Cortical Dysplasia Secondary to a Mosaic Somatic Mutation in MTOR. Neurology 2015, 84, 2029–2032. [Google Scholar] [CrossRef]
- Lim, J.S.; Kim, W.; Kang, H.-C.; Kim, S.H.; Park, A.H.; Park, E.K.; Cho, Y.-W.; Kim, S.; Kim, H.M.; Kim, J.A.; et al. Brain Somatic Mutations in MTOR Cause Focal Cortical Dysplasia Type II Leading to Intractable Epilepsy. Nat. Med. 2015, 21, 395–400. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, M.; Saitsu, H.; Takei, N.; Tohyama, J.; Kato, M.; Kitaura, H.; Shiina, M.; Shirozu, H.; Masuda, H.; Watanabe, K.; et al. Somatic Mutations in the MTOR Gene Cause Focal Cortical Dysplasia Type IIb. Ann. Neurol. 2015, 78, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Park, S.M.; Lim, J.S.; Ramakrishina, S.; Kim, S.H.; Kim, W.K.; Lee, J.; Kang, H.-C.; Reiter, J.F.; Kim, D.S.; Kim, H.; et al. Brain Somatic Mutations in MTOR Disrupt Neuronal Ciliogenesis, Leading to Focal Cortical Dyslamination. Neuron 2018, 99, 83–97.e7. [Google Scholar] [CrossRef]
- Margolin, D.H.; Kousi, M.; Chan, Y.-M.; Lim, E.T.; Schmahmann, J.D.; Hadjivassiliou, M.; Hall, J.E.; Adam, I.; Dwyer, A.; Plummer, L.; et al. Ataxia, Dementia, and Hypogonadotropism Caused by Disordered Ubiquitination. N. Engl. J. Med. 2013, 368, 1992–2003. [Google Scholar] [CrossRef]
- Garza-Lombó, C.; Gonsebatt, M.E. Mammalian Target of Rapamycin: Its Role in Early Neural Development and in Adult and Aged Brain Function. Front. Cell. Neurosci. 2016, 10, 157. [Google Scholar] [CrossRef]
- Yang, K.; Yu, B.; Cheng, C.; Cheng, T.; Yuan, B.; Li, K.; Xiao, J.; Qiu, Z.; Zhou, Y. Mir505–3p Regulates Axonal Development via Inhibiting the Autophagy Pathway by Targeting Atg12. Autophagy 2017, 13, 1679–1696. [Google Scholar] [CrossRef]
- Courchet, J.; Lewis, T.L.; Lee, S.; Courchet, V.; Liou, D.-Y.; Aizawa, S.; Polleux, F. Terminal Axon Branching Is Regulated by the LKB1-NUAK1 Kinase Pathway via Presynaptic Mitochondrial Capture. Cell 2013, 153, 1510–1525. [Google Scholar] [CrossRef] [PubMed]
- Spillane, M.; Ketschek, A.; Merianda, T.T.; Twiss, J.L.; Gallo, G. Mitochondria Coordinate Sites of Axon Branching through Localized Intra-Axonal Protein Synthesis. Cell Rep. 2013, 5, 1564–1575. [Google Scholar] [CrossRef] [PubMed]
- Ban, B.-K.; Jun, M.-H.; Ryu, H.-H.; Jang, D.-J.; Ahmad, S.T.; Lee, J.-A. Autophagy Negatively Regulates Early Axon Growth in Cortical Neurons. Mol. Cell. Biol. 2013, 33, 3907–3919. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Fang, Y.; Zhao, X.; Zheng, Y.; Ma, Y.; Li, S.; Huang, Z.; Li, L. BRUCE Silencing Leads to Axonal Dystrophy by Repressing Autophagosome-Lysosome Fusion in Alzheimer’s Disease. Transl. Psychiatry 2021, 11, 421. [Google Scholar] [CrossRef]
- Yamaguchi, J.; Suzuki, C.; Nanao, T.; Kakuta, S.; Ozawa, K.; Tanida, I.; Saitoh, T.; Sunabori, T.; Komatsu, M.; Tanaka, K.; et al. Atg9a Deficiency Causes Axon-Specific Lesions Including Neuronal Circuit Dysgenesis. Autophagy 2018, 14, 764–777. [Google Scholar] [CrossRef] [PubMed]
- Clarke, J.; Mearow, K. Autophagy Inhibition in Endogenous and Nutrient-deprived Conditions Reduces Dorsal Root Ganglia Neuron Survival and Neurite Growth in Vitro. J. Neurosci. Res. 2016, 94, 653–670. [Google Scholar] [CrossRef]
- Wojnacki, J.; Nola, S.; Bun, P.; Cholley, B.; Filippini, F.; Pressé, M.T.; Lipecka, J.; Man Lam, S.; N’guyen, J.; Simon, A.; et al. Role of VAMP7-Dependent Secretion of Reticulon 3 in Neurite Growth. Cell Rep. 2020, 33, 108536. [Google Scholar] [CrossRef]
- Coupé, B.; Ishii, Y.; Dietrich, M.O.; Komatsu, M.; Horvath, T.L.; Bouret, S.G. Loss of Autophagy in Pro-Opiomelanocortin Neurons Perturbs Axon Growth and Causes Metabolic Dysregulation. Cell Metab. 2012, 15, 247–255. [Google Scholar] [CrossRef]
- Popovic, D.; Dikic, I. TBC1D5 and the AP2 Complex Regulate ATG9 Trafficking and Initiation of Autophagy. EMBO Rep. 2014, 15, 392–401. [Google Scholar] [CrossRef]
- Kononenko, N.L.; Claßen, G.A.; Kuijpers, M.; Puchkov, D.; Maritzen, T.; Tempes, A.; Malik, A.R.; Skalecka, A.; Bera, S.; Jaworski, J.; et al. Retrograde Transport of TrkB-Containing Autophagosomes via the Adaptor AP-2 Mediates Neuronal Complexity and Prevents Neurodegeneration. Nat. Commun. 2017, 8, 14819. [Google Scholar] [CrossRef]
- Koscielny, A.; Malik, A.R.; Liszewska, E.; Zmorzynska, J.; Tempes, A.; Tarkowski, B.; Jaworski, J. Adaptor Complex 2 Controls Dendrite Morphology via mTOR-Dependent Expression of GluA2. Mol. Neurobiol. 2018, 55, 1590–1606. [Google Scholar] [CrossRef] [PubMed]
- Kijak, E.; Pyza, E. TOR Signaling Pathway and Autophagy Are Involved in the Regulation of Circadian Rhythms in Behavior and Plasticity of L2 Interneurons in the Brain of Drosophila Melanogaster. PLoS ONE 2017, 12, e0171848. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.G.; Graybeal, L.L.; Bhattacharjee, S.; Thomas, C.; Bhattacharya, S.; Cox, D.N. Basal Autophagy Is Required for Promoting Dendritic Terminal Branching in Drosophila Sensory Neurons. PLoS ONE 2018, 13, e0206743. [Google Scholar] [CrossRef] [PubMed]
- Hol, F.A.; Schepens, M.T.; van Beersum, S.E.; Redolfi, E.; Affer, M.; Vezzoni, P.; Hamel, B.C.; Karnes, P.S.; Mariman, E.C.; Zucchi, I. Identification and Characterization of an Xq26-Q27 Duplication in a Family with Spina Bifida and Panhypopituitarism Suggests the Involvement of Two Distinct Genes. Genomics 2000, 69, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Abou Jamra, R.; Philippe, O.; Raas-Rothschild, A.; Eck, S.H.; Graf, E.; Buchert, R.; Borck, G.; Ekici, A.; Brockschmidt, F.F.; Nöthen, M.M.; et al. Adaptor Protein Complex 4 Deficiency Causes Severe Autosomal-Recessive Intellectual Disability, Progressive Spastic Paraplegia, Shy Character, and Short Stature. Am. J. Hum. Genet. 2011, 88, 788–795. [Google Scholar] [CrossRef]
- Moreno-De-Luca, A.; Helmers, S.L.; Mao, H.; Burns, T.G.; Melton, A.M.A.; Schmidt, K.R.; Fernhoff, P.M.; Ledbetter, D.H.; Martin, C.L. Adaptor Protein Complex-4 (AP-4) Deficiency Causes a Novel Autosomal Recessive Cerebral Palsy Syndrome with Microcephaly and Intellectual Disability. J. Med. Genet. 2011, 48, 141–144. [Google Scholar] [CrossRef]
- Suleiman, J.; Allingham-Hawkins, D.; Hashem, M.; Shamseldin, H.E.; Alkuraya, F.S.; El-Hattab, A.W. WDR45B -Related Intellectual Disability, Spastic Quadriplegia, Epilepsy, and Cerebral Hypoplasia: A Consistent Neurodevelopmental Syndrome: SULEIMAN et Al. Clin. Genet. 2018, 93, 360–364. [Google Scholar] [CrossRef]
- Bhukel, A.; Beuschel, C.B.; Maglione, M.; Lehmann, M.; Juhász, G.; Madeo, F.; Sigrist, S.J. Autophagy within the Mushroom Body Protects from Synapse Aging in a Non-Cell Autonomous Manner. Nat. Commun. 2019, 10, 1318. [Google Scholar] [CrossRef]
- Shen, W.; Ganetzky, B. Autophagy Promotes Synapse Development in Drosophila. J. Cell Biol. 2009, 187, 71–79. [Google Scholar] [CrossRef]
- Gupta, V.K.; Pech, U.; Bhukel, A.; Fulterer, A.; Ender, A.; Mauermann, S.F.; Andlauer, T.F.M.; Antwi-Adjei, E.; Beuschel, C.; Thriene, K.; et al. Spermidine Suppresses Age-Associated Memory Impairment by Preventing Adverse Increase of Presynaptic Active Zone Size and Release. PLoS Biol. 2016, 14, e1002563. [Google Scholar] [CrossRef]
- Rudnick, N.D.; Griffey, C.J.; Guarnieri, P.; Gerbino, V.; Wang, X.; Piersaint, J.A.; Tapia, J.C.; Rich, M.M.; Maniatis, T. Distinct Roles for Motor Neuron Autophagy Early and Late in the SOD1 G93A Mouse Model of ALS. Proc. Natl. Acad. Sci. USA 2017, 114, E8294–E8303. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, D.; Torres, C.A.; Setlik, W.; Cebrián, C.; Mosharov, E.V.; Tang, G.; Cheng, H.-C.; Kholodilov, N.; Yarygina, O.; Burke, R.E.; et al. Regulation of Presynaptic Neurotransmission by Macroautophagy. Neuron 2012, 74, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Binotti, B.; Pavlos, N.J.; Riedel, D.; Wenzel, D.; Vorbrüggen, G.; Schalk, A.M.; Kühnel, K.; Boyken, J.; Erck, C.; Martens, H.; et al. The GTPase Rab26 Links Synaptic Vesicles to the Autophagy Pathway. eLife 2015, 4, e05597. [Google Scholar] [CrossRef] [PubMed]
- Nikoletopoulou, V.; Sidiropoulou, K.; Kallergi, E.; Dalezios, Y.; Tavernarakis, N. Modulation of Autophagy by BDNF Underlies Synaptic Plasticity. Cell Metab. 2017, 26, 230–242.e5. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Porch, M.W.; Court-Vazquez, B.; Bennett, M.V.L.; Zukin, R.S. Activation of Autophagy Rescues Synaptic and Cognitive Deficits in Fragile X Mice. Proc. Natl. Acad. Sci. USA 2018, 115, E9707–E9716. [Google Scholar] [CrossRef]
- Tang, G.; Gudsnuk, K.; Kuo, S.-H.; Cotrina, M.L.; Rosoklija, G.; Sosunov, A.; Sonders, M.S.; Kanter, E.; Castagna, C.; Yamamoto, A.; et al. Loss of mTOR-Dependent Macroautophagy Causes Autistic-like Synaptic Pruning Deficits. Neuron 2014, 83, 1131–1143. [Google Scholar] [CrossRef] [PubMed]
- Bateup, H.S.; Takasaki, K.T.; Saulnier, J.L.; Denefrio, C.L.; Sabatini, B.L. Loss of Tsc1 in Vivo Impairs Hippocampal mGluR-LTD and Increases Excitatory Synaptic Function. J. Neurosci. Off. J. Soc. Neurosci. 2011, 31, 8862–8869. [Google Scholar] [CrossRef]
- Amegandjin, C.A.; Choudhury, M.; Jadhav, V.; Carriço, J.N.; Quintal, A.; Berryer, M.; Snapyan, M.; Chattopadhyaya, B.; Saghatelyan, A.; Di Cristo, G. Sensitive Period for Rescuing Parvalbumin Interneurons Connectivity and Social Behavior Deficits Caused by TSC1 Loss. Nat. Commun. 2021, 12, 3653. [Google Scholar] [CrossRef]
- Matsuda, S.; Miura, E.; Matsuda, K.; Kakegawa, W.; Kohda, K.; Watanabe, M.; Yuzaki, M. Accumulation of AMPA Receptors in Autophagosomes in Neuronal Axons Lacking Adaptor Protein AP-4. Neuron 2008, 57, 730–745. [Google Scholar] [CrossRef]
- Shehata, M.; Abdou, K.; Choko, K.; Matsuo, M.; Nishizono, H.; Inokuchi, K. Autophagy Enhances Memory Erasure through Synaptic Destabilization. J. Neurosci. 2018, 38, 3809–3822. [Google Scholar] [CrossRef]
- Lee, J.L.C.; Nader, K.; Schiller, D. An Update on Memory Reconsolidation Updating. Trends Cogn. Sci. 2017, 21, 531–545. [Google Scholar] [CrossRef] [PubMed]
- Glatigny, M.; Moriceau, S.; Rivagorda, M.; Ramos-Brossier, M.; Nascimbeni, A.C.; Lante, F.; Shanley, M.R.; Boudarene, N.; Rousseaud, A.; Friedman, A.K.; et al. Autophagy Is Required for Memory Formation and Reverses Age-Related Memory Decline. Curr. Biol. 2019, 29, 435–448.e8. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Wang, H.; Gao, J.; Yu, M.; Wang, R.; Yang, Z.; Zhang, T. Rapamycin Effectively Impedes Melamine-Induced Impairments of Cognition and Synaptic Plasticity in Wistar Rats. Mol. Neurobiol. 2017, 54, 819–832. [Google Scholar] [CrossRef] [PubMed]
- Zhai, B.; Shang, X.; Fu, J.; Li, F.; Zhang, T. Rapamycin Relieves Anxious Emotion and Synaptic Plasticity Deficits Induced by Hindlimb Unloading in Mice. Neurosci. Lett. 2018, 677, 44–48. [Google Scholar] [CrossRef] [PubMed]
- Rowland, A.M.; Richmond, J.E.; Olsen, J.G.; Hall, D.H.; Bamber, B.A. Presynaptic Terminals Independently Regulate Synaptic Clustering and Autophagy of GABAA Receptors in Caenorhabditis elegans. J. Neurosci. 2006, 26, 1711–1720. [Google Scholar] [CrossRef] [PubMed]
- Lalo, U.; Nezis, I.P.; Pankratov, Y. Impact of Autophagy Impairment on Experience- and Diet-Related Synaptic Plasticity. Int. J. Mol. Sci. 2022, 23, 9228. [Google Scholar] [CrossRef] [PubMed]
- Sumitomo, A.; Yukitake, H.; Hirai, K.; Horike, K.; Ueta, K.; Chung, Y.; Warabi, E.; Yanagawa, T.; Kitaoka, S.; Furuyashiki, T.; et al. Ulk2 Controls Cortical Excitatory–Inhibitory Balance via Autophagic Regulation of P62 and GABAA Receptor Trafficking in Pyramidal Neurons. Hum. Mol. Genet. 2018, 27, 3165–3176. [Google Scholar] [CrossRef]
- Hui, K.K.; Takashima, N.; Watanabe, A.; Chater, T.E.; Matsukawa, H.; Nekooki-Machida, Y.; Nilsson, P.; Endo, R.; Goda, Y.; Saido, T.C.; et al. GABARAPs Dysfunction by Autophagy Deficiency in Adolescent Brain Impairs GABA A Receptor Trafficking and Social Behavior. Sci. Adv. 2019, 5, eaau8237. [Google Scholar] [CrossRef]
- Kim, H.-J.; Cho, M.-H.; Shim, W.H.; Kim, J.K.; Jeon, E.-Y.; Kim, D.-H.; Yoon, S.-Y. Deficient Autophagy in Microglia Impairs Synaptic Pruning and Causes Social Behavioral Defects. Mol. Psychiatry 2017, 22, 1576–1584. [Google Scholar] [CrossRef]
- Bassetti, D.; Luhmann, H.J.; Kirischuk, S. Effects of Mutations in TSC Genes on Neurodevelopment and Synaptic Transmission. Int. J. Mol. Sci. 2021, 22, 7273. [Google Scholar] [CrossRef]
- Barnes, M.R.; Huxley-Jones, J.; Maycox, P.R.; Lennon, M.; Thornber, A.; Kelly, F.; Bates, S.; Taylor, A.; Reid, J.; Jones, N.; et al. Transcription and Pathway Analysis of the Superior Temporal Cortex and Anterior Prefrontal Cortex in Schizophrenia. J. Neurosci. Res. 2011, 89, 1218–1227. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, C.M.B.; Vasanth, S.; Shinawi, M.; Russell, C.; Ramocki, M.B.; Brown, C.W.; Graakjaer, J.; Skytte, A.-B.; Vianna-Morgante, A.M.; Krepischi, A.C.V.; et al. Dosage Changes of a Segment at 17p13.1 Lead to Intellectual Disability and Microcephaly as a Result of Complex Genetic Interaction of Multiple Genes. Am. J. Hum. Genet. 2014, 95, 565–578. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liénard, C.; Pintart, A.; Bomont, P. Neuronal Autophagy: Regulations and Implications in Health and Disease. Cells 2024, 13, 103. https://doi.org/10.3390/cells13010103
Liénard C, Pintart A, Bomont P. Neuronal Autophagy: Regulations and Implications in Health and Disease. Cells. 2024; 13(1):103. https://doi.org/10.3390/cells13010103
Chicago/Turabian StyleLiénard, Caroline, Alexandre Pintart, and Pascale Bomont. 2024. "Neuronal Autophagy: Regulations and Implications in Health and Disease" Cells 13, no. 1: 103. https://doi.org/10.3390/cells13010103