
Redefining Autoimmune Disorders’ Pathoetiology: Implications for Mood and Psychotic Disorders’ Association with Neurodegenerative and Classical Autoimmune Disorders





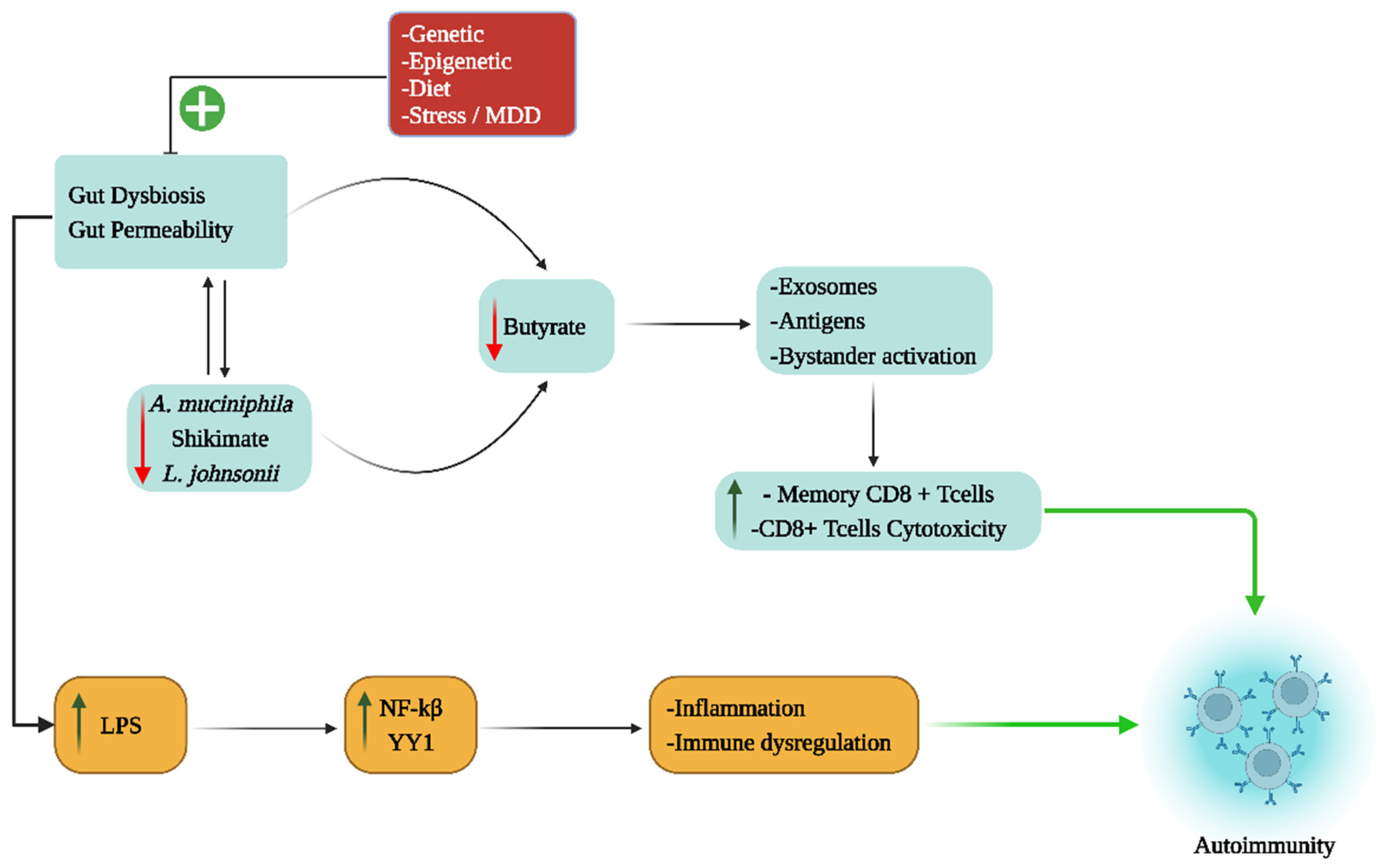{kind=link}
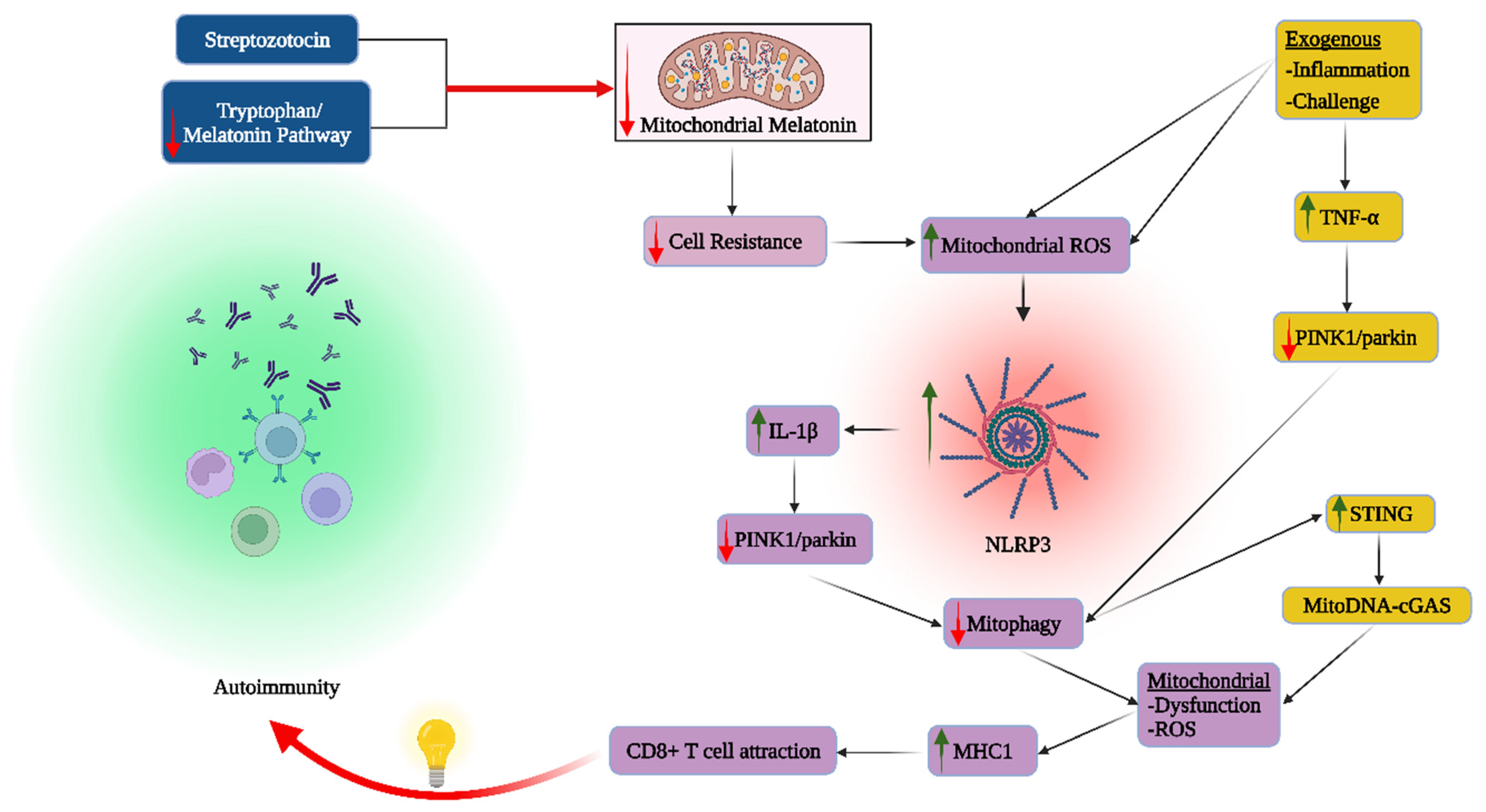{kind=link}
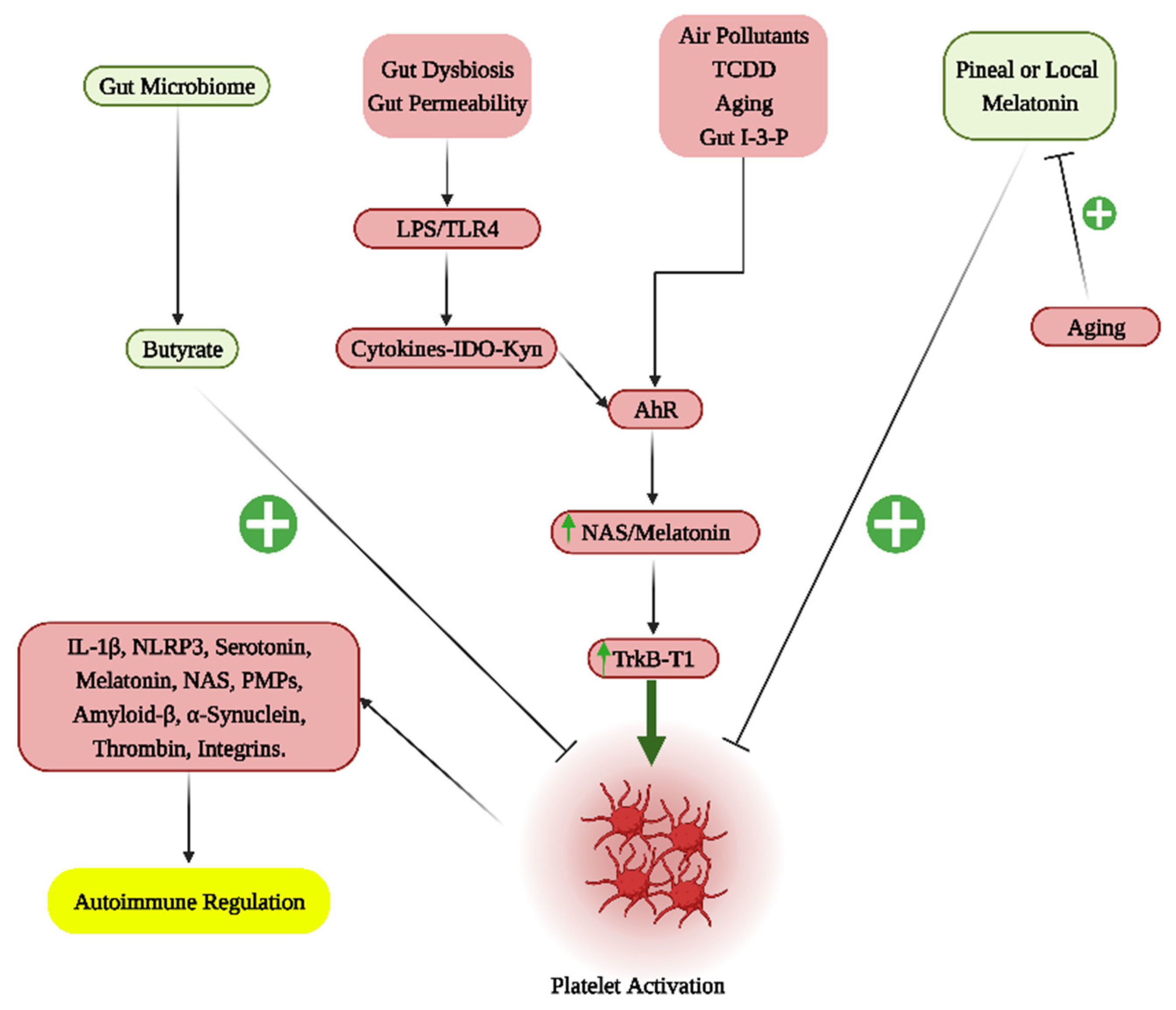{kind=link}
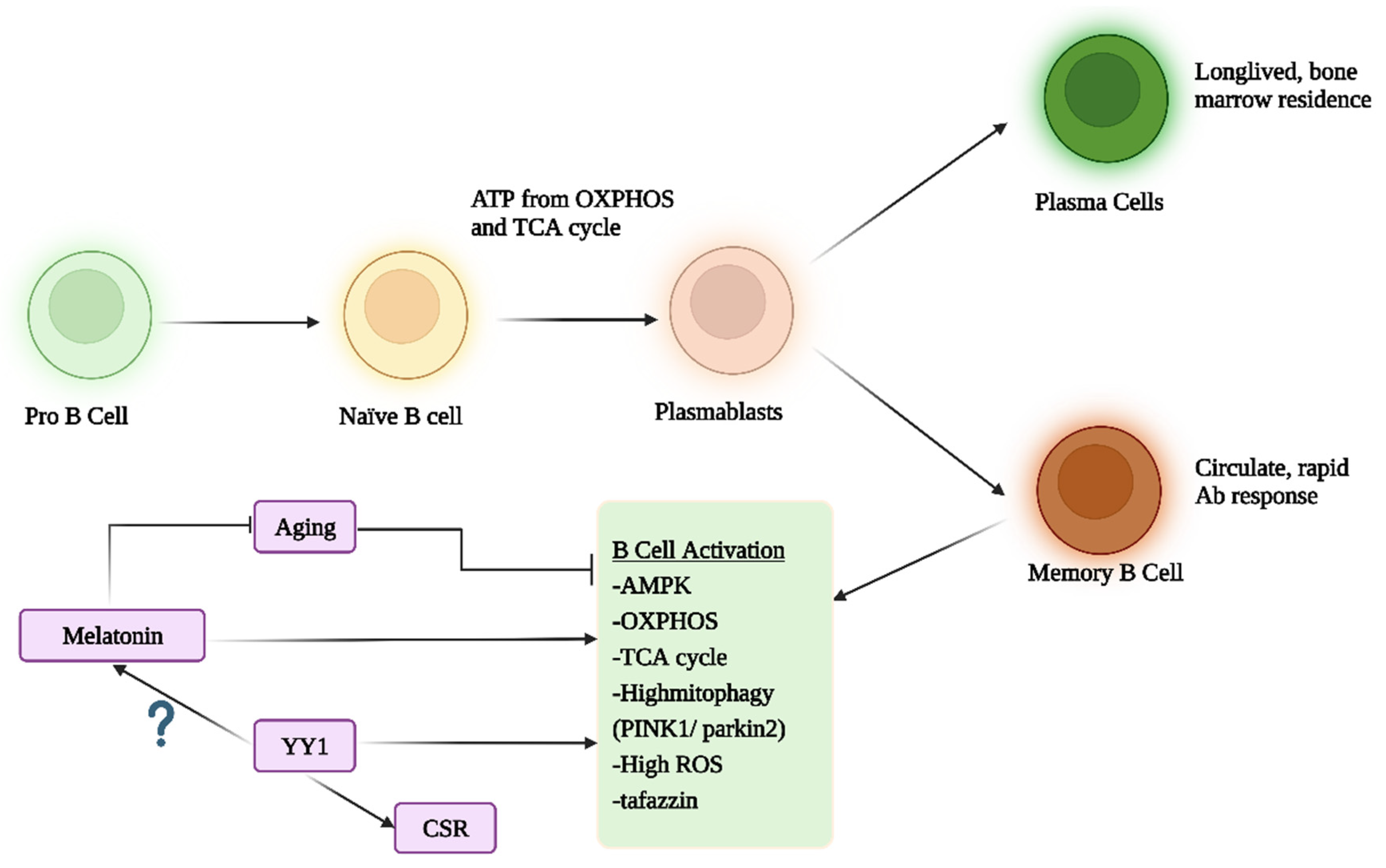{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Classical ‘Autoimmunity’ and Mitochondria
2.1. Classical Autoimmunity
2.2. Mitochondrial Metabolism and ‘Autoimmunity’
2.3. B-Cells, Antibodies and Mitochondria
3. Gut Microbiome and Systemic Mitochondria
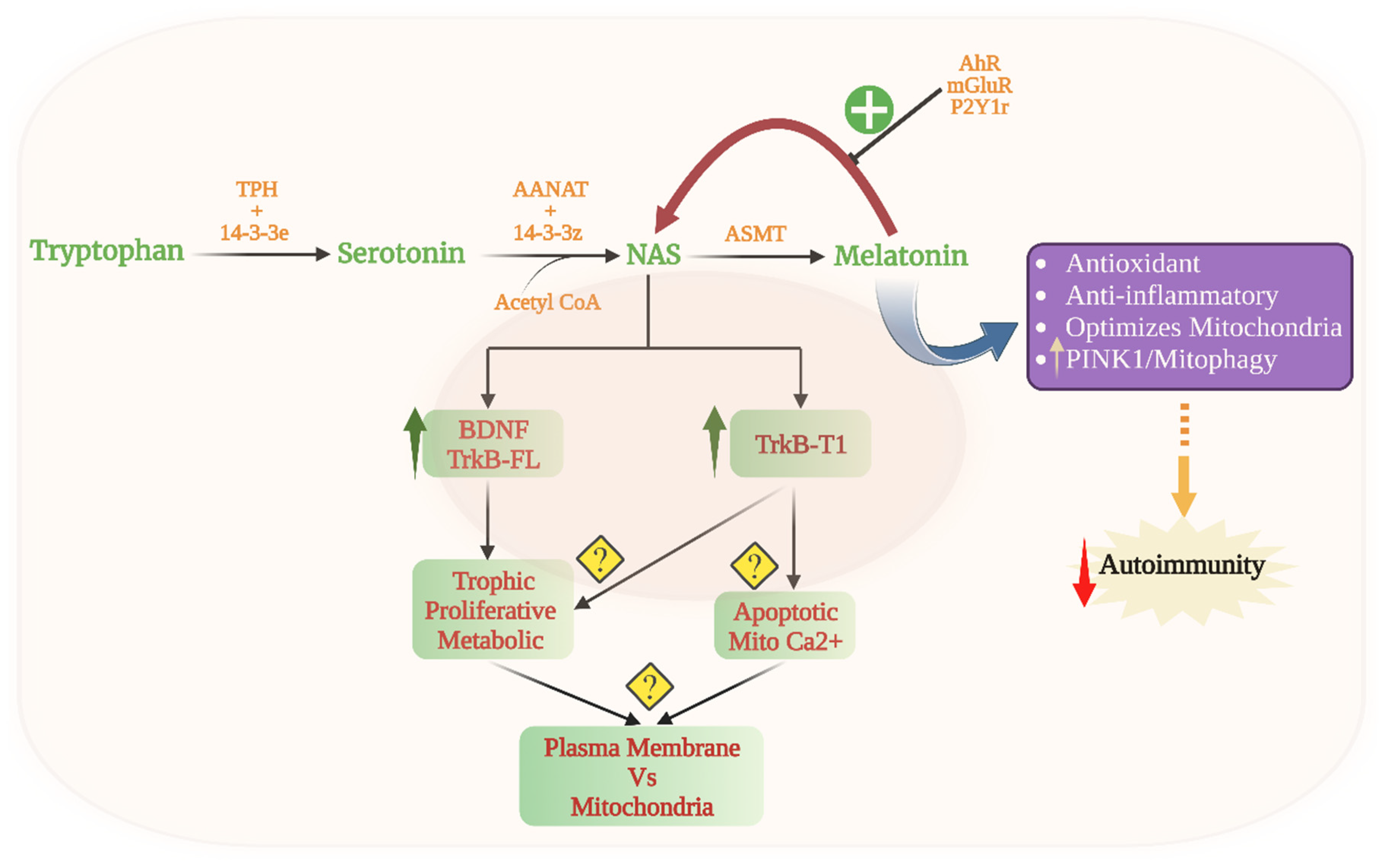
3.1. Tryptophan and the Melatonergic Pathway
3.2. Gut Microbiome, Melatonergic Pathway and Mitochondria
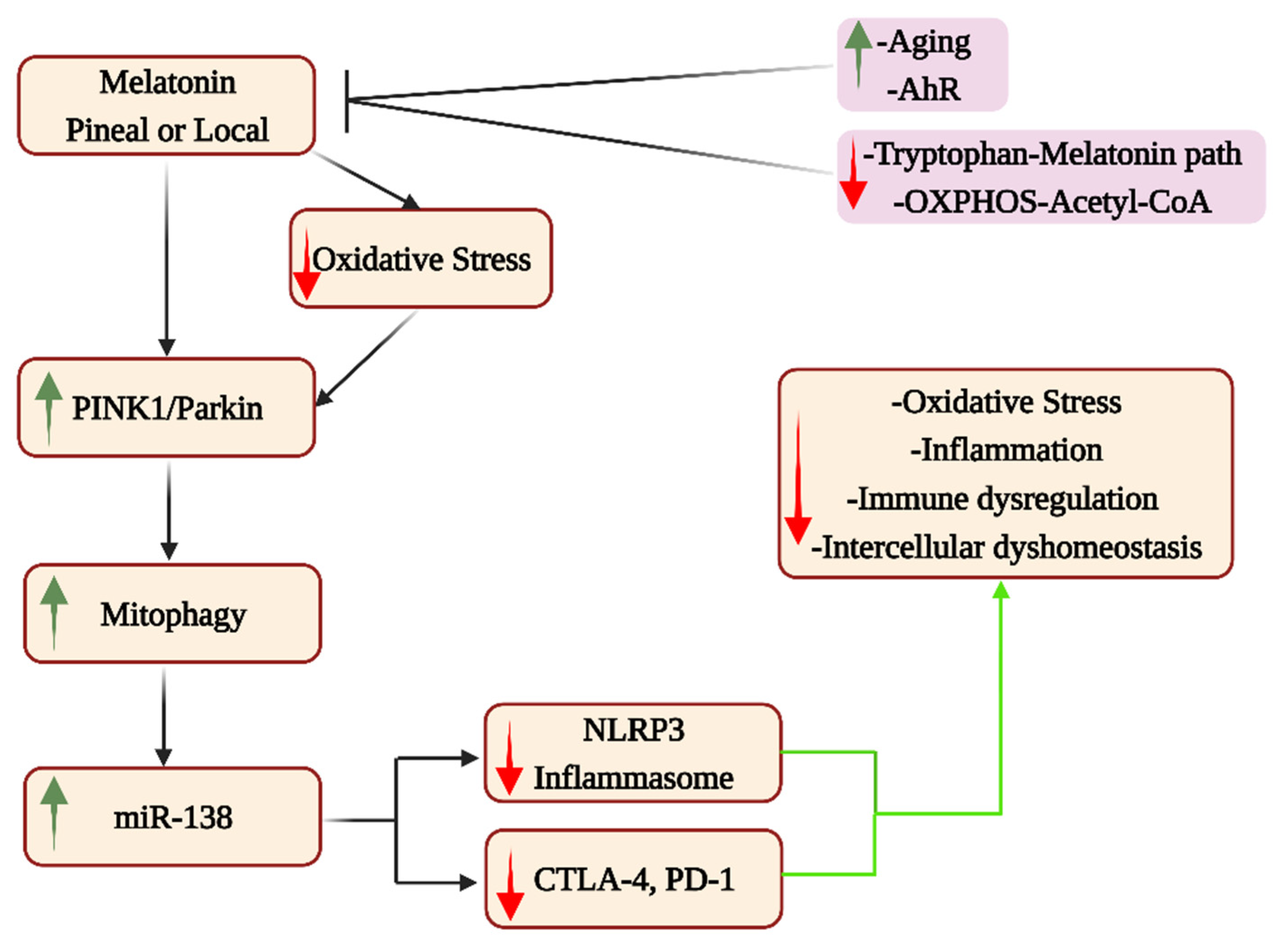
4. Pineal Melatonin and Systemic Mitochondria
5. Local Intercellular Interactions, Mitochondrial Melatonin and Cell Elimination
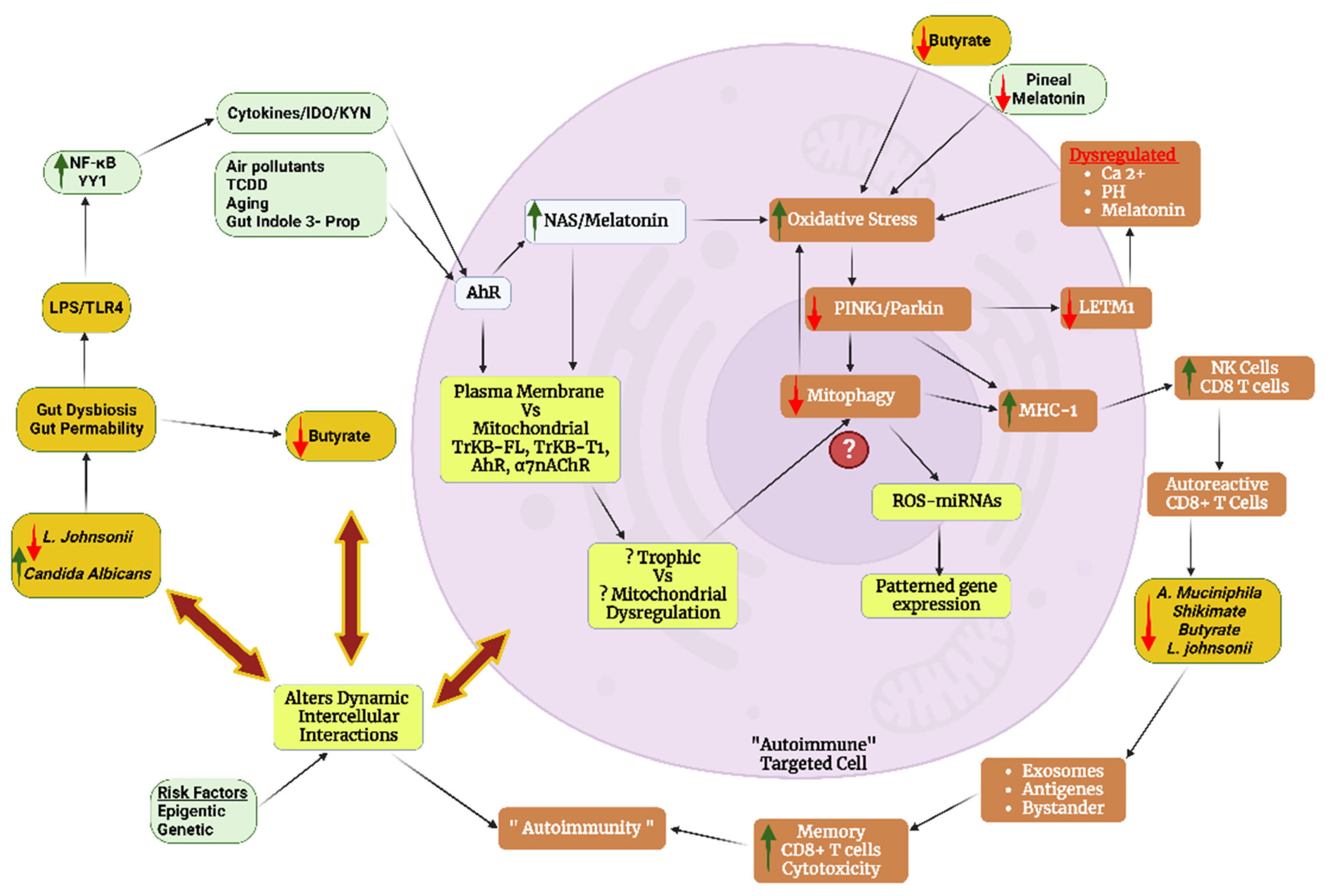
6. Integrating Mitochondrial Function across ‘Autoimmunity’
6.1. Intercellular Interactions and Dyshomeostasis
6.2. Cytolytic Cells and Mitochondrial Dysfunction
6.3. B-Cells and Metabolism
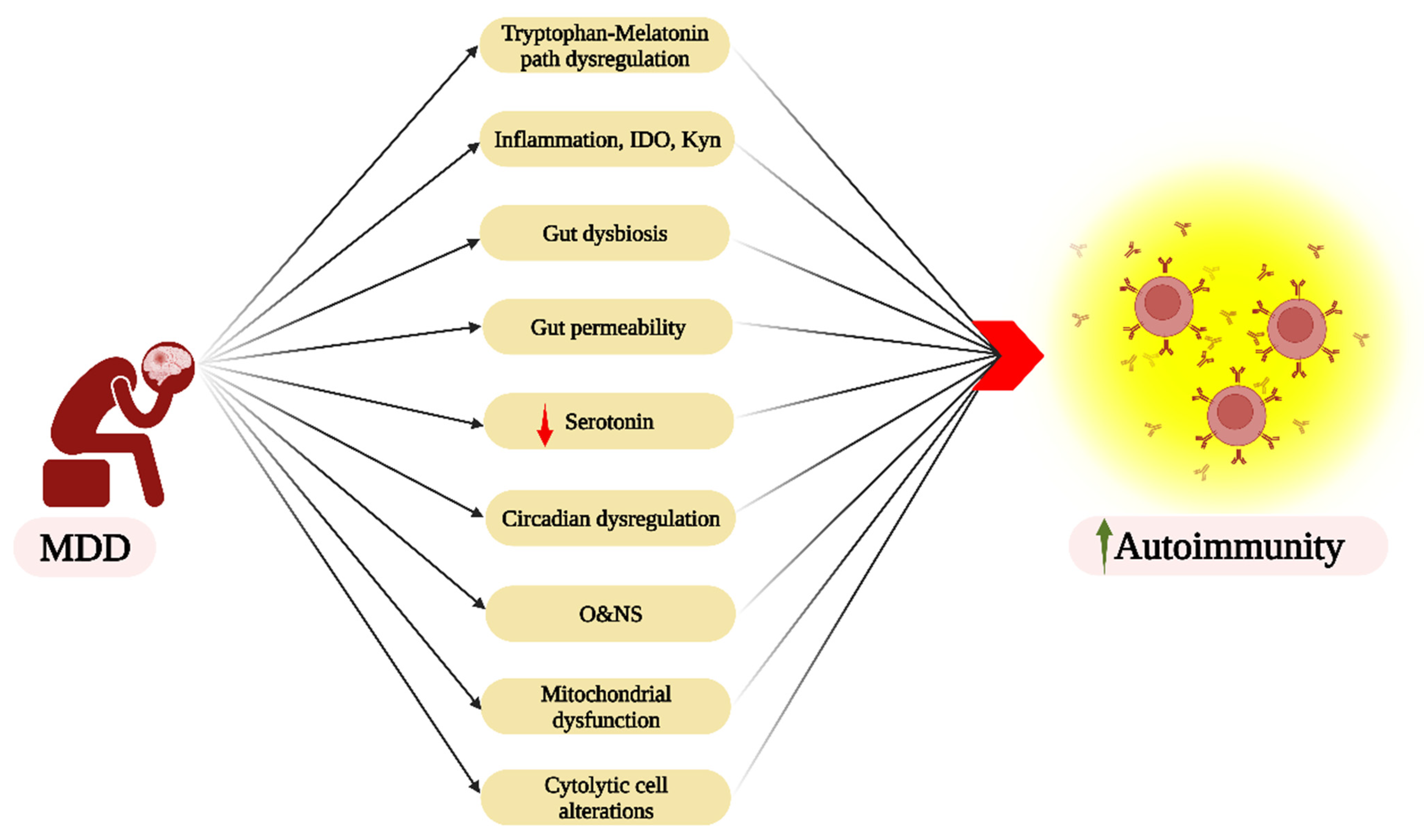
7. Autoimmunity and Psychiatric Disorders
8. Future Research Directions
9. Treatment Implications
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AANAT | aralkylamine N-acetyltransferase |
| AhR | aryl hydrocarbon receptor |
| APAS | antiphospholipid antibody syndrome |
| ASMT | acetylserotonin methytransferase |
| BACE1 | beta-site amyloid precursor protein cleaving enzyme 1 |
| BDNF | rain-derived neurotrophic factor |
| Bmal1 | brain and muscle ARNT-like 1 |
| CSR | class switch recombination |
| CTLA-4 | cytotoxic T-lymphocyte-associated molecule 4 |
| CYP | cytochrome P450 |
| DC | dendritic cell |
| EAAT | excitatory amino acid transporter |
| GBH | glyphosate-based herbicides |
| HDAC | histone deacetylase |
| HMGB | high-mobility group box |
| hsp | heat shock protein |
| IDO | indoleamine 2,3-dioxygenase |
| IFN | interferon |
| KYNA | kynurenic acid |
| LETM1 | leucine zipper-EF hand-containing transmembrane protein 1 |
| LPS | lipopolysaccharide |
| LUBAC | linear ubiquitin chain assembly complex |
| MDSC | myeloid-derived suppressor cell |
| mGluR5 | metabotropic glutamate receptor |
| MHC-1 | major histocompatibility complex, class 1 |
| NAS | N-acetylserotonin |
| NF-kB | nuclear factor kappa-light-chain-enhancer of activated B-cells |
| NK | natural killer cell |
| NMDA | N-methyl-d-aspartate |
| O&NS | oxidative and nitrosative stress |
| OXPHOS | oxidative phosphorylation |
| PD-1 | programmed cell-death 1 |
| PDC | pyruvate dehydrogenase complex |
| PDK | pyruvate dehydrogenase kinase |
| PD-L1 | programmed cell-death ligand 1 |
| PEPT | peptide transporter |
| PINK1 | PTEN-induced kinase 1 |
| PMP | platelet microparticles |
| ROS | reactive oxygen species |
| SNP | single nucleotide polymorphisms |
| SOD | superoxide dismutase |
| TCA | tricarboxylic acid |
| TCDD | 2,3,7,8-tetrachlorodibenzo-p-dioxin |
| TDO | tryptophan 2,3-dioxygenase |
| TFAM | mitochondrial transcription factor A |
| TGF | transforming growth factor |
| TLR | toll-like receptor |
| TMAO | trimethylamine N-oxide |
| TNF | tumor necrosis factor |
| TPH2 | tryptophan hydroxylase 2 |
| Treg | regulatory T cell |
| TrkB | tyrosine receptor kinase B |
| VDAC1 | voltage-dependent anion channel |
References
- Warburg, O.; Wind, F.; Negelein, E. The Metabolism of Tumors in the Body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef]
- Atlante, A.; Amadoro, G.; Latina, V.; Valenti, D. Therapeutic Potential of Targeting Mitochondria for Alzheimer’s Disease Treatment. J. Clin. Med. 2022, 11, 6742. [Google Scholar] [CrossRef]
- Huang, X.; Zeng, Z.; Li, S.; Xie, Y.; Tong, X. The Therapeutic Strategies Targeting Mitochondrial Metabolism in Cardiovascular Disease. Pharmaceutics 2022, 14, 2760. [Google Scholar] [CrossRef]
- Anderson, G. Depression Pathophysiology: Astrocyte Mitochondrial Melatonergic Pathway as Crucial Hub. Int. J. Mol. Sci. 2023, 24, 350. [Google Scholar] [CrossRef]
- Das, S.C.; Hjelm, B.E.; Rollins, B.L.; Sequeira, A.; Morgan, L.; Omidsalar, A.A.; Schatzberg, A.F.; Barchas, J.D.; Lee, F.S.; Myers, R.M.; et al. Mitochondria DNA copy number, mitochondria DNA total somatic deletions, Complex I activity, synapse number, and synaptic mitochondria number are altered in schizophrenia and bipolar disorder. Transl. Psychiatry 2022, 12, 353. [Google Scholar] [CrossRef]
- Fang, L.; Zhang, M.; Li, J.; Zhou, L.; Tamm, M.; Roth, M. Airway Smooth Muscle Cell Mitochondria Damage and Mitophagy in COPD via ERK1/2 MAPK. Int. J. Mol. Sci. 2022, 23, 13987. [Google Scholar] [CrossRef] [PubMed]
- Swalsingh, G.; Pani, P.; Bal, N.C. Structural functionality of skeletal muscle mitochondria and its correlation with metabolic diseases. Clin. Sci. 2022, 136, 1851–1871. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G.; Rodriguez, M.; Reiter, R.J. Multiple Sclerosis: Melatonin, Orexin, and Ceramide Interact with Platelet Activation Coagulation Factors and Gut-Microbiome-Derived Butyrate in the Circadian Dysregulation of Mitochondria in Glia and Immune Cells. Int. J. Mol. Sci. 2019, 20, 5500. [Google Scholar] [CrossRef] [PubMed]
- Sonkar, V.K.; Eustes, A.S.; Ahmed, A.; Jensen, M.; Solanki, M.V.; Swamy, J.; Kumar, R.; Fidler, T.P.; Houtman, J.C.D.; Allen, B.G.; et al. Endogenous SOD2 (Superoxide Dismutase) Regulates Platelet-Dependent Thrombin Generation and Thrombosis During Aging. Arter. Thromb. Vasc. Biol. 2023, 43, 79–91. [Google Scholar] [CrossRef]
- Tiwari-Heckler, S.; Robson, S.C.; Longhi, M.S. Mitochondria Drive Immune Responses in Critical Disease. Cells 2022, 11, 4113. [Google Scholar] [CrossRef]
- Klerman, E.B.; Brager, A.; Carskadon, M.A.; Depner, C.M.; Foster, R.; Goel, N.; Harrington, M.; Holloway, P.M.; Knauert, M.P.; LeBourgeois, M.K.; et al. Keeping an eye on circadian time in clinical research and medicine. Clin. Transl. Med. 2022, 12, e1131. [Google Scholar] [CrossRef] [PubMed]
- Parkinson, J. An Essay on the Shaking Palsy; Sherwood, Neely, and Jones: London, UK, 1817. [Google Scholar]
- Anderson, G. Why Do Anti-Amyloid Beta Antibodies not work? Time to reconceptualize Dementia Pathophysiology by incorporating astrocyte melatonergic pathway desynchronization from amyloid-beta production. Braz. J. Psychiatry 2022. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Deng, F.; Chen, J.; Chen, F.; Wu, Z.; Li, L.; Hou, K. Fecal microbiota transplantation treatment of autoimmune-mediated type 1 diabetes: A systematic review. Front. Cell Infect. Microbiol. 2022, 12, 1075201. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G. Amyotrophic Lateral Sclerosis Pathoetiology and Pathophysiology: Roles of Astrocytes, Gut microbiome and muscle interactions via the Mitochondrial melatonergic pathway, with disruption by Glyphosate-based herbicides. Int. J. Mol. Sci. 2023, 24, 587. [Google Scholar] [CrossRef]
- Lu, G.; Wen, Q.; Cui, B.; Li, Q.; Zhang, F. Washed microbiota transplantation stopped the deterioration of amyotrophic lateral sclerosis: The first case report and narrative review. J. Biomed. Res. 2022, 37, 69–76. [Google Scholar] [CrossRef]
- Anderson, G. Type I diabetes pathoetiology and pathophysiology: Roles of the gut microbiome, pancreatic cellular interactions, and the ‘bystander’ activation of memory CD8+ T cells. Int. J. Mol. Sci. 2023, 24, 3300. [Google Scholar] [CrossRef]
- Fan, H.H.; Cui, L.; Jiang, X.X.; Song, Y.D.; Liu, S.S.; Wu, K.Y.; Dong, H.J.; Mao, M.; Ovlyakulov, B.; Wu, H.M.; et al. Autoimmune Disease Associated CLEC16A Variants Convey Risk of Parkinson’s Disease in Han Chinese. Front. Genet. 2022, 13, 856493. [Google Scholar] [CrossRef]
- Cui, C.; Longinetti, E.; Larsson, H.; Andersson, J.; Pawitan, Y.; Piehl, F.; Fang, F. Associations between autoimmune diseases and amyotrophic lateral sclerosis: A register-based study. Amyotroph. Lateral Scler Front. Degener. 2021, 22, 211–219. [Google Scholar] [CrossRef]
- Weaver, D.F. Alzheimer’s disease as an innate autoimmune disease (AD2): A new molecular paradigm. Alzheimer’s Dement. 2022, 19, 1086–1098. [Google Scholar] [CrossRef]
- Ashraf, H.; Solla, P.; Sechi, L.A. Current Advancement of Immunomodulatory Drugs as Potential Pharmacotherapies for Autoimmunity Based Neurological Diseases. Pharmaceuticals 2022, 15, 1077. [Google Scholar] [CrossRef]
- de Gruijter, N.M.; Jebson, B.; Rosser, E.C. Cytokine production by human B cells: Role in health and autoimmune disease. Clin. Exp. Immunol. 2022, 210, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Sanders, J.M.; Jeyamogan, S.; Mathew, J.M.; Leventhal, J.R. Foxp3+ regulatory T cell therapy for tolerance in autoimmunity and solid organ transplantation. Front. Immunol. 2022, 13, 1055466. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Li, C.; Xu, Y.; Huang, M.; Cui, D.; Xie, J. Myeloid-derived suppressor cell: A crucial player in autoimmune diseases. Front. Immunol. 2022, 13, 1021612. [Google Scholar] [CrossRef] [PubMed]
- Tizaoui, K.; Terrazzino, S.; Cargnin, S.; Lee, K.H.; Gauckler, P.; Li, H.; Shin, J.I.; Kronbichler, A. The role of PTPN22 in the pathogenesis of autoimmune diseases: A comprehensive review. Semin. Arthritis Rheum. 2021, 51, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Mei, X.; Zhang, B.; Zhao, M.; Lu, Q. An update on epigenetic regulation in autoimmune diseases. J. Transl. Autoimmun. 2022, 5, 100176. [Google Scholar] [CrossRef] [PubMed]
- Schiweck, C.; Edwin Thanarajah, S.; Aichholzer, M.; Matura, S.; Reif, A.; Vrieze, E.; Weigert, A.; Visekruna, A. Regulation of CD4+ and CD8+ T Cell Biology by Short-Chain Fatty Acids and Its Relevance for Autoimmune Pathology. Int. J. Mol. Sci 2022, 23, 8272. [Google Scholar] [CrossRef]
- Hu, M.; Alashkar Alhamwe, B.; Santner-Nanan, B.; Miethe, S.; Harb, H.; Renz, H.; Potaczek, D.P.; Nanan, R.K. Short-Chain Fatty Acids Augment Differentiation and Function of Human Induced Regulatory T Cells. Int. J. Mol. Sci 2022, 23, 5740. [Google Scholar] [CrossRef]
- Lewkowicz, P.; Cwiklińska, H.; Mycko, M.P.; Cichalewska, M.; Domowicz, M.; Lewkowicz, N.; Jurewicz, A.; Selmaj, K.W. Dysregulated RNA-Induced Silencing Complex (RISC) Assembly within CNS Corresponds with Abnormal miRNA Expression during Autoimmune Demyelination. J. Neurosci. 2015, 35, 7521–7537. [Google Scholar] [CrossRef]
- Huang, Y.; Xue, Q.; Cheng, C.; Wang, Y.; Wang, X.; Chang, J.; Miao, C. Circular RNA in autoimmune diseases: Special emphasis on regulation mechanism in RA and SLE. J. Pharm. Pharmacol. 2023, 75, 370–384. [Google Scholar] [CrossRef]
- Yang, J.; Li, Z.; Wang, L.; Yun, X.; Zeng, Y.; Ng, J.P.L.; Lo, H.; Wang, Y.; Zhang, K.; Law, B.Y.K.; et al. The role of non-coding RNAs (miRNA and lncRNA) in the clinical management of rheumatoid arthritis. Pharmacol. Res. 2022, 186, 106549. [Google Scholar] [CrossRef]
- Saban, M.; Costilla, M.; Klecha, A.J.; Di Cugno, M.; Curria, M.I.; Cremaschi, G.; Barreiro Arcos, M.L. Regulation of the cellular redox state and the expression of DNA methyltransferase-1 in peripheral blood mononuclear cells from patients with Graves’ disease. Endocrinol. Diabetes Nutr. 2022, 69, 409–417. [Google Scholar] [CrossRef]
- Pandey, S.P.; Bender, M.J.; McPherson, A.C.; Phelps, C.M.; Sanchez, L.M.; Rana, M.; Hedden, L.; Sangani, K.A.; Chen, L.; Shapira, J.H.; et al. Tet2 deficiency drives liver microbiome dysbiosis triggering Tc1 cell autoimmune hepatitis. Cell Host Microbe 2022, 30, 1003–1019.e10. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, L.; Luo, S.; Sun, X.; Li, J.; Pang, H.; Zhou, J.; Zhou, Y.; Shi, X.; Li, X.; et al. The m6A methylation profiles of immune cells in type 1 diabetes mellitus. Front. Immunol. 2022, 13, 1030728. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, X.; Jia, H.; An, G.; Ni, J. The m6A methyltransferase METTL3 modifies PGC-1α mRNA promoting mitochondrial dysfunction and oxLDL-induced inflammation in monocytes. J. Biol. Chem. 2021, 297, 101058. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.Y.; Hsieh, S.C.; Lu, C.S.; Wu, T.H.; Liao, H.T.; Wu, C.H.; Li, K.J.; Kuo, Y.M.; Lee, H.T.; Shen, C.Y.; et al. Cross-Talk between Mitochondrial Dysfunction-Provoked Oxidative Stress and Aberrant Noncoding RNA Expression in the Pathogenesis and Pathophysiology of SLE. Int. J. Mol. Sci. 2019, 20, 5183. [Google Scholar] [CrossRef]
- Ferraz, R.S.; Santos, L.C.B.; da-Silva-Cruz, R.L.; Braga-da-Silva, C.H.; Magalhães, L.; Ribeiro-Dos-Santos, A.; Vidal, A.; Vinasco-Sandoval, T.; Reis-das-Mercês, L.; Sena-Dos-Santos, C.; et al. Global miRNA expression reveals novel nuclear and mitochondrial interactions in Type 1 diabetes mellitus. Front. Endocrinol. 2022, 13, 1033809. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, G.; Radhakrishnan, R.; Kowluru, R.A. Epigenetic Modifications Compromise Mitochondrial DNA Quality Control in the Development of Diabetic Retinopathy. Investig. Ophthalmol. Vis. Sci. 2019, 60, 3943–3951. [Google Scholar] [CrossRef]
- Matheoud, D.; Cannon, T.; Voisin, A.; Penttinen, A.M.; Ramet, L.; Fahmy, A.M.; Ducrot, C.; Laplante, A.; Bourque, M.J.; Zhu, L.; et al. Intestinal infection triggers Parkinson’s disease-like symptoms in Pink1−/− mice. Nature 2019, 571, 565–569. [Google Scholar] [CrossRef]
- Anderson, G.; Seo, M.; Berk, M.; Carvalho, A.F.; Maes, M. Gut Permeability and Microbiota in Parkinson’s Disease: Role of Depression, Tryptophan Catabolites, Oxidative and Nitrosative Stress and Melatonergic Pathways. Curr. Pharm. Des. 2016, 22, 6142–6151. [Google Scholar] [CrossRef]
- Matheoud, D.; Sugiura, A.; Bellemare-Pelletier, A.; Laplante, A.; Rondeau, C.; Chemali, M.; Fazel, A.; Bergeron, J.J.; Trudeau, L.E.; Burelle, Y.; et al. Parkinson’s Disease-Related Proteins PINK1 and Parkin Repress Mitochondrial Antigen Presentation. Cell 2016, 166, 314–327. [Google Scholar] [CrossRef]
- Cannon, T.; Sinha, A.; Trudeau, L.E.; Maurice, C.F.; Gruenheid, S. Characterization of the intestinal microbiota during Citrobacter rodentium infection in a mouse model of infection-triggered Parkinson’s disease. Gut Microbes 2020, 12, 1830694. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Kim, J.; Chun, J.; Han, K.; Soh, H.; Kang, E.A.; Lee, H.J.; Im, J.P.; Kim, J.S. Patients with Inflammatory Bowel Disease Are at an Increased Risk of Parkinson’s Disease: A South Korean Nationwide Population-Based Study. J. Clin. Med. 2019, 8, 1191. [Google Scholar] [CrossRef]
- Okada, M.; Zhang, V.; Loaiza Naranjo, J.D.; Tillett, B.J.; Wong, F.S.; Steptoe, R.J.; Bergot, A.S.; Hamilton-Williams, E.E. Islet-specific CD8+ T cells gain effector function in the gut lymphoid tissues via bystander activation not molecular mimicry. Immunol. Cell Biol. 2023, 101, 36–48. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Hou, L.; Liu, Z.; Huang, S.; Meng, Z.; Liang, L. A mitophagic response to iron overload-induced oxidative damage associated with the PINK1/Parkin pathway in pancreatic beta cells. J. Trace Elem. Med. Biol. 2020, 60, 126493. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.Y.; Ye, Y.Y.; Qian, C.; Zhang, H.B.; Mao, H.X.; Yao, L.P.; Sun, X.; Lu, G.H.; Zhang, S.Z. Stress increases MHC-I expression in dopaminergic neurons and induces autoimmune activation in Parkinson’s disease. Neural Regen Res. 2021, 16, 2521–2527. [Google Scholar] [CrossRef] [PubMed]
- do Carmo Buonfiglio, D.; Peliciari-Garcia, R.A.; do Amaral, F.G.; Peres, R.; Nogueira, T.C.; Afeche, S.C.; Cipolla-Neto, J. Early-stage retinal melatonin synthesis impairment in streptozotocin-induced diabetic wistar rats. Investig. Ophthalmol. Vis. Sci. 2011, 52, 7416–7422. [Google Scholar] [CrossRef] [PubMed]
- Yoon, Y.M.; Go, G.; Yoon, S.; Lim, J.H.; Lee, G.; Lee, J.H.; Lee, S.H. Melatonin Treatment Improves Renal Fibrosis via miR-4516/SIAH3/PINK1 Axis. Cells 2021, 10, 1682. [Google Scholar] [CrossRef]
- Adi, N.; Mash, D.C.; Ali, Y.; Singer, C.; Shehadeh, L.; Papapetropoulos, S. Melatonin MT1 and MT2 receptor expression in Parkinson’s disease. Med. Sci. Monit. 2010, 16, BR61-7. [Google Scholar]
- Jiang, J.M.; Mo, M.L.; Long, X.P.; Xie, L.H. MiR-144-3p induced by SP1 promotes IL-1β-induced pyroptosis in chondrocytes via PTEN/PINK1/Parkin axis. Autoimmunity 2022, 55, 21–31. [Google Scholar] [CrossRef]
- Zhang, Y.; Xi, Y.; Yang, C.; Gong, W.; Wang, C.; Wu, L.; Wang, D. Short-Chain Fatty Acids Attenuate 5-Fluorouracil-Induced THP-1 Cell Inflammation through Inhibiting NF-κB/NLRP3 Signaling via Glycerolphospholipid and Sphingolipid Metabolism. Molecules 2023, 28, 494. [Google Scholar] [CrossRef]
- Willemsen, J.; Neuhoff, M.T.; Hoyler, T.; Noir, E.; Tessier, C.; Sarret, S.; Thorsen, T.N.; Littlewood-Evans, A.; Zhang, J.; Hasan, M.; et al. TNF leads to mtDNA release and cGAS/STING-dependent interferon responses that support inflammatory arthritis. Cell Rep. 2021, 37, 109977. [Google Scholar] [CrossRef]
- Patergnani, S.; Bonora, M.; Ingusci, S.; Previati, M.; Marchi, S.; Zucchini, S.; Perrone, M.; Wieckowski, M.R.; Castellazzi, M.; Pugliatti, M.; et al. Antipsychotic drugs counteract autophagy and mitophagy in multiple sclerosis. Proc. Natl. Acad. Sci. USA 2021, 118, e2020078118. [Google Scholar] [CrossRef] [PubMed]
- Nam, J.H.; Lee, J.H.; Choi, H.J.; Choi, S.Y.; Noh, K.E.; Jung, N.C.; Song, J.Y.; Choi, J.; Seo, H.G.; Jung, S.Y.; et al. TNF-α Induces Mitophagy in Rheumatoid Arthritis Synovial Fibroblasts, and Mitophagy Inhibition Alleviates Synovitis in Collagen Antibody-Induced Arthritis. Int. J. Mol. Sci. 2022, 23, 5650. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Hu, X.; Xiao, F.; Zhang, X.; Zhao, L.; Wang, M. Mitochondrial impairment and repair in the pathogenesis of systemic lupus erythematosus. Front. Immunol. 2022, 13, 929520. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.Y.; Gong, D.J.; Zhang, J.J.; Liu, X.H.; Wang, L. Diabetes aggravates renal ischemia-reperfusion injury by repressing mitochondrial function and PINK1/Parkin-mediated mitophagy. Am. J. Physiol. Renal. Physiol. 2019, 317, F852–F864. [Google Scholar] [CrossRef]
- Wang, N.; Yuan, J.; Karim, M.R.; Zhong, P.; Sun, Y.P.; Zhang, H.Y.; Wang, Y.F. Effects of Mitophagy on Regulatory T Cell Function in Patients With Myasthenia Gravis. Front. Neurol. 2020, 11, 238. [Google Scholar] [CrossRef]
- Szekerczés, T.; Gógl, A.; Illyés, I.; Mandl, J.; Borka, K.; Kiss, A.; Schaff, Z.; Lendvai, G.; Werling, K. Autophagy, Mitophagy and MicroRNA Expression in Chronic Hepatitis C and Autoimmune Hepatitis. Pathol. Oncol. Res. 2020, 26, 2143–2151. [Google Scholar] [CrossRef]
- Siracusa, R.; D’Amico, R.; Impellizzeri, D.; Cordaro, M.; Peritore, A.F.; Gugliandolo, E.; Crupi, R.; Salinaro, A.T.; Raffone, E.; Genovese, T.; et al. Autophagy and Mitophagy Promotion in a Rat Model of Endometriosis. Int. J. Mol. Sci. 2021, 22, 5074. [Google Scholar] [CrossRef]
- Li, J.; Yang, D.; Li, Z.; Zhao, M.; Wang, D.; Sun, Z.; Wen, P.; Dai, Y.; Gou, F.; Ji, Y.; et al. PINK1/Parkin-mediated mitophagy in neurodegenerative diseases. Ageing Res. Rev. 2022, 84, 101817. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G. Tumour microenvironment and metabolism: Role of the mitochondrial melatonergic pathway in determining Intercellular Interactions in a new dynamic homeostasis. Int. J. Mol. Sci. 2023, 24, 311. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G. Tumour Microenvironment: Roles of the Aryl Hydrocarbon Receptor, O-GlcNAcylation, Acetyl-CoA and Melatonergic Pathway in Regulating Dynamic Metabolic Interactions across Cell Types-Tumour Microenvironment and Metabolism. Int. J. Mol. Sci. 2020, 22, 141. [Google Scholar] [CrossRef]
- Markus, R.P.; Fernandes, P.A.; Kinker, G.S.; Cruz-Machado, S.D.S.; Marçola, M. Immune-pineal axis—Acute inflammatory responses coordinate melatonin synthesis by pinealocytes and phagocytes. Br. J. Pharmacol. 2017, 175, 3239–3250. [Google Scholar] [CrossRef] [PubMed]
- Muxel, S.M.; Pires-Lapa, M.A.; Monteiro, A.W.A.; Cecon, E.; Tamura, E.K.; Floeter-Winter, L.M.; Markus, R.P. NF-κB Drives the Synthesis of Melatonin in RAW 264.7 Macrophages by Inducing the Transcription of the Arylalkylamine-N-Acetyltransferase (AA-NAT) Gene. PLoS ONE 2012, 7, e52010. [Google Scholar] [CrossRef]
- Nocella, C.; Bartimoccia, S.; Cammisotto, V.; D’Amico, A.; Pastori, D.; Frati, G.; Sciarretta, S.; Rosa, P.; Felici, C.; Riggio, O.; et al. Oxidative Stress in the Pathogenesis of Antiphospholipid Syndrome: Implications for the Atherothrombotic Process. Antioxidants 2021, 10, 1790. [Google Scholar] [CrossRef] [PubMed]
- Butt, A.; Erkan, D.; Lee, A.I. COVID-19 and antiphospholipid antibodies. Best Pract. Res. Clin. Haematol. 2022, 35, 101402. [Google Scholar] [CrossRef] [PubMed]
- Kits, A.; Pantalone, M.R.; Illies, C.; Antovic, A.; Landtblom, A.M.; Iacobaeus, E. Fatal Acute Hemorrhagic Encephalomyelitis and Antiphospholipid Antibodies following SARS-CoV-2 Vaccination: A Case Report. Vaccines 2022, 10, 2046. [Google Scholar] [CrossRef]
- Ames, P.R.J.; Bucci, T.; Merashli, M.; Arcaro, A.; Gentile, F. Thrombocytopenia in antiphospholipid syndrome: A free radical perspective. Rheumatology 2022, keac650. [Google Scholar] [CrossRef]
- Jiang, S.Z.; To, J.L.; Hughes, M.R.; McNagny, K.M.; Kim, H. Platelet signaling at the nexus of innate immunity and rheumatoid arthritis. Front. Immunol. 2022, 13, 977828. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.; Bison, E.; Pontara, E.; Cattini, M.G.; Tonello, M.; Denas, G.; Pengo, V. Platelet- and endothelial-derived microparticles in the context of different antiphospholipid antibody profiles. Lupus 2022, 31, 1328–1334. [Google Scholar] [CrossRef]
- Chen, E.; Chen, Z.; Chen, L.; Hu, X. Platelet-derived respiratory-competent mitochondria transfer to mesenchymal stem cells to promote wound healing via metabolic reprogramming. Platelets 2022, 33, 171–173. [Google Scholar] [CrossRef]
- Léger, J.L.; Soucy, M.N.; Veilleux, V.; Foulem, R.D.; Robichaud, G.A.; Surette, M.E.; Allain, E.P.; Boudreau, L.H. Functional platelet-derived mitochondria induce the release of human neutrophil microvesicles. EMBO Rep. 2022, 23, e54910. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Hu, W.; Song, X.; Zhao, Y. Immune Modulation of Platelet-Derived Mitochondria on Memory CD4+ T Cells in Humans. Int. J. Mol. Sci. 2020, 21, 6295. [Google Scholar] [CrossRef] [PubMed]
- Di, L.; Zha, C.; Liu, Y. Platelet-derived microparticles stimulated by anti-β2GPI/β2GPI complexes induce pyroptosis of endothelial cells in antiphospholipid syndrome. Platelets 2023, 34, 2156492. [Google Scholar] [CrossRef] [PubMed]
- Ferrer-Raventós, P.; Beyer, K. Alternative platelet activation pathways and their role in neurodegenerative diseases. Neurobiol. Dis. 2021, 159, 105512. [Google Scholar] [CrossRef] [PubMed]
- Waters, L.R.; Ahsan, F.M.; Wolf, D.M.; Shirihai, O.; Teitell, M.A. Initial B Cell Activation Induces Metabolic Reprogramming and Mitochondrial Remodeling. iScience 2018, 5, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Bystrom, J.; Taher, T.E.; Henson, S.M.; Gould, D.J.; Mageed, R.A. Metabolic requirements of Th17 cells and of B cells: Regulation and defects in health and in inflammatory diseases. Front. Immunol. 2022, 13, 990794. [Google Scholar] [CrossRef]
- Oishi, K.; Horiuchi, S.; Frere, J.; Schwartz, R.E.; tenOever, B.R. A diminished immune response underlies age-related SARS-CoV-2 pathologies. Cell Rep. 2022, 39, 111002. [Google Scholar] [CrossRef]
- Brookens, S.K.; Cho, S.H.; Basso, P.J.; Boothby, M.R. AMPKα1 in B Cells Dampens Primary Antibody Responses yet Promotes Mitochondrial Homeostasis and Persistence of B Cell Memory. J. Immunol. 2020, 205, 3011–3022. [Google Scholar] [CrossRef]
- Srivastava, S. The Mitochondrial Basis of Aging and Age-Related Disorders. Genes 2017, 8, 398. [Google Scholar] [CrossRef]
- Liang, H.; Fu, W.; Yu, W.; Cao, Z.; Liu, E.; Sun, F.; Kong, X.; Gao, Y.; Zhou, Y. Elucidating the mitochondrial function of murine lymphocyte subsets and the heterogeneity of the mitophagy pathway inherited from hematopoietic stem cells. Front. Immunol. 2022, 13, 1061448. [Google Scholar] [CrossRef]
- Zegallai, H.M.; Abu-El-Rub, E.; Cole, L.K.; Field, J.; Mejia, E.M.; Gordon, J.W.; Marshall, A.J.; Hatch, G.M. Tafazzin deficiency impairs mitochondrial metabolism and function of lipopolysaccharide activated B lymphocytes in mice. FASEB J. 2021, 35, e22023. [Google Scholar] [CrossRef] [PubMed]
- Wigton, E.J.; Mikami, Y.; McMonigle, R.J.; Castellanos, C.A.; Wade-Vallance, A.K.; Zhou, S.K.; Kageyama, R.; Litterman, A.; Roy, S.; Kitamura, D.; et al. MicroRNA-directed pathway discovery elucidates an miR-221/222-mediated regulatory circuit in class switch recombination. J. Exp. Med. 2021, 218, e20201422. [Google Scholar] [CrossRef]
- Seo, M.; Anderson, G. Gut-Amygdala Interactions in Autism Spectrum Disorders: Developmental Roles via regulating Mitochondria, Exosomes, Immunity and microRNAs. Curr. Pharm. Des. 2019, 25, 4344–4356. [Google Scholar] [CrossRef]
- Pagan, C.; Goubran-Botros, H.; Delorme, R.; Benabou, M.; Lemière, N.; Murray, K.; Amsellem, F.; Callebert, J.; Chaste, P.; Jamain, S.; et al. Disruption of melatonin synthesis is associated with impaired 14-3-3 and miR-451 levels in patients with autism spectrum disorders. Sci. Rep. 2017, 7, 2096. [Google Scholar] [CrossRef]
- Jang, S.W.; Liu, X.; Pradoldej, S.; Tosini, G.; Chang, Q.; Iuvone, P.M.; Ye, K. N-acetylserotonin activates TrkB receptor in a circadian rhythm. Proc. Natl. Acad. Sci. USA 2010, 107, 3876–3881. [Google Scholar] [CrossRef]
- Portich, J.P.; Gil, M.S.; Dos Santos, R.P.; Goulart, B.K.; Ferreira, M.B.; Loss, J.F.; Gregianin, L.J.; Brunetto, A.L.; Brunetto, A.T.; Roesler, R.; et al. Low brain-derived neurotrophic factor levels are associated with active disease and poor prognosis in childhood acute leukemia. Cancer Biomark 2016, 17, 347–352. [Google Scholar] [CrossRef] [PubMed]
- Hillis, J.; O’Dwyer, M.; Gorman, A.M. Neurotrophins and B-cell malignancies. Cell Mol. Life Sci. 2016, 73, 41–56. [Google Scholar] [CrossRef] [PubMed]
- Kerschensteiner, M.; Gallmeier, E.; Behrens, L.; Leal, V.V.; Misgeld, T.; Klinkert, W.E.; Kolbeck, R.; Hoppe, E.; Oropeza-Wekerle, R.L.; Bartke, I.; et al. Activated human T cells, B cells, and monocytes produce brain-derived neurotrophic factor in vitro and in inflammatory brain lesions: A neuroprotective role of inflammation? J. Exp. Med. 1999, 189, 865–870. [Google Scholar] [CrossRef]
- Yu, Q.; Miller, S.C.; Osmond, D.G. Melatonin inhibits apoptosis during early B-cell development in mouse bone marrow. J. Pineal Res. 2000, 29, 86–93. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, Z.; Dong, Y.; Cao, J.; Chen, Y. Melatonin Nuclear Receptors Mediate Green-and-Blue-Monochromatic-Light-Combinations-Inhibited B Lymphocyte Apoptosis in the Bursa of Chickens via Reducing Oxidative Stress and Nfκb Expression. Antioxidants 2022, 11, 748. [Google Scholar] [CrossRef]
- Guo, X.; Xu, W.; Zhang, W.; Pan, C.; Thalacker-Mercer, A.E.; Zheng, H.; Gu, Z. High-frequency and functional mitochondrial DNA mutations at the single-cell level. Proc. Natl. Acad. Sci. USA 2023, 120, e2201518120. [Google Scholar] [CrossRef] [PubMed]
- Nandi, S.; Liang, G.; Sindhava, V.; Angireddy, R.; Basu, A.; Banerjee, S.; Hodawadekar, S.; Zhang, Y.; Avadhani, N.G.; Sen, R.; et al. YY1 control of mitochondrial-related genes does not account for regulation of immunoglobulin class switch recombination in mice. Eur. J. Immunol. 2020, 50, 822–838. [Google Scholar] [CrossRef] [PubMed]
- Bernard, M.; Voisin, P. Photoreceptor-specific expression, light-dependent localization, and transcriptional targets of the zinc-finger protein Yin Yang 1 in the chicken retina. J. Neurochem. 2008, 105, 595–604. [Google Scholar] [CrossRef] [PubMed]
- Catalán, D.; Mansilla, M.A.; Ferrier, A.; Soto, L.; Oleinika, K.; Aguillón, J.C.; Aravena, O. Immunosuppressive Mechanisms of Regulatory B Cells. Front. Immunol. 2021, 12, 611795. [Google Scholar] [CrossRef] [PubMed]
- Su, J.; Wang, K.; Zhou, X.; Wang, Y.; Xu, J.; Tao, L.; Zeng, X.; Chen, N.; Bai, X.; Li, X. B-cell-specific-peroxisome proliferator-activated receptor γ deficiency augments contact hypersensitivity with impaired regulatory B cells. Immunology 2019, 156, 282–296. [Google Scholar] [CrossRef] [PubMed]
- Zegallai, H.M.; Abu-El-Rub, E.; Olayinka-Adefemi, F.; Cole, L.K.; Sparagna, G.C.; Marshall, A.J.; Hatch, G.M. Tafazzin deficiency in mouse mesenchymal stem cells promote reprogramming of activated B lymphocytes toward immunosuppressive phenotypes. FASEB J. 2022, 36, e22443. [Google Scholar] [CrossRef]
- Cao, J.; Ni, Y.; Ning, X.; Zhang, H. Wedelolactone ameliorates synovial inflammation and cardiac complications in a murine model of collagen-induced arthritis by inhibiting NF-κB/NLRP3 inflammasome activation. Folia Histochem. Cytobiol. 2022, 60, 301–310. [Google Scholar] [CrossRef]
- Kosasih, F.R.; Bonavida, B. Involvement of Yin Yang 1 (YY1) Expression in T-Cell Subsets Differentiation and Their Functions: Implications in T Cell-Mediated Diseases. Crit. Rev. Immunol. 2019, 39, 491–510. [Google Scholar] [CrossRef]
- Anderson, G.; Maes, M. Gut Dysbiosis Dysregulates Central and Systemic Homeostasis via Suboptimal Mitochondrial Function: Assessment, Treatment and Classification Implications. Curr. Top. Med. Chem. 2020, 20, 524–539. [Google Scholar] [CrossRef]
- Kobayashi, M.; Mikami, D.; Kimura, H.; Kamiyama, K.; Morikawa, Y.; Yokoi, S.; Kasuno, K.; Takahashi, N.; Taniguchi, T.; Iwano, M. Short-chain fatty acids, GPR41 and GPR43 ligands, inhibit TNF-α-induced MCP-1 expression by modulating p38 and JNK signaling pathways in human renal cortical epithelial cells. Biochem. Biophys Res. Commun. 2017, 486, 499–505. [Google Scholar] [CrossRef]
- Mesnage, R.; Antoniou, M.N. Computational modelling provides insight into the effects of glyphosate on the shikimate pathway in the human gut microbiome. Curr. Res. Toxicol. 2020, 1, 25–33. [Google Scholar] [CrossRef]
- Anderson, G.; Maes, M. How Immune-inflammatory Processes Link CNS and Psychiatric Disorders: Classification and Treatment Implications. In CNS & Neurological Disorders—Drug Targets; Bentham Science Publishers: Sharjah, United Arab Emirates, 2017; Volume 16, pp. 266–278. [Google Scholar] [CrossRef]
- Tan, D.X.; Manchester, L.C.; Liu, X.; Rosales-Corral, S.A.; Acuna-Castroviejo, D.; Reiter, R.J. Mitochondria and chloroplasts as the original sites of melatonin synthesis: A hypothesis related to melatonin’s primary function and evolution in eukaryotes. J. Pineal Res. 2013, 54, 127–138. [Google Scholar] [CrossRef]
- Anderson, G.; Maes, M. Reconceptualizing adult neurogenesis: Role for sphingosine-1-phosphate and fibroblast growth factor-1 in co-ordinating astrocyte-neuronal precursor interactions. CNS Neurol. Disord. Drug Targets. 2014, 13, 126–136. [Google Scholar] [CrossRef]
- Moriwaki, K.; Wada, M.; Kuwabara, H.; Ayani, Y.; Terada, T.; Higashino, M.; Kawata, R.; Asahi, M. BDNF/TRKB axis provokes EMT progression to induce cell aggressiveness via crosstalk with cancer-associated fibroblasts in human parotid gland cancer. Sci. Rep. 2022, 12, 17553. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G. Breast cancer: Occluded role of mitochondria N-acetylserotonin/melatonin ratio in co-ordinating pathophysiology. Biochem. Pharmacol. 2019, 168, 259–268. [Google Scholar] [CrossRef]
- Wang, S.; Duan, H.; Li, B.; Hong, W.; Li, X.; Wang, Y.; Guo, Z.C. BDNF and TrKB expression levels in patients with endometriosis and their associations with dysmenorrhoea. J. Ovarian Res. 2022, 15, 35. [Google Scholar] [CrossRef]
- Anderson, G. Endometriosis Pathoetiology and Pathophysiology: Roles of Vitamin A, Estrogen, Immunity, Adipocytes, Gut Microbiome and Melatonergic Pathway on Mitochondria Regulation. Biomol. Concepts 2019, 10, 133–149. [Google Scholar] [CrossRef] [PubMed]
- Dubey, S.; Ghosh, S.; Goswami, D.; Ghatak, D.; De, R. Immunometabolic attributes and mitochondria-associated signaling of Tumor-Associated Macrophages in tumor microenvironment modulate cancer progression. Biochem. Pharmacol. 2022, 208, 115369. [Google Scholar] [CrossRef]
- Prokopidis, K.; Giannos, P.; Witard, O.C.; Peckham, D.; Ispoglou, T. Aberrant mitochondrial homeostasis at the crossroad of musculoskeletal ageing and non-small cell lung cancer. PLoS ONE 2022, 17, e0273766. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liang, X.; Dong, W.; Fang, Y.; Lv, J.; Zhang, T.; Fiskesund, R.; Xie, J.; Liu, J.; Yin, X.; et al. Tumor-Repopulating Cells Induce PD-1 Expression in CD8+ T Cells by Transferring Kynurenine and AhR Activation. Cancer Cell 2018, 33, 480–494.e7. [Google Scholar] [CrossRef]
- Anquetil, F.; Mondanelli, G.; Gonzalez, N.; Rodriguez Calvo, T.; Zapardiel Gonzalo, J.; Krogvold, L.; Dahl-Jørgensen, K.; Van den Eynde, B.; Orabona, C.; Grohmann, U.; et al. Loss of IDO1 Expression From Human Pancreatic β-Cells Precedes Their Destruction During the Development of Type 1 Diabetes. Diabetes 2018, 67, 1858–1866. [Google Scholar] [CrossRef] [PubMed]
- Souza-Teodoro, L.H.; Dargenio-Garcia, L.; Petrilli-Lapa, C.L.; Souza Eda, S.; Fernandes, P.A.; Markus, R.P.; Ferreira, Z.S. Adenosine triphosphate inhibits melatonin synthesis in the rat pineal gland. J. Pineal Res. 2016, 60, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Idle, J.R.; Krausz, K.W.; Gonzalez, F.J. Metabolism of melatonin by human cytochromes p450. Drug Metab. Dispos. 2005, 33, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G.; Reiter, R.J. Glioblastoma: Role of Mitochondria N-acetylserotonin/Melatonin Ratio in Mediating Effects of miR-451, Aryl Hydrocarbon Receptor and in Co-ordinating Wider Biochemical Changes. Int. J. Tryptophan. Res. 2019, 2, 44–66. [Google Scholar] [CrossRef]
- Kang, L.L.; Zhang, D.M.; Jiao, R.Q.; Pan, S.M.; Zhao, X.J.; Zheng, Y.J.; Chen, T.Y.; Kong, L.D. Pterostilbene Attenuates Fructose-Induced Myocardial Fibrosis by Inhibiting ROS-Driven Pitx2c/miR-15b Pathway. Oxid. Med. Cell. Longev. 2019, 2019, 1243215. [Google Scholar] [CrossRef]
- Jin, C.J.; Engstler, A.J.; Sellmann, C.; Ziegenhardt, D.; Landmann, M.; Kanuri, G.; Lounis, H.; Schröder, M.; Vetter, W.; Bergheim, I. Sodium butyrate protects mice from the development of the early signs of non-alcoholic fatty liver disease: Role of melatonin and lipid peroxidation. Br. J. Nutr. 2016, 116, 1682–1693. [Google Scholar] [CrossRef]
- Pontes, G.N.; Cardoso, E.C.; Carneiro-Sampaio, M.M.; Markus, R.P. Pineal melatonin and the innate immune response: The TNF-alpha increase after cesarean section suppresses nocturnal melatonin production. J. Pineal Res. 2007, 43, 365–371. [Google Scholar] [CrossRef] [PubMed]
- García, J.J.; Piñol-Ripoll, G.; Martínez-Ballarín, E.; Fuentes-Broto, L.; Miana-Mena, F.J.; Venegas, C.; Caballero, B.; Escames, G.; Coto-Montes, A.; Acuña-Castroviejo, D. Melatonin reduces membrane rigidity and oxidative damage in the brain of SAMP8 mice. Neurobiol. Aging 2011, 32, 2045–2054. [Google Scholar] [CrossRef]
- Wu, Z.; Berlemann, L.A.; Bader, V.; Sehr, D.A.; Dawin, E.; Covallero, A.; Meschede, J.; Angersbach, L.; Showkat, C.; Michaelis, J.B.; et al. LUBAC assembles a ubiquitin signaling platform at mitochondria for signal amplification and transport of NF-κB to the nucleus. EMBO J. 2022, 41, e112006. [Google Scholar] [CrossRef]
- Reiter, R.J.; Sharma, R.; Rodriguez, C.; Martin, V.; Rosales-Corral, S.; Zuccari, D.A.P.C.; Chuffa, L.G.A. Part-time cancers and role of melatonin in determining their metabolic phenotype. Life Sci. 2021, 278, 119597. [Google Scholar] [CrossRef]
- Milán-Tomás, Á.; Shapiro, C.M. Circadian Rhythms Disturbances in Alzheimer Disease: Current Concepts, Diagnosis, and Management. Alzheimer Dis. Assoc. Disord. 2018, 32, 162–171. [Google Scholar] [CrossRef] [PubMed]
- Verma, A.K.; Singh, S.; Rizvi, S.I. Aging, circadian disruption and neurodegeneration: Interesting interplay. Exp. Gerontol. 2022, 172, 112076. [Google Scholar] [CrossRef] [PubMed]
- Palagini, L.; Miniati, M.; Marazziti, D.; Massa, L.; Grassi, L.; Geoffroy, P.A. Circadian Rhythm Alterations May be Related to Impaired Resilience, Emotional Dysregulation and to the Severity of Mood Features in Bipolar I and II Disorders. Clin. Neuropsychiatry 2022, 19, 174–186. [Google Scholar] [CrossRef] [PubMed]
- Karasek, M.; Reiter, R.J. Melatonin and aging. Neuroendocrinol. Lett. 2002, 23 (Suppl. 1), 14–16. [Google Scholar]
- Yao, J.K.; Dougherty, G.G., Jr.; Reddy, R.D.; Keshavan, M.S.; Montrose, D.M.; Matson, W.R.; Rozen, S.; Krishnan, R.R.; McEvoy, J.; Kaddurah-Daouk, R. Altered interactions of tryptophan metabolites in first-episode neuroleptic-naive patients with schizophrenia. Mol. Psychiatry 2010, 15, 938–953. [Google Scholar] [CrossRef]
- Reiter, R.J.; Guerrero, J.M.; Garcia, J.J.; Acuña-Castroviejo, D. Reactive oxygen intermediates, molecular damage, and aging. Relation to melatonin. Ann. N. Y. Acad. Sci. 1998, 854, 410–424. [Google Scholar] [CrossRef]
- Huo, X.; Wang, C.; Yu, Z.; Peng, Y.; Wang, S.; Feng, S.; Zhang, S.; Tian, X.; Sun, C.; Liu, K.; et al. Human transporters, PEPT1/2, facilitate melatonin transportation into mitochondria of cancer cells: An implication of the therapeutic potential. J. Pineal Res. 2017, 62, e12390. [Google Scholar] [CrossRef]
- Cucielo, M.S.; Cesário, R.C.; Silveira, H.S.; Gaiotte, L.B.; Dos Santos, S.A.A.; de Campos Zuccari, D.A.P.; Seiva, F.R.F.; Reiter, R.J.; de Almeida Chuffa, L.G. Melatonin Reverses the Warburg-Type Metabolism and Reduces Mitochondrial Membrane Potential of Ovarian Cancer Cells Independent of MT1 Receptor Activation. Molecules 2022, 27, 4350. [Google Scholar] [CrossRef]
- Ning, L.; Rui, X.; Guorui, L.; Tinglv, F.; Donghang, L.; Chenzhen, X.; Xiaojing, W.; Qing, G. A novel mechanism for the protection against acute lung injury by melatonin: Mitochondrial quality control of lung epithelial cells is preserved through SIRT3-dependent deacetylation of SOD2. Cell Mol. Life Sci. 2022, 79, 610. [Google Scholar] [CrossRef]
- Anderson, G. Daytime orexin and night-time melatonin regulation of mitochondria melatonin::roles in circadian oscillations systemically and centrally in breast cancer symptomatology. Melatonin Res. 2019, 2, 1–8. [Google Scholar] [CrossRef]
- Reiter, R.J.; Sharma, R.; Ma, Q.; Rosales-Corral, S.; Acuna-Castroviejo, D.; Escames, G. Inhibition of mitochondrial pyruvate dehydrogenase kinase: A proposed mechanism by which melatonin causes cancer cells to overcome cytosolic glycolysis, reduce tumor biomass and reverse insensitivity to chemotherapy. Melatonin Res. 2019, 2, 105–119. [Google Scholar] [CrossRef]
- Anderson, G.; Reiter, R.J. Melatonin: Roles in influenza, Covid-19, and other viral infections. Rev. Med. Virol. 2020, 30, e2109. [Google Scholar] [CrossRef]
- Cela, O.; Scrima, R.; Pazienza, V.; Merla, G.; Benegiamo, G.; Augello, B.; Fugetto, S.; Menga, M.; Rubino, R.; Fuhr, L.; et al. Clock genes-dependent acetylation of complex I sets rhythmic activity of mitochondrial OxPhos. Biochim. Biophys Acta 2016, 1863, 596–606. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Zhang, J.; Zhou, Y.; Li, X.; Li, H.; Liu, J.; Gou, K.; Zhao, J.; Cui, S. MicroRNA-7 inhibits melatonin synthesis by acting as a linking molecule between leptin and norepinephrine signaling pathways in pig pineal gland. J. Pineal Res. 2019, 66, e12552. [Google Scholar] [CrossRef]
- Verma, A.K.; Singh, S.; Rizvi, S.I. Therapeutic potential of melatonin and its derivatives in aging and neurodegenerative diseases. Biogerontology 2023, 24, 183–206. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Zhang, S.; Zhang, H.; Gao, Z.; Wang, X.; Liu, X.; Xue, C.; Yao, L.; Lu, G. Mechanisms of Autoimmune Cell in DA Neuron Apoptosis of Parkinson’s Disease: Recent Advancement. Oxid. Med. Cell. Longev. 2022, 2022, 7965433. [Google Scholar] [CrossRef]
- Liu, D.; Wu, L.; Wei, H.; Zhu, C.; Tian, R.; Zhu, W.; Xu, Q. The SFT2D2 gene is associated with the autoimmune pathology of schizophrenia in a Chinese population. Front. Neurol. 2022, 13, 1037777. [Google Scholar] [CrossRef]
- Elgayar, S.A.M.; Abdel-Hafez, A.A.M.; Gomaa, A.M.S.; Elsherif, R. Vulnerability of glia and vessels of rat substantia nigra in rotenone Parkinson model. Ultrastruct. Pathol. 2018, 42, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Xie, Y.Z.; Liu, Y.S. Accelerated aging-related transcriptome alterations in neurovascular unit cells in the brain of Alzheimer’s disease. Front. Aging Neurosci. 2022, 14, 949074. [Google Scholar] [CrossRef] [PubMed]
- Haissaguerre, M.; Vantyghem, M.C. What an endocrinologist should know for patients receiving lithium therapy. Ann. Endocrinol. 2022, 83, 219–225. [Google Scholar] [CrossRef]
- Maussion, G.; Yang, J.; Yerko, V.; Barker, P.; Mechawar, N.; Ernst, C.; Turecki, G. Regulation of a truncated form of tropomyosin-related kinase B (TrkB) by Hsa-miR-185* in frontal cortex of suicide completers. PLoS ONE 2012, 7, e39301. [Google Scholar] [CrossRef]
- Cao, W.; Lu, J.; Li, L.; Qiu, C.; Qin, X.; Wang, T.; Li, S.; Zhang, J.; Xu, J. Activation of the Aryl Hydrocarbon Receptor Ameliorates Acute Rejection of Rat Liver Transplantation by Regulating Treg Proliferation and PD-1 Expression. Transplantation 2022, 106, 2172–2181. [Google Scholar] [CrossRef] [PubMed]
- Amobi-McCloud, A.; Muthuswamy, R.; Battaglia, S.; Yu, H.; Liu, T.; Wang, J.; Putluri, V.; Singh, P.K.; Qian, F.; Huang, R.Y.; et al. IDO1 Expression in Ovarian Cancer Induces PD-1 in T Cells via Aryl Hydrocarbon Receptor Activation. Front. Immunol. 2021, 12, 678999. [Google Scholar] [CrossRef] [PubMed]
- Campesato, L.F.; Budhu, S.; Tchaicha, J.; Weng, C.H.; Gigoux, M.; Cohen, I.J.; Redmond, D.; Mangarin, L.; Pourpe, S.; Liu, C.; et al. Blockade of the AHR restricts a Treg-macrophage suppressive axis induced by L-Kynurenine. Nat. Commun. 2020, 11, 4011. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.T.; Chu, C.I.; Wang, F.Y.; Yang, H.Y.; Tseng, W.S.; Chang, C.R.; Chang, C.C. A change of PD-1/PD-L1 expression on peripheral T cell subsets correlates with the different stages of Alzheimer’s Disease. Cell Biosci. 2022, 12, 162. [Google Scholar] [CrossRef]
- Yang, C.E.; Wang, Y.N.; Hua, M.R.; Miao, H.; Zhao, Y.Y.; Cao, G. Aryl hydrocarbon receptor: From pathogenesis to therapeutic targets in aging-related tissue fibrosis. Ageing Res. Rev. 2022, 79, 101662. [Google Scholar] [CrossRef]
- Wei, J.; Nduom, E.K.; Kong, L.Y.; Hashimoto, Y.; Xu, S.; Gabrusiewicz, K.; Ling, X.; Huang, N.; Qiao, W.; Zhou, S.; et al. MiR-138 exerts anti-glioma efficacy by targeting immune checkpoints. Neuro Oncol. 2016, 18, 639–648. [Google Scholar] [CrossRef]
- Liu, F.; Yang, Y.; Peng, W.; Zhao, N.; Chen, J.; Xu, Z.; Cui, Y.; Qian, K. Mitophagy-promoting miR-138-5p promoter demethylation inhibits pyroptosis in sepsis-associated acute lung injury. Inflamm. Res. 2023, 72, 329–346. [Google Scholar] [CrossRef]
- Fan, L.; Zhaohong, X.; Xiangxue, W.; Yingying, X.; Xiao, Z.; Xiaoyan, Z.; Jieke, Y.; Chao, L. Melatonin Ameliorates the Progression of Alzheimer’s Disease by Inducing TFEB Nuclear Translocation, Promoting Mitophagy, and Regulating NLRP3 Inflammasome Activity. BioMed Res. Int. 2022, 2022, 8099459. [Google Scholar] [CrossRef]
- Hänninen, A.; Toivonen, R.; Pöysti, S.; Belzer, C.; Plovier, H.; Ouwerkerk, J.P.; Emani, R.; Cani, P.D.; De Vos, W.M. Akkermansia muciniphila induces gut microbiota remodelling and controls islet autoimmunity in NOD mice. Gut 2018, 67, 1445–1453. [Google Scholar] [CrossRef]
- Liu, S.; Rezende, R.M.; Moreira, T.G.; Tankou, S.K.; Cox, L.M.; Wu, M.; Song, A.; Dhang, F.H.; Wei, Z.; Costamagna, G.; et al. Oral Administration of miR-30d from Feces of MS Patients Suppresses MS-like Symptoms in Mice by Expanding Akkermansia muciniphila. Cell Host Microbe 2019, 26, 779–794.e8. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Yan, C.; Zhao, S.; Zhao, Y.; Huang, R.; Li, Y. The preventive effects of probiotic Akkermansia muciniphila on D-galactose/AlCl3 mediated Alzheimer’s disease-like rats. Exp. Gerontol. 2022, 170, 111959. [Google Scholar] [CrossRef] [PubMed]
- Amorim Neto, D.P.; Bosque, B.P.; Pereira de Godoy, J.V.; Rodrigues, P.V.; Meneses, D.D.; Tostes, K.; Costa Tonoli, C.C.; Faustino de Carvalho, H.; González-Billault, C.; de Castro Fonseca, M. Akkermansia muciniphila induces mitochondrial calcium overload and α -synuclein aggregation in an enteroendocrine cell line. iScience 2022, 25, 103908. [Google Scholar] [CrossRef]
- Wang, C.H.; Yen, H.R.; Lu, W.L.; Ho, H.H.; Lin, W.Y.; Kuo, Y.W.; Huang, Y.Y.; Tsai, S.Y.; Lin, H.C. Adjuvant Probiotics of Lactobacillus salivarius subsp. salicinius AP-32, L. johnsonii MH-68, and Bifidobacterium animalis subsp. lactis CP-9 Attenuate Glycemic Levels and Inflammatory Cytokines in Patients with Type 1 Diabetes Mellitus. Front. Endocrinol. 2022, 13, 754401. [Google Scholar] [CrossRef]
- He, T.; Zhu, Y.H.; Yu, J.; Xia, B.; Liu, X.; Yang, G.Y.; Su, J.H.; Guo, L.; Wang, M.L.; Wang, J.F. Lactobacillus johnsonii L531 reduces pathogen load and helps maintain short-chain fatty acid levels in the intestines of pigs challenged with Salmonella enterica Infantis. Vet. Microbiol. 2019, 230, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Vazquez-Munoz, R.; Thompson, A.; Russell, J.T.; Sobue, T.; Zhou, Y.; Dongari-Bagtzoglou, A. Insights from the Lactobacillus johnsonii Genome Suggest the Production of Metabolites With Antibiofilm Activity Against the Pathobiont Candida albicans. Front. Microbiol. 2022, 13, 853762. [Google Scholar] [CrossRef]
- Gargano, F.; Guerrera, G.; Piras, E.; Serafini, B.; Di Paola, M.; Rizzetto, L.; Buscarinu, M.C.; Annibali, V.; Vuotto, C.; De Bardi, M.; et al. Proinflammatory mucosal-associated invariant CD8+ T cells react to gut flora yeasts and infiltrate multiple sclerosis brain. Front. Immunol. 2022, 13, 890298. [Google Scholar] [CrossRef]
- Gürsoy, S.; Koçkar, T.; Atik, S.U.; Önal, Z.; Önal, H.; Adal, E. Autoimmunity and intestinal colonization by Candida albicans in patients with type 1 diabetes at the time of the diagnosis. Korean J. Pediatr. 2018, 61, 217–220. [Google Scholar] [CrossRef]
- Lau, K.; Benitez, P.; Ardissone, A.; Wilson, T.D.; Collins, E.L.; Lorca, G.; Li, N.; Sankar, D.; Wasserfall, C.; Neu, J.; et al. Inhibition of type 1 diabetes correlated to a Lactobacillus johnsonii N6.2-mediated Th17 bias. J. Immunol. 2011, 186, 3538–3546. [Google Scholar] [CrossRef]
- Luu, M.; Weigand, K.; Wedi, F.; Breidenbend, C.; Leister, H.; Pautz, S.; Adhikary, T.; Visekruna, A. Regulation of the effector function of CD8+ T cells by gut microbiota-derived metabolite butyrate. Sci. Rep. 2018, 8, 14430. [Google Scholar] [CrossRef]
- Bachem, A.; Makhlouf, C.; Binger, K.J.; de Souza, D.P.; Tull, D.; Hochheiser, K.; Whitney, P.G.; Fernandez-Ruiz, D.; Dähling, S.; Kastenmüller, W.; et al. Microbiota-Derived Short-Chain Fatty Acids Promote the Memory Potential of Antigen-Activated CD8+ T Cells. Immunity 2019, 51, 285–297.e5. [Google Scholar] [CrossRef]
- Heuberger, C.; Pott, J.; Maloy, K.J. Why do intestinal epithelial cells express MHC class II? Immunology 2021, 162, 357–367. [Google Scholar] [CrossRef]
- Nicoli, F.; Cabral-Piccin, M.P.; Papagno, L.; Gallerani, E.; Fusaro, M.; Folcher, V.; Dubois, M.; Clave, E.; Vallet, H.; Frere, J.J.; et al. Altered Basal Lipid Metabolism Underlies the Functional Impairment of Naive CD8+ T Cells in Elderly Humans. J. Immunol. 2022, 208, 562–570. [Google Scholar] [CrossRef]
- Brinkmann, V.; Ale-Agha, N.; Haendeler, J.; Ventura, N. The Aryl Hydrocarbon Receptor (AhR) in the Aging Process: Another Puzzling Role for This Highly Conserved Transcription Factor. Front. Physiol. 2020, 10, 1561. [Google Scholar] [CrossRef]
- Ting, L.; Mingjun, S.; Yuanyan, C.; Jingyu, Y.; Jiang, L.; Miao, X.; He, D.; Juan, L.; Haitao, Y. Association between AhR in B cells and systemic lupus erythematosus with renal damage. Int. Immunopharmacol. 2022, 113 Pt A, 109381. [Google Scholar] [CrossRef]
- Nicaise, A.J.; McDonald, A.; Sears, E.R.; Sturgis, T.; Kaplan, B.L.F. TCDD Inhibition of IgG1 Production in Experimental Autoimmune Encephalomyelitis (EAE) and In Vitro. Antibodies 2022, 11, 4. [Google Scholar] [CrossRef] [PubMed]
- McDonald, A.; Nicaise, A.; Sears, E.R.; Bell, A.; Kummari, E.; Kaplan, B.L.F. Potential for TCDD to induce regulatory functions in B cells as part of the mechanism for T cell suppression in EAE. Toxicol. Appl. Pharmacol. 2022, 454, 116259. [Google Scholar] [CrossRef]
- Tousif, S.; Wang, Y.; Jackson, J.; Hough, K.P.; Strenkowski, J.G.; Athar, M.; Thannickal, V.J.; McCusker, R.H.; Ponnazhagan, S.; Deshane, J.S. Indoleamine 2, 3-Dioxygenase Promotes Aryl Hydrocarbon Receptor-Dependent Differentiation of Regulatory B Cells in Lung Cancer. Front. Immunol. 2021, 12, 747780. [Google Scholar] [CrossRef] [PubMed]
- Lightman, S.M.; Peresie, J.L.; Carlson, L.M.; Holling, G.A.; Honikel, M.M.; Chavel, C.A.; Nemeth, M.J.; Olejniczak, S.H.; Lee, K.P. Indoleamine 2,3-dioxygenase 1 is essential for sustaining durable antibody responses. Immunity 2021, 54, 2772–2783.e5. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G.; Maes, M. Interactions of Tryptophan and Its Catabolites with Melatonin and the Alpha 7 Nicotinic Receptor in Central Nervous System and Psychiatric Disorders: Role of the Aryl Hydrocarbon Receptor and Direct Mitochondria Regulation. Int. J. Tryptophan Res. 2017, 10, 1178646917691738. [Google Scholar] [CrossRef]
- Hwang, H.J.; Dornbos, P.; Steidemann, M.; Dunivin, T.K.; Rizzo, M.; LaPres, J.J. Mitochondrial-targeted aryl hydrocarbon receptor and the impact of 2,3,7,8-tetrachlorodibenzo-p-dioxin on cellular respiration and the mitochondrial proteome. Toxicol. Appl. Pharmacol. 2016, 304, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Jakob, C.; Götz, H.; Hellmann, T.; Hellmann, K.P.; Reymann, S.; Flörke, H.; Thinnes, F.P.; Hilschmann, N. Studies on human porin: XIII. The type-1 VDAC ‘porin 31HL’ biotinylated at the plasmalemma of trypan blue excluding human B lymphocytes. FEBS Lett. 1995, 368, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.; Sominsky, L.; Walder, K.R.; Berk, M.; Marx, W.; Carvalho, A.F.; Bortolasci, C.C.; Maes, M.; Puri, B.K. Inflammation and Nitro-oxidative Stress as Drivers of Endocannabinoid System Aberrations in Mood Disorders and Schizophrenia. Mol. Neurobiol. 2022, 59, 3485–3503. [Google Scholar] [CrossRef] [PubMed]
- Moylan, S.; Berk, M.; Dean, O.M.; Samuni, Y.; Williams, L.J.; O’Neil, A.; Hayley, A.C.; Pasco, J.A.; Anderson, G.; Jacka, F.N.; et al. Oxidative & nitrosative stress in depression: Why so much stress? Neurosci. Biobehav. Rev. 2014, 45, 46–62. [Google Scholar] [CrossRef] [PubMed]
- Maruani, J.; Anderson, G.; Etain, B.; Lejoyeux, M.; Bellivier, F.; Geoffroy, P.A. The neurobiology of adaptation to seasons: Relevance and correlations in bipolar disorders. Chronobiol. Int. 2018, 35, 1335–1353. [Google Scholar] [CrossRef] [PubMed]
- MahmoudianDehkordi, S.; Bhattacharyya, S.; Brydges, C.R.; Jia, W.; Fiehn, O.; Rush, A.J.; Dunlop, B.W.; Kaddurah-Daouk, R. Gut Microbiome-Linked Metabolites in the Pathobiology of Major Depression with or Without Anxiety-A Role for Bile Acids. Front. Neurosci. 2022, 16, 937906. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G.; Maes, M. Oxidative/nitrosative stress and immuno-inflammatory pathways in depression: Treatment implications. Curr. Pharm. Des. 2014, 20, 3812–3847. [Google Scholar] [CrossRef]
- Suzuki, H.; Savitz, J.; Kent Teague, T.; Gandhapudi, S.K.; Tan, C.; Misaki, M.; McKinney, B.A.; Irwin, M.R.; Drevets, W.C.; Bodurka, J. Altered populations of natural killer cells, cytotoxic T lymphocytes, and regulatory T cells in major depressive disorder: Association with sleep disturbance. Brain Behav. Immun. 2017, 66, 193–200. [Google Scholar] [CrossRef]
- Jones, C.D.; Motl, R.; Sandroff, B.M. Depression in multiple sclerosis: Is one approach for its management enough? Mult. Scler. Relat. Disord. 2021, 51, 102904. [Google Scholar] [CrossRef]
- Hsu, T.W.; Chen, M.H.; Bai, Y.M.; Chang, W.H.; Cheng, C.M.; Su, T.P.; Chen, T.J.; Tsai, S.J.; Liang, C.S. Family coaggregation of type 1 diabetes mellitus, major depressive disorder, attention-deficiency hyperactivity disorder and autism spectrum disorder in affected families: A nationwide study. Acta Diabetol. 2023, 60, 517–525. [Google Scholar] [CrossRef]
- Vallerand, I.A.; Lewinson, R.T.; Frolkis, A.D.; Lowerison, M.W.; Kaplan, G.G.; Swain, M.G.; Bulloch, A.G.M.; Patten, S.B.; Barnabe, C. Depression as a risk factor for the development of rheumatoid arthritis: A population-based cohort study. RMD Open 2018, 4, e000670. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, M.; Wootla, B.; Anderson, G. Multiple Sclerosis, Gut Microbiota and Permeability: Role of Tryptophan Catabolites, Depression and the Driving Down of Local Melatonin. Curr. Pharm. Des. 2016, 22, 6134–6141. [Google Scholar] [CrossRef] [PubMed]
- Rastad, S.; Barjaste, N.; Lanjanian, H.; Moeini, A.; Kiani, F.; Masoudi-Nejad, A. Parallel molecular alteration between Alzheimer’s disease and major depressive disorder in the human brain dorsolateral prefrontal cortex: An insight from gene expression and methylation profile analyses. Genes Genet. Syst. 2023, 97, 311–324. [Google Scholar] [CrossRef]
- Meng, D.; Jin, Z.; Wang, Y.; Fang, B. Longitudinal cognitive changes in patients with early Parkinson’s disease and neuropsychiatric symptoms. CNS Neurosci. Ther. 2023. Online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Kan, H.; Zhang, W.; Zhong, Y.; Liao, W.; Huang, G.; Wu, P. Mendelian randomization study on the causal effects of systemic lupus erythematosus on major depressive disorder. J. Hum. Genet. 2023, 68, 11–16. [Google Scholar] [CrossRef]
- Anderson, G. Linking the biological underpinnings of depression: Role of mitochondria interactions with melatonin, inflammation, sirtuins, tryptophan catabolites, DNA repair and oxidative and nitrosative stress, with consequences for classification and cognition. Prog. Neuropsychopharmacol. Biol. Psychiatry 2018, 80, 255–266. [Google Scholar] [CrossRef]
- Maes, M.; Moraes, J.B.; Bonifacio, K.L.; Barbosa, D.S.; Vargas, H.O.; Michelin, A.P.; Nunes, S.O.V. Towards a new model and classification of mood disorders based on risk resilience, neuro-affective toxicity, staging, and phenome features using the nomothetic network psychiatry approach. Metab. Brain Dis. 2021, 36, 509–521. [Google Scholar] [CrossRef]
- Anderson, G.; Maes, M. Role of Opioidergic System in Regulating Depression Pathophysiology. Curr. Pharm. Des. 2020, 26, 5317–5334. [Google Scholar] [CrossRef]
- Dai, Y.X.; Tai, Y.H.; Chang, Y.T.; Chen, T.J.; Chen, M.H. Association between major depressive disorder and subsequent autoimmune skin diseases: A nationwide population-based cohort study. J. Affect. Disord. 2020, 274, 334–338. [Google Scholar] [CrossRef]
- Slominski, A.; Pisarchik, A.; Semak, I.; Sweatman, T.; Wortsman, J.; Szczesniewski, A.; Slugocki, G.; McNulty, J.; Kauser, S.; Tobin, D.J.; et al. Serotoninergic and melatoninergic systems are fully expressed in human skin. FASEB J. 2002, 16, 896–898. [Google Scholar] [CrossRef]
- Zawada, A.E.; Naskręt, D.; Piłaciński, S.; Adamska, A.; Grzymisławski, M.; Eder, P.; Grzelka-Woźniak, A.; Zozulińska-Ziółkiewicz, D.; Dobrowolska, A. Helicobacter pylori infection is associated with increased accumulation of advanced glycation end products in the skin in patients with type 1 diabetes: A preliminary study. Adv. Clin. Exp. Med. 2023. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Uaratanawong, R.; Choudhary, V.; Hardin, M.; Zhang, C.; Melnyk, S.; Chen, X.; Bollag, W.B. Advanced Glycation End Products and Activation of Toll-like Receptor-2 and -4 Induced Changes in Aquaporin-3 Expression in Mouse Keratinocytes. Int. J. Mol. Sci. 2023, 24, 1376. [Google Scholar] [CrossRef] [PubMed]
- Hügle, T.; Gratzl, S.; Daikeler, T.; Frey, D.; Tyndall, A.; Walker, U.A. Sclerosing skin disorders in association with multiple sclerosis. Coincidence, underlying autoimmune pathology or interferon induced? Ann. Rheum. Dis. 2009, 68, 47–50. [Google Scholar] [CrossRef] [PubMed]
- Kulcsarova, K.; Baloghova, J.; Necpal, J.; Skorvanek, M. Skin Conditions and Movement Disorders: Hiding in Plain Sight. Mov. Disord. Clin. Pract. 2022, 9, 566–583. [Google Scholar] [CrossRef]
- Wu, S.; Wang, H.; Zhou, Y.; Xia, X.; Yue, Y.; Wu, Y.; Peng, R.; Yang, R.; Li, R.; Yuan, N.; et al. Clinical correlates of autoimmune thyroiditis and non-autoimmune hypothyroidism in treatment-naïve patients with major depressive disorders. J. Affect. Disord. 2023, 323, 755–761. [Google Scholar] [CrossRef]
- Garcia-Marin, R.; Fernandez-Santos, J.M.; Morillo-Bernal, J.; Gordillo-Martinez, F.; Vazquez-Roman, V.; Utrilla, J.C.; Carrillo-Vico, A.; Guerrero, J.M.; Martin-Lacave, I. Melatonin in the thyroid gland: Regulation by thyroid-stimulating hormone and role in thyroglobulin gene expression. J. Physiol. Pharmacol. 2015, 66, 643–652. [Google Scholar] [PubMed]
- Yang, C.; Lin, X.; Wang, X.; Liu, H.; Huang, J.; Wang, S. The schizophrenia and gut microbiota: A bibliometric and visual analysis. Front. Psychiatry 2022, 13, 1022472. [Google Scholar] [CrossRef]
- Liu, J.; Fan, Y.; Zeng, L.-L.; Liu, B.; Ju, Y.; Wang, M.; Dong, Q.; Lu, X.; Sun, J.; Zhang, L.; et al. The neuroprogressive nature of major depressive disorder: Evidence from an intrinsic connectome analysis. Transl. Psychiatry 2021, 11, 102. [Google Scholar] [CrossRef]
- Harrison, P.J.; Luciano, S. Incidence of Parkinson’s disease, dementia, cerebrovascular disease and stroke in bipolar disorder compared to other psychiatric disorders: An electronic health records network study of 66 million people. Bipolar. Disord. 2021, 23, 454–462. [Google Scholar] [CrossRef]
- Khurshid, K.A. Comorbid Insomnia and Psychiatric Disorders: An Update. Innov. Clin. Neurosci. 2018, 15, 28–32. [Google Scholar]
- Akkaoui, M.A.; Palagini, L.; Geoffroy, P.A. Sleep Immune Cross Talk and Insomnia. Adv. Exp. Med. Biol. 2023, 1411, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Hansen, J. Night Shift Work and Risk of Breast Cancer. Curr. Environ. Health Rep. 2017, 4, 325–339. [Google Scholar] [CrossRef] [PubMed]
- Pan, S.; Wu, Y.J.; Zhang, S.S.; Cheng, X.P.; Olatunji, O.J.; Yin, Q.; Zuo, J. The Effect of α7nAChR Signaling on T Cells and Macrophages and Their Clinical Implication in the Treatment of Rheumatic Diseases. Neurochem. Res. 2022, 47, 531–544. [Google Scholar] [CrossRef]
- Zinglersen, A.H.; Drange, I.L.; Myhr, K.A.; Fuchs, A.; Pfeiffer-Jensen, M.; Brock, C.; Jacobsen, S. Vagus nerve stimulation as a novel treatment for systemic lupus erythematous: Study protocol for a randomised, parallel-group, sham-controlled investigator-initiated clinical trial, the SLE-VNS study. BMJ Open 2022, 12, e064552. [Google Scholar] [CrossRef]
- Markus, R.P.; Silva, C.L.; Franco, D.G.; Barbosa, E.M., Jr.; Ferreira, Z.S. Is modulation of nicotinic acetylcholine receptors by melatonin relevant for therapy with cholinergic drugs? Pharmacol. Ther. 2010, 126, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Sommansson, A.; Nylander, O.; Sjöblom, M. Melatonin decreases duodenal epithelial paracellular permeability via a nicotinic receptor-dependent pathway in rats in vivo. J. Pineal Res. 2013, 54, 282–291. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.P.; Carreno, F.R.; Wu, H.; Chung, Y.A.; Frazer, A. Role of TrkB in the anxiolytic-like and antidepressant-like effects of vagal nerve stimulation: Comparison with desipramine. Neuroscience 2016, 322, 273–286. [Google Scholar] [CrossRef]
- Yanpallewar, S.; Fulgenzi, G.; Tomassoni-Ardori, F.; Barrick, C.; Tessarollo, L. Delayed onset of inherited ALS by deletion of the BDNF receptor TrkB.T1 is non-cell autonomous. Exp. Neurol. 2021, 337, 113576. [Google Scholar] [CrossRef]
- Fulgenzi, G.; Hong, Z.; Tomassoni-Ardori, F.; Barella, L.F.; Becker, J.; Barrick, C.; Swing, D.; Yanpallewar, S.; Croix, B.S.; Wess, J.; et al. Novel metabolic role for BDNF in pancreatic β-cell insulin secretion. Nat. Commun. 2020, 11, 1950. [Google Scholar] [CrossRef]
- Wiedemann, F.R.; Siemen, D.; Mawrin, C.; Horn, T.F.; Dietzmann, K. The neurotrophin receptor TrkB is colocalized to mitochondrial membranes. Int. J. Biochem. Cell Biol. 2006, 38, 610–620. [Google Scholar] [CrossRef]
- Sun, T.; Li, Y.; Wu, J.; Cao, Y.; Yang, Y.; He, Y.; Huang, W.; Liu, B.; Yang, W. Downregulation of exosomal MHC-I promotes glioma cells escaping from systemic immunosurveillance. Nanomedicine 2022, 46, 102605. [Google Scholar] [CrossRef] [PubMed]
- Taylor, B.C.; Balko, J.M. Mechanisms of MHC-I Downregulation and Role in Immunotherapy Response. Front. Immunol. 2022, 13, 844866. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Chatterjee, A.; Jana, S.; Dey, S.; Roy, H.; Das, M.K.; Alam, J.; Adhikary, A.; Chowdhury, A.; Biswas, A.; et al. Transforming growth factor beta orchestrates PD-L1 enrichment in tumor-derived exosomes and mediates CD8 T-cell dysfunction regulating early phosphorylation of TCR signalome in breast cancer. Carcinogenesis 2021, 42, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Lu, J.; Zhou, H.; Wang, Y.; Zhang, Q.; Li, S.; Wang, S. Role of B cell-derived exosomes in immunoregulation: Review. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi 2021, 37, 174–177. [Google Scholar] [PubMed]
- Sobue, A.; Ito, N.; Nagai, T.; Shan, W.; Hada, K.; Nakajima, A.; Murakami, Y.; Mouri, A.; Yamamoto, Y.; Nabeshima, T.; et al. Astroglial major histocompatibility complex class I following immune activation leads to behavioral and neuropathological changes. Glia 2018, 66, 1034–1052. [Google Scholar] [CrossRef]
- Moraes, J.B.; Maes, M.; Roomruangwong, C.; Bonifacio, K.L.; Barbosa, D.S.; Vargas, H.O.; Anderson, G.; Kubera, M.; Carvalho, A.F.; Nunes, S.O.V. In major affective disorders, early life trauma predict increased nitro-oxidative stress, lipid peroxidation and protein oxidation and recurrence of major affective disorders, suicidal behaviors and a lowered quality of life. Metab. Brain Dis. 2018, 33, 1081–1096. [Google Scholar] [CrossRef]
- Maes, M.; Vasupanrajit, A.; Jirakran, K.; Klomkliew, P.; Chanchaem, P.; Tunvirachaisakul, C.; Plaimas, K.; Suratanee, A.; Payungporn, S. Adverse childhood experiences and reoccurrence of illness impact the gut microbiome, which affects suicidal behaviors and the phenome of major depression: Towards enterotypic-phenotypes. Acta Neuropsychiatr. 2023, in press. [Google Scholar] [CrossRef]
- Bransfield, R.C. Adverse Childhood Events, Post-Traumatic Stress Disorder, Infectious Encephalopathies and Immune-Mediated Disease. Healthcare 2022, 10, 1127. [Google Scholar] [CrossRef]
- Makinde, E.; Ma, L.; Mellick, G.D.; Feng, Y. Mitochondrial Modulators: The Defender. Biomolecules 2023, 13, 226. [Google Scholar] [CrossRef]
- Goicoechea, L.; Conde de la Rosa, L.; Torres, S.; García-Ruiz, C.; Fernández-Checa, J.C. Mitochondrial cholesterol: Metabolism and impact on redox biology and disease. Redox Biol. 2023, 61, 102643. [Google Scholar] [CrossRef]
- Chen, L.; Li, Y.; Sottas, C.; Lazaris, A.; Petrillo, S.K.; Metrakos, P.; Li, L.; Ishida, Y.; Saito, T.; Garza, S.; et al. Loss of mitochondrial ATPase ATAD3A contributes to nonalcoholic fatty liver disease through accumulation of lipids and damaged mitochondria. J. Biol. Chem. 2022, 298, 102008. [Google Scholar] [CrossRef] [PubMed]
- Baumann, A.; Jin, C.J.; Brandt, A.; Sellmann, C.; Nier, A.; Burkard, M.; Venturelli, S.; Bergheim, I. Oral Supplementation of Sodium Butyrate Attenuates the Progression of Non-Alcoholic Steatohepatitis. Nutrients 2020, 12, 951. [Google Scholar] [CrossRef] [PubMed]
- Ou, T.H.; Tung, Y.T.; Yang, T.H.; Chien, Y.W. Melatonin Improves Fatty Liver Syndrome by Inhibiting the Lipogenesis Pathway in Hamsters with High-Fat Diet-Induced Hyperlipidemia. Nutrients 2019, 11, 748. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anderson, G.; Almulla, A.F.; Reiter, R.J.; Maes, M. Redefining Autoimmune Disorders’ Pathoetiology: Implications for Mood and Psychotic Disorders’ Association with Neurodegenerative and Classical Autoimmune Disorders. Cells 2023, 12, 1237. https://doi.org/10.3390/cells12091237
Anderson G, Almulla AF, Reiter RJ, Maes M. Redefining Autoimmune Disorders’ Pathoetiology: Implications for Mood and Psychotic Disorders’ Association with Neurodegenerative and Classical Autoimmune Disorders. Cells. 2023; 12(9):1237. https://doi.org/10.3390/cells12091237
Chicago/Turabian StyleAnderson, George, Abbas F. Almulla, Russel J. Reiter, and Michael Maes. 2023. "Redefining Autoimmune Disorders’ Pathoetiology: Implications for Mood and Psychotic Disorders’ Association with Neurodegenerative and Classical Autoimmune Disorders" Cells 12, no. 9: 1237. https://doi.org/10.3390/cells12091237