

Corneal Regeneration Using Gene Therapy Approaches
 , , , and
, , , and
Abstract
:1. Introduction
2. Genes of Corneal Diseases
2.1. Genes and Genetics of Inherited Diseases
2.2. Genes Associated with Acquired Corneal Conditions
2.2.1. Corneal Wound Healing
2.2.2. Corneal Neovascularization
2.2.3. Corneal Graft Survival
2.2.4. Multifactorial and Polygenic Diseases
3. Clinical Presentation of Various Corneal Diseases
4. Clinical Treatment Options Currently Available
5. Current Challenges in the Treatment of Corneal Diseases
6. Gene Therapy for Corneal Diseases
6.1. Viral Vectors
6.1.1. Adeno-Associated Virus (AAV)
6.1.2. Lentivirus (LV)
6.1.3. Adenovirus (Ad)
6.2. Non-Viral Vectors
6.2.1. Electroporation
6.2.2. Nanoparticles

{kind=link}
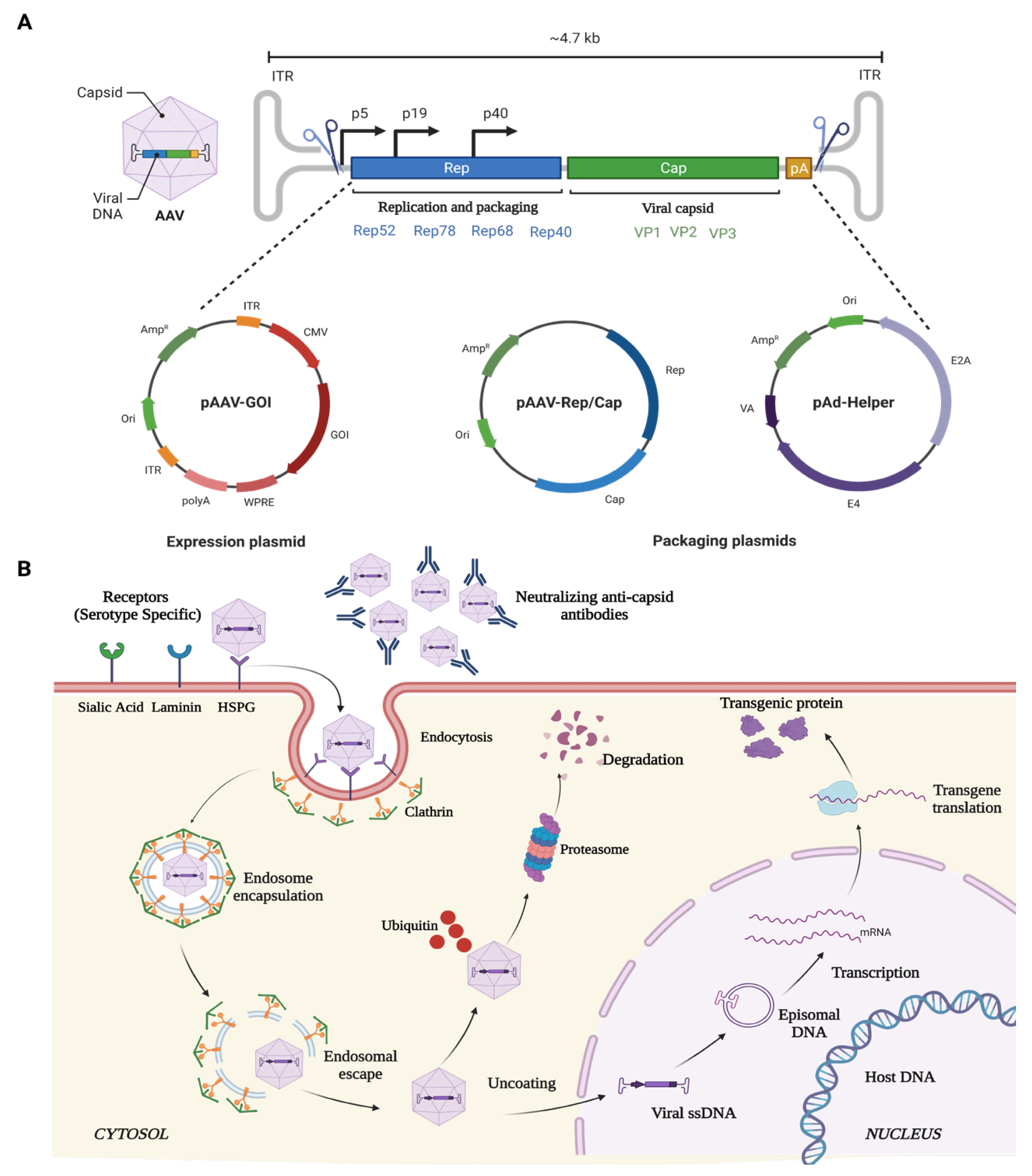{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Non-Viral Mediated Corneal Gene Therapy | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Vector | Disease | Gene | Promoter | Dosage | Model | Species | Mode of Administration | Outcome | Reference |
| Electroporation | Stromal Keratitis | IL-10 | CMV, UbC | 1 µL Plasmid solution (5 µg Plasmid DNA in 10 mM Tris, pH 8.0; 1 mM EDTA; and 140 mM NaCl) | In Vivo | Female Balb/c mice weighing 16 to 24 g | Stromal injection of plasmid DNA, followed by gold-plated Genetrode electrodes were placed on the cornea on either side of the area injected. An ECM 830 square wave electroporator was used to deliver eight pulses of 10-msec duration at a field strength of 200 V/cm. | Gene expression driven by the CMV promoter remained high for three days, which started to reduce by 2-fold each day thereafter. Replacing the promoter with UbC surprisingly showed similar half-life gene expression. Also, an adverse effect was observed when using DNA nuclear-targeting sequences in vectors. | [148] |
| CRISPR/dCas9 | Corneal Endothelial wound healing | SOX2 | Not mentioned | 0.1 nmol | In Vivo | 6–8 weeks old Sprague–Dawley rats; a 12-h light/dark cycle at 25 °C for 7 days before initiating experiments. | Anterior chamber injection of plasmid DNA, followed by 7-mm Tweezertrodes were placed on each cornea, with the positive electrode on the plasmid-injected eye. The parameters were set at 140 V, 100 milliseconds length, 950 milliseconds interval, five pulses, and 100 V/cm2 | SOX2 activation promoted the reduction of central corneal thickness and corneal opacity in comparison to the control group. Additionally, an increase in Cell viability, proliferation rate, and the number of cells in the S-phase was observed after SOX2 overexpression. | [147] |
| CRISPR-Cas9 | Granular corneal dystrophy (GCD 2) | TGFBI | Not mentioned | CRISPR-Cas9 constructs (2.5 µg per well) and ssODN (1 µg per well) | In Vitro | Human, GCD 2 patient-derived corneal keratocytes | Transfection | Effective gene correction efficiency of R124H mutation associated with GCD2 disorder was observed without any off-target effects. In heterozygous cells, the correction efficiency was found to be 20.6% and in homozygous 41.3% respectively. | [156] |
| Lipofection | Meesmann’s Epithelial Corneal Dystrophy (MECD) | KRT12 | Not mentioned | 200 ng plasmid (1 well of 12 well plate) | In Vitro | Corneal limbal epithelial cell derived from limbal biopsy of MECD patients | Lipofectamine 2000 | Potent and specific knockdown of K12-Leu132Pro at both the mRNA and protein levels. An allele-specific knockdown of 63% of the endogenous mutant allele was observed. | [157] |
| Lipofection | lattice corneal dystrophy type I (LCDI) | TGFBI-Arg124Cys | CMV | 200 ng plasmid (1 well of 12 well plate) | Ex Vivo | Corneal limbal epithelial cell derived from limbal biopsy of LCD1 patients | Lipofectamine 2000 | The siRNA specific to TGFBI-Arg124Cys, efficiently suppressed the mutant allele | [158] |
| PEI gold nanoparticle | Corneal Fibrosis | BMP7 | CAG | 37.5 µL of 150 mM PEI2-GNPs with 10 µg of plasmid DNA | In Vivo | Female New Zealand White rabbit weighing 2.0 to 3.0 kg | Topical | Significant inhibition of fibrosis post-PRK was observed. | [36] |
| PEI nanoparticle | Corneal Fibrosis | Decorin | CAG | 150 mM linear 22 kDa PEI+ 2 µg of plasmid | In Vitro | Horse | Topical | 22 KDa PEI nanoparticle effectively inhibited TGFb-mediated fibrosis | [159] |
6.3. Gene Therapy Strategies for Precise and Targeted Therapeutics
6.3.1. Gene Augmentation
6.3.2. Gene Editing
6.3.3. Gene Silencing
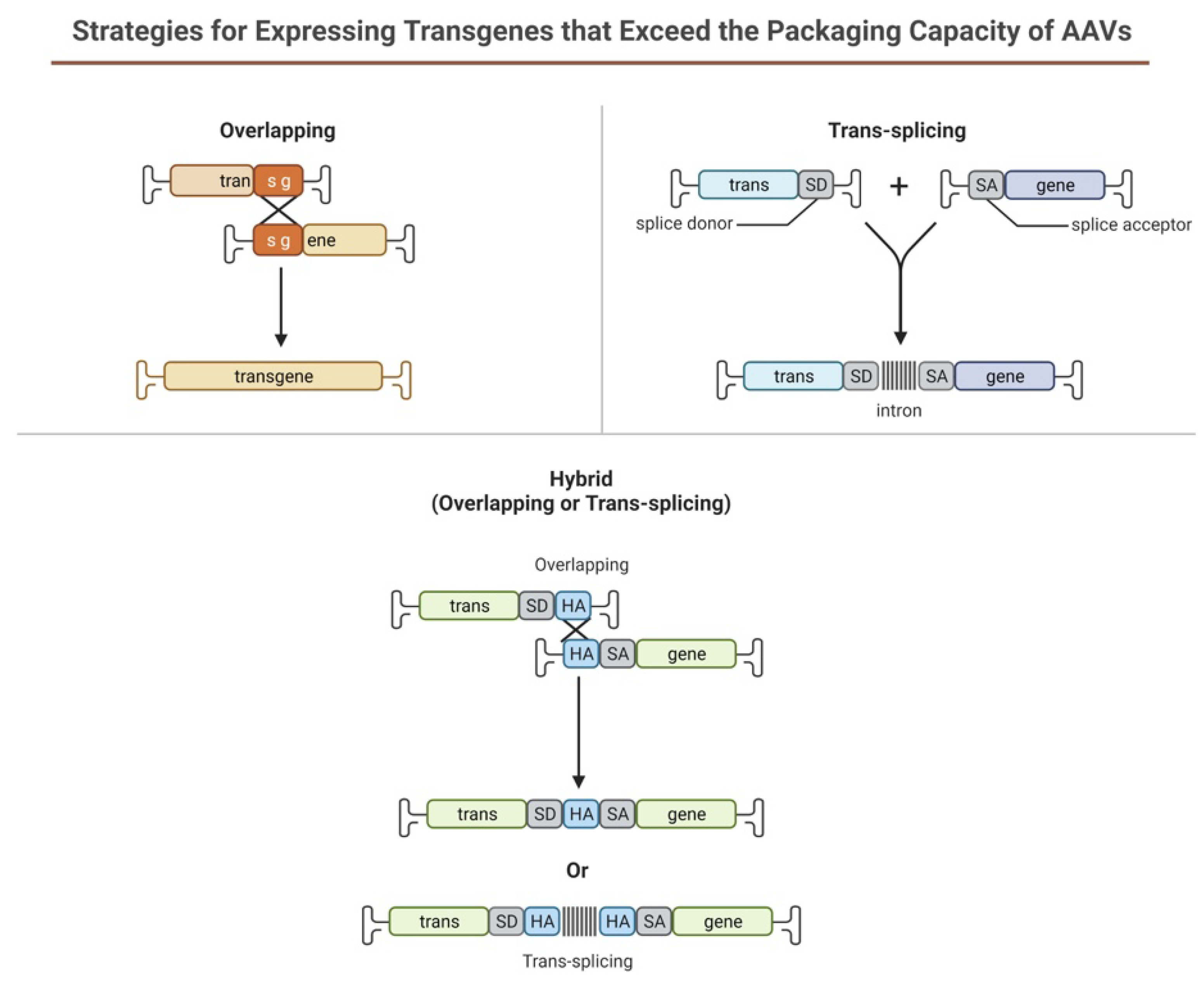
6.3.4. Dual Vectors
6.4. Current Scenario of Corneal Gene Therapy
7. Tailored Therapeutics Using Gene Therapy
7.1. Promoter Selection for Targeted Therapeutics
7.2. Capsid Engineering
8. Challenges and Safety Aspect of Gene Therapy for Corneal Diseases
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Whitcher, J.P.; Srinivasan, M.; Upadhyay, M.P. Corneal blindness: A global perspective. Bull. World Health Organ. 2001, 79, 214–221. [Google Scholar] [PubMed]
- Prado, D.A.; Acosta-Acero, M.; Maldonado, R.S. Gene therapy beyond luxturna: A new horizon of the treatment for inherited retinal disease. Curr. Opin. Ophthalmol. 2020, 31, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Mashhour, B.; Couton, D.; Perricaudet, M.; Briand, P. In vivo adenovirus-mediated gene transfer into ocular tissues. Gene Ther. 1994, 1, 122–126. [Google Scholar] [PubMed]
- Constantin, C. Corneal dystrophies: Pathophysiological, genetic, clinical, and therapeutic considerations. Rom. J. Ophthalmol. 2021, 65, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Lisch, W.; Weiss, J.S. Early and late clinical landmarks of corneal dystrophies. Exp. Eye Res. 2020, 198, 108139. [Google Scholar] [CrossRef]
- Loukovitis, E.; Sfakianakis, K.; Syrmakesi, P.; Tsotridou, E.; Orfanidou, M.; Bakaloudi, D.R.; Stoila, M.; Kozei, A.; Koronis, S.; Zachariadis, Z.; et al. Genetic Aspects of Keratoconus: A Literature Review Exploring Potential Genetic Contributions and Possible Genetic Relationships with Comorbidities. Ophthalmol. Ther. 2018, 7, 263–292. [Google Scholar] [CrossRef]
- Sonoda, S.; Tachibana, K.; Uchino, E.; Okubo, A.; Yamamoto, M.; Sakoda, K.; Hisatomi, T.; Sonoda, K.H.; Negishi, Y.; Izumi, Y.; et al. Gene transfer to corneal epithelium and keratocytes mediated by ultrasound with microbubbles. Investig. Ophthalmol. Vis. Sci. 2006, 47, 558–564. [Google Scholar] [CrossRef]
- Stechschulte, S.U.; Joussen, A.M.; von Recum, H.A.; Poulaki, V.; Moromizato, Y.; Yuan, J.; D’Amato, R.J.; Kuo, C.; Adamis, A.P. Rapid ocular angiogenic control via naked DNA delivery to cornea. Investig. Ophthalmol. Vis. Sci. 2001, 42, 1975–1979. [Google Scholar]
- Huang, Y.-X.; Li, Q.-H. An active artificial cornea with the function of inducing new corneal tissue generation in vivo—A new approach to corneal tissue engineering. Biomed. Mater. 2007, 2, S121. [Google Scholar] [CrossRef]
- Jhanji, V.; Mehta, J.S.; Sharma, N.; Sharma, B.; Vajpayee, R.B. Targeted corneal transplantation. Curr. Opin. Ophthalmol. 2012, 23, 324–329. [Google Scholar] [CrossRef]
- Chamberlain, W.D. Femtosecond laser-assisted deep anterior lamellar keratoplasty. Curr. Opin. Ophthalmol. 2019, 30, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Luengo-Gimeno, F.; Tan, D.T.; Mehta, J.S. Evolution of deep anterior lamellar keratoplasty (DALK). Ocul. Surf. 2011, 9, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Klausner, E.A.; Peer, D.; Chapman, R.L.; Multack, R.F.; Andurkar, S.V. Corneal gene therapy. J. Control. Release 2007, 124, 107–133. [Google Scholar] [CrossRef] [PubMed]
- Gain, P.; Jullienne, R.; He, Z.; Aldossary, M.; Acquart, S.; Cognasse, F.; Thuret, G. Global Survey of Corneal Transplantation and Eye Banking. JAMA Ophthalmol. 2016, 134, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Fuest, M.; Yam, G.H.; Peh, G.S.; Mehta, J.S. Advances in corneal cell therapy. Regen. Med. 2016, 11, 601–615. [Google Scholar] [CrossRef]
- Mobaraki, M.; Abbasi, R.; Omidian Vandchali, S.; Ghaffari, M.; Moztarzadeh, F.; Mozafari, M. Corneal Repair and Regeneration: Current Concepts and Future Directions. Front. Bioeng. Biotechnol. 2019, 7, 135. [Google Scholar] [CrossRef]
- Mohan, R.R.; Rodier, J.T.; Sharma, A. Corneal gene therapy: Basic science and translational perspective. Ocul. Surf. 2013, 11, 150–164. [Google Scholar] [CrossRef]
- Torrecilla, J.; Del Pozo-Rodriguez, A.; Vicente-Pascual, M.; Solinis, M.A.; Rodriguez-Gascon, A. Targeting corneal inflammation by gene therapy: Emerging strategies for keratitis. Exp. Eye Res. 2018, 176, 130–140. [Google Scholar] [CrossRef]
- Sakamoto, T.; Oshima, Y.; Nakagawa, K.; Ishibashi, T.; Inomata, H.; Sueishi, K. Target gene transfer of tissue plasminogen activator to cornea by electric pulse inhibits intracameral fibrin formation and corneal cloudiness. Hum. Gene Ther. 1999, 10, 2551–2557. [Google Scholar] [CrossRef]
- Oshima, Y.; Sakamoto, T.; Yamanaka, I.; Nishi, T.; Ishibashi, T.; Inomata, H. Targeted gene transfer to corneal endothelium in vivo by electric pulse. Gene Ther. 1998, 5, 1347–1354. [Google Scholar] [CrossRef]
- Williams, K.A.; Jessup, C.F.; Coster, D.J. Gene therapy approaches to prolonging corneal allograft survival. Expert Opin. Biol. Ther. 2004, 4, 1059–1071. [Google Scholar] [CrossRef] [PubMed]
- Lakshminarayanan, R.; Chaurasia, S.S.; Anandalakshmi, V.; Chai, S.M.; Murugan, E.; Vithana, E.N.; Beuerman, R.W.; Mehta, J.S. Clinical and genetic aspects of the TGFBI-associated corneal dystrophies. Ocul. Surf. 2014, 12, 234–251. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, F.; Bystrom, B.; Davidson, A.E.; Backman, L.J.; Kellgren, T.G.; Tuft, S.J.; Koskela, T.; Ryden, P.; Sandgren, O.; Danielson, P.; et al. Mutations in collagen, type XVII, alpha 1 (COL17A1) cause epithelial recurrent erosion dystrophy (ERED). Hum. Mutat. 2015, 36, 463–473. [Google Scholar] [CrossRef] [PubMed]
- Irvine, A.D.; Corden, L.D.; Swensson, O.; Swensson, B.; Moore, J.E.; Frazer, D.G.; Smith, F.J.; Knowlton, R.G.; Christophers, E.; Rochels, R.; et al. Mutations in cornea-specific keratin K3 or K12 genes cause Meesmann’s corneal dystrophy. Nat. Genet. 1997, 16, 184–187. [Google Scholar] [CrossRef] [PubMed]
- Omary, M.B.; Coulombe, P.A.; McLean, W.H. Intermediate filament proteins and their associated diseases. N. Engl. J. Med. 2004, 351, 2087–2100. [Google Scholar] [CrossRef]
- Hassan, H.; Thaung, C.; Ebenezer, N.D.; Larkin, G.; Hardcastle, A.J.; Tuft, S.J. Severe Meesmann’s epithelial corneal dystrophy phenotype due to a missense mutation in the helix-initiation motif of keratin 12. Eye 2013, 27, 367–373. [Google Scholar] [CrossRef]
- Nishida, K.; Honma, Y.; Dota, A.; Kawasaki, S.; Adachi, W.; Nakamura, T.; Quantock, A.J.; Hosotani, H.; Yamamoto, S.; Okada, M.; et al. Isolation and chromosomal localization of a cornea-specific human keratin 12 gene and detection of four mutations in Meesmann corneal epithelial dystrophy. Am. J. Hum. Genet. 1997, 61, 1268–1275. [Google Scholar] [CrossRef]
- Szaflik, J.P.; Oldak, M.; Maksym, R.B.; Kaminska, A.; Pollak, A.; Udziela, M.; Ploski, R.; Szaflik, J. Genetics of Meesmann corneal dystrophy: A novel mutation in the keratin 3 gene in an asymptomatic family suggests genotype-phenotype correlation. Mol. Vis. 2008, 14, 1713–1718. [Google Scholar]
- Wieben, E.D.; Aleff, R.A.; Tang, X.; Butz, M.L.; Kalari, K.R.; Highsmith, E.W.; Jen, J.; Vasmatzis, G.; Patel, S.V.; Maguire, L.J.; et al. Trinucleotide Repeat Expansion in the Transcription Factor 4 (TCF4) Gene Leads to Widespread mRNA Splicing Changes in Fuchs’ Endothelial Corneal Dystrophy. Investig. Ophthalmol. Vis. Sci. 2017, 58, 343–352. [Google Scholar] [CrossRef]
- Jordan, T.; Hanson, I.; Zaletayev, D.; Hodgson, S.; Prosser, J.; Seawright, A.; Hastie, N.; van Heyningen, V. The human PAX6 gene is mutated in two patients with aniridia. Nat. Genet. 1992, 1, 328–332. [Google Scholar] [CrossRef]
- Tomatsu, S.; Montano, A.M.; Dung, V.C.; Grubb, J.H.; Sly, W.S. Mutations and polymorphisms in GUSB gene in mucopolysaccharidosis VII (Sly Syndrome). Hum. Mutat. 2009, 30, 511–519. [Google Scholar] [CrossRef] [PubMed]
- Garrido, E.; Cormand, B.; Hopwood, J.J.; Chabas, A.; Grinberg, D.; Vilageliu, L. Maroteaux-Lamy syndrome: Functional characterization of pathogenic mutations and polymorphisms in the arylsulfatase B gene. Mol. Genet. Metab. 2008, 94, 305–312. [Google Scholar] [CrossRef]
- Massague, J.; Wotton, D. Transcriptional control by the TGF-beta/Smad signaling system. EMBO J. 2000, 19, 1745–1754. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.J.; Mohan, R.R.; Mohan, R.R.; Wilson, S.E. Effect of PDGF, IL-1alpha, and BMP2/4 on corneal fibroblast chemotaxis: Expression of the platelet-derived growth factor system in the cornea. Investig. Ophthalmol. Vis. Sci. 1999, 40, 1364–1372. [Google Scholar]
- Dudley, A.T.; Lyons, K.M.; Robertson, E.J. A requirement for bone morphogenetic protein-7 during development of the mammalian kidney and eye. Genes Dev. 1995, 9, 2795–2807. [Google Scholar] [CrossRef]
- Tandon, A.; Sharma, A.; Rodier, J.T.; Klibanov, A.M.; Rieger, F.G.; Mohan, R.R. BMP7 gene transfer via gold nanoparticles into stroma inhibits corneal fibrosis in vivo. PLoS ONE 2013, 8, e66434. [Google Scholar] [CrossRef] [PubMed]
- Saika, S.; Ikeda, K.; Yamanaka, O.; Miyamoto, T.; Ohnishi, Y.; Sato, M.; Muragaki, Y.; Ooshima, A.; Nakajima, Y.; Kao, W.W.; et al. Expression of Smad7 in mouse eyes accelerates healing of corneal tissue after exposure to alkali. Am. J. Pathol. 2005, 166, 1405–1418. [Google Scholar] [CrossRef]
- Wilson, S.E.; Chen, L.; Mohan, R.R.; Liang, Q.; Liu, J. Expression of HGF, KGF, EGF and receptor messenger RNAs following corneal epithelial wounding. Exp. Eye Res. 1999, 68, 377–397. [Google Scholar] [CrossRef]
- Li, J.F.; Duan, H.F.; Wu, C.T.; Zhang, D.J.; Deng, Y.; Yin, H.L.; Han, B.; Gong, H.C.; Wang, H.W.; Wang, Y.L. HGF accelerates wound healing by promoting the dedifferentiation of epidermal cells through beta1-integrin/ILK pathway. Biomed. Res. Int. 2013, 2013, 470418. [Google Scholar] [CrossRef]
- Chang, J.H.; Gabison, E.E.; Kato, T.; Azar, D.T. Corneal neovascularization. Curr. Opin. Ophthalmol. 2001, 12, 242–249. [Google Scholar] [CrossRef]
- Kvanta, A.; Sarman, S.; Fagerholm, P.; Seregard, S.; Steen, B. Expression of matrix metalloproteinase-2 (MMP-2) and vascular endothelial growth factor (VEGF) in inflammation-associated corneal neovascularization. Exp. Eye Res. 2000, 70, 419–428. [Google Scholar] [CrossRef] [PubMed]
- Klettner, A.; Roider, J. Treating age-related macular degeneration—Interaction of VEGF-antagonists with their target. Mini Rev. Med. Chem. 2009, 9, 1127–1135. [Google Scholar] [CrossRef] [PubMed]
- Penn, J.S.; Madan, A.; Caldwell, R.B.; Bartoli, M.; Caldwell, R.W.; Hartnett, M.E. Vascular endothelial growth factor in eye disease. Prog. Retin. Eye Res. 2008, 27, 331–371. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, W.; Cheng, S.F.; Dastjerdi, M.H.; Ferrari, G.; Dana, R. Corneal neovascularization and the utility of topical VEGF inhibition: Ranibizumab (Lucentis) vs bevacizumab (Avastin). Ocul. Surf. 2012, 10, 67–83. [Google Scholar] [CrossRef] [PubMed]
- Yoon, K.C.; Ahn, K.Y.; Lee, J.H.; Chun, B.J.; Park, S.W.; Seo, M.S.; Park, Y.G.; Kim, K.K. Lipid-mediated delivery of brain-specific angiogenesis inhibitor 1 gene reduces corneal neovascularization in an in vivo rabbit model. Gene Ther. 2005, 12, 617–624. [Google Scholar] [CrossRef]
- Cho, Y.K.; Uehara, H.; Young, J.R.; Tyagi, P.; Kompella, U.B.; Zhang, X.; Luo, L.; Singh, N.; Archer, B.; Ambati, B.K. Flt23k nanoparticles offer additive benefit in graft survival and anti-angiogenic effects when combined with triamcinolone. Investig. Ophthalmol. Vis. Sci. 2012, 53, 2328–2336. [Google Scholar] [CrossRef]
- Lai, C.M.; Shen, W.Y.; Brankov, M.; Lai, Y.K.; Barnett, N.L.; Lee, S.Y.; Yeo, I.Y.; Mathur, R.; Ho, J.E.; Pineda, P.; et al. Long-term evaluation of AAV-mediated sFlt-1 gene therapy for ocular neovascularization in mice and monkeys. Mol. Ther. 2005, 12, 659–668. [Google Scholar] [CrossRef]
- Li, Z.; He, T.; Du, K.; Xing, Y.Q.; Run, Y.M.; Yan, Y.; Shen, Y. Inhibition of oxygen-induced ischemic retinal neovascularization with adenoviral 15-lipoxygenase-1 gene transfer via up-regulation of PPAR-gamma and down-regulation of VEGFR-2 expression. PLoS ONE 2014, 9, e85824. [Google Scholar] [CrossRef]
- Qazi, Y.; Stagg, B.; Singh, N.; Singh, S.; Zhang, X.; Luo, L.; Simonis, J.; Kompella, U.B.; Ambati, B.K. Nanoparticle-mediated delivery of shRNA.VEGF-a plasmids regresses corneal neovascularization. Investig. Ophthalmol. Vis. Sci. 2012, 53, 2837–2844. [Google Scholar] [CrossRef]
- Torrecilla, J.; Gomez-Aguado, I.; Vicente-Pascual, M.; Del Pozo-Rodriguez, A.; Solinis, M.A.; Rodriguez-Gascon, A. MMP-9 Downregulation with Lipid Nanoparticles for Inhibiting Corneal Neovascularization by Gene Silencing. Nanomaterials 2019, 9, 631. [Google Scholar] [CrossRef]
- Yu, H.; Chen, L.; Jiang, J. Administration of pigment epithelium-derived factor delivered by adeno-associated virus inhibits blood-retinal barrier breakdown in diabetic rats. Mol. Vis. 2010, 16, 2384–2394. [Google Scholar] [PubMed]
- Coster, D.J.; Williams, K.A. The impact of corneal allograft rejection on the long-term outcome of corneal transplantation. Am. J. Ophthalmol. 2005, 140, 1112–1122. [Google Scholar] [CrossRef] [PubMed]
- Parker, D.G.; Brereton, H.M.; Coster, D.J.; Williams, K.A. The potential of viral vector-mediated gene transfer to prolong corneal allograft survival. Curr. Gene Ther. 2009, 9, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Beutelspacher, S.C.; Pillai, R.; Watson, M.P.; Tan, P.H.; Tsang, J.; McClure, M.O.; George, A.J.; Larkin, D.F. Function of indoleamine 2,3-dioxygenase in corneal allograft rejection and prolongation of allograft survival by over-expression. Eur. J. Immunol. 2006, 36, 690–700. [Google Scholar] [CrossRef] [PubMed]
- Gong, N.; Pleyer, U.; Volk, H.D.; Ritter, T. Effects of local and systemic viral interleukin-10 gene transfer on corneal allograft survival. Gene Ther. 2007, 14, 484–490. [Google Scholar] [CrossRef] [PubMed]
- Gong, N.; Pleyer, U.; Yang, J.; Vogt, K.; Hill, M.; Anegon, I.; Volk, H.D.; Ritter, T. Influence of local and systemic CTLA4Ig gene transfer on corneal allograft survival. J. Gene Med. 2006, 8, 459–467. [Google Scholar] [CrossRef]
- Pillai, R.G.; Beutelspacher, S.C.; Larkin, D.F.; George, A.J. Expression of the chemokine antagonist vMIP II using a non-viral vector can prolong corneal allograft survival. Transplantation 2008, 85, 1640–1647. [Google Scholar] [CrossRef]
- Williams, K.A.; Coster, D.J. Gene therapy for diseases of the cornea—A review. Clin. Exp. Ophthalmol. 2010, 38, 93–103. [Google Scholar] [CrossRef]
- Murthy, R.C.; McFarland, T.J.; Yoken, J.; Chen, S.; Barone, C.; Burke, D.; Zhang, Y.; Appukuttan, B.; Stout, J.T. Corneal transduction to inhibit angiogenesis and graft failure. Investig. Ophthalmol. Vis. Sci. 2003, 44, 1837–1842. [Google Scholar] [CrossRef]
- Comer, R.M.; King, W.J.; Ardjomand, N.; Theoharis, S.; George, A.J.; Larkin, D.F. Effect of administration of CTLA4-Ig as protein or cDNA on corneal allograft survival. Investig. Ophthalmol. Vis. Sci. 2002, 43, 1095–1103. [Google Scholar]
- Tang, X.L.; Sun, J.F.; Wang, X.Y.; Du, L.L.; Liu, P. Blocking neuropilin-2 enhances corneal allograft survival by selectively inhibiting lymphangiogenesis on vascularized beds. Mol. Vis. 2010, 16, 2354–2361. [Google Scholar] [PubMed]
- Cho, Y.K.; Zhang, X.; Uehara, H.; Young, J.R.; Archer, B.; Ambati, B. Vascular Endothelial Growth Factor Receptor 1 morpholino increases graft survival in a murine penetrating keratoplasty model. Investig. Ophthalmol. Vis. Sci. 2012, 53, 8458–8471. [Google Scholar] [CrossRef] [PubMed]
- Swierkowska, J.; Gajecka, M. Genetic factors influencing the reduction of central corneal thickness in disorders affecting the eye. Ophthalmic Genet. 2017, 38, 501–510. [Google Scholar] [CrossRef]
- Dudakova, L.; Sasaki, T.; Liskova, P.; Palos, M.; Jirsova, K. The presence of lysyl oxidase-like enzymes in human control and keratoconic corneas. Histol. Histopathol. 2016, 31, 63–71. [Google Scholar] [CrossRef]
- Shetty, R.; Sathyanarayanamoorthy, A.; Ramachandra, R.A.; Arora, V.; Ghosh, A.; Srivatsa, P.R.; Pahuja, N.; Nuijts, R.M.; Sinha-Roy, A.; Mohan, R.R.; et al. Attenuation of lysyl oxidase and collagen gene expression in keratoconus patient corneal epithelium corresponds to disease severity. Mol. Vis. 2015, 21, 12–25. [Google Scholar]
- Smith, V.A.; Matthews, F.J.; Majid, M.A.; Cook, S.D. Keratoconus: Matrix metalloproteinase-2 activation and TIMP modulation. Biochim. Biophys. Acta 2006, 1762, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Shetty, R.; Ghosh, A.; Lim, R.R.; Subramani, M.; Mihir, K.; Reshma, A.R.; Ranganath, A.; Nagaraj, S.; Nuijts, R.M.; Beuerman, R.; et al. Elevated expression of matrix metalloproteinase-9 and inflammatory cytokines in keratoconus patients is inhibited by cyclosporine A. Investig. Ophthalmol. Vis. Sci. 2015, 56, 738–750. [Google Scholar] [CrossRef] [PubMed]
- Shetty, R.; D’Souza, S.; Khamar, P.; Ghosh, A.; Nuijts, R.; Sethu, S. Biochemical Markers and Alterations in Keratoconus. Asia Pac. J. Ophthalmol. 2020, 9, 533–540. [Google Scholar] [CrossRef]
- Brancati, F.; Valente, E.M.; Sarkozy, A.; Feher, J.; Castori, M.; Del Duca, P.; Mingarelli, R.; Pizzuti, A.; Dallapiccola, B. A locus for autosomal dominant keratoconus maps to human chromosome 3p14-q13. J. Med. Genet. 2004, 41, 188–192. [Google Scholar] [CrossRef]
- Burdon, K.P.; Coster, D.J.; Charlesworth, J.C.; Mills, R.A.; Laurie, K.J.; Giunta, C.; Hewitt, A.W.; Latimer, P.; Craig, J.E. Apparent autosomal dominant keratoconus in a large Australian pedigree accounted for by digenic inheritance of two novel loci. Hum. Genet. 2008, 124, 379–386. [Google Scholar] [CrossRef]
- Hameed, A.; Khaliq, S.; Ismail, M.; Anwar, K.; Ebenezer, N.D.; Jordan, T.; Mehdi, S.Q.; Payne, A.M.; Bhattacharya, S.S. A novel locus for Leber congenital amaurosis (LCA4) with anterior keratoconus mapping to chromosome 17p13. Investig. Ophthalmol. Vis. Sci. 2000, 41, 629–633. [Google Scholar]
- Hutchings, H.; Ginisty, H.; Le Gallo, M.; Levy, D.; Stoesser, F.; Rouland, J.F.; Arne, J.L.; Lalaux, M.H.; Calvas, P.; Roth, M.P.; et al. Identification of a new locus for isolated familial keratoconus at 2p24. J. Med. Genet. 2005, 42, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, J.; Hauser, M.A.; Afshari, N.A.; Allingham, R.R.; Liu, Y. The Genetics of Keratoconus: A Review. Reprod. Syst. Sex Disord. 2012. [Google Scholar] [CrossRef]
- Czugala, M.; Karolak, J.A.; Nowak, D.M.; Polakowski, P.; Pitarque, J.; Molinari, A.; Rydzanicz, M.; Bejjani, B.A.; Yue, B.Y.; Szaflik, J.P.; et al. Novel mutation and three other sequence variants segregating with phenotype at keratoconus 13q32 susceptibility locus. Eur. J. Hum. Genet. 2012, 20, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Droitcourt, C.; Touboul, D.; Ged, C.; Ezzedine, K.; Cario-Andre, M.; de Verneuil, H.; Colin, J.; Taieb, A. A prospective study of filaggrin null mutations in keratoconus patients with or without atopic disorders. Dermatology 2011, 222, 336–341. [Google Scholar] [CrossRef] [PubMed]
- Guan, T.; Liu, C.; Ma, Z.; Ding, S. The point mutation and polymorphism in keratoconus candidate gene TGFBI in Chinese population. Gene 2012, 503, 137–139. [Google Scholar] [CrossRef]
- Heon, E.; Greenberg, A.; Kopp, K.K.; Rootman, D.; Vincent, A.L.; Billingsley, G.; Priston, M.; Dorval, K.M.; Chow, R.L.; McInnes, R.R.; et al. VSX1: A gene for posterior polymorphous dystrophy and keratoconus. Hum. Mol. Genet. 2002, 11, 1029–1036. [Google Scholar] [CrossRef]
- Lechner, J.; Dash, D.P.; Muszynska, D.; Hosseini, M.; Segev, F.; George, S.; Frazer, D.G.; Moore, J.E.; Kaye, S.B.; Young, T.; et al. Mutational spectrum of the ZEB1 gene in corneal dystrophies supports a genotype-phenotype correlation. Investig. Ophthalmol. Vis. Sci. 2013, 54, 3215–3223. [Google Scholar] [CrossRef]
- Udar, N.; Atilano, S.R.; Brown, D.J.; Holguin, B.; Small, K.; Nesburn, A.B.; Kenney, M.C. SOD1: A candidate gene for keratoconus. Investig. Ophthalmol. Vis. Sci. 2006, 47, 3345–3351. [Google Scholar] [CrossRef]
- Gupta, N.; Vashist, P.; Ganger, A.; Tandon, R.; Gupta, S.K. Eye donation and eye banking in India. Natl. Med. J. India 2018, 31, 283–286. [Google Scholar] [CrossRef]
- Weiss, J.S.; Moller, H.U.; Aldave, A.J.; Seitz, B.; Bredrup, C.; Kivela, T.; Munier, F.L.; Rapuano, C.J.; Nischal, K.K.; Kim, E.K.; et al. IC3D classification of corneal dystrophies--edition 2. Cornea 2015, 34, 117–159. [Google Scholar] [CrossRef] [PubMed]
- Bourges, J.L. Corneal dystrophies. J. Fr. Ophtalmol. 2017, 40, e177–e192. [Google Scholar] [CrossRef]
- Soh, Y.Q.; Kocaba, V.; Weiss, J.S.; Jurkunas, U.V.; Kinoshita, S.; Aldave, A.J.; Mehta, J.S. Corneal dystrophies. Nat. Rev. Dis. Primers 2020, 6, 46. [Google Scholar] [CrossRef] [PubMed]
- Hamill, C.E.; Schmedt, T.; Jurkunas, U. Fuchs endothelial cornea dystrophy: A review of the genetics behind disease development. Semin. Ophthalmol. 2013, 28, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Adamis, A.P.; Filatov, V.; Tripathi, B.J.; Tripathi, R.C. Fuchs’ endothelial dystrophy of the cornea. Surv. Ophthalmol. 1993, 38, 149–168. [Google Scholar] [CrossRef]
- Nishtala, K.; Pahuja, N.; Shetty, R.; Nuijts, R.M.; Ghosh, A. Tear biomarkers for keratoconus. Eye Vis. 2016, 3, 19. [Google Scholar] [CrossRef] [PubMed]
- Fan Gaskin, J.C.; Patel, D.V.; McGhee, C.N. Acute corneal hydrops in keratoconus—New perspectives. Am. J. Ophthalmol. 2014, 157, 921–928. [Google Scholar] [CrossRef]
- Hassan, O.M.; Farooq, A.V.; Soin, K.; Djalilian, A.R.; Hou, J.H. Management of Corneal Scarring Secondary to Herpes Zoster Keratitis. Cornea 2017, 36, 1018–1023. [Google Scholar] [CrossRef] [PubMed]
- Menda, S.A.; Das, M.; Panigrahi, A.; Prajna, N.V.; Acharya, N.R.; Lietman, T.M.; McLeod, S.D.; Keenan, J.D. Association of Postfungal Keratitis Corneal Scar Features with Visual Acuity. JAMA Ophthalmol. 2020, 138, 113–118. [Google Scholar] [CrossRef]
- Maharana, P.K.; Sharma, N.; Vajpayee, R.B. Acute corneal hydrops in keratoconus. Indian J. Ophthalmol. 2013, 61, 461–464. [Google Scholar] [CrossRef]
- Hashemi, H.; Heydarian, S.; Hooshmand, E.; Saatchi, M.; Yekta, A.; Aghamirsalim, M.; Valadkhan, M.; Mortazavi, M.; Hashemi, A.; Khabazkhoob, M. The Prevalence and Risk Factors for Keratoconus: A Systematic Review and Meta-Analysis. Cornea 2020, 39, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Rebenitsch, R.L.; Kymes, S.M.; Walline, J.J.; Gordon, M.O. The lifetime economic burden of keratoconus: A decision analysis using a markov model. Am. J. Ophthalmol. 2011, 151, 768–773. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.T.; Ang, L.P. Automated lamellar therapeutic keratoplasty for post-PRK corneal scarring and thinning. Am. J. Ophthalmol. 2004, 138, 1067–1069. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, R.; Reddy, J.C.; Rapuano, C.J.; Vaddavalli, P.K. Phototherapeutic keratectomy: Indications, methods and decision making. Indian J. Ophthalmol. 2020, 68, 2856–2866. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.T.; Mehta, J.S. Future directions in lamellar corneal transplantation. Cornea 2007, 26, S21–S28. [Google Scholar] [CrossRef]
- Saini, J.S. Realistic targets and strategies in eye banking. Indian J. Ophthalmol. 1997, 45, 141–142. [Google Scholar]
- Dandona, L.; Naduvilath, T.J.; Janarthanan, M.; Ragu, K.; Rao, G.N. Survival analysis and visual outcome in a large series of corneal transplants in India. Br. J. Ophthalmol. 1997, 81, 726–731. [Google Scholar] [CrossRef]
- Sinha, R.; Vanathi, M.; Sharma, N.; Titiyal, J.S.; Vajpayee, R.B.; Tandon, R. Outcome of penetrating keratoplasty in patients with bilateral corneal blindness. Eye 2005, 19, 451–454. [Google Scholar] [CrossRef]
- Hos, D.; Matthaei, M.; Bock, F.; Maruyama, K.; Notara, M.; Clahsen, T.; Hou, Y.; Le, V.N.H.; Salabarria, A.C.; Horstmann, J.; et al. Immune reactions after modern lamellar (DALK, DSAEK, DMEK) versus conventional penetrating corneal transplantation. Prog. Retin. Eye Res. 2019, 73, 100768. [Google Scholar] [CrossRef]
- Alio Del Barrio, J.L.; Bhogal, M.; Ang, M.; Ziaei, M.; Robbie, S.; Montesel, A.; Gore, D.M.; Mehta, J.S.; Alio, J.L. Corneal transplantation after failed grafts: Options and outcomes. Surv. Ophthalmol. 2021, 66, 20–40. [Google Scholar] [CrossRef]
- Di Zazzo, A.; Kheirkhah, A.; Abud, T.B.; Goyal, S.; Dana, R. Management of high-risk corneal transplantation. Surv. Ophthalmol. 2017, 62, 816–827. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.T.; Dart, J.K.; Holland, E.J.; Kinoshita, S. Corneal transplantation. Lancet 2012, 379, 1749–1761. [Google Scholar] [CrossRef] [PubMed]
- de By, T.M. Shortage in the face of plenty: Improving the allocation of corneas for transplantation. Dev. Ophthalmol. 2003, 36, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Atchison, R.W.; Casto, B.C.; Hammon, W.M. Adenovirus-Associated Defective Virus Particles. Science 1965, 149, 754–756. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, J.; Taylor, K.A.; Chapman, M.S. Adeno-associated virus-2 and its primary cellular receptor--Cryo-EM structure of a heparin complex. Virology 2009, 385, 434–443. [Google Scholar] [CrossRef]
- Qing, K.; Mah, C.; Hansen, J.; Zhou, S.; Dwarki, V.; Srivastava, A. Human fibroblast growth factor receptor 1 is a co-receptor for infection by adeno-associated virus 2. Nat. Med. 1999, 5, 71–77. [Google Scholar] [CrossRef]
- Summerford, C.; Bartlett, J.S.; Samulski, R.J. AlphaVbeta5 integrin: A co-receptor for adeno-associated virus type 2 infection. Nat. Med. 1999, 5, 78–82. [Google Scholar] [CrossRef]
- Bartlett, J.S.; Wilcher, R.; Samulski, R.J. Infectious entry pathway of adeno-associated virus and adeno-associated virus vectors. J. Virol. 2000, 74, 2777–2785. [Google Scholar] [CrossRef]
- Douar, A.M.; Poulard, K.; Stockholm, D.; Danos, O. Intracellular trafficking of adeno-associated virus vectors: Routing to the late endosomal compartment and proteasome degradation. J. Virol. 2001, 75, 1824–1833. [Google Scholar] [CrossRef]
- Mohan, R.R.; Schultz, G.S.; Hong, J.W.; Mohan, R.R.; Wilson, S.E. Gene transfer into rabbit keratocytes using AAV and lipid-mediated plasmid DNA vectors with a lamellar flap for stromal access. Exp. Eye Res. 2003, 76, 373–383. [Google Scholar] [CrossRef]
- Chiorini, J.A.; Kim, F.; Yang, L.; Kotin, R.M. Cloning and characterization of adeno-associated virus type 5. J. Virol. 1999, 73, 1309–1319. [Google Scholar] [CrossRef] [PubMed]
- Zabner, J.; Seiler, M.; Walters, R.; Kotin, R.M.; Fulgeras, W.; Davidson, B.L.; Chiorini, J.A. Adeno-associated virus type 5 (AAV5) but not AAV2 binds to the apical surfaces of airway epithelia and facilitates gene transfer. J. Virol. 2000, 74, 3852–3858. [Google Scholar] [CrossRef]
- Gupta, S.; Rodier, J.T.; Sharma, A.; Giuliano, E.A.; Sinha, P.R.; Hesemann, N.P.; Ghosh, A.; Mohan, R.R. Targeted AAV5-Smad7 gene therapy inhibits corneal scarring in vivo. PLoS ONE 2017, 12, e0172928. [Google Scholar] [CrossRef] [PubMed]
- Mohan, R.R.; Tandon, A.; Sharma, A.; Cowden, J.W.; Tovey, J.C. Significant inhibition of corneal scarring in vivo with tissue-selective, targeted AAV5 decorin gene therapy. Investig. Ophthalmol. Vis. Sci. 2011, 52, 4833–4841. [Google Scholar] [CrossRef] [PubMed]
- Hippert, C.; Ibanes, S.; Serratrice, N.; Court, F.; Malecaze, F.; Kremer, E.J.; Kalatzis, V. Corneal transduction by intra-stromal injection of AAV vectors in vivo in the mouse and ex vivo in human explants. PLoS ONE 2012, 7, e35318. [Google Scholar] [CrossRef]
- Sharma, A.; Tovey, J.C.; Ghosh, A.; Mohan, R.R. AAV serotype influences gene transfer in corneal stroma in vivo. Exp. Eye Res. 2010, 91, 440–448. [Google Scholar] [CrossRef] [PubMed]
- Vance, M.; Llanga, T.; Bennett, W.; Woodard, K.; Murlidharan, G.; Chungfat, N.; Asokan, A.; Gilger, B.; Kurtzberg, J.; Samulski, R.J.; et al. AAV Gene Therapy for MPS1-associated Corneal Blindness. Sci. Rep. 2016, 6, 22131. [Google Scholar] [CrossRef] [PubMed]
- O’Callaghan, J.; Crosbie, D.E.; Cassidy, P.S.; Sherwood, J.M.; Flugel-Koch, C.; Lutjen-Drecoll, E.; Humphries, M.M.; Reina-Torres, E.; Wallace, D.; Kiang, A.S.; et al. Therapeutic potential of AAV-mediated MMP-3 secretion from corneal endothelium in treating glaucoma. Hum. Mol. Genet. 2017, 26, 1230–1246. [Google Scholar] [CrossRef]
- Bastola, P.; Song, L.; Gilger, B.C.; Hirsch, M.L. Adeno-Associated Virus Mediated Gene Therapy for Corneal Diseases. Pharmaceutics 2020, 12, 767. [Google Scholar] [CrossRef]
- Miyadera, K.; Conatser, L.; Llanga, T.A.; Carlin, K.; O’Donnell, P.; Bagel, J.; Song, L.; Kurtzberg, J.; Samulski, R.J.; Gilger, B.; et al. Intrastromal Gene Therapy Prevents and Reverses Advanced Corneal Clouding in a Canine Model of Mucopolysaccharidosis I. Mol. Ther. 2020, 28, 1455–1463. [Google Scholar] [CrossRef]
- Gilger, B.C.; Hirsch, M.L. Therapeutic Applications of Adeno-Associated Virus (AAV) Gene Transfer of HLA-G in the Eye. Int. J. Mol. Sci. 2022, 23, 3465. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.C.; Yeh, S.I.; Tsao, Y.P.; Kuo, P.C. Subconjunctival injection of recombinant AAV-angiostatin ameliorates alkali burn induced corneal angiogenesis. Mol. Vis. 2007, 13, 2344–2352. [Google Scholar] [PubMed]
- Lai, Y.K.; Shen, W.Y.; Brankov, M.; Lai, C.M.; Constable, I.J.; Rakoczy, P.E. Potential long-term inhibition of ocular neovascularisation by recombinant adeno-associated virus-mediated secretion gene therapy. Gene Ther. 2002, 9, 804–813. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Tai, P.W.L.; Ai, J.; Gessler, D.J.; Su, Q.; Yao, X.; Zheng, Q.; Zamore, P.D.; Xu, X.; Gao, G. Transcriptome Profiling of Neovascularized Corneas Reveals miR-204 as a Multi-target Biotherapy Deliverable by rAAVs. Mol. Ther. Nucleic Acids 2018, 10, 349–360. [Google Scholar] [CrossRef]
- Gupta, S.; Fink, M.K.; Kempuraj, D.; Sinha, N.R.; Martin, L.M.; Keele, L.M.; Sinha, P.R.; Giuliano, E.A.; Hesemann, N.P.; Raikwar, S.P.; et al. Corneal fibrosis abrogation by a localized AAV-mediated inhibitor of differentiation 3 (Id3) gene therapy in rabbit eyes in vivo. Mol. Ther. 2022, 30, 3257–3269. [Google Scholar] [CrossRef]
- Maddon, P.J.; McDougal, J.S.; Clapham, P.R.; Dalgleish, A.G.; Jamal, S.; Weiss, R.A.; Axel, R. HIV infection does not require endocytosis of its receptor, CD4. Cell 1988, 54, 865–874. [Google Scholar] [CrossRef]
- Gonda, M.A.; Wong-Staal, F.; Gallo, R.C.; Clements, J.E.; Narayan, O.; Gilden, R.V. Sequence homology and morphologic similarity of HTLV-III and visna virus, a pathogenic lentivirus. Science 1985, 227, 173–177. [Google Scholar] [CrossRef]
- Mohan, R.R.; Martin, L.M.; Sinha, N.R. Novel insights into gene therapy in the cornea. Exp. Eye Res. 2021, 202, 108361. [Google Scholar] [CrossRef]
- Mohan, R.R.; Tovey, J.C.; Sharma, A.; Schultz, G.S.; Cowden, J.W.; Tandon, A. Targeted decorin gene therapy delivered with adeno-associated virus effectively retards corneal neovascularization in vivo. PLoS ONE 2011, 6, e26432. [Google Scholar] [CrossRef]
- Barcia, R.N.; Dana, M.R.; Kazlauskas, A. Corneal graft rejection is accompanied by apoptosis of the endothelium and is prevented by gene therapy with bcl-xL. Am. J. Transplant. 2007, 7, 2082–2089. [Google Scholar] [CrossRef]
- Nosov, M.; Wilk, M.; Morcos, M.; Cregg, M.; O’Flynn, L.; Treacy, O.; Ritter, T. Role of lentivirus-mediated overexpression of programmed death-ligand 1 on corneal allograft survival. Am. J. Transplant. 2012, 12, 1313–1322. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Zhou, X.T.; Yu, Y.; Zhu, J.Y.; Dai, J.H.; Qu, X.M.; Le, Q.H.; Chu, R.Y. Inhibition of corneal fibrosis by Smad7 in rats after photorefractive keratectomy. Chin. Med. J. (Engl.) 2013, 126, 1445–1450. [Google Scholar] [PubMed]
- Parker, D.G.; Kaufmann, C.; Brereton, H.M.; Anson, D.S.; Francis-Staite, L.; Jessup, C.F.; Marshall, K.; Tan, C.; Koldej, R.; Coster, D.J.; et al. Lentivirus-mediated gene transfer to the rat, ovine and human cornea. Gene Ther. 2007, 14, 760–767. [Google Scholar] [CrossRef] [PubMed]
- Pastak, M.; Kleff, V.; Saban, D.R.; Czugala, M.; Steuhl, K.P.; Ergun, S.; Singer, B.B.; Fuchsluger, T.A. Gene Therapy for Modulation of T-Cell-Mediated Immune Response Provoked by Corneal Transplantation. Hum. Gene Ther. 2018, 29, 467–479. [Google Scholar] [CrossRef] [PubMed]
- Petrie, N.C.; Yao, F.; Eriksson, E. Gene therapy in wound healing. Surg. Clin. N. Am. 2003, 83, 597–616. [Google Scholar] [CrossRef]
- Borras, T.; Gabelt, B.T.; Klintworth, G.K.; Peterson, J.C.; Kaufman, P.L. Non-invasive observation of repeated adenoviral GFP gene delivery to the anterior segment of the monkey eye in vivo. J. Gene Med. 2001, 3, 437–449. [Google Scholar] [CrossRef]
- Fisher, K.D.; Stallwood, Y.; Green, N.K.; Ulbrich, K.; Mautner, V.; Seymour, L.W. Polymer-coated adenovirus permits efficient retargeting and evades neutralising antibodies. Gene Ther. 2001, 8, 341–348. [Google Scholar] [CrossRef]
- Saika, S.; Ikeda, K.; Yamanaka, O.; Flanders, K.C.; Nakajima, Y.; Miyamoto, T.; Ohnishi, Y.; Kao, W.W.; Muragaki, Y.; Ooshima, A. Therapeutic effects of adenoviral gene transfer of bone morphogenic protein-7 on a corneal alkali injury model in mice. Lab. Investig. 2005, 85, 474–486. [Google Scholar] [CrossRef]
- Nguyen, P.; Yiu, S.C. Strategies for local gene therapy of corneal allograft rejection. Middle East Afr. J. Ophthalmol. 2013, 20, 11–25. [Google Scholar] [CrossRef]
- Zhou, S.Y.; Xie, Z.L.; Xiao, O.; Yang, X.R.; Heng, B.C.; Sato, Y. Inhibition of mouse alkali burn induced-corneal neovascularization by recombinant adenovirus encoding human vasohibin-1. Mol. Vis. 2010, 16, 1389–1398. [Google Scholar]
- Yu, H.; Wu, J.; Li, H.; Wang, Z.; Chen, X.; Tian, Y.; Yi, M.; Ji, X.; Ma, J.; Huang, Q. Inhibition of corneal neovascularization by recombinant adenovirus-mediated sFlk-1 expression. Biochem. Biophys. Res. Commun. 2007, 361, 946–952. [Google Scholar] [CrossRef] [PubMed]
- Saika, S.; Yamanaka, O.; Okada, Y.; Miyamoto, T.; Kitano, A.; Flanders, K.C.; Ohnishi, Y.; Nakajima, Y.; Kao, W.W.; Ikeda, K. Effect of overexpression of PPARgamma on the healing process of corneal alkali burn in mice. Am. J. Physiol. Cell Physiol. 2007, 293, C75–C86. [Google Scholar] [CrossRef] [PubMed]
- Saghizadeh, M.; Kramerov, A.A.; Yu, F.S.; Castro, M.G.; Ljubimov, A.V. Normalization of wound healing and diabetic markers in organ cultured human diabetic corneas by adenoviral delivery of c-Met gene. Investig. Ophthalmol. Vis. Sci. 2010, 51, 1970–1980. [Google Scholar] [CrossRef] [PubMed]
- Klebe, S.; Sykes, P.J.; Coster, D.J.; Krishnan, R.; Williams, K.A. Prolongation of sheep corneal allograft survival by ex vivo transfer of the gene encoding interleukin-10. Transplantation 2001, 71, 1214–1220. [Google Scholar] [CrossRef]
- Klebe, S.; Coster, D.J.; Sykes, P.J.; Swinburne, S.; Hallsworth, P.; Scheerlinck, J.P.; Krishnan, R.; Williams, K.A. Prolongation of sheep corneal allograft survival by transfer of the gene encoding ovine IL-12-p40 but not IL-4 to donor corneal endothelium. J. Immunol. 2005, 175, 2219–2226. [Google Scholar] [CrossRef]
- Trapani, I.; Puppo, A.; Auricchio, A. Vector platforms for gene therapy of inherited retinopathies. Prog. Retin. Eye Res. 2014, 43, 108–128. [Google Scholar] [CrossRef]
- Chang, Y.K.; Hwang, J.S.; Chung, T.Y.; Shin, Y.J. SOX2 Activation Using CRISPR/dCas9 Promotes Wound Healing in Corneal Endothelial Cells. Stem Cells 2018, 36, 1851–1862. [Google Scholar] [CrossRef]
- Zhou, R.; Dean, D.A. Gene transfer of interleukin 10 to the murine cornea using electroporation. Exp. Biol. Med. (Maywood) 2007, 232, 362–369. [Google Scholar]
- Oshima, Y.; Sakamoto, T.; Hisatomi, T.; Tsutsumi, C.; Sassa, Y.; Ishibashi, T.; Inomata, H. Targeted gene transfer to corneal stroma in vivo by electric pulses. Exp. Eye Res. 2002, 74, 191–198. [Google Scholar] [CrossRef]
- Rosazza, C.; Meglic, S.H.; Zumbusch, A.; Rols, M.P.; Miklavcic, D. Gene Electrotransfer: A Mechanistic Perspective. Curr. Gene Ther. 2016, 16, 98–129. [Google Scholar] [CrossRef]
- de la Fuente, M.; Seijo, B.; Alonso, M.J. Bioadhesive hyaluronan-chitosan nanoparticles can transport genes across the ocular mucosa and transfect ocular tissue. Gene Ther. 2008, 15, 668–676. [Google Scholar] [CrossRef] [PubMed]
- Jain, G.K.; Pathan, S.A.; Akhter, S.; Jayabalan, N.; Talegaonkar, S.; Khar, R.K.; Ahmad, F.J. Microscopic and spectroscopic evaluation of novel PLGA-chitosan Nanoplexes as an ocular delivery system. Colloids Surf. B Biointerfaces 2011, 82, 397–403. [Google Scholar] [CrossRef]
- Nagarwal, R.C.; Singh, P.N.; Kant, S.; Maiti, P.; Pandit, J.K. Chitosan nanoparticles of 5-fluorouracil for ophthalmic delivery: Characterization, in-vitro and in-vivo study. Chem. Pharm. Bull. 2011, 59, 272–278. [Google Scholar] [CrossRef] [PubMed]
- Felgner, P.L.; Gadek, T.R.; Holm, M.; Roman, R.; Chan, H.W.; Wenz, M.; Northrop, J.P.; Ringold, G.M.; Danielsen, M. Lipofection: A highly efficient, lipid-mediated DNA-transfection procedure. Proc. Natl. Acad. Sci. USA 1987, 84, 7413–7417. [Google Scholar] [CrossRef] [PubMed]
- Farhood, H.; Serbina, N.; Huang, L. The role of dioleoyl phosphatidylethanolamine in cationic liposome mediated gene transfer. Biochim. Biophys. Acta 1995, 1235, 289–295. [Google Scholar] [CrossRef]
- Taketani, Y.; Kitamoto, K.; Sakisaka, T.; Kimakura, M.; Toyono, T.; Yamagami, S.; Amano, S.; Kuroda, M.; Moore, T.; Usui, T.; et al. Repair of the TGFBI gene in human corneal keratocytes derived from a granular corneal dystrophy patient via CRISPR/Cas9-induced homology-directed repair. Sci. Rep. 2017, 7, 16713. [Google Scholar] [CrossRef]
- Courtney, D.G.; Atkinson, S.D.; Allen, E.H.; Moore, J.E.; Walsh, C.P.; Pedrioli, D.M.; MacEwen, C.J.; Pellegrini, G.; Maurizi, E.; Serafini, C.; et al. siRNA silencing of the mutant keratin 12 allele in corneal limbal epithelial cells grown from patients with Meesmann’s epithelial corneal dystrophy. Investig. Ophthalmol. Vis. Sci. 2014, 55, 3352–3360. [Google Scholar] [CrossRef]
- Courtney, D.G.; Atkinson, S.D.; Moore, J.E.; Maurizi, E.; Serafini, C.; Pellegrini, G.; Black, G.C.; Manson, F.D.; Yam, G.H.; Macewen, C.J.; et al. Development of allele-specific gene-silencing siRNAs for TGFBI Arg124Cys in lattice corneal dystrophy type I. Investig. Ophthalmol. Vis. Sci. 2014, 55, 977–985. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, K.S.; Giuliano, E.A.; Sharma, A.; Tandon, A.; Rodier, J.T.; Mohan, R.R. Decorin-PEI nanoconstruct attenuates equine corneal fibroblast differentiation. Vet. Ophthalmol. 2014, 17, 162–169. [Google Scholar] [CrossRef]
- Hu, M.L.; Edwards, T.L.; O’Hare, F.; Hickey, D.G.; Wang, J.H.; Liu, Z.; Ayton, L.N. Gene therapy for inherited retinal diseases: Progress and possibilities. Clin. Exp. Optom. 2021, 104, 444–454. [Google Scholar] [CrossRef]
- Lee, J.H.; Wang, J.H.; Chen, J.; Li, F.; Edwards, T.L.; Hewitt, A.W.; Liu, G.S. Gene therapy for visual loss: Opportunities and concerns. Prog. Retin. Eye Res. 2019, 68, 31–53. [Google Scholar] [CrossRef] [PubMed]
- Chtarto, A.; Bockstael, O.; Gebara, E.; Vermoesen, K.; Melas, C.; Pythoud, C.; Levivier, M.; De Witte, O.; Luthi-Carter, R.; Clinkers, R.; et al. An adeno-associated virus-based intracellular sensor of pathological nuclear factor-kappaB activation for disease-inducible gene transfer. PLoS ONE 2013, 8, e53156. [Google Scholar] [CrossRef] [PubMed]
- Burnight, E.R.; Giacalone, J.C.; Cooke, J.A.; Thompson, J.R.; Bohrer, L.R.; Chirco, K.R.; Drack, A.V.; Fingert, J.H.; Worthington, K.S.; Wiley, L.A.; et al. CRISPR-Cas9 genome engineering: Treating inherited retinal degeneration. Prog. Retin. Eye Res. 2018, 65, 28–49. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Kanasty, R.L.; Eltoukhy, A.A.; Vegas, A.J.; Dorkin, J.R.; Anderson, D.G. Non-viral vectors for gene-based therapy. Nat. Rev. Genet. 2014, 15, 541–555. [Google Scholar] [CrossRef] [PubMed]
- Raikwar, S.P.; Raikwar, A.S.; Chaurasia, S.S.; Mohan, R.R. Gene editing for corneal disease management. World J. Transl. Med. 2016, 5, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Ma, E.; Harrington, L.B.; O’Connell, M.R.; Zhou, K.; Doudna, J.A. Single-Stranded DNA Cleavage by Divergent CRISPR-Cas9 Enzymes. Mol. Cell 2015, 60, 398–407. [Google Scholar] [CrossRef]
- Yu, W.; Wu, Z. Ocular delivery of CRISPR/Cas genome editing components for treatment of eye diseases. Adv. Drug Deliv. Rev. 2021, 168, 181–195. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, L.; Bell, P.; McMenamin, D.; He, Z.; White, J.; Yu, H.; Xu, C.; Morizono, H.; Musunuru, K.; et al. A dual AAV system enables the Cas9-mediated correction of a metabolic liver disease in newborn mice. Nat. Biotechnol. 2016, 34, 334–338. [Google Scholar] [CrossRef]
- Courtney, D.G.; Moore, J.E.; Atkinson, S.D.; Maurizi, E.; Allen, E.H.; Pedrioli, D.M.; McLean, W.H.; Nesbit, M.A.; Moore, C.B. CRISPR/Cas9 DNA cleavage at SNP-derived PAM enables both in vitro and in vivo KRT12 mutation-specific targeting. Gene Ther. 2016, 23, 108–112. [Google Scholar] [CrossRef]
- Elbashir, S.M.; Harborth, J.; Lendeckel, W.; Yalcin, A.; Weber, K.; Tuschl, T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 2001, 411, 494–498. [Google Scholar] [CrossRef]
- Hammond, S.M.; Bernstein, E.; Beach, D.; Hannon, G.J. An RNA-directed nuclease mediates post-transcriptional gene silencing in Drosophila cells. Nature 2000, 404, 293–296. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Wu, K.; Qiu, X.; Yang, Y.; Lin, X.; Yu, M. siRNA silencing of gene expression in trabecular meshwork: RhoA siRNA reduces IOP in mice. Curr. Mol. Med. 2012, 12, 1015–1027. [Google Scholar] [CrossRef] [PubMed]
- Supe, S.; Upadhya, A.; Singh, K. Role of small interfering RNA (siRNA) in targeting ocular neovascularization: A review. Exp. Eye Res. 2021, 202, 108329. [Google Scholar] [CrossRef] [PubMed]
- Bennett, C.F.; Krainer, A.R.; Cleveland, D.W. Antisense Oligonucleotide Therapies for Neurodegenerative Diseases. Annu. Rev. Neurosci. 2019, 42, 385–406. [Google Scholar] [CrossRef]
- Cursiefen, C.; Viaud, E.; Bock, F.; Geudelin, B.; Ferry, A.; Kadlecova, P.; Levy, M.; Al Mahmood, S.; Colin, S.; Thorin, E.; et al. Aganirsen antisense oligonucleotide eye drops inhibit keratitis-induced corneal neovascularization and reduce need for transplantation: The I-CAN study. Ophthalmology 2014, 121, 1683–1692. [Google Scholar] [CrossRef]
- Cursiefen, C.; Bock, F.; Horn, F.K.; Kruse, F.E.; Seitz, B.; Borderie, V.; Fruh, B.; Thiel, M.A.; Wilhelm, F.; Geudelin, B.; et al. GS-101 antisense oligonucleotide eye drops inhibit corneal neovascularization: Interim results of a randomized phase II trial. Ophthalmology 2009, 116, 1630–1637. [Google Scholar] [CrossRef]
- Ghosh, A.; Duan, D. Expanding adeno-associated viral vector capacity: A tale of two vectors. Biotechnol. Genet. Eng. Rev. 2007, 24, 165–177. [Google Scholar] [CrossRef]
- Ghosh, A.; Yue, Y.; Duan, D. Efficient transgene reconstitution with hybrid dual AAV vectors carrying the minimized bridging sequences. Hum. Gene Ther. 2011, 22, 77–83. [Google Scholar] [CrossRef]
- Ginn, S.L.; Amaya, A.K.; Alexander, I.E.; Edelstein, M.; Abedi, M.R. Gene therapy clinical trials worldwide to 2017: An update. J. Gene Med. 2018, 20, e3015. [Google Scholar] [CrossRef]
- Di Iorio, E.; Barbaro, V.; Alvisi, G.; Trevisan, M.; Ferrari, S.; Masi, G.; Nespeca, P.; Ghassabian, H.; Ponzin, D.; Palu, G. New Frontiers of Corneal Gene Therapy. Hum. Gene Ther. 2019, 30, 923–945. [Google Scholar] [CrossRef]
- Mohan, R.R.; Tripathi, R.; Sharma, A.; Sinha, P.R.; Giuliano, E.A.; Hesemann, N.P.; Chaurasia, S.S. Decorin antagonizes corneal fibroblast migration via caveolae-mediated endocytosis of epidermal growth factor receptor. Exp. Eye Res. 2019, 180, 200–207. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Fink, M.K.; Ghosh, A.; Tripathi, R.; Sinha, P.R.; Sharma, A.; Hesemann, N.P.; Chaurasia, S.S.; Giuliano, E.A.; Mohan, R.R. Novel Combination BMP7 and HGF Gene Therapy Instigates Selective Myofibroblast Apoptosis and Reduces Corneal Haze In Vivo. Investig. Ophthalmol. Vis. Sci. 2018, 59, 1045–1057. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, K.; Moore, H.; Tandon, A.; Gupta, S.; Khanna, R.; Mohan, R.R. Nanotechnology and adeno-associated virus-based decorin gene therapy ameliorates peritoneal fibrosis. Am. J. Physiol. Renal. Physiol. 2014, 307, F777–F782. [Google Scholar] [CrossRef] [PubMed]
- Lai, L.J.; Xiao, X.; Wu, J.H. Inhibition of corneal neovascularization with endostatin delivered by adeno-associated viral (AAV) vector in a mouse corneal injury model. J. Biomed. Sci. 2007, 14, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Parker, D.G.; Coster, D.J.; Brereton, H.M.; Hart, P.H.; Koldej, R.; Anson, D.S.; Williams, K.A. Lentivirus-mediated gene transfer of interleukin 10 to the ovine and human cornea. Clin. Exp. Ophthalmol. 2010, 38, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Fuchsluger, T.A.; Jurkunas, U.; Kazlauskas, A.; Dana, R. Anti-apoptotic gene therapy prolongs survival of corneal endothelial cells during storage. Gene Ther. 2011, 18, 778–787. [Google Scholar] [CrossRef]
- Watson, Z.L.; Washington, S.D.; Phelan, D.M.; Lewin, A.S.; Tuli, S.S.; Schultz, G.S.; Neumann, D.M.; Bloom, D.C. In Vivo Knockdown of the Herpes Simplex Virus 1 Latency-Associated Transcript Reduces Reactivation from Latency. J. Virol. 2018, 92. [Google Scholar] [CrossRef]
- Elbadawy, H.M.; Gailledrat, M.; Desseaux, C.; Salvalaio, G.; Di Iorio, E.; Ferrari, B.; Bertolin, M.; Barbaro, V.; Parekh, M.; Gayon, R.; et al. Gene transfer of integration defective anti-HSV-1 meganuclease to human corneas ex vivo. Gene Ther. 2014, 21, 272–281. [Google Scholar] [CrossRef]
- Sawamoto, K.; Chen, H.H.; Almeciga-Diaz, C.J.; Mason, R.W.; Tomatsu, S. Gene therapy for Mucopolysaccharidoses. Mol. Genet. Metab. 2018, 123, 59–68. [Google Scholar] [CrossRef]
- Song, L.; Bower, J.J.; Llanga, T.; Salmon, J.H.; Hirsch, M.L.; Gilger, B.C. Ocular Tolerability and Immune Response to Corneal Intrastromal AAV-IDUA Gene Therapy in New Zealand White Rabbits. Mol. Ther. Methods Clin. Dev. 2020, 18, 24–32. [Google Scholar] [CrossRef]
- Kamata, Y.; Okuyama, T.; Kosuga, M.; O’Hira, A.; Kanaji, A.; Sasaki, K.; Yamada, M.; Azuma, N. Adenovirus-mediated gene therapy for corneal clouding in mice with mucopolysaccharidosis type VII. Mol. Ther. 2001, 4, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Serratrice, N.; Cubizolle, A.; Ibanes, S.; Mestre-Frances, N.; Bayo-Puxan, N.; Creyssels, S.; Gennetier, A.; Bernex, F.; Verdier, J.M.; Haskins, M.E.; et al. Corrective GUSB transfer to the canine mucopolysaccharidosis VII cornea using a helper-dependent canine adenovirus vector. J. Control. Release 2014, 181, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Cotugno, G.; Annunziata, P.; Tessitore, A.; O’Malley, T.; Capalbo, A.; Faella, A.; Bartolomeo, R.; O’Donnell, P.; Wang, P.; Russo, F.; et al. Long-term amelioration of feline Mucopolysaccharidosis VI after AAV-mediated liver gene transfer. Mol. Ther. 2011, 19, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Carlson, E.C.; Liu, C.Y.; Yang, X.; Gregory, M.; Ksander, B.; Drazba, J.; Perez, V.L. In vivo gene delivery and visualization of corneal stromal cells using an adenoviral vector and keratocyte-specific promoter. Investig. Ophthalmol. Vis. Sci. 2004, 45, 2194–2200. [Google Scholar] [CrossRef]
- Liu, J.J.; Kao, W.W.; Wilson, S.E. Corneal epithelium-specific mouse keratin K12 promoter. Exp. Eye Res. 1999, 68, 295–301. [Google Scholar] [CrossRef]
- Hamilton, A.I. Keratoblast and keratocyte, not keratinocyte. Nature 1972, 238, 98. [Google Scholar] [CrossRef]
- Liu, C.; Arar, H.; Kao, C.; Kao, W.W. Identification of a 3.2 kb 5′-flanking region of the murine keratocan gene that directs beta-galactosidase expression in the adult corneal stroma of transgenic mice. Gene 2000, 250, 85–96. [Google Scholar] [CrossRef]
- Korecki, A.J.; Cueva-Vargas, J.L.; Fornes, O.; Agostinone, J.; Farkas, R.A.; Hickmott, J.W.; Lam, S.L.; Mathelier, A.; Zhou, M.; Wasserman, W.W.; et al. Human MiniPromoters for ocular-rAAV expression in ON bipolar, cone, corneal, endothelial, Muller glial, and PAX6 cells. Gene Ther. 2021, 28, 351–372. [Google Scholar] [CrossRef]
- Sakai, R.; Kinouchi, T.; Kawamoto, S.; Dana, M.R.; Hamamoto, T.; Tsuru, T.; Okubo, K.; Yamagami, S. Construction of human corneal endothelial cDNA library and identification of novel active genes. Investig. Ophthalmol. Vis. Sci. 2002, 43, 1749–1756. [Google Scholar]
- Verkman, A.S. Role of aquaporin water channels in eye function. Exp. Eye Res. 2003, 76, 137–143. [Google Scholar] [CrossRef]
- Johari, Y.B.; Mercer, A.C.; Liu, Y.; Brown, A.J.; James, D.C. Design of synthetic promoters for controlled expression of therapeutic genes in retinal pigment epithelial cells. Biotechnol. Bioeng. 2021, 118, 2001–2015. [Google Scholar] [CrossRef] [PubMed]
- Croze, R.H.; Kotterman, M.; Burns, C.H.; Schmitt, C.E.; Quezada, M.; Schaffer, D.; Kirn, D.; Francis, P. Viral Vector Technologies and Strategies: Improving on Nature. Int. Ophthalmol. Clin. 2021, 61, 59–89. [Google Scholar] [CrossRef] [PubMed]
- Lerch, T.F.; Xie, Q.; Chapman, M.S. The structure of adeno-associated virus serotype 3B (AAV-3B): Insights into receptor binding and immune evasion. Virology 2010, 403, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Meyer, N.L.; Hu, G.; Davulcu, O.; Xie, Q.; Noble, A.J.; Yoshioka, C.; Gingerich, D.S.; Trzynka, A.; David, L.; Stagg, S.M.; et al. Structure of the gene therapy vector, adeno-associated virus with its cell receptor, AAVR. Elife 2019, 8, e44707. [Google Scholar] [CrossRef] [PubMed]
- Ng, R.; Govindasamy, L.; Gurda, B.L.; McKenna, R.; Kozyreva, O.G.; Samulski, R.J.; Parent, K.N.; Baker, T.S.; Agbandje-McKenna, M. Structural characterization of the dual glycan binding adeno-associated virus serotype 6. J. Virol. 2010, 84, 12945–12957. [Google Scholar] [CrossRef]
- Shen, S.; Troupes, A.N.; Pulicherla, N.; Asokan, A. Multiple roles for sialylated glycans in determining the cardiopulmonary tropism of adeno-associated virus 4. J. Virol. 2013, 87, 13206–13213. [Google Scholar] [CrossRef]
- Mays, L.E.; Wang, L.; Tenney, R.; Bell, P.; Nam, H.J.; Lin, J.; Gurda, B.; Van Vliet, K.; Mikals, K.; Agbandje-McKenna, M.; et al. Mapping the structural determinants responsible for enhanced T cell activation to the immunogenic adeno-associated virus capsid from isolate rhesus 32.33. J. Virol. 2013, 87, 9473–9485. [Google Scholar] [CrossRef]
- Gurda, B.L.; DiMattia, M.A.; Miller, E.B.; Bennett, A.; McKenna, R.; Weichert, W.S.; Nelson, C.D.; Chen, W.J.; Muzyczka, N.; Olson, N.H.; et al. Capsid antibodies to different adeno-associated virus serotypes bind common regions. J. Virol. 2013, 87, 9111–9124. [Google Scholar] [CrossRef]
- Tseng, Y.S.; Gurda, B.L.; Chipman, P.; McKenna, R.; Afione, S.; Chiorini, J.A.; Muzyczka, N.; Olson, N.H.; Baker, T.S.; Kleinschmidt, J.; et al. Adeno-associated virus serotype 1 (AAV1)- and AAV5-antibody complex structures reveal evolutionary commonalities in parvovirus antigenic reactivity. J. Virol. 2015, 89, 1794–1808. [Google Scholar] [CrossRef]
- Wobus, C.E.; Hugle-Dorr, B.; Girod, A.; Petersen, G.; Hallek, M.; Kleinschmidt, J.A. Monoclonal antibodies against the adeno-associated virus type 2 (AAV-2) capsid: Epitope mapping and identification of capsid domains involved in AAV-2-cell interaction and neutralization of AAV-2 infection. J. Virol. 2000, 74, 9281–9293. [Google Scholar] [CrossRef]
- Mah, C.; Qing, K.; Khuntirat, B.; Ponnazhagan, S.; Wang, X.S.; Kube, D.M.; Yoder, M.C.; Srivastava, A. Adeno-associated virus type 2-mediated gene transfer: Role of epidermal growth factor receptor protein tyrosine kinase in transgene expression. J. Virol. 1998, 72, 9835–9843. [Google Scholar] [CrossRef] [PubMed]
- Qing, K.; Wang, X.S.; Kube, D.M.; Ponnazhagan, S.; Bajpai, A.; Srivastava, A. Role of tyrosine phosphorylation of a cellular protein in adeno-associated virus 2-mediated transgene expression. Proc. Natl. Acad. Sci. USA 1997, 94, 10879–10884. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; Zhao, W.; Wu, J.; Li, B.; Zolotukhin, S.; Govindasamy, L.; Agbandje-McKenna, M.; Srivastava, A. A dual role of EGFR protein tyrosine kinase signaling in ubiquitination of AAV2 capsids and viral second-strand DNA synthesis. Mol. Ther. 2007, 15, 1323–1330. [Google Scholar] [CrossRef] [PubMed]
- Markusic, D.M.; Herzog, R.W.; Aslanidi, G.V.; Hoffman, B.E.; Li, B.; Li, M.; Jayandharan, G.R.; Ling, C.; Zolotukhin, I.; Ma, W.; et al. High-efficiency transduction and correction of murine hemophilia B using AAV2 vectors devoid of multiple surface-exposed tyrosines. Mol. Ther. 2010, 18, 2048–2056. [Google Scholar] [CrossRef] [PubMed]
- Kay, C.N.; Ryals, R.C.; Aslanidi, G.V.; Min, S.H.; Ruan, Q.; Sun, J.; Dyka, F.M.; Kasuga, D.; Ayala, A.E.; Van Vliet, K.; et al. Targeting photoreceptors via intravitreal delivery using novel, capsid-mutated AAV vectors. PLoS ONE 2013, 8, e62097. [Google Scholar] [CrossRef]







| Region | Name of Dystrophy | Gene | Gene Locus | Mode of Inheritance | IC3D Category | Age of Onset | Symptoms | Visual Acuity | Clinical Appearance of the Cornea |
|---|---|---|---|---|---|---|---|---|---|
| Epithelial and Subepithelial Dystrophies | Epithelial basement membrane dystrophy (EBMD) | TGFBI | 5q31 | Sporadic | 1 | Adult | Corneal erosion, slightly distorted vision | Mild visual reduction | Thickening of the epithelium, round or oval opacities, and lines. |
| Epithelial recurrent erosion dystrophy (ERED) | COL17E1 | 10q23 | Autosomal Dominant | 3 | 1st Decade | Stinging, burning, painful corneal erosion, photophobia | Sometimes impaired | Epithelial erosion | |
| Subepithelial mucinous corneal dystrophy (SMCD) | Unknown | unknown | Autosomal Dominant | 4 | 1st Decade | Painful incidence of recurrent corneal erosion | Progressive loss of vision | Bilateral subepithelial opacities and haze, mostly denser centrally, involving the entire cornea | |
| Meesmann corneal dystrophy (MECD) | KRT3, KRT12, | 12q13, 17q12 | Autosomal Dominant | 1 | Early childhood | Mild erosion and reduced sensation of the cornea | Rarely blurred vision | Multiple, tiny, distinct epithelial vesicles extend to the limbus and are mostly cumulated in the interpalpebral area. | |
| Lisch epithelial corneal dystrophy (LECD) | Unknown | Xp22.3 | X-linked Dominant | 2 | Childhood | symptomatic or blurred vision if the pupillary zone is involved | Sometimes impaired | Localized epithelial opacities of various patterns: whorls, bands, flame, feather shaped. | |
| Gelatinous drop-like corneal dystrophy (GDLD) | TACSTD2 (M1S1) | 1p32 | Autosomal Recessive | 1 | 1st to 2nd Decade | Distorted vision, photophobia, scratchy sensation, redness, tearing | Marked visual impairment | Appearance of subepithelial lesions, indicating extremely hyperpermeable corneal epithelium, Superficial vascularization, stromal opacification | |
| Bowman Layer Dystrophies | Reis–Buckler’s corneal dystrophy (RBCD) | TGFBI | 5q31 | Autosomal Dominant | 1 | Childhood | Painful incidence of recurrent corneal erosion | Progressive deuteriation of vision | Replacement of bowman layer by sheet like connective tissue with granular deposits, which extends to subepithelial stroma. |
| Thiel–Behnke corneal dystrophy (TBCD) | TGFBI | 5q31 | Autosomal Dominant | 2 | Childhood | Painful incidence of recurrent corneal erosion | Gradual visual impairment | Symmetrical subepithelial reticular (honeycomb) opacities in the central cornea; which can progress to deep stromal layers and corneal periphery. | |
| Stromal Dystrophies | Lattice corneal dystrophy (LCD) | TGFBI | 5q31 | Autosomal Dominant | 1 | 1st Decade | Stinging, burning, Painful incidence of recurrent corneal erosion | Progressive visual impairment | Thin branching refractile lines or subepithelial ovoid dots in the central cornea, diffuse stromal, ground-glass haze develops later |
| Granular corneal dystrophy (Type 1 and 2) | TGFBI | 5q31 | Autosomal Dominant | 1 | Childhood; early as 2 years of age | Frequent corneal erosion, photophobia, glare. | Decrease in visual acuity as opacification progresses with age. | Well-defined granular opacities are observed that don’t extend to the limbus. Type 2 can add snowflakes and lattice lines between the granules. | |
| Macular corneal dystrophy (MCD) | CHST6 | 16q22 | Autosomal Recessive | 1 | Childhood | Painful incidence of recurrent corneal erosion, reduced corneal sensitivity, photophobia | Severe visual impairment between 10–30 years. | Thinning of the cornea, in advanced stage corneal endothelium is affected and the Descemet membrane develops guttate excrescences. Limbus to limbus stromal haze, which later spreads to superficial, central, elevated white opacities. | |
| Schnyder Corneal Dystrophy (SCD) | UBIAD1 | 1p36 | Autosomal Dominant | 1 | Childhood to 2nd or 3rd decade | Reduced corneal sensitivity, glare increases, disproportionate decrease of photopic vision, may have hyperlipoproteinemia (type IIa, III, or IV) | Visual acuity decreases with age | Initial signs include central corneal haze and/or subepithelial crystals (>23 years), arcus lipoids (23–38 years), mid-peripheral panstromal haze (after 38 years) | |
| Congenital Stromal Corneal Dystrophy (CSCD) | DCN | 12q21.33 | Autosomal Dominant | 1 | Congenital | Irregular and cloudy appearance of the cornea, reduced visual acuity, increased glare | Moderate to severe visual loss | Diffuse, bilateral, corneal clouding with flake-like, whitish stromal opacities throughout the stroma, pachymetry demonstrates increase in thickness. | |
| Fleck Corneal Dystrophy (FCD) | PIKFYVE | 2q34 | Autosomal Dominant | 1 | Congenital | Asymptomatic | Normal | Small, translucent, discoid opacities that are scattered sparsely throughout without affecting the central cornea. Involvement of the asymmetric or unilateral corneal. | |
| Posterior Amorphous Corneal Dystrophy (PACD) | KERA, LUM, DCN, EPYC | 12q21.33 | Autosomal Dominant | 3 | 1st Decade, early as 16 weeks, possibly congenital nature | Mildly effected visual acuity | Mild visual reduction | Diffused grayish-white sheet like opacities in the posterior part of stroma, corneal thinning (>380 µm), flat corneal topography (<41.00 D) and hyperopia in the centroperipheral form. | |
| Descemets Membrane and Endothelial Dystrophies | Fuchs Endothelial Corneal Dystrophy (FECD); early and late-onset | COL8A2 | 1p34.3–p32, 13pTel–13q12.13, 15q, 18q21.2-q21.32 | Autosomal Dominant | 1, 2 | 4th decade or later | Epiphora due to recurrent corneal erosion, photophobia, pain, epithelial/stromal edema. | Progressive visual impairment | Diffuse thickening of Descemet membrane with excrescences (guttae). Endothelial cells sparse and atrophic |
| Posterior Polymorphous Corneal Dystrophy (PPCD); Type 1, 2, 3 and 4 | OVOL2, COL8A2, ZEB1, GRHL2 | 20p11.23, 1p34.3–p32.3, 10p11.2, 8q22.3 | Autosomal Dominant | 2, 1 | Childhood | Stromal clouding, endothelial decomposition that is often asymptomatic | Rarely extensive and progressive visual impairment | Deep corneal lesions that can be nodular, vesicular or blister-like. Edema of the stromal and epithelial layer, endothelial decomposition | |
| Congenital hereditary endothelial dystrophy | SLC4A11 | 20p13 | Autosomal Recessive | 1 | Congenital | Stromal clouding, blurred vision, photophobia | Blurring vision | Corneal thickening, clouding of the cornea, elevated IOP | |
| X-linked Endothelial Corneal Dystrophy | Unknown | Xq25 | X-chromosomal Dominant | 2 | Congenital | Blurring vision | Blurred vision in males | Cloudy cornea only in males, moon crater–like endothelial changes |
| Vectors |  AdenoVirus |  Lenti Virus |  AAV Virus |  Non-Viral |
|---|---|---|---|---|
| Advantages |
|
|
|
|
| Disadvantages |
|
potential
|
|
|
| Adeno-Associated Virus Mediated Corneal Gene Therapy | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Vector | Disease | Gene | Promoter | Serotype | Dosage | Model | Species | Mode of Administration | Outcome | Reference |
| rAAV | Glaucoma | MMP3 | CMV | AAV-2/9 | 1 × 1011 vgc | In Vivo | WT mouse | Intracameral | Efficient transduction resulted in an increase in aqueous concentration and MMP3 activity, also increased outflow facility and decreased IOP | [118] |
| rAAV | MPS1-associated Corneal Blindness | IDUA | CMV | AAV8G9 | 1 × 1010 vgc | Ex Vivo | Human | Intrastromal | Efficient widespread transduction resulted in a >10-fold supraphysiological increase in IDUA activity. No significant apoptosis due to AAV or IDUA Overexpression was observed. | [119] |
| rAAV | MPS1-associated Corneal Blindness | IDUA | CMV | AAV8G9 | Increasing volume (50–80 µL); escalating viral dose ranging from (5 × 1010–8 × 1010) per cornea | In Vivo | MPS I canine model | Intrastromal | Resolution of corneal clouding as early as 1st week, followed by sustained corneal transparency until the end of 25 weeks in eyes of MPS I canines with advanced disease whereas, prevention against the development of advanced corneal changes while restoring clarity was observed in MPS I canines with early corneal disease. | [120] |
| rAAV | Corneal scarring | Smad7 | CAG | AAV5 | 75 μL; (2.67 × 1013 μg/mL; n = 6) | In Vivo | 2–3-month-old New Zealand White female rabbits | Topical drops | Single topical application of AAV vectors in rabbit cornea post-PRK led to a significant decrease in corneal fibrosis and corneal haze. | [113] |
| rAAV | Corneal scarring | Decorin | CAG | AAV5 | 100 μL; 6.5 × 1012 μg/mL | In Vivo | 2–3-month-old New Zealand White female rabbits | Topical drops | The stromal haze and fibrosis were decreased in the corneas infected with rAAV. Decorin virions. No immunogenic or toxic response was observed. | [113] |
| rAAV | corneal inflammation and prevent corneal graft rejection | HLA-G1 | JET | AAV8G9 | 2.4 × 1010 vgc total dose | In Vivo | Naive Lewis rats | Intrastromal | Corneal intrastromal delivery of AAV.HLA-G subsequently reduced corneal inflammation, vascularization and fibrosis post-corneal injury. | [121] |
| rAAV | Corneal Neovascularization | Angiostatin | CMV | - | 5 µL; 1 × 1010 viral particles | In Vivo | 10–12 weeks old, Male Sprague-Dawley rats | Subconjunctival | Subconjunctival delivery of AAV. Angiostatin showed a significant reduction of alkali burn-induced corneal angiogenesis. | [122] |
| rAAV | Corneal Neovascularization | Flt-1 | CMV | AAV9 | 4 × 1011 particles/mL | In Vivo | Rats | Anterior chamber | The subsequent reduction in the development of corneal neovascularization in the stroma of cauterised rats by 36% in comparison to the control group. | [123] |
| rAAV | Corneal Neovascularization | miR-204 | CMV | rAAVrh.10 | 3.6 × 1010 GCs (Intrastromal), 3.6 × 1010 GCs (Subconjunctival) | In Vivo | 6- to 8-week-old female C57BL/6J mice | Intrastromal injection; subconjunctival injection | Attenuation of corneal neovascularization was observed in the alkali-burned cornea | [124] |
| rAAV | Corneal scarring/ fibrosis | Id3 | CAG | AAV5 | 100 μL; 6.5 × 1012 μg/mL | In Vivo | 2–3-month-old New Zealand White female rabbits | Topical drops | Attenuation of corneal fibrosis and restoration of corneal transparency was observed. No cellular toxicity was reported. | [125] |
| Lentivirus Mediated Corneal Gene Therapy | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Vector | Disease | Gene | Promoter | Dosage | Model | Species | Mode of Administration | Outcome | Reference |
| rLV | Corneal Neovascularization | Endostatin/Kringle 5 | CMV | 50 µL, approximately 108 virus particles/mL | In Vivo | New Zealand White rabbits | Subconjunctival | Corneas transduced with rLV.E-K-5 vector demonstrated inhibition of neovascularization and graft failure, whereas in control animals an early onset and profound neovascularization was observed. 5/6 animals in the control group had graft failure. | [59] |
| rLV | Corneal Graft Rejection | Bcl-xL | CMV | 5.5 × 106 IU/mL | In Vivo | BALB/c mice were used as recipients, and C57BL/6 mice (MHC and multiple minor H disparate) or BALB/c (syngeneic) corneas were used as donors | Topically transducing the cultured corneas ex vivo before transplantation. | Delivery of rLV. Bcl-xL to the corneal endothelium of donor corneas significantly improved and promoted allografts’ survival by preventing the endothelium’s apoptosis. | [130] |
| rLV | Corneal Graft Rejection | PD-L1 | Ubi-1 | Between 3.4 × 107 and 1 × 108 titration units (TU)/mL | In Vivo | Male Lewis (LEW, RT-1) rats served as recipients of male Dark Agouti (DA, RT-1avl) grafts | Topically transducing the cultured corneas ex vivo before transplantation. | A subsequent increase in the expression of PD-L1 levels in corneal cells helped prolong allograft survival with minimal proinflammatory cytokine expression. | [131] |
| rLV | Corneal Fibrosis | Smad7 | CMV | 1 × 104 IFU/μL | In Vivo | Sprague-Dawley rats, approximately 8 weeks of age | Topical drops | Exogenous expression of Smad7 gene expression resulted in reduced activation of the TGFβ/Smad signalling caused by the downregulation of phosphorylation of Smad2. Cell proliferation and fibrotic markers were also inhibited by Smad7. | [132] |
| rLV | Corneal Fibrosis | IL-10 | SV40 | 6.3 × 106 TU/mL | In Vivo | Ovine | Topically transducing the cultured corneas ex vivo before transplantation. | Prolonged survival of corneal allograft by a median of 7 days in recipient corneas transduced with lentivirus containing the therapeutic gene, compared to the control group. | [133] |
| rLV | Corneal Fibrosis | p35 | CMV | 3 × 105 IU/mL | In Vivo | 8 to 12 weeks old male C57BL/6 (B6) and (B/C) mice | Topically transducing the cultured corneas ex vivo before transplantation | Exogenous expression of the p35 gene reduced the graft-mediated immune response due to decreased CD4+ T-cells expression in the cornea. | [134] |
| Adenovirus Mediated Corneal Gene Therapy | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Vector | Disease | Gene | Promoter | Dosage | Model | Species | Mode of Administration | Outcome | Reference |
| Ad | Corneal scarring | BMP-7 | CAG | 3 µL, 2 × 107 PFU/mL | In Vivo | C57BL/6 Mice | Topical | Overexpression of BMP-7 subsequently reduced the scarring of the alkali burn corneas post-20 days of infection. | [138] |
| Ad | Corneal scarring | Smad7 | CAG | 1 × 107 PFU/μL | In Vivo | C57BL/6 Mice | Topical | Exogenous expression of Smad7 in the burned corneal tissue resulted in reduced activation or blocking of the Smad3 signalling and nuclear factor-κB signalling via RelA/p65. | [139] |
| Ad | Corneal Neovascularization | Vasohibin-1 | MMP-1 | 1 × 109 PFU/μL | In Vivo | 6 to 8 weeks old female BALB/c mice | Subconjunctival | Subconjunctival delivery of the therapeutic gene significantly reduced the scarring of the alkali burn corneas | [140] |
| Ad | Corneal Neovascularization | Flk-1 | CMV | 2 µL, 3 × 108 PFU/μL | In Vivo | 6–8 weeks old Sprague–Dawley rats | Anterior chamber | Significant inhibition of neovascularization was observed in the cauterized rat in comparison to the control group. | [141] |
| Ad | Corneal Fibrosis | PPAR-γ | CAG | 1 × 107 PFU/μL | In Vivo | Not specified, possibly topical | Upon overexpression of the transgene in alkali-burned mouse cornea, subsequent suppression of the pro-fibrotic factors and promotion of epithelial healing was observed. | [142] | |
| Ad | Corneal wound healing | c-met | CMV | 1 × 108 PFU/μL | Ex Vivo | Human | Topically transducing the cultured corneas ex vivo | Transduction of diabetic corneas c-met with c-met restored the HGF signalling, normalized the diabetic marker patterns, and accelerated the wound healing process | [143] |
| Ad | Corneal Graft Rejection | CMV | 1 × 108 PFU/μL | In Vivo | Lewis rats served as recipients of female rats of Dark Agouti | Topically transducing the cultured corneas ex vivo before transplantation. | Successful prevention of allogeneic graft rejection in corneal transplantation | [56] | |
| Ad | Corneal Graft Rejection | IL-10 | CMV | 6.6 × 102–6.6 × 108 PFU/μL | In Vivo | Sheep | Topically transducing the cultured corneas ex vivo before transplantation. | Overexpression of the immunomodulatory cytokine IL-10 showed significantly prolonged corneal allograft survival and reduced incidence of graft rejection. | [144] |
| Ad | Corneal Graft Rejection | IL-12 p40 | CMV | 0.8–1 × 107 PFU/cornea | In Vivo | Sheep | Topically transducing the cultured corneas ex vivo before transplantation. | Local intraocular production of p40 IL-12 increased the corneal graft survival in comparison to the control group which got rejected at the median of 20 days. | [145] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sarkar, S.; Panikker, P.; D’Souza, S.; Shetty, R.; Mohan, R.R.; Ghosh, A. Corneal Regeneration Using Gene Therapy Approaches. Cells 2023, 12, 1280. https://doi.org/10.3390/cells12091280
Sarkar S, Panikker P, D’Souza S, Shetty R, Mohan RR, Ghosh A. Corneal Regeneration Using Gene Therapy Approaches. Cells. 2023; 12(9):1280. https://doi.org/10.3390/cells12091280
Chicago/Turabian StyleSarkar, Subhradeep, Priyalakshmi Panikker, Sharon D’Souza, Rohit Shetty, Rajiv R. Mohan, and Arkasubhra Ghosh. 2023. "Corneal Regeneration Using Gene Therapy Approaches" Cells 12, no. 9: 1280. https://doi.org/10.3390/cells12091280