
Amyloid Beta Oligomers Activate Death Receptors and Mitochondria-Mediated Apoptotic Pathways in Cerebral Vascular Smooth Muscle Cells; Protective Effects of Carbonic Anhydrase Inhibitors

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Aβ Preparation and Oligomerization
2.2. Cell Culture
2.3. Methazolamide and Acetazolamide Preparation
2.4. Cell Death ELISA
2.5. Cell Event Fluorescent Caspase 3/7 Assay
2.6. Mitochondrial Stress Test
2.7. Western Blot
2.8. Quantitative PCR
2.9. Immunocytochemistry
2.10. Caspase 9 Activity Assay
2.11. DR4 and DR5 Gene Silencing
2.12. Mitotracker CMHX2ROS
2.13. TgSwDI Animals
2.14. TgSwDI Treatment with CAIs
2.15. Immunohistochemistry in TgSwDI
3. Results
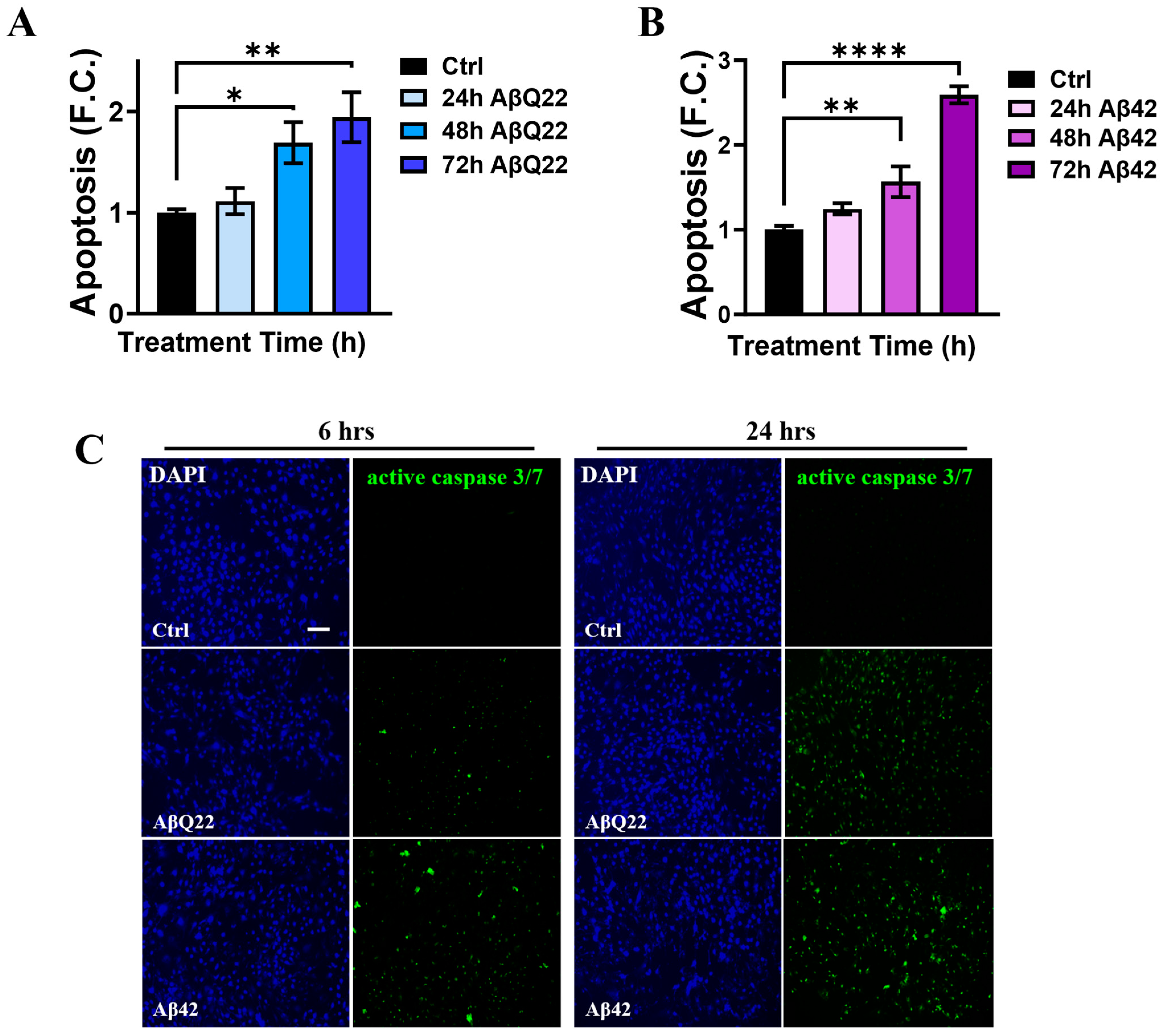
3.1. Prolonged Challenge with Aβ42 and AβQ22 Oligomers Induces Apoptosis in Human Brain Vascular Smooth Muscle Cells
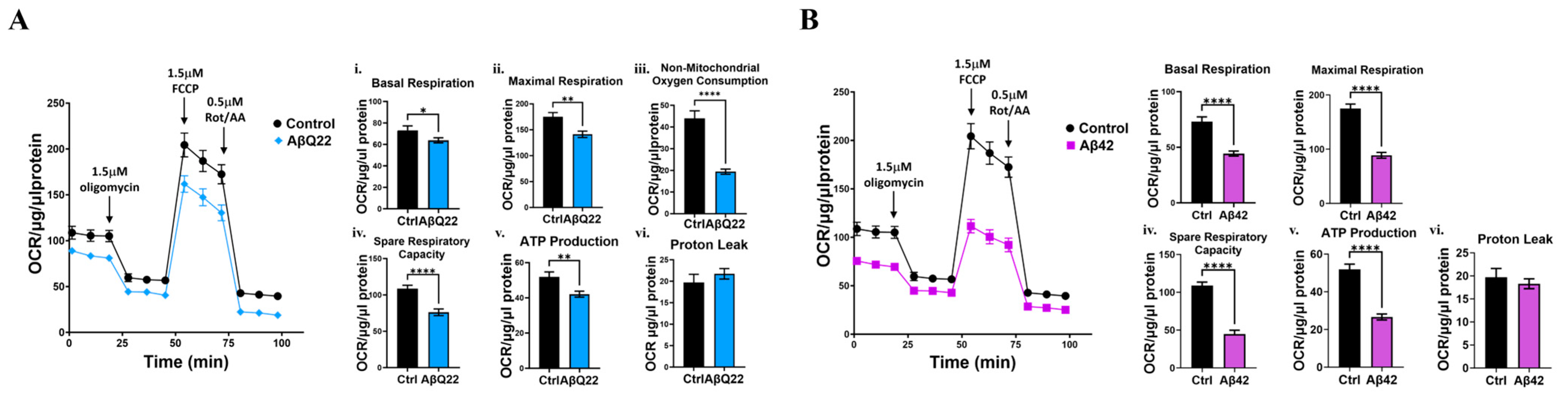
3.2. Aβ Oligomers Impair Mitochondrial Respiration in BVSMCs
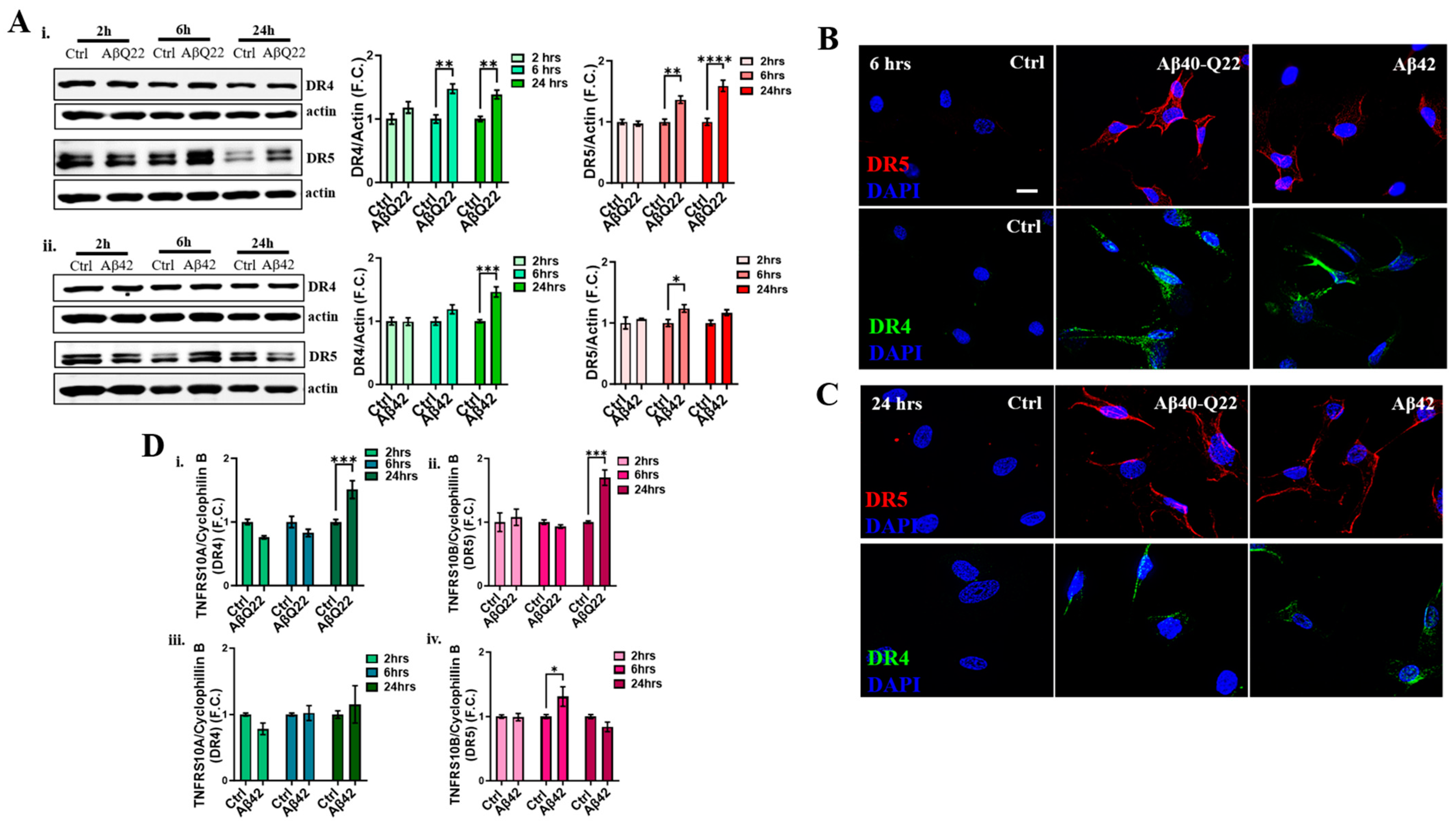
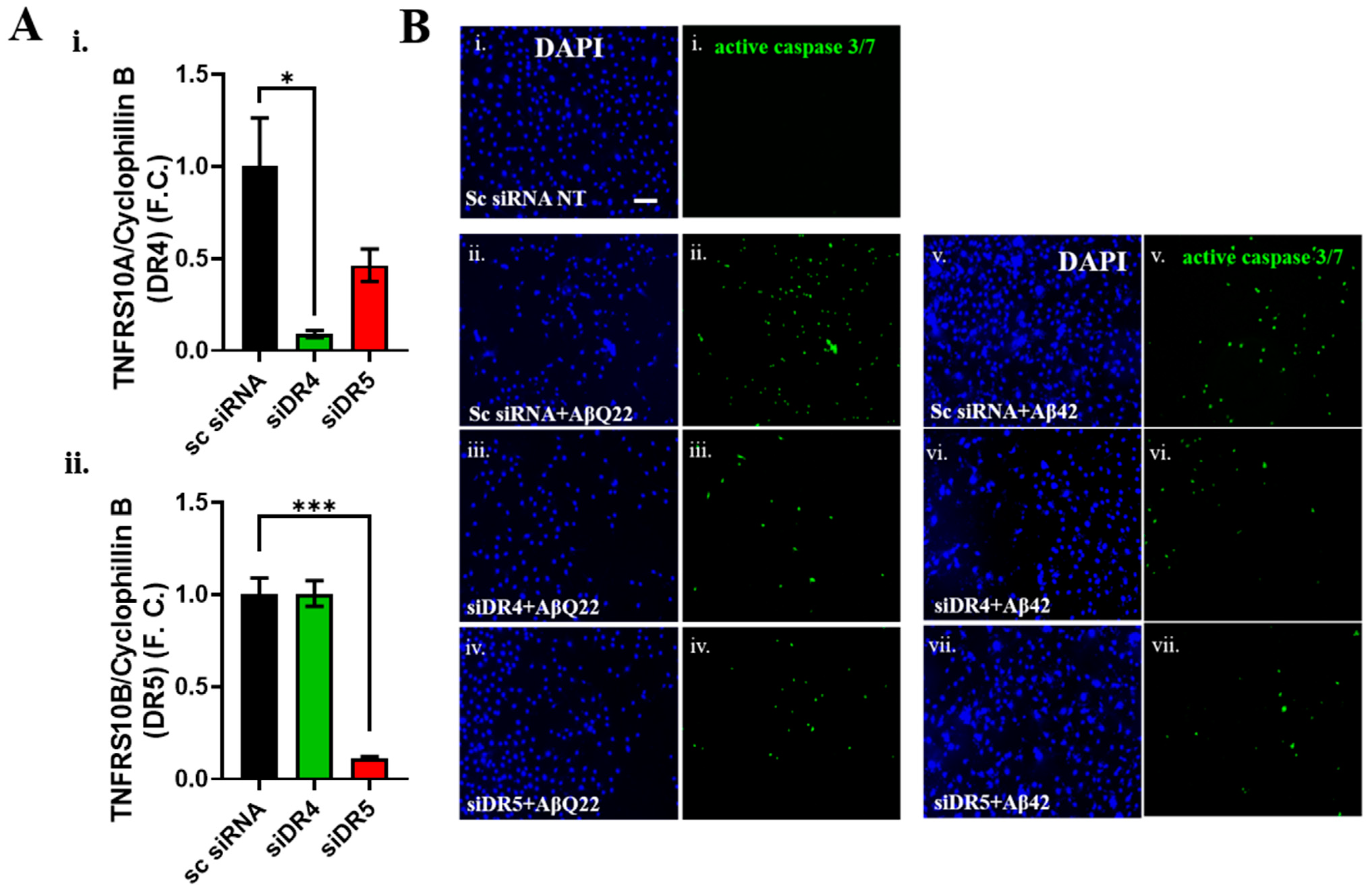
3.3. DR4 and DR5 Mediate Aβ Oligomers’ Toxicity in BVSMCs
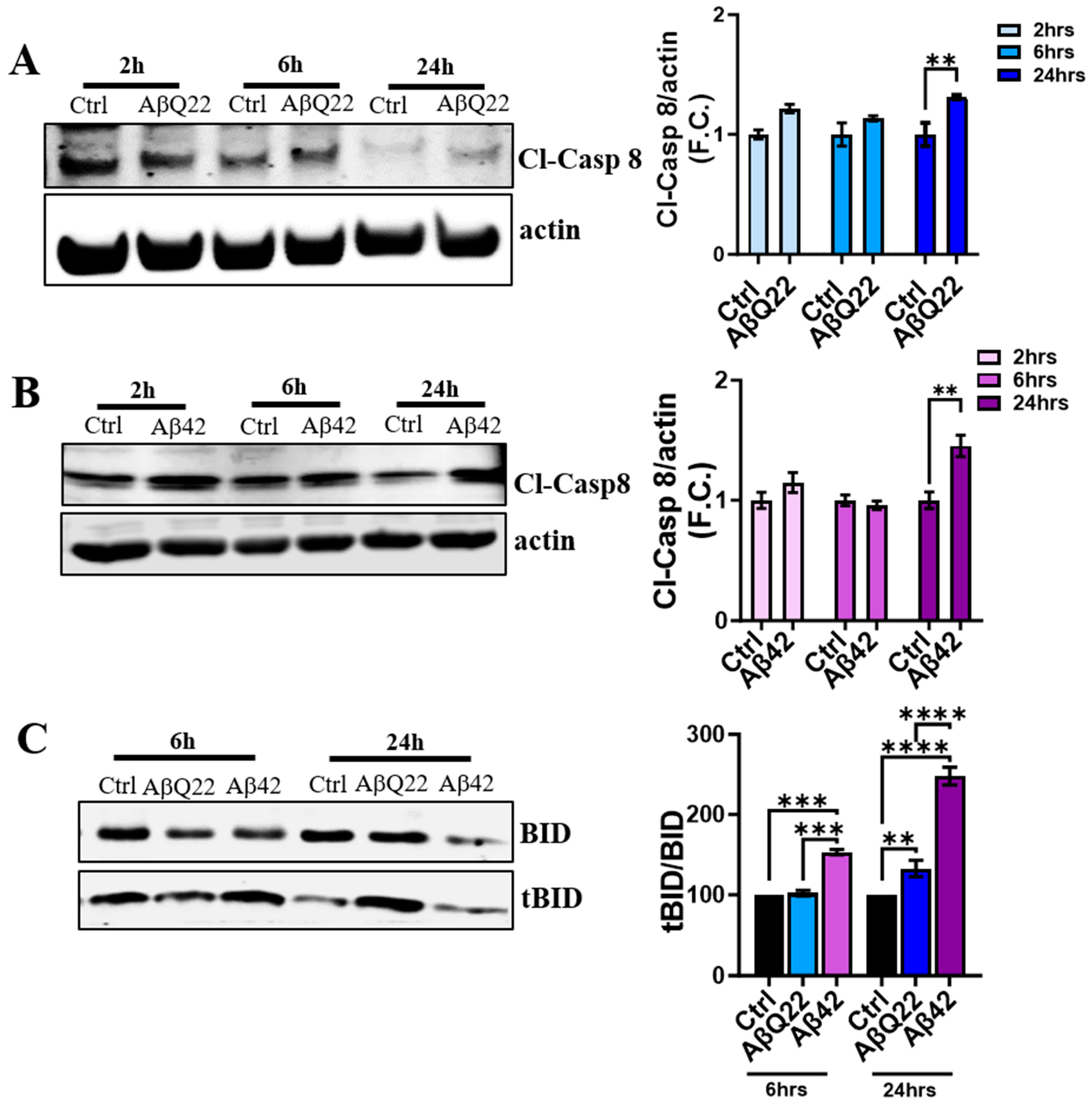
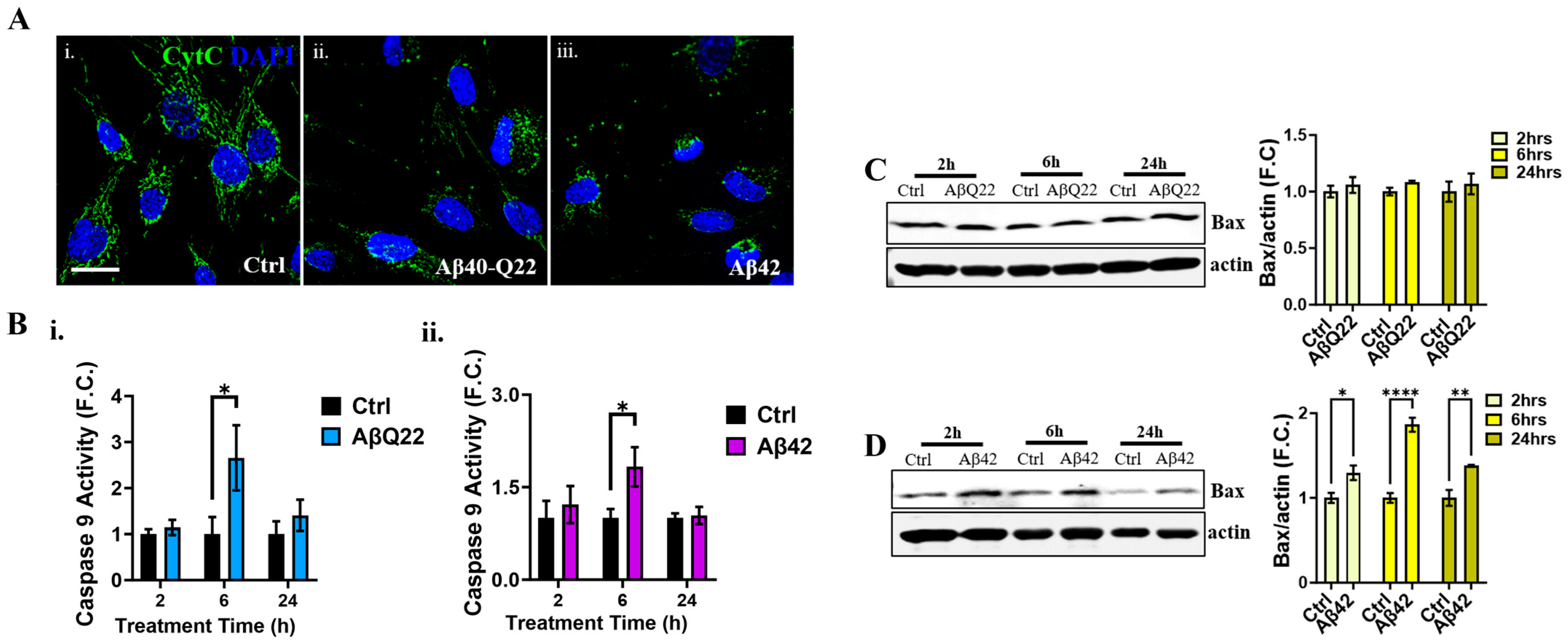
3.4. Aβ Oligomers Induce Caspase 8 Activation, CytC Release from the Mitochondria, and Caspase 9 Activation in Human BVSMCs
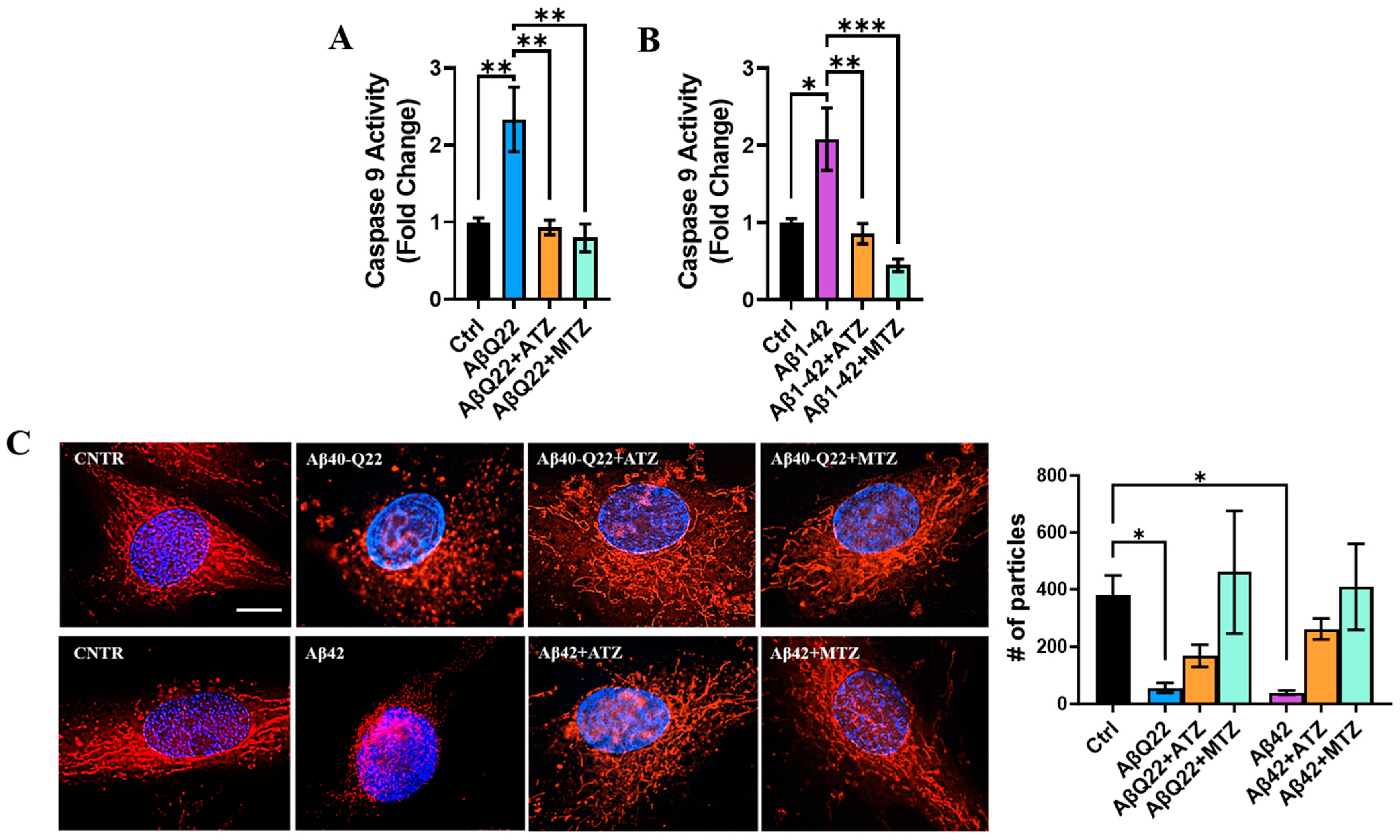
3.5. Carbonic Anhydrase Inhibition Attenuates Caspase 9 Activation and Loss of Mitochondrial in BVSMCs
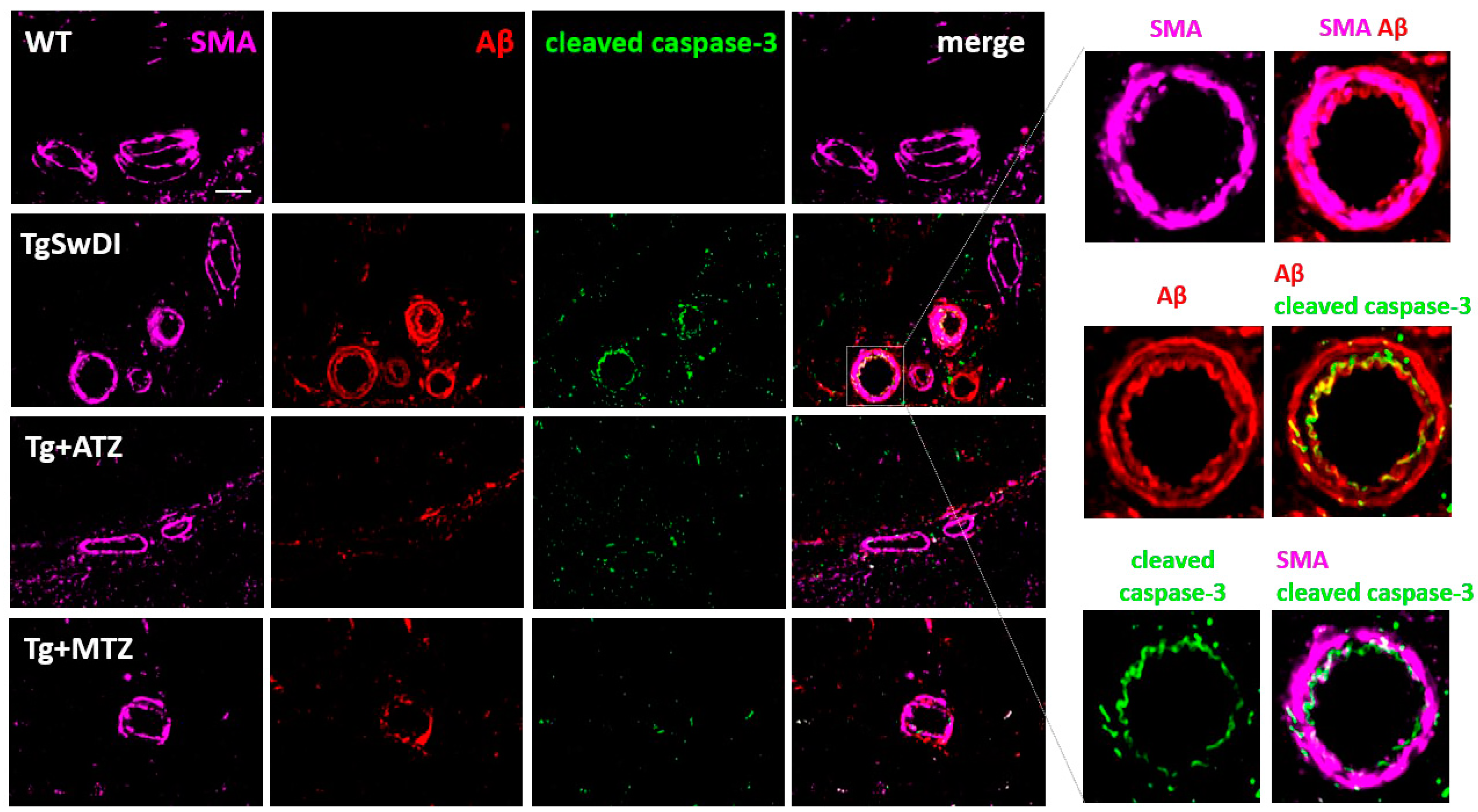
3.6. Carbonic Anhydrase Inhibitors Reduce Caspase 3 Activation in Brain Vascular Smooth Muscle Cells In Vivo in a Mouse Model of CAA
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Abbreviation | Keywords |
| Aβ | Amyloid beta |
| Aβ42 | Amyloid beta 1-42 |
| AβQ22 | Amyloid beta 1-40, Dutch mutation |
| CAA | Cerebral Amyloid Angiopathy |
| AD | Alzheimer’s Disease |
| BVSMCs | Brain Vascular Smooth Muscle Cells |
| TRAILs | TNF-Related Apoptosis-Inducing Ligands |
| DRs | Death Receptors |
| IPAD | Intramural Peri-Arterial Drainage |
| ARIA | Amyloid-Related Imaging Abnormalities |
| FADD | Fas-Associated protein with Death Domain |
| DISC | Death-Inducing Signaling Complex |
| BID | BH3-Interacting Domain death agonist |
| MOMP | Mitochondrial Outer Membrane Permeabilization |
| CytC | Cytochrome C |
| ROS | Reactive Oxygen Species |
| CAs | Carbonic Anhydrases |
| MTZ | Methazolamide |
| ATZ | Acetazolamide |
| OCR | Oxygen Consumption Rate |
| hAPP | Human Amyloid beta Precursor Protein |
| ADRD | AD and related dementia |
References
- Cortes-Canteli, M.; Iadecola, C. Alzheimer’s Disease and Vascular Aging: JACC Focus Seminar. J. Am. Coll. Cardiol. 2020, 75, 942–951. [Google Scholar] [CrossRef]
- Haase, N.; Herse, F.; Spallek, B.; Haase, H.; Morano, I.; Qadri, F.; Szijártó, I.A.; Rohm, I.; Yilmaz, A.; Warrington, J.P. Amyloid-β peptides activate α1-adrenergic cardiovascular receptors. Hypertension 2013, 62, 966–972. [Google Scholar] [CrossRef]
- Kanekiyo, T.; Liu, C.-C.; Shinohara, M.; Li, J.; Bu, G. LRP1 in Brain Vascular Smooth Muscle Cells Mediates Local Clearance of Alzheimer’s Amyloid-β. J. Neurosci. 2012, 32, 16458–16465. [Google Scholar] [CrossRef] [PubMed]
- Carare, R.O.; Aldea, R.; Agarwal, N.; Bacskai, B.J.; Bechman, I.; Boche, D.; Bu, G.; Bulters, D.; Clemens, A.; Counts, S.E.; et al. Clearance of interstitial fluid (ISF) and CSF (CLIC) group-part of Vascular Professional Interest Area (PIA): Cerebrovascular disease and the failure of elimination of Amyloid-beta from the brain and retina with age and Alzheimer’s disease-Opportunities for Therapy. Alzheimers Dement. 2020, 12, e12053. [Google Scholar] [CrossRef]
- Aldea, R.; Weller, R.O.; Wilcock, D.M.; Carare, R.O.; Richardson, G. Cerebrovascular Smooth Muscle Cells as the Drivers of Intramural Periarterial Drainage of the Brain. Front. Aging Neurosci. 2019, 11, 1. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P.; LeVine, H., III. Alzheimer’s Disease and the Amyloid-β Peptide. J. Alzheimer’s Dis. 2010, 19, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Fossati, S.; Cam, J.; Meyerson, J.; Mezhericher, E.; Romero, I.; Couraud, P.; Weksler, B.; Ghiso, J.; Rostagno, A. Differential activation of mitochondrial apoptotic pathways by vasculotropic amyloid-β variants in cells composing the cerebral vessel walls. FASEB J. 2010, 24, 229–241. [Google Scholar] [CrossRef] [PubMed]
- Fossati, S.; Ghiso, J.; Rostagno, A. TRAIL death receptors DR4 and DR5 mediate cerebral microvascular endothelial cell apoptosis induced by oligomeric Alzheimer’s Abeta. Cell Death Dis. 2012, 3, e321. [Google Scholar] [CrossRef] [PubMed]
- Kelly, L.; Brown, C.; Michalik, D.; Hawkes, C.A.; Aldea, R.; Agarwal, N.; Salib, R.; Alzetani, A.; Ethell, D.W.; Counts, S.E.; et al. Clearance of interstitial fluid (ISF) and CSF (CLIC) group-part of Vascular Professional Interest Area (PIA), updates in 2022–2023. Cerebrovascular disease and the failure of elimination of Amyloid-beta from the brain and retina with age and Alzheimer’s disease: Opportunities for therapy. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2023. [Google Scholar] [CrossRef]
- Aldea, S.; Gaillard, S. Multiple Aneurysms Clipping Through the Eyebrow Approach: 2-Dimensional Operative Video. Oper. Neurosurg. 2019, 16, E78. [Google Scholar] [CrossRef]
- Hawkes, C.A.; Hartig, W.; Kacza, J.; Schliebs, R.; Weller, R.O.; Nicoll, J.A.; Carare, R.O. Perivascular drainage of solutes is impaired in the ageing mouse brain and in the presence of cerebral amyloid angiopathy. Acta Neuropathol. 2011, 121, 431–443. [Google Scholar] [CrossRef]
- Hampel, H.; Elhage, A.; Cho, M.; Apostolova, L.G.; Nicoll, J.A.R.; Atri, A. Amyloid-related imaging abnormalities (ARIA): Radiological, biological and clinical characteristics. Brain J. Neurol. 2023, 146, 4414–4424. [Google Scholar] [CrossRef]
- Guicciardi, M.E.; Gores, G.J. Life and death by death receptors. FASEB J. 2009, 23, 1625–1637. [Google Scholar] [CrossRef]
- Locksley, R.M.; Killeen, N.; Lenardo, M.J. The TNF and TNF receptor superfamilies: Integrating mammalian biology. Cell 2001, 104, 487–501. [Google Scholar] [CrossRef]
- Billen, L.P.; Shamas-Din, A.; Andrews, D.W. Bid: A Bax-like BH3 protein. Oncogene 2008, 27 (Suppl. S1), S93–S104. [Google Scholar] [CrossRef]
- Green, D.R.; Kroemer, G. The pathophysiology of mitochondrial cell death. Science 2004, 305, 626–629. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Abrams, J.M.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; Dawson, T.M.; Dawson, V.L.; El-Deiry, W.S.; Fulda, S.; et al. Molecular definitions of cell death subroutines: Recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012, 19, 107–120. [Google Scholar] [CrossRef]
- Daugas, E.; Nochy, D.; Ravagnan, L.; Loeffler, M.; Susin, S.A.; Zamzami, N.; Kroemer, G. Apoptosis-inducing factor (AIF): A ubiquitous mitochondrial oxidoreductase involved in apoptosis. FEBS Lett. 2000, 476, 118–123. [Google Scholar] [CrossRef]
- Suzuki, Y.; Imai, Y.; Nakayama, H.; Takahashi, K.; Takio, K.; Takahashi, R. A serine protease, HtrA2, is released from the mitochondria and interacts with XIAP, inducing cell death. Mol. Cell 2001, 8, 613–621. [Google Scholar] [CrossRef]
- van Loo, G.; Saelens, X.; van Gurp, M.; MacFarlane, M.; Martin, S.J.; Vandenabeele, P. The role of mitochondrial factors in apoptosis: A Russian roulette with more than one bullet. Cell Death Differ. 2002, 9, 1031–1042. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Chandel, N.S.; Li, X.X.; Schumacker, P.T.; Colombini, M.; Thompson, C.B. Outer mitochondrial membrane permeability can regulate coupled respiration and cell survival. Proc. Natl. Acad. Sci. USA 2000, 97, 4666–4671. [Google Scholar] [CrossRef]
- Treulen, F.; Uribe, P.; Boguen, R.; Villegas, J.V. Mitochondrial outer membrane permeabilization increases reactive oxygen species production and decreases mean sperm velocity but is not associated with DNA fragmentation in human sperm. Mol. Hum. Reprod. 2016, 22, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Solesio, M.E.; Peixoto, P.M.; Debure, L.; Madamba, S.M.; de Leon, M.J.; Wisniewski, T.; Pavlov, E.V.; Fossati, S. Carbonic anhydrase inhibition selectively prevents amyloid beta neurovascular mitochondrial toxicity. Aging Cell 2018, 17, e12787. [Google Scholar] [CrossRef] [PubMed]
- Canepa, E.; Parodi-Rullan, R.; Vazquez-Torres, R.; Gamallo-Lana, B.; Guzman-Hernandez, R.; Lemon, N.L.; Angiulli, F.; Debure, L.; Ilies, M.A.; Ostergaard, L.; et al. FDA-approved carbonic anhydrase inhibitors reduce amyloid beta pathology and improve cognition, by ameliorating cerebrovascular health and glial fitness. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2023, 19, 5048–5073. [Google Scholar] [CrossRef] [PubMed]
- Lemon, N.; Canepa, E.; Ilies, M.A.; Fossati, S. Carbonic Anhydrases as Potential Targets Against Neurovascular Unit Dysfunction in Alzheimer’s Disease and Stroke. Front. Aging Neurosci. 2021, 13, 772278. [Google Scholar] [CrossRef]
- Provensi, G.; Carta, F.; Nocentini, A.; Supuran, C.T.; Casamenti, F.; Passani, M.B.; Fossati, S. A New Kid on the Block? Carbonic Anhydrases as Possible New Targets in Alzheimer’s Disease. Int. J. Mol. Sci. 2019, 20, 4724. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.; Dunkerly-Eyring, B.L.; Keceli, G.; Ranek, M.J. Phosphorylation Modifications Regulating Cardiac Protein Quality Control Mechanisms. Front. Physiol. 2020, 11, 593585. [Google Scholar] [CrossRef]
- Zamanova, S.; Shabana, A.M.; Mondal, U.K.; Ilies, M.A. Carbonic anhydrases as disease markers. Expert Opin. Ther. Pat. 2019, 29, 509–533. [Google Scholar] [CrossRef]
- Mishra, C.B.; Tiwari, M.; Supuran, C.T. Progress in the development of human carbonic anhydrase inhibitors and their pharmacological applications: Where are we today? Med. Res. Rev. 2020, 40, 2485–2565. [Google Scholar] [CrossRef]
- Supuran, C.T. Applications of carbonic anhydrases inhibitors in renal and central nervous system diseases. Expert Opin. Ther. Pat. 2018, 28, 713–721. [Google Scholar] [CrossRef]
- Fossati, S.; Giannoni, P.; Solesio, M.E.; Cocklin, S.L.; Cabrera, E.; Ghiso, J.; Rostagno, A. The carbonic anhydrase inhibitor methazolamide prevents amyloid beta-induced mitochondrial dysfunction and caspase activation protecting neuronal and glial cells in vitro and in the mouse brain. Neurobiol. Dis. 2016, 86, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Parodi-Rullan, R.; Ghiso, J.; Cabrera, E.; Rostagno, A.; Fossati, S. Alzheimer’s amyloid beta heterogeneous species differentially affect brain endothelial cell viability, blood-brain barrier integrity, and angiogenesis. Aging Cell 2020, 19, e13258. [Google Scholar] [CrossRef] [PubMed]
- Stine, W.B., Jr.; Dahlgren, K.N.; Krafft, G.A.; LaDu, M.J. In vitro characterization of conditions for amyloid-beta peptide oligomerization and fibrillogenesis. J. Biol. Chem. 2003, 278, 11612–11622. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Yang, Y.; Song, S.S.; Na, J.H.; Oh, K.J.; Jeong, C.; Yu, Y.G.; Shin, Y.K. Beta-amyloid oligomers activate apoptotic BAK pore for cytochrome c release. Biophys. J. 2014, 107, 1601–1608. [Google Scholar] [CrossRef]
- Qosa, H.; LeVine, H., 3rd; Keller, J.N.; Kaddoumi, A. Mixed oligomers and monomeric amyloid-beta disrupts endothelial cells integrity and reduces monomeric amyloid-beta transport across hCMEC/D3 cell line as an in vitro blood-brain barrier model. Biochim. Biophys. Acta 2014, 1842, 1806–1815. [Google Scholar] [CrossRef] [PubMed]
- Fossati, S.; Ghiso, J.; Rostagno, A. Insights into Caspase-Mediated Apoptotic Pathways Induced by Amyloid-beta in Cerebral Microvascular Endothelial Cells. Neurodegener. Dis. 2012, 10, 324–328. [Google Scholar] [CrossRef] [PubMed]
- Carrero, I.; Gonzalo, M.R.; Martin, B.; Sanz-Anquela, J.M.; Arevalo-Serrano, J.; Gonzalo-Ruiz, A. Oligomers of beta-amyloid protein (Abeta1-42) induce the activation of cyclooxygenase-2 in astrocytes via an interaction with interleukin-1beta, tumour necrosis factor-alpha, and a nuclear factor kappa-B mechanism in the rat brain. Exp. Neurol. 2012, 236, 215–227. [Google Scholar] [CrossRef]
- Ono, K.; Condron, M.M.; Teplow, D.B. Structure-neurotoxicity relationships of amyloid beta-protein oligomers. Proc. Natl. Acad. Sci. USA 2009, 106, 14745–14750. [Google Scholar] [CrossRef]
- Prasad, S.; Yadav, V.R.; Kannappan, R.; Aggarwal, B.B. Ursolic acid, a pentacyclin triterpene, potentiates TRAIL-induced apoptosis through p53-independent up-regulation of death receptors: Evidence for the role of reactive oxygen species and JNK. J. Biol. Chem. 2011, 286, 5546–5557. [Google Scholar] [CrossRef]
- Mahalingam, D.; Szegezdi, E.; Keane, M.; de Jong, S.; Samali, A. TRAIL receptor signalling and modulation: Are we on the right TRAIL? Cancer Treat. Rev. 2009, 35, 280–288. [Google Scholar] [CrossRef]
- Son, Y.G.; Kim, E.H.; Kim, J.Y.; Kim, S.U.; Kwon, T.K.; Yoon, A.R.; Yun, C.O.; Choi, K.S. Silibinin sensitizes human glioma cells to TRAIL-mediated apoptosis via DR5 up-regulation and down-regulation of c-FLIP and survivin. Cancer Res. 2007, 67, 8274–8284. [Google Scholar] [CrossRef]
- Psahoulia, F.H.; Drosopoulos, K.G.; Doubravska, L.; Andera, L.; Pintzas, A. Quercetin enhances TRAIL-mediated apoptosis in colon cancer cells by inducing the accumulation of death receptors in lipid rafts. Mol. Cancer Ther. 2007, 6, 2591–2599. [Google Scholar] [CrossRef] [PubMed]
- Alber, J.; Alladi, S.; Bae, H.J.; Barton, D.A.; Beckett, L.A.; Bell, J.M.; Berman, S.E.; Biessels, G.J.; Black, S.E.; Bos, I.; et al. White matter hyperintensities in vascular contributions to cognitive impairment and dementia (VCID): Knowledge gaps and opportunities. Alzheimers Dement. 2019, 5, 107–117. [Google Scholar] [CrossRef]
- Davis, J.; Cribbs, D.H.; Cotman, C.W.; Van Nostrand, W.E. Pathogenic amyloid beta-protein induces apoptosis in cultured human cerebrovascular smooth muscle cells. Amyloid 1999, 6, 157–164. [Google Scholar] [CrossRef]
- Christie, R.; Yamada, M.; Moskowitz, M.; Hyman, B. Structural and functional disruption of vascular smooth muscle cells in a transgenic mouse model of amyloid angiopathy. Am. J. Pathol. 2001, 158, 1065–1071. [Google Scholar] [CrossRef] [PubMed]
- Folin, M.; Baiguera, S.; Fioravanzo, L.; Conconi, M.T.; Grandi, C.; Nussdorfer, G.G.; Parnigotto, P.P. Caspase-8 activation and oxidative stress are involved in the cytotoxic effect of beta-amyloid on rat brain microvascular endothelial cells. Int. J. Mol. Med. 2006, 17, 431–435. [Google Scholar]
- Knowles, J.K.; Rajadas, J.; Nguyen, T.V.; Yang, T.; LeMieux, M.C.; Vander Griend, L.; Ishikawa, C.; Massa, S.M.; Wyss-Coray, T.; Longo, F.M. The p75 neurotrophin receptor promotes amyloid-beta(1-42)-induced neuritic dystrophy in vitro and in vivo. J. Neurosci. 2009, 29, 10627–10637. [Google Scholar] [CrossRef]
- Sotthibundhu, A.; Li, Q.X.; Thangnipon, W.; Coulson, E.J. Abeta(1-42) stimulates adult SVZ neurogenesis through the p75 neurotrophin receptor. Neurobiol. Aging 2009, 30, 1975–1985. [Google Scholar] [CrossRef] [PubMed]
- Sotthibundhu, A.; Sykes, A.M.; Fox, B.; Underwood, C.K.; Thangnipon, W.; Coulson, E.J. Beta-amyloid(1-42) induces neuronal death through the p75 neurotrophin receptor. J. Neurosci. 2008, 28, 3941–3946. [Google Scholar] [CrossRef]
- Yaar, M.; Zhai, S.; Fine, R.E.; Eisenhauer, P.B.; Arble, B.L.; Stewart, K.B.; Gilchrest, B.A. Amyloid beta binds trimers as well as monomers of the 75-kDa neurotrophin receptor and activates receptor signaling. J. Biol. Chem. 2002, 277, 7720–7725. [Google Scholar] [CrossRef]
- Dhage, P.A.; Sharbidre, A.A.; Magdum, S.M. Interlacing the relevance of caspase activation in the onset and progression of Alzheimer’s disease. Brain Res. Bull. 2023, 192, 83–92. [Google Scholar] [CrossRef]
- Nonaka, S.; Nakanishi, H. Microglial clearance of focal apoptotic synapses. Neurosci. Lett. 2019, 707, 134317. [Google Scholar] [CrossRef]
- Kim, S.H.; Ahn, J.H.; Yang, H.; Lee, P.; Koh, G.Y.; Jeong, Y. Cerebral amyloid angiopathy aggravates perivascular clearance impairment in an Alzheimer’s disease mouse model. Acta Neuropathol. Commun. 2020, 8, 181. [Google Scholar] [CrossRef]
- Parodi-Rullan, R.M.; Javadov, S.; Fossati, S. Dissecting the Crosstalk between Endothelial Mitochondrial Damage, Vascular Inflammation, and Neurodegeneration in Cerebral Amyloid Angiopathy and Alzheimer’s Disease. Cells 2021, 10, 2903. [Google Scholar] [CrossRef]
- Parodi-Rullan, R.; Sone, J.Y.; Fossati, S. Endothelial Mitochondrial Dysfunction in Cerebral Amyloid Angiopathy and Alzheimer’s Disease. J. Alzheimers Dis. 2019, 72, 1019–1039. [Google Scholar] [CrossRef]
- Swerdlow, R.H. Mitochondria and Mitochondrial Cascades in Alzheimer’s Disease. J. Alzheimers Dis. 2018, 62, 1403–1416. [Google Scholar] [CrossRef]
- Swerdlow, R.H.; Burns, J.M.; Khan, S.M. The Alzheimer’s disease mitochondrial cascade hypothesis. J. Alzheimers Dis. 2010, 20 (Suppl. S2), S265–S279. [Google Scholar] [CrossRef]
- Wang, W.; Zhao, F.; Ma, X.; Perry, G.; Zhu, X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: Recent advances. Mol. Neurodegener. 2020, 15, 30. [Google Scholar] [CrossRef]
- Adav, S.S.; Park, J.E.; Sze, S.K. Quantitative profiling brain proteomes revealed mitochondrial dysfunction in Alzheimer’s disease. Mol. Brain 2019, 12, 8. [Google Scholar] [CrossRef]
- Hu, H.; Tan, C.C.; Tan, L.; Yu, J.T. A Mitocentric View of Alzheimer’s Disease. Mol. Neurobiol. 2016, 54, 6046–6060. [Google Scholar] [CrossRef]
- Xie, H.; Guan, J.; Borrelli, L.A.; Xu, J.; Serrano-Pozo, A.; Bacskai, B.J. Mitochondrial alterations near amyloid plaques in an Alzheimer’s disease mouse model. J. Neurosci. 2013, 33, 17042–17051. [Google Scholar] [CrossRef] [PubMed]
- Trushina, E.; Nemutlu, E.; Zhang, S.; Christensen, T.; Camp, J.; Mesa, J.; Siddiqui, A.; Tamura, Y.; Sesaki, H.; Wengenack, T.M.; et al. Defects in mitochondrial dynamics and metabolomic signatures of evolving energetic stress in mouse models of familial Alzheimer’s disease. PLoS ONE 2012, 7, e32737. [Google Scholar] [CrossRef] [PubMed]
- Calkins, M.J.; Manczak, M.; Mao, P.; Shirendeb, U.; Reddy, P.H. Impaired mitochondrial biogenesis, defective axonal transport of mitochondria, abnormal mitochondrial dynamics and synaptic degeneration in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 2011, 20, 4515–4529. [Google Scholar] [CrossRef]
- Du, H.; Guo, L.; Yan, S.; Sosunov, A.A.; McKhann, G.M.; Yan, S.S. Early deficits in synaptic mitochondria in an Alzheimer’s disease mouse model. Proc. Natl. Acad. Sci. USA 2010, 107, 18670–18675. [Google Scholar] [CrossRef]
- Dewanjee, S.; Chakraborty, P.; Bhattacharya, H.; Chacko, L.; Singh, B.; Chaudhary, A.; Javvaji, K.; Pradhan, S.R.; Vallamkondu, J.; Dey, A.; et al. Altered glucose metabolism in Alzheimer’s disease: Role of mitochondrial dysfunction and oxidative stress. Free Radic. Biol. Med. 2022, 193, 134–157. [Google Scholar] [CrossRef]
- Fani, G.; La Torre, C.E.; Cascella, R.; Cecchi, C.; Vendruscolo, M.; Chiti, F. Misfolded protein oligomers induce an increase of intracellular Ca2+ causing an escalation of reactive oxidative species. Cell Mol. Life Sci. 2022, 79, 500. [Google Scholar] [CrossRef]
- Owen, J.B.; Sultana, R.; Aluise, C.D.; Erickson, M.A.; Price, T.O.; Bu, G.; Banks, W.A.; Butterfield, D.A. Oxidative modification to LDL receptor-related protein 1 in hippocampus from subjects with Alzheimer disease: Implications for Abeta accumulation in AD brain. Free Radic. Biol. Med. 2010, 49, 1798–1803. [Google Scholar] [CrossRef]
- Leuner, K.; Schutt, T.; Kurz, C.; Eckert, S.H.; Schiller, C.; Occhipinti, A.; Mai, S.; Jendrach, M.; Eckert, G.P.; Kruse, S.E.; et al. Mitochondrion-derived reactive oxygen species lead to enhanced amyloid beta formation. Antioxid. Redox Signal. 2012, 16, 1421–1433. [Google Scholar] [CrossRef]
- Moreira, P.I.; Santos, M.S.; Moreno, A.; Oliveira, C. Amyloid beta-peptide promotes permeability transition pore in brain mitochondria. Biosci. Rep. 2001, 21, 789–800. [Google Scholar] [CrossRef]
- Rodrigues, C.M.; Sola, S.; Brito, M.A.; Brondino, C.D.; Brites, D.; Moura, J.J. Amyloid beta-peptide disrupts mitochondrial membrane lipid and protein structure: Protective role of tauroursodeoxycholate. Biochem. Biophys. Res. Commun. 2001, 281, 468–474. [Google Scholar] [CrossRef]
- Devi, L.; Prabhu, B.M.; Galati, D.F.; Avadhani, N.G.; Anandatheerthavarada, H.K. Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer’s disease brain is associated with mitochondrial dysfunction. J. Neurosci. 2006, 26, 9057–9068. [Google Scholar] [CrossRef]
- Murphy, K.M.; Ranganathan, V.; Farnsworth, M.L.; Kavallaris, M.; Lock, R.B. Bcl-2 inhibits Bax translocation from cytosol to mitochondria during drug-induced apoptosis of human tumor cells. Cell Death Differ. 2000, 7, 102–111. [Google Scholar] [CrossRef]
- Moreira, P.I.; Carvalho, C.; Zhu, X.; Smith, M.A.; Perry, G. Mitochondrial dysfunction is a trigger of Alzheimer’s disease pathophysiology. Biochim. Biophys. Acta 2010, 1802, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Settakis, G.; Molnar, C.; Kerenyi, L.; Kollar, J.; Legemate, D.; Csiba, L.; Fulesdi, B. Acetazolamide as a vasodilatory stimulus in cerebrovascular diseases and in conditions affecting the cerebral vasculature. Eur. J. Neurol. 2003, 10, 609–620. [Google Scholar] [CrossRef]
- Wright, A.; Brearey, S.; Imray, C. High hopes at high altitudes: Pharmacotherapy for acute mountain sickness and high-altitude cerebral and pulmonary oedema. Expert Opin. Pharmacother. 2008, 9, 119–127. [Google Scholar] [CrossRef]
- Supuran, C.T. Carbonic anhydrases: Novel therapeutic applications for inhibitors and activators. Nat. Rev. Drug Discov. 2008, 7, 168–181. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anzovino, A.; Canepa, E.; Alves, M.; Lemon, N.L.; Carare, R.O.; Fossati, S. Amyloid Beta Oligomers Activate Death Receptors and Mitochondria-Mediated Apoptotic Pathways in Cerebral Vascular Smooth Muscle Cells; Protective Effects of Carbonic Anhydrase Inhibitors. Cells 2023, 12, 2840. https://doi.org/10.3390/cells12242840
Anzovino A, Canepa E, Alves M, Lemon NL, Carare RO, Fossati S. Amyloid Beta Oligomers Activate Death Receptors and Mitochondria-Mediated Apoptotic Pathways in Cerebral Vascular Smooth Muscle Cells; Protective Effects of Carbonic Anhydrase Inhibitors. Cells. 2023; 12(24):2840. https://doi.org/10.3390/cells12242840
Chicago/Turabian StyleAnzovino, Amy, Elisa Canepa, Micaelly Alves, Nicole L. Lemon, Roxana O. Carare, and Silvia Fossati. 2023. "Amyloid Beta Oligomers Activate Death Receptors and Mitochondria-Mediated Apoptotic Pathways in Cerebral Vascular Smooth Muscle Cells; Protective Effects of Carbonic Anhydrase Inhibitors" Cells 12, no. 24: 2840. https://doi.org/10.3390/cells12242840