
Five Inhibitory Receptors Display Distinct Vesicular Distributions in Murine T Cells
 , , , , , , ,
, , , , , , ,
Abstract
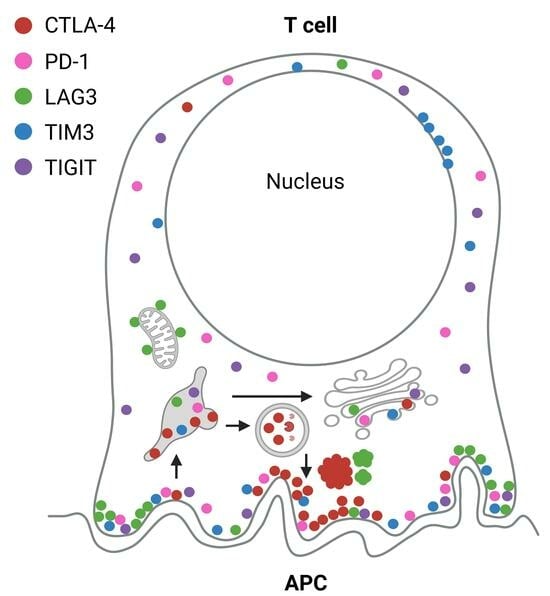
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Cells and Media
2.3. Antibodies
2.4. Plasmids
2.5. T Cell Isolation and Stimulation
2.6. T Cell Retroviral Transduction
2.7. Spinning Disk Confocal Imaging
2.7.1. Imaging of Cell Couples
2.7.2. Determination of Fluorescence at the Cell Edge Versus Interior and of Distribution Heterogeneity
Vesicular Reorientation towards Interface
Imaging of Individual T Cells to Estimate Association of Inhibitory Receptors with the Cell Edge
2.8. TIRF Imaging
2.9. APEX Proteomics and Electron Microscopy
2.10. Immunostaining
2.11. Immunoprecipitation
3. Results
3.1. Inhibitory Receptors Are Enriched in the Cell Interior with Varying Degrees of Clustering


3.2. Vesicles Transporting Inhibitory Receptors Are Inserted into the T Cell Plasma Membrane upon T Cell Activation with Distinct Frequency and Kinetics Resulting in Distinct Interface Distributions

3.3. The Proteomes of Vesicles Harboring CTLA-4, LAG3, and TIM3 Are Distinct
3.4. CTLA-4 Associates More Extensively with Lysosomes
3.5. Association with Regulators of Receptor Transport Is Largely Shared across the Five Inhibitory Receptors

4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Andrews, L.P.; Yano, H.; Vignali, D.A.A. Inhibitory receptors and ligands beyond PD-1, PD-L1 and CTLA-4: Breakthroughs or backups. Nat. Immunol. 2019, 20, 1425–1434. [Google Scholar] [CrossRef] [PubMed]
- Anderson, A.C.; Joller, N.; Kuchroo, V.K. Lag-3, Tim-3, and TIGIT: Co-inhibitory Receptors with Specialized Functions in Immune Regulation. Immunity 2016, 44, 989–1004. [Google Scholar] [CrossRef] [PubMed]
- Schildberg, F.A.; Klein, S.R.; Freeman, G.J.; Sharpe, A.H. Coinhibitory Pathways in the B7-CD28 Ligand-Receptor Family. Immunity 2016, 44, 955–972. [Google Scholar] [CrossRef] [PubMed]
- Honda, T.; Egen, J.G.; Lammermann, T.; Kastenmuller, W.; Torabi-Parizi, P.; Germain, R.N. Tuning of antigen sensitivity by T cell receptor-dependent negative feedback controls T cell effector function in inflamed tissues. Immunity 2014, 40, 235–247. [Google Scholar] [CrossRef]
- Davis, S.J.; Ikemizu, S.; Evans, E.J.; Fugger, L.; Bakker, T.R.; van der Merwe, P.A. The nature of molecular recognition by T cells. Nat. Immunol. 2003, 4, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Valk, E.; Rudd, C.E.; Schneider, H. CTLA-4 trafficking and surface expression. Trends Immunol. 2008, 29, 272–279. [Google Scholar] [CrossRef]
- Khailaie, S.; Rowshanravan, B.; Robert, P.A.; Waters, E.; Halliday, N.; Badillo Herrera, J.D.; Walker, L.S.K.; Sansom, D.M.; Meyer-Hermann, M. Characterization of CTLA4 Trafficking and Implications for Its Function. Biophys. J. 2018, 115, 1330–1343. [Google Scholar] [CrossRef]
- Leung, H.T.; Bradshaw, J.; Cleaveland, J.S.; Linsley, P.S. Cytotoxic T lymphocyte-associated molecule-4, a high-avidity receptor for CD80 and CD86, contains an intracellular localization motif in its cytoplasmic tail. J. Biol. Chem. 1995, 270, 25107–25114. [Google Scholar] [CrossRef]
- Lo, B.; Zhang, K.; Lu, W.; Zheng, L.; Zhang, Q.; Kanellopoulou, C.; Zhang, Y.; Liu, Z.; Fritz, J.M.; Marsh, R.; et al. AUTOIMMUNE DISEASE. Patients with LRBA deficiency show CTLA4 loss and immune dysregulation responsive to abatacept therapy. Science 2015, 349, 436–440. [Google Scholar] [CrossRef]
- Valk, E.; Leung, R.; Kang, H.; Kaneko, K.; Rudd, C.E.; Schneider, H. T cell receptor-interacting molecule acts as a chaperone to modulate surface expression of the CTLA-4 coreceptor. Immunity 2006, 25, 807–821. [Google Scholar] [CrossRef]
- Banton, M.C.; Inder, K.L.; Valk, E.; Rudd, C.E.; Schneider, H. Rab8 binding to immune cell-specific adaptor LAX facilitates formation of trans-Golgi network-proximal CTLA-4 vesicles for surface expression. Mol. Cell Biol. 2014, 34, 1486–1499. [Google Scholar] [CrossRef] [PubMed]
- Iida, T.; Ohno, H.; Nakaseko, C.; Sakuma, M.; Takeda-Ezaki, M.; Arase, H.; Kominami, E.; Fujisawa, T.; Saito, T. Regulation of cell surface expression of CTLA-4 by secretion of CTLA-4-containing lysosomes upon activation of CD4+ T cells. J. Immunol. 2000, 165, 5062–5068. [Google Scholar] [CrossRef]
- Esposito, L.; Hunter, K.M.; Clark, J.; Rainbow, D.B.; Stevens, H.; Denesha, J.; Duley, S.; Dawson, S.; Coleman, G.; Nutland, S.; et al. Investigation of soluble and transmembrane CTLA-4 isoforms in serum and microvesicles. J. Immunol. 2014, 193, 889–900. [Google Scholar] [CrossRef] [PubMed]
- Janman, D.; Hinze, C.; Kennedy, A.; Halliday, N.; Waters, E.; Williams, C.; Rowshanravan, B.; Hou, T.Z.; Minogue, S.; Qureshi, O.S.; et al. Regulation of CTLA-4 recycling by LRBA and Rab11. Immunology 2021, 164, 106–119. [Google Scholar] [CrossRef] [PubMed]
- Egen, J.G.; Allison, J.P. Cytotoxic T Lymphocyte Antigen-4 Accumulation in the Immunological Synapse Is Regulated by TCR Signal Strength. Immunity 2002, 16, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Yokosuka, T.; Kobayashi, W.; Takamatsu, M.; Sakata-Sogawa, K.; Zeng, H.; Hashimoto-Tane, A.; Yagita, H.; Tokunaga, M.; Saito, T. Spatiotemporal basis of CTLA-4 costimulatory molecule-mediated negative regulation of T cell activation. Immunity 2010, 33, 326–339. [Google Scholar] [CrossRef]
- Qureshi, O.S.; Zheng, Y.; Nakamura, K.; Attridge, K.; Manzotti, C.; Schmidt, E.M.; Baker, J.; Jeffery, L.E.; Kaur, S.; Briggs, Z.; et al. Trans-endocytosis of CD80 and CD86: A molecular basis for the cell-extrinsic function of CTLA-4. Science 2011, 332, 600–603. [Google Scholar] [CrossRef]
- Pentcheva-Hoang, T.; Chen, L.; Pardoll, D.M.; Allison, J.P. Programmed death-1 concentration at the immunological synapse is determined by ligand affinity and availability. Proc. Natl. Acad. Sci. USA 2007, 104, 17765–17770. [Google Scholar] [CrossRef]
- Raimondi, G.; Shufesky, W.J.; Tokita, D.; Morelli, A.E.; Thomson, A.W. Regulated compartmentalization of programmed cell death-1 discriminates CD4+CD25+ resting regulatory T cells from activated T cells. J. Immunol. 2006, 176, 2808–2816. [Google Scholar] [CrossRef]
- Bricogne, C.; Fine, M.; Pereira, P.M.; Sung, J.; Tijani, M.; Wang, Y.; Henriques, R.; Collins, M.K.; Hilgemann, D.W. TMEM16F activation by Ca2+ triggers plasma membrane expansion and directs PD-1 trafficking. Sci. Rep. 2019, 9, 619. [Google Scholar] [CrossRef]
- Woo, S.R.; Li, N.; Bruno, T.C.; Forbes, K.; Brown, S.; Workman, C.; Drake, C.G.; Vignali, D.A. Differential subcellular localization of the regulatory T-cell protein LAG-3 and the coreceptor CD4. Eur. J. Immunol. 2010, 40, 1768–1777. [Google Scholar] [CrossRef] [PubMed]
- Bae, J.; Lee, S.J.; Park, C.G.; Lee, Y.S.; Chun, T. Trafficking of LAG-3 to the surface on activated T cells via its cytoplasmic domain and protein kinase C signaling. J. Immunol. 2014, 193, 3101–3112. [Google Scholar] [CrossRef] [PubMed]
- Clayton, K.L.; Haaland, M.S.; Douglas-Vail, M.B.; Mujib, S.; Chew, G.M.; Ndhlovu, L.C.; Ostrowski, M.A. T cell Ig and mucin domain-containing protein 3 is recruited to the immune synapse, disrupts stable synapse formation, and associates with receptor phosphatases. J. Immunol. 2014, 192, 782–791. [Google Scholar] [CrossRef] [PubMed]
- Prevost, J.; Edgar, C.R.; Richard, J.; Trothen, S.M.; Jacob, R.A.; Mumby, M.J.; Pickering, S.; Dube, M.; Kaufmann, D.E.; Kirchhoff, F.; et al. HIV-1 Vpu Downregulates Tim-3 from the Surface of Infected CD4(+) T Cells. J. Virol. 2020, 94, e01999-19. [Google Scholar] [CrossRef]
- Jenkinson, S.R.; Williams, N.A.; Morgan, D.J. The role of intercellular adhesion molecule-1/LFA-1 interactions in the generation of tumor-specific CD8+ T cell responses. J. Immunol. 2005, 174, 3401–3407. [Google Scholar] [CrossRef]
- Seder, R.A.; Paul, W.E.; Davis, M.M.; Fazekas de St Groth, B. The presence of interleukin 4 during in vitro priming determines the lymphokine-producing potential of CD4+ T cells from T cell receptor transgenic mice. J. Exp. Med. 1992, 176, 1091–1098. [Google Scholar] [CrossRef]
- Alamir, H.; Wong, C.C.W.; Alsubaiti, A.; Edmunds, G.L.; Grant, T.; Alsulaimani, S.; Boyd, J.; Holland, C.J.; Morgan, D.J.; Gallimore, A.M.; et al. TIM3 is a context-dependent co-regulator of cytotoxic T cell function. BioRxiv 2023. [Google Scholar] [CrossRef]
- Ambler, R.; Edmunds, G.L.; Tan, S.L.; Cirillo, S.; Pernes, J.I.; Ruan, X.; Huete-Carrasco, J.; Wong, C.C.W.; Lu, J.; Ward, J.; et al. PD-1 suppresses the maintenance of cell couples between cytotoxic T cells and target tumor cells within the tumor. Sci. Signal 2020, 13, eaau4518. [Google Scholar] [CrossRef]
- Ambler, R.; Ruan, X.; Murphy, R.F.; Wulfing, C. Systems Imaging of the Immune Synapse. Methods Mol. Biol. 2017, 1584, 409–421. [Google Scholar] [CrossRef]
- Roybal, K.T.; Mace, E.M.; Mantell, J.M.; Verkade, P.; Orange, J.S.; Wulfing, C. Early Signaling in Primary T Cells Activated by Antigen Presenting Cells Is Associated with a Deep and Transient Lamellal Actin Network. PLoS ONE 2015, 10, e0133299. [Google Scholar] [CrossRef]
- Singleton, K.L.; Roybal, K.T.; Sun, Y.; Fu, G.; Gascoigne, N.R.; van Oers, N.S.; Wülfing, C. Spatiotemporal patterning during T cell activation is highly diverse. Sci. Signal 2009, 2, ra15. [Google Scholar] [CrossRef] [PubMed]
- Cross, S.J. ModularImageAnalysis (MIA), version 1.1.1; Zenodo: Geneva, Switzerland, 2022. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, U.; Weigert, M.; Broaddus, C.; Myers, G. Cell Detection with Star-Convex Polygons. In Medical Image Computing and Computer Assisted Intervention–MICCAI 2018, Proceedings of the 21st International Conference; Granada, Spain, 16–20 September 2018, Springer International Publishing: New York, NY, USA, 2018. [Google Scholar]
- Amgad, M.; Itoh, A.; Tsui, M.M. Extending Ripley’s K-Function to Quantify Aggregation in 2-D Grayscale Images. PLoS ONE 2015, 10, e0144404. [Google Scholar] [CrossRef] [PubMed]
- Cross, S.J. ModularImageAnalysis (MIA), version 1.2.7; Zenodo: Geneve, Switzerland, 2023. [CrossRef]
- Ronneberger, O.; Fischer, P.; Brox, T. U-Net: Convolutional Networks for Biomedical Image Segmentation. In Medical Image Computing and Computer-Assisted Intervention–MICCAI 2015, Proceedings of the 18th International Conference; Munich, Germany, 5–9 October 2015, Springer International Publishing: New York, NY, USA, 2015. [Google Scholar] [CrossRef]
- Gomez-de-Mariscal, E.; Garcia-Lopez-de-Haro, C.; Ouyang, W.; Donati, L.; Lundberg, E.; Unser, M.; Munoz-Barrutia, A.; Sage, D. DeepImageJ: A user-friendly environment to run deep learning models in ImageJ. Nat. Methods 2021, 18, 1192–1195. [Google Scholar] [CrossRef]
- Legland, D.; Arganda-Carreras, I.; Andrey, P. MorphoLibJ: Integrated library and plugins for mathematical morphology with ImageJ. Bioinformatics 2016, 32, 3532–3534. [Google Scholar] [CrossRef]
- Hearst, M.A.; Dumais, S.T.; Osuna, E.; Platt, J.; Scholkopf, B. Support vector machines. IEEE Intell. Syst. Their Appl. 1998, 13, 18–28. [Google Scholar] [CrossRef]
- Otsu, N. A threshold selection method from gray-level histograms. IEEE Trans. Syst. Man Cybern. 1979, 9, 62–66. [Google Scholar] [CrossRef]
- Daubechies, I. Ten Lectures on Wavelets; SIAM: Philadelphia, PA, USA, 1992. [Google Scholar]
- Cross, S.J. Miaanalysis/Mia, version 0.21.13; Zenodo: Geneva, Switzerland, 2022. [CrossRef]
- Sauvola, J.; Pietikäinen, M. Adaptive document image binarization. Pattern Recognit. 2000, 33, 225–236. [Google Scholar] [CrossRef]
- Tinevez, J.Y.; Perry, N.; Schindelin, J.; Hoopes, G.M.; Reynolds, G.D.; Laplantine, E.; Bednarek, S.Y.; Shorte, S.L.; Eliceiri, K.W. TrackMate: An open and extensible platform for single-particle tracking. Methods 2017, 115, 80–90. [Google Scholar] [CrossRef]
- Loh, K.H.; Stawski, P.S.; Draycott, A.S.; Udeshi, N.D.; Lehrman, E.K.; Wilton, D.K.; Svinkina, T.; Deerinck, T.J.; Ellisman, M.H.; Stevens, B.; et al. Proteomic Analysis of Unbounded Cellular Compartments: Synaptic Clefts. Cell 2016, 166, 1295–1307.e21. [Google Scholar] [CrossRef]
- Rauniyar, N.; Yates, J.R., 3rd. Isobaric labeling-based relative quantification in shotgun proteomics. J. Proteome Res. 2014, 13, 5293–5309. [Google Scholar] [CrossRef]
- Phillips, A.M.; Unwin, R.D.; Hubbard, S.J.; Dowsey, A.W. Uncertainty-Aware Protein-Level Quantification and Differential Expression Analysis of Proteomics Data with seaMass. Methods Mol. Biol. 2023, 2426, 141–162. [Google Scholar] [CrossRef]
- Hadfield, J.D. MCMC Methods for Multi-Response Generalized Linear Mixed Models: The MCMCglmm R Package. J. Stat. Softw. 2010, 33, 1–22. [Google Scholar] [CrossRef]
- van der Maaten, L.; Hinton, G. Visualizing data using t-SNE. J. Mach. Learn. Res. 2008, 9, 2579–2605. [Google Scholar]
- Wagner, T. Ridge Detection; Zenodo; Geneve, Switzerland, 2017. [CrossRef]
- Arganda-Carreras, I.; Kaynig, V.; Rueden, C.; Schindelin, J.; Cardona, A.; Seung, H.S. Trainable Segmentation, v3.1.2; Zenodo: Geneva, Switzerland, 2016. [CrossRef]
- Cross, S.J. sjcross/MIA, version 0.18.15; Zenodo: Geneva, Switzerland, 2021. [CrossRef]
- Huang, L.K.; Wang, M.J. Image thresholding by minimizing the measures of fuzziness. Pattern Recognit. 1995, 28, 41–51. [Google Scholar] [CrossRef]
- Cross, S.J. sjcross/MIA_MATLAB, version 1.2.3; Zenodo: Geneva, Switzerland, 2021. [CrossRef]
- Zhang, Y.; Du, X.; Liu, M.; Tang, F.; Zhang, P.; Ai, C.; Fields, J.K.; Sundberg, E.J.; Latinovic, O.S.; Devenport, M.; et al. Hijacking antibody-induced CTLA-4 lysosomal degradation for safer and more effective cancer immunotherapy. Cell Res. 2019, 29, 609–627. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, M.; Akiba, H.; Takeda, K.; Kojima, Y.; Hashiguchi, M.; Azuma, M.; Yagita, H.; Okumura, K. Tim-3 mediates phagocytosis of apoptotic cells and cross-presentation. Blood 2009, 113, 3821–3830. [Google Scholar] [CrossRef]
- Banta, K.L.; Xu, X.; Chitre, A.S.; Au-Yeung, A.; Takahashi, C.; O’Gorman, W.E.; Wu, T.D.; Mittman, S.; Cubas, R.; Comps-Agrar, L.; et al. Mechanistic convergence of the TIGIT and PD-1 inhibitory pathways necessitates co-blockade to optimize anti-tumor CD8(+) T cell responses. Immunity 2022, 55, 512–526.e9. [Google Scholar] [CrossRef]
- Das, V.; Nal, B.; Dujeancourt, A.; Thoulouze, M.I.; Galli, T.; Roux, P.; Dautry-Varsat, A.; Alcover, A. Activation-induced polarized recycling targets T cell antigen receptors to the immunological synapse; involvement of SNARE complexes. Immunity 2004, 20, 577–588. [Google Scholar] [CrossRef]
- Alcover, A.; Alarcon, B. Internalization and intracellular fate of TCR-CD3 complexes. Crit. Rev. Immunol. 2000, 20, 325–346. [Google Scholar] [CrossRef] [PubMed]
- Pesini, C.; Hidalgo, S.; Arias, M.A.; Santiago, L.; Calvo, C.; Ocariz-Diez, M.; Isla, D.; Lanuza, P.M.; Agustin, M.J.; Galvez, E.M.; et al. PD-1 is expressed in cytotoxic granules of NK cells and rapidly mobilized to the cell membrane following recognition of tumor cells. Oncoimmunology 2022, 11, 2096359. [Google Scholar] [CrossRef] [PubMed]
- Ritter, A.T.; Asano, Y.; Stinchcombe, J.C.; Dieckmann, N.M.; Chen, B.C.; Gawden-Bone, C.; van Engelenburg, S.; Legant, W.; Gao, L.; Davidson, M.W.; et al. Actin depletion initiates events leading to granule secretion at the immunological synapse. Immunity 2015, 42, 864–876. [Google Scholar] [CrossRef]
- Tskvitaria-Fuller, I.; Rozelle, A.L.; Yin, H.L.; Wülfing, C. Regulation of sustained actin dynamics by the TCR and costimulation as a mechanism of receptor localization. J. Immunol. 2003, 171, 2287–2295. [Google Scholar] [CrossRef] [PubMed]
- Rhee, H.W.; Zou, P.; Udeshi, N.D.; Martell, J.D.; Mootha, V.K.; Carr, S.A.; Ting, A.Y. Proteomic mapping of mitochondria in living cells via spatially restricted enzymatic tagging. Science 2013, 339, 1328–1331. [Google Scholar] [CrossRef]
- Lam, S.S.; Martell, J.D.; Kamer, K.J.; Deerinck, T.J.; Ellisman, M.H.; Mootha, V.K.; Ting, A.Y. Directed evolution of APEX2 for electron microscopy and proximity labeling. Nat. Methods 2015, 12, 51–54. [Google Scholar] [CrossRef]
- Simonetti, B.; Danson, C.M.; Heesom, K.J.; Cullen, P.J. Sequence-dependent cargo recognition by SNX-BARs mediates retromer-independent transport of CI-MPR. J. Cell Biol. 2017, 216, 3695–3712. [Google Scholar] [CrossRef]
- Chen, K.E.; Healy, M.D.; Collins, B.M. Towards a molecular understanding of endosomal trafficking by Retromer and Retriever. Traffic 2019, 20, 465–478. [Google Scholar] [CrossRef]
- Vermassen, E.; Parys, J.B.; Mauger, J.P. Subcellular distribution of the inositol 1,4,5-trisphosphate receptors: Functional relevance and molecular determinants. Biol. Cell 2004, 96, 3–17. [Google Scholar] [CrossRef]
- Fendl, S.; Vieira, R.M.; Borst, A. Conditional protein tagging methods reveal highly specific subcellular distribution of ion channels in motion-sensing neurons. eLife 2020, 9, e62953. [Google Scholar] [CrossRef]
- Clark, D.J.; McMillan, L.E.; Tan, S.L.; Bellomo, G.; Massoue, C.; Thompson, H.; Mykhaylechko, L.; Alibhai, D.; Ruan, X.; Singleton, K.L.; et al. Transient protein accumulation at the center of the T cell antigen-presenting cell interface drives efficient IL-2 secretion. eLife 2019, 8, e45789. [Google Scholar] [CrossRef] [PubMed]
- Roybal, K.T.; Buck, T.E.; Ruan, X.; Cho, B.H.; Clark, D.J.; Ambler, R.; Tunbridge, H.M.; Zhang, J.; Verkade, P.; Wulfing, C.; et al. Computational spatiotemporal analysis identifies WAVE2 and cofilin as joint regulators of costimulation-mediated T cell actin dynamics. Sci. Signal 2016, 9, rs3. [Google Scholar] [CrossRef] [PubMed]
- Yost, K.E.; Satpathy, A.T.; Wells, D.K.; Qi, Y.; Wang, C.; Kageyama, R.; McNamara, K.L.; Granja, J.M.; Sarin, K.Y.; Brown, R.A.; et al. Clonal replacement of tumor-specific T cells following PD-1 blockade. Nat. Med. 2019, 25, 1251–1259. [Google Scholar] [CrossRef] [PubMed]
- Valm, A.M.; Cohen, S.; Legant, W.R.; Melunis, J.; Hershberg, U.; Wait, E.; Cohen, A.R.; Davidson, M.W.; Betzig, E.; Lippincott-Schwartz, J. Applying systems-level spectral imaging and analysis to reveal the organelle interactome. Nature 2017, 546, 162–167. [Google Scholar] [CrossRef] [PubMed]
- Loconte, V.; Singla, J.; Li, A.; Chen, J.H.; Ekman, A.; McDermott, G.; Sali, A.; Le Gros, M.; White, K.L.; Larabell, C.A. Soft X-ray tomography to map and quantify organelle interactions at the mesoscale. Structure 2022, 30, 510–521.e3. [Google Scholar] [CrossRef] [PubMed]
- Muller, A.; Schmidt, D.; Xu, C.S.; Pang, S.; D’Costa, J.V.; Kretschmar, S.; Munster, C.; Kurth, T.; Jug, F.; Weigert, M.; et al. 3D FIB-SEM reconstruction of microtubule-organelle interaction in whole primary mouse beta cells. J. Cell Biol. 2021, 220, e202010039. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, C.; Li, M.; Zhao, C.; Zheng, Y. A System-Wide Spatiotemporal Characterization of ErbB Receptor Complexes by Subcellular Fractionation Integrated Quantitative Mass Spectrometry. Anal. Chem. 2021, 93, 7933–7941. [Google Scholar] [CrossRef]
- Heinrich, L.; Bennett, D.; Ackerman, D.; Park, W.; Bogovic, J.; Eckstein, N.; Petruncio, A.; Clements, J.; Pang, S.; Xu, C.S.; et al. Whole-cell organelle segmentation in volume electron microscopy. Nature 2021, 599, 141–146. [Google Scholar] [CrossRef]
- Huang, Y.; Yin, H.; Li, B.; Wu, Q.; Liu, Y.; Poljak, K.; Maldutyte, J.; Tang, X.; Wang, M.; Wu, Z.; et al. An in vitro vesicle formation assay reveals cargo clients and factors that mediate vesicular trafficking. Proc. Natl. Acad. Sci. USA 2021, 118, e2101287118. [Google Scholar] [CrossRef]
- Liberali, P.; Snijder, B.; Pelkmans, L. A hierarchical map of regulatory genetic interactions in membrane trafficking. Cell 2014, 157, 1473–1487. [Google Scholar] [CrossRef]
- Altman, J.B.; Taft, J.; Wedeking, T.; Gruber, C.N.; Holtmannspotter, M.; Piehler, J.; Bogunovic, D. Type I IFN is siloed in endosomes. Proc. Natl. Acad. Sci. USA 2020, 117, 17510–17512. [Google Scholar] [CrossRef] [PubMed]
- Weng, L.; Enomoto, A.; Miyoshi, H.; Takahashi, K.; Asai, N.; Morone, N.; Jiang, P.; An, J.; Kato, T.; Kuroda, K.; et al. Regulation of cargo-selective endocytosis by dynamin 2 GTPase-activating protein girdin. EMBO J. 2014, 33, 2098–2112. [Google Scholar] [CrossRef] [PubMed]
- Grossier, J.P.; Xouri, G.; Goud, B.; Schauer, K. Cell adhesion defines the topology of endocytosis and signaling. EMBO J. 2014, 33, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Monks, C.R.; Freiberg, B.A.; Kupfer, H.; Sciaky, N.; Kupfer, A. Three-dimensional segregation of supramolecular activation clusters in T cells. Nature 1998, 395, 82–86. [Google Scholar] [CrossRef] [PubMed]
- Lakadamyali, M.; Rust, M.J.; Zhuang, X. Ligands for clathrin-mediated endocytosis are differentially sorted into distinct populations of early endosomes. Cell 2006, 124, 997–1009. [Google Scholar] [CrossRef]
- Varandas, K.C.; Irannejad, R.; von Zastrow, M. Retromer Endosome Exit Domains Serve Multiple Trafficking Destinations and Regulate Local G Protein Activation by GPCRs. Curr. Biol. 2016, 26, 3129–3142. [Google Scholar] [CrossRef]
- Zhai, Y.; Celis-Gutierrez, J.; Voisinne, G.; Mori, D.; Girard, L.; Burlet-Schiltz, O.; de Peredo, A.G.; Roncagalli, R.; Malissen, B. Opposing regulatory functions of the TIM3 (HAVCR2) signalosome in primary effector T cells as revealed by quantitative interactomics. Cell Mol. Immunol. 2021, 18, 1581–1583. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, J.; Veler, A.; Simonetti, B.; Raj, T.; Chou, P.H.; Cross, S.J.; Phillips, A.M.; Ruan, X.; Huynh, L.; Dowsey, A.W.; et al. Five Inhibitory Receptors Display Distinct Vesicular Distributions in Murine T Cells. Cells 2023, 12, 2558. https://doi.org/10.3390/cells12212558
Lu J, Veler A, Simonetti B, Raj T, Chou PH, Cross SJ, Phillips AM, Ruan X, Huynh L, Dowsey AW, et al. Five Inhibitory Receptors Display Distinct Vesicular Distributions in Murine T Cells. Cells. 2023; 12(21):2558. https://doi.org/10.3390/cells12212558
Chicago/Turabian StyleLu, Jiahe, Alisa Veler, Boris Simonetti, Timsse Raj, Po Han Chou, Stephen J. Cross, Alexander M. Phillips, Xiongtao Ruan, Lan Huynh, Andrew W. Dowsey, and et al. 2023. "Five Inhibitory Receptors Display Distinct Vesicular Distributions in Murine T Cells" Cells 12, no. 21: 2558. https://doi.org/10.3390/cells12212558