
LRRK2 Attenuates Antioxidant Response in Familial Parkinson’s Disease Derived Neural Stem Cells

Abstract
: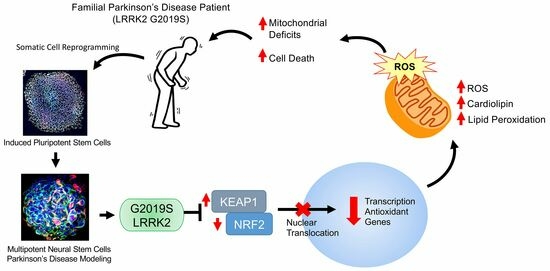
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Induced Pluripotent Stem Cell (iPSC) Cultures
2.2. Isolation of NSCs from iPSCs
2.3. Differentiation of NSCs into Dopaminergic Neurons
2.4. Drug Testing Assay Compounds
2.5. Western Blotting
2.6. Gene Expression Analysis
2.7. Immunocytochemistry
2.8. Single Cell RNA-Sequencing Pipeline
2.9. Measuring Superoxide Content with MitoSOX Red Staining
2.10. Electron Paramagnetic Resonance Spectroscopy
2.11. Measuring Cardiolipin Content
2.12. Measuring Lipid Peroxidation
2.13. Seahorse Extracellular Flux Analyzer: Mito Stress Assay
2.14. Measuring Intracellular Glutathione Concentration
2.15. Statistical Analysis
3. Results
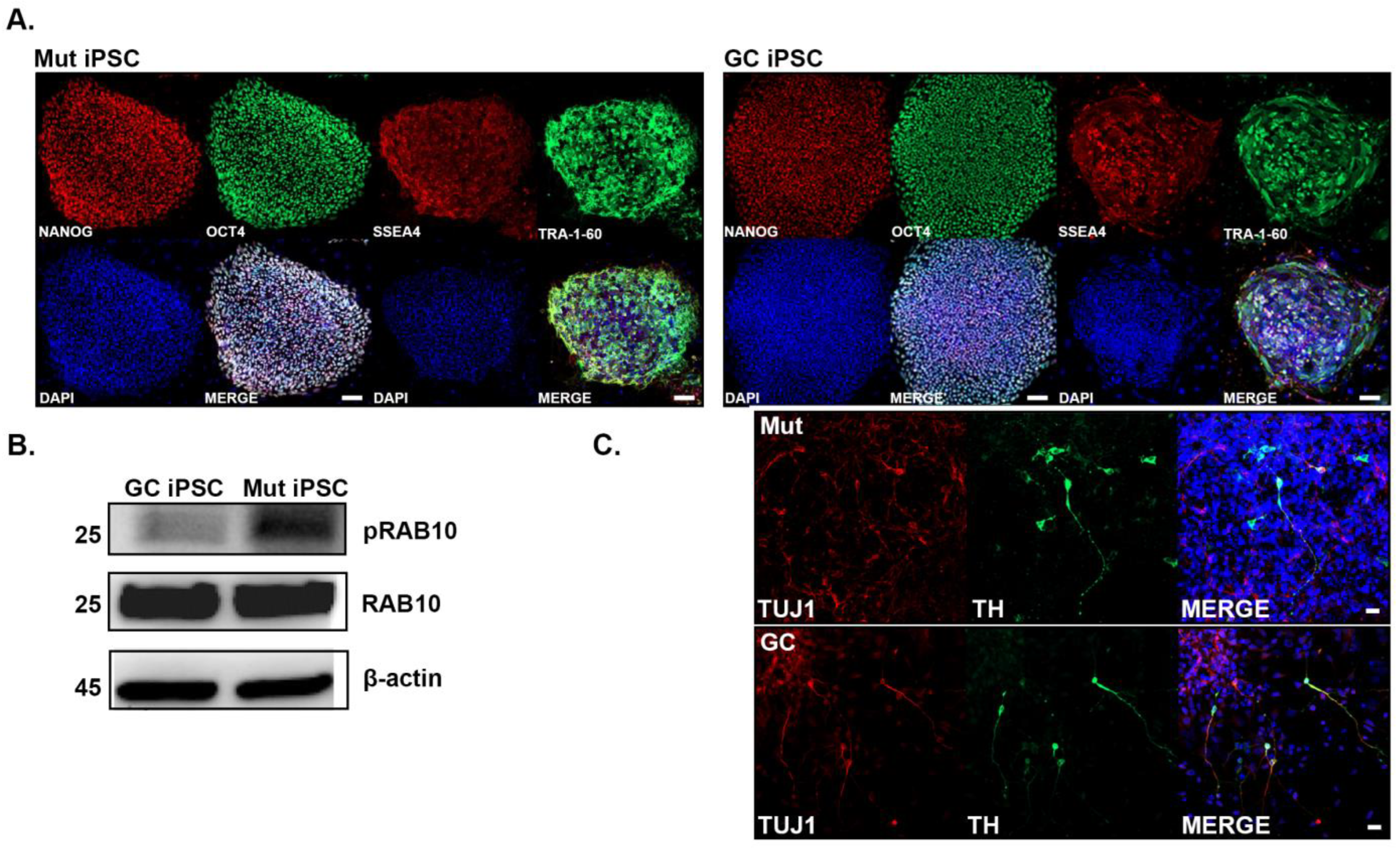
3.1. Characterization of iPSCs Derived from G2019S LRRK2 PD Patient
3.2. Single-Cell Transcriptomic Analysis of Mut iPSCs and GC iPSCs Revealed Defect in the NRF2-Mediated Oxidative Stress Response among Other Pathways
3.3. LRRK2 G2019S Cells Exhibit Increased Basal Oxidative Stress
3.4. Mitochondrial Deficits in G2019S LRRK2 Cells
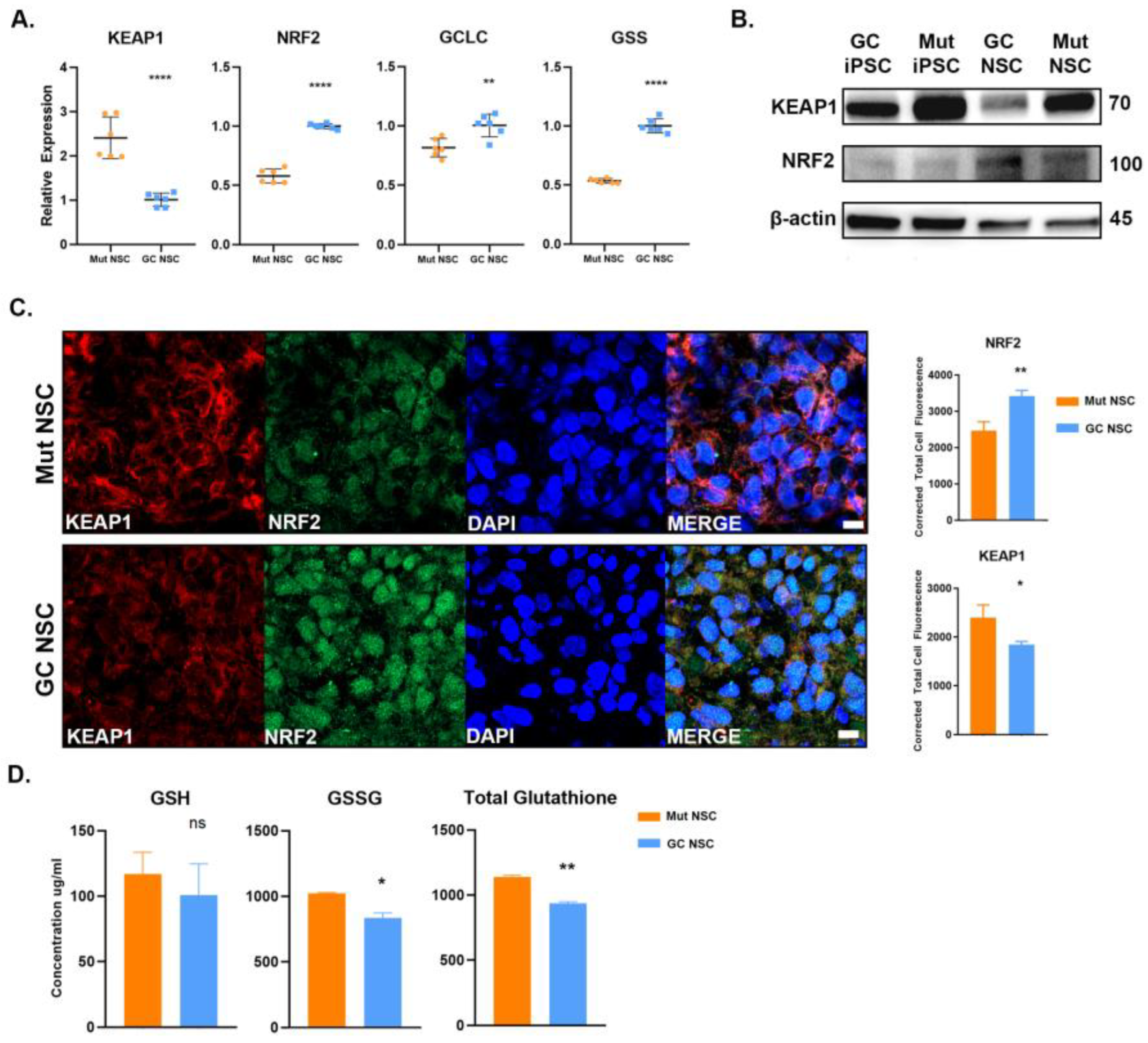
3.5. Antioxidant Response Is Perturbed in G2019S LRRK2 Cells
3.6. Antioxidant Effects of PRC-210 and Edaravone on the LRRK2 Compromised Oxidative Stress Response
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Braak, H.; Braak, E.; Yilmazer, D.; Schultz, C.; de Vos, R.A.; Jansen, E.N. Nigral and extranigral pathology in Parkinson’s disease. J. Neural Transm. Suppl. 1995, 46, 15–31. [Google Scholar]
- Jellinger, K.A. Pathology of Parkinson’s disease. Changes other than the nigrostriatal pathway. Mol. Chem. Neuropathol. 1991, 14, 153–197. [Google Scholar] [CrossRef] [PubMed]
- Emre, M.; Aarsland, D.; Brown, R.; Burn, D.J.; Duyckaerts, C.; Mizuno, Y.; Broe, G.A.; Cummings, J.; Dickson, D.W.; Gauthier, S.; et al. Clinical diagnostic criteria for dementia associated with Parkinson’s disease. Mov. Disord. 2007, 22, 1689–1707. [Google Scholar] [CrossRef] [PubMed]
- Gelb, D.J.; Oliver, E.; Gilman, S. Diagnostic criteria for Parkinson Disease. Arch. Neurol. 1999, 56, 7. [Google Scholar] [CrossRef]
- Hughes, A.J.; Daniel, S.E.; Kilford, L.; Lees, A.J. Accuracy of clinical diagnosis of idiopathic Parkinson’s disease: A clinico-pathological study of 100 cases. J. Neurol. Neurosurg. Psychiatry 1992, 55, 181–184. [Google Scholar] [CrossRef]
- Jankovic, J. Parkinson’s disease: Clinical features and diagnosis. J. Neurol. Neurosurg. Psychiatry 2008, 79, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Remy, P.; Doder, M.; Lees, A.; Turjanski, N.; Brooks, D. Depression in Parkinson’s disease: Loss of dopamine and noradrenaline innervation in the limbic system. Brain 2005, 128, 1314–1322. [Google Scholar] [CrossRef] [PubMed]
- de Lau, L.M.; Breteler, M.M. Epidemiology of Parkinson’s disease. Lancet Neurol. 2006, 5, 525–535. [Google Scholar] [CrossRef]
- Gilks, W.P.; Abou-Sleiman, P.M.; Gandhi, S.; Jain, S.; Singleton, A.; Lees, A.J.; Shaw, K.; Bhatia, K.P.; Bonifati, V.; Quinn, N.P.; et al. A common LRRK2 mutation in idiopathic Parkinson’s disease. Lancet 2005, 365, 415–416. [Google Scholar] [CrossRef] [PubMed]
- Healy, D.G.; Falchi, M.; O’Sullivan, S.S.; Bonifati, V.; Durr, A.; Bressman, S.; Brice, A.; Aasly, J.; Zabetian, C.P.; Goldwurm, S.; et al. Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: A case-control study. Lancet Neurol. 2008, 7, 583–590. [Google Scholar] [CrossRef]
- Paisan-Ruiz, C.; Jain, S.; Evans, E.W.; Gilks, W.P.; Simon, J.; van der Brug, M.; Lopez de Munain, A.; Aparicio, S.; Gil, A.M.; Khan, N.; et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron 2004, 44, 595–600. [Google Scholar] [CrossRef]
- Zimprich, A.; Biskup, S.; Leitner, P.; Lichtner, P.; Farrer, M.; Lincoln, S.; Kachergus, J.; Hulihan, M.; Uitti, R.J.; Calne, D.B.; et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 2004, 44, 601–607. [Google Scholar] [CrossRef] [PubMed]
- Cookson, M.R. The role of leucine-rich repeat kinase 2 (LRRK2) in Parkinson’s disease. Nat. Rev. Neurosci. 2010, 11, 791–797. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.C.-y.; Rideout, H.J.; Ribe, E.; Troy, C.M.; Dauer, W.T. The Parkinson Disease Protein LRRK2 Transduces Death Signals Via FADD and Caspase-8 in a Cellular Model of Neurodegeneration. J. Neurosci. 2009, 29, 1011–1016. [Google Scholar] [CrossRef]
- Imai, Y.; Gehrke, S.; Wang, H.Q.; Takahashi, R.; Hasegawa, K.; Oota, E.; Lu, B. Phosphorylation of 4E-BP by LRRK2 affects the maintenance of dopaminergic neurons in Drosophila. EMBO J. 2008, 27, 2432–2443. [Google Scholar] [CrossRef]
- Kanao, T.; Venderova, K.; Park, D.S.; Unterman, T.; Lu, B.; Imai, Y. Activation of FoxO by LRRK2 induces expression of proapoptotic proteins and alters survival of postmitotic dopaminergic neuron in Drosophila. Hum. Mol. Genet. 2010, 19, 3747–3758. [Google Scholar] [CrossRef] [PubMed]
- Migheli, R.; Del Giudice, M.G.; Spissu, Y.; Sanna, G.; Xiong, Y.; Dawson, T.M.; Dawson, V.L.; Galioto, M.; Rocchitta, G.; Biosa, A.; et al. LRRK2 Affects Vesicle Trafficking, Neurotransmitter Extracellular Level and Membrane Receptor Localization. PLoS ONE 2013, 8, e77198. [Google Scholar] [CrossRef]
- Smith, W.W.; Pei, Z.; Jiang, H.; Moore, D.J.; Liang, Y.; West, A.B.; Dawson, V.L.; Dawson, T.M.; Ross, C.A. Leucine-rich repeat kinase 2 (LRRK2) interacts with parkin, and mutant LRRK2 induces neuronal degeneration. PNAS 2005, 102, 18676–18681. [Google Scholar] [CrossRef]
- Dachsel, J.C.; Farrer, M.J. LRRK2 and Parkinson disease. Arch. Neurol. 2010, 67, 542–547. [Google Scholar] [CrossRef]
- Lesage, S.; Durr, A.; Tazir, M.; Lohmann, E.; Leutenegger, A.L.; Janin, S.; Pollak, P.; Brice, A.; French Parkinson’s Disease Genetics Study, G. LRRK2 G2019S as a cause of Parkinson’s disease in North African Arabs. N. Engl. J. Med. 2006, 354, 422–423. [Google Scholar] [CrossRef]
- Ozelius, L.J.; Senthil, G.; Saunders-Pullman, R.; Ohmann, E.; Deligtisch, A.; Tagliati, M.; Hunt, A.L.; Klein, C.; Henick, B.; Hailpern, S.M.; et al. LRRK2 G2019S as a cause of Parkinson’s disease in Ashkenazi Jews. N. Engl. J. Med. 2006, 354, 424–425. [Google Scholar] [CrossRef]
- Fung, H.C.; Chen, C.M.; Hardy, J.; Hernandez, D.; Singleton, A.; Wu, Y.R. Lack of G2019S LRRK2 mutation in a cohort of Taiwanese with sporadic Parkinson’s disease. Mov. Disord. 2006, 21, 880–881. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, P.N.; Chen, S.G.; Wilson-Delfosse, A.L. Leucine-rich repeat kinase 2 (LRRK2): A key player in the pathogenesis of Parkinson’s disease. J. Neurosci. Res. 2009, 87, 1283–1295. [Google Scholar] [CrossRef] [PubMed]
- Kalinderi, K.; Fidani, L.; Bostantjopoulou, S.; Katsarou, Z.; Kotsis, A. The G2019S LRRK2 mutation is uncommon amongst Greek patients with sporadic Parkinson’s disease. Eur. J. Neurol. 2007, 14, 1088–1090. [Google Scholar] [CrossRef]
- Tan, E.K.; Shen, H.; Tan, L.C.; Farrer, M.; Yew, K.; Chua, E.; Jamora, R.D.; Puvan, K.; Puong, K.Y.; Zhao, Y.; et al. The G2019S LRRK2 mutation is uncommon in an Asian cohort of Parkinson’s disease patients. Neurosci. Lett. 2005, 384, 327–329. [Google Scholar] [CrossRef] [PubMed]
- Tan, E.K.; Zhao, Y.; Skipper, L.; Tan, M.G.; Di Fonzo, A.; Sun, L.; Fook-Chong, S.; Tang, S.; Chua, E.; Yuen, Y.; et al. The LRRK2 Gly2385Arg variant is associated with Parkinson’s disease: Genetic and functional evidence. Hum. Genet. 2007, 120, 857–863. [Google Scholar] [CrossRef]
- Tomiyama, H.; Li, Y.; Funayama, M.; Hasegawa, K.; Yoshino, H.; Kubo, S.; Sato, K.; Hattori, T.; Lu, C.S.; Inzelberg, R.; et al. Clinicogenetic study of mutations in LRRK2 exon 41 in Parkinson’s disease patients from 18 countries. Mov. Disord. 2006, 21, 1102–1108. [Google Scholar] [CrossRef]
- Greggio, E.; Jain, S.; Kingsbury, A.; Bandopadhyay, R.; Lewis, P.; Kaganovich, A.; van der Brug, M.P.; Beilina, A.; Blackinton, J.; Thomas, K.J.; et al. Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol. Dis. 2006, 23, 329–341. [Google Scholar] [CrossRef]
- Luzón-Toro, B.; de la Torre, E.R.; Delgado, A.; Pérez-Tur, J.; Hilfiker, S. Mechanistic insight into the dominant mode of the Parkinson’s disease-associated G2019S LRRK2 mutation. Hum. Mol. Genet. 2007, 16, 2031–2039. [Google Scholar] [CrossRef]
- Smith, W.W.; Pei, Z.; Jiang, H.; Dawson, V.L.; Dawson, T.M.; Ross, C.A. Kinase activity of mutant LRRK2 mediates neuronal toxicity. Nat. Neurosci. 2006, 9, 1231–1233. [Google Scholar] [CrossRef]
- West, A.B.; Moore, D.J.; Biskup, S.; Bugayenko, A.; Smith, W.W.; Ross, C.A.; Dawson, V.L.; Dawson, T.M. Parkinson’s disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc. Natl. Acad. Sci. USA 2005, 102, 16842–16847. [Google Scholar] [CrossRef]
- Beilina, A.; Rudenko, I.N.; Kaganovich, A.; Civiero, L.; Chau, H.; Kalia, S.K.; Kalia, L.V.; Lobbestael, E.; Chia, R.; Ndukwe, K.; et al. Unbiased screen for interactors of leucine-rich repeat kinase 2 supports a common pathway for sporadic and familial Parkinson disease. Proc. Natl. Acad. Sci. USA 2014, 111, 2626–2631. [Google Scholar] [CrossRef] [PubMed]
- Marras, C.; Alcalay, R.N.; Caspell-Garcia, C.; Coffey, C.; Chan, P.; Duda, J.E.; Facheris, M.F.; Fernandez-Santiago, R.; Ruiz-Martinez, J.; Mestre, T.; et al. Motor and nonmotor heterogeneity of LRRK2-related and idiopathic Parkinson’s disease. Mov. Disord. 2016, 31, 1192–1202. [Google Scholar] [CrossRef] [PubMed]
- Klein, C.; Westenberger, A. Genetics of Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a008888. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Sances, S.; Workman, M.J.; Svendsen, C.N. Multi-lineage Human iPSC-Derived Platforms for Disease Modeling and Drug Discovery. Cell Stem Cell 2020, 26, 309–329. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Daadi, E.W.; Oh, T.; Daadi, E.S.; Daadi, M.M. Human Induced Pluripotent Stem Cell Phenotyping and Preclinical Modeling of Familial Parkinson’s Disease. Genes. 2022, 13, 1937. [Google Scholar] [CrossRef]
- Kim, J.; Daadi, M.M. Non-cell autonomous mechanism of Parkinson’s disease pathology caused by G2019S LRRK2 mutation in Ashkenazi Jewish patient: Single cell analysis. Brain Res. 2019, 1722, 146342. [Google Scholar] [CrossRef]
- Reinhardt, P.; Schmid, B.; Burbulla, L.F.; Schondorf, D.C.; Wagner, L.; Glatza, M.; Hoing, S.; Hargus, G.; Heck, S.A.; Dhingra, A.; et al. Genetic correction of a LRRK2 mutation in human iPSCs links parkinsonian neurodegeneration to ERK-dependent changes in gene expression. Cell Stem Cell 2013, 12, 354–367. [Google Scholar] [CrossRef]
- Daadi, M.M. Generation of Neural Stem Cells from Induced Pluripotent Stem Cells. Methods Mol. Biol. 2019, 1919, 1–7. [Google Scholar] [CrossRef]
- Daadi, M.M. Differentiation of Neural Stem Cells Derived from Induced Pluripotent Stem Cells into Dopaminergic Neurons. Methods Mol. Biol. 2019, 1919, 89–96. [Google Scholar] [CrossRef]
- Sahni, S.K.; Rydkina, E.; Sahni, A. The proteasome inhibitor MG132 induces nuclear translocation of erythroid transcription factor Nrf2 and cyclooxygenase-2 expression in human vascular endothelial cells. Thromb. Res. 2008, 122, 820–825. [Google Scholar] [CrossRef]
- McCloy, R.A.; Rogers, S.; Caldon, C.E.; Lorca, T.; Castro, A.; Burgess, A. Partial inhibition of Cdk1 in G 2 phase overrides the SAC and decouples mitotic events. Cell Cycle 2014, 13, 1400–1412. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Daadi, M.M. Single-Cell Library Preparation of iPSC-Derived Neural Stem Cells. Methods Mol. Biol. 2019, 1919, 129–143. [Google Scholar] [CrossRef]
- Kim, J.; Daadi, M.M. Bioinformatics Analysis of Single-Cell RNA-Seq Raw Data from iPSC-Derived Neural Stem Cells. Methods Mol. Biol. 2019, 1919, 145–159. [Google Scholar] [CrossRef]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef] [PubMed]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef]
- Zerbino, D.R.; Achuthan, P.; Akanni, W.; Amode, M.R.; Barrell, D.; Bhai, J.; Billis, K.; Cummins, C.; Gall, A.; Giron, C.G.; et al. Ensembl 2018. Nucleic Acids Res 2018, 46, D754–D761. [Google Scholar] [CrossRef] [PubMed]
- Pimentel, H.; Bray, N.L.; Puente, S.; Melsted, P.; Pachter, L. Differential analysis of RNA-seq incorporating quantification uncertainty. Nat. Methods 2017, 14, 687–690. [Google Scholar] [CrossRef]
- Huang da, W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2008, 4, 44–57. [Google Scholar] [CrossRef]
- Ito, G.; Katsemonova, K.; Tonelli, F.; Lis, P.; Baptista, M.A.; Shpiro, N.; Duddy, G.; Wilson, S.; Ho, P.W.; Ho, S.L.; et al. Phos-tag analysis of Rab10 phosphorylation by LRRK2: A powerful assay for assessing kinase function and inhibitors. Biochem. J. 2016, 473, 2671–2685. [Google Scholar] [CrossRef]
- Wu, S.; Zhou, F.; Zhang, Z.; Xing, D. Mitochondrial oxidative stress causes mitochondrial fragmentation via differential modulation of mitochondrial fission-fusion proteins. FEBS J. 2011, 278, 941–954. [Google Scholar] [CrossRef]
- Paradies, G.; Petrosillo, G.; Paradies, V.; Ruggiero, F.M. Role of cardiolipin peroxidation and Ca2+ in mitochondrial dysfunction and disease. Cell Calcium 2009, 45, 643–650. [Google Scholar] [CrossRef]
- Liu, Z.; Wang, X.; Yu, Y.; Li, X.; Wang, T.; Jiang, H.; Ren, Q.; Jiao, Y.; Sawa, A.; Moran, T.; et al. A Drosophila model for LRRK2-linked parkinsonism. Proc. Natl. Acad. Sci. USA 2008, 105, 2693–2698. [Google Scholar] [CrossRef]
- Jermusek, F., Jr.; Benedict, C.; Dreischmeier, E.; Brand, M.; Uder, M.; Jeffery, J.J.; Ranallo, F.N.; Fahl, W.E. Significant Suppression of CT Radiation-Induced DNA Damage in Normal Human Cells by the PrC-210 Radioprotector. Radiat. Res. 2018, 190, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Sison, S.L.; Vermilyea, S.C.; Emborg, M.E.; Ebert, A.D. Using Patient-Derived Induced Pluripotent Stem Cells to Identify Parkinson’s Disease-Relevant Phenotypes. Curr. Neurol. Neurosci. Rep. 2018, 18, 84. [Google Scholar] [CrossRef]
- Walter, J.; Bolognin, S.; Antony, P.M.A.; Nickels, S.L.; Poovathingal, S.K.; Salamanca, L.; Magni, S.; Perfeito, R.; Hoel, F.; Qing, X.; et al. Neural Stem Cells of Parkinson’s Disease Patients Exhibit Aberrant Mitochondrial Morphology and Functionality. Stem Cell Rep. 2019, 12, 878–889. [Google Scholar] [CrossRef] [PubMed]
- Ebadi, M.; Srinivasan, S.K.; Baxi, M.D. Oxidative stress and antioxidant therapy in Parkinson’s disease. Prog. Neurobiol. 1996, 48, 1–19. [Google Scholar] [CrossRef] [PubMed]
- De Lazzari, F.; Sandrelli, F.; Whitworth, A.J.; Bisaglia, M. Antioxidant Therapy in Parkinson’s Disease: Insights from Drosophila melanogaster. Antioxidants 2020, 9, 52. [Google Scholar] [CrossRef]
- Oyewole, A.O.; Birch-Machin, M.A. Mitochondria-targeted antioxidants. FASEB J. 2015, 29, 4766–4771. [Google Scholar] [CrossRef]
- Sian, J.; Dexter, D.T.; Lees, A.J.; Daniel, S.; Agid, Y.; Javoy-Agid, F.; Jenner, P.; Marsden, C.D. Alterations in glutathione levels in Parkinson’s disease and other neurodegenerative disorders affecting basal ganglia. Ann. Neurol. 1994, 36, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Sian, J.; Dexter, D.T.; Lees, A.J.; Daniel, S.; Jenner, P.; Marsden, C.D. Glutathione-related enzymes in brain in Parkinson’s disease. Ann. Neurol. 1994, 36, 356–361. [Google Scholar] [CrossRef] [PubMed]
- Sofic, E.; Lange, K.W.; Jellinger, K.; Riederer, P. Reduced and oxidized glutathione in the substantia nigra of patients with Parkinson’s disease. Neurosci. Lett. 1992, 142, 128–130. [Google Scholar] [CrossRef]
- Uhlig, S.; Wendel, A. The physiological consequences of glutathione variations. Life Sci. 1992, 51, 1083–1094. [Google Scholar] [CrossRef] [PubMed]
- Park, H.A.; Khanna, S.; Rink, C.; Gnyawali, S.; Roy, S.; Sen, C.K. Glutathione disulfide induces neural cell death via a 12-lipoxygenase pathway. Cell Death Differ. 2009, 16, 1167–1179. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.; Daadi, E.W.; Daadi, E.S.; Oh, T.; Deleidi, M.; Daadi, M.M. LRRK2 Attenuates Antioxidant Response in Familial Parkinson’s Disease Derived Neural Stem Cells. Cells 2023, 12, 2550. https://doi.org/10.3390/cells12212550
Kim J, Daadi EW, Daadi ES, Oh T, Deleidi M, Daadi MM. LRRK2 Attenuates Antioxidant Response in Familial Parkinson’s Disease Derived Neural Stem Cells. Cells. 2023; 12(21):2550. https://doi.org/10.3390/cells12212550
Chicago/Turabian StyleKim, Jeffrey, Etienne W. Daadi, Elyas Sebastien Daadi, Thomas Oh, Michela Deleidi, and Marcel M. Daadi. 2023. "LRRK2 Attenuates Antioxidant Response in Familial Parkinson’s Disease Derived Neural Stem Cells" Cells 12, no. 21: 2550. https://doi.org/10.3390/cells12212550