
Identification of Small Molecules for Prevention of Lens Epithelium-Derived Cataract Using Zebrafish

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Fish Lines
2.2. Small Molecule Screen and Treatments
2.3. Histology and Immunohistochemistry
2.4. Imaging
2.5. Data Quantification and Statistical Analyses
3. Results
3.1. Evidence for Rough Endoplasmic Reticulum Abnormalities in Lens Epithelium and Presumptive Cornea of plod3 Mutants
3.2. 4-PBA Rescues the Lens Phenotype of plod3vu222 Mutants
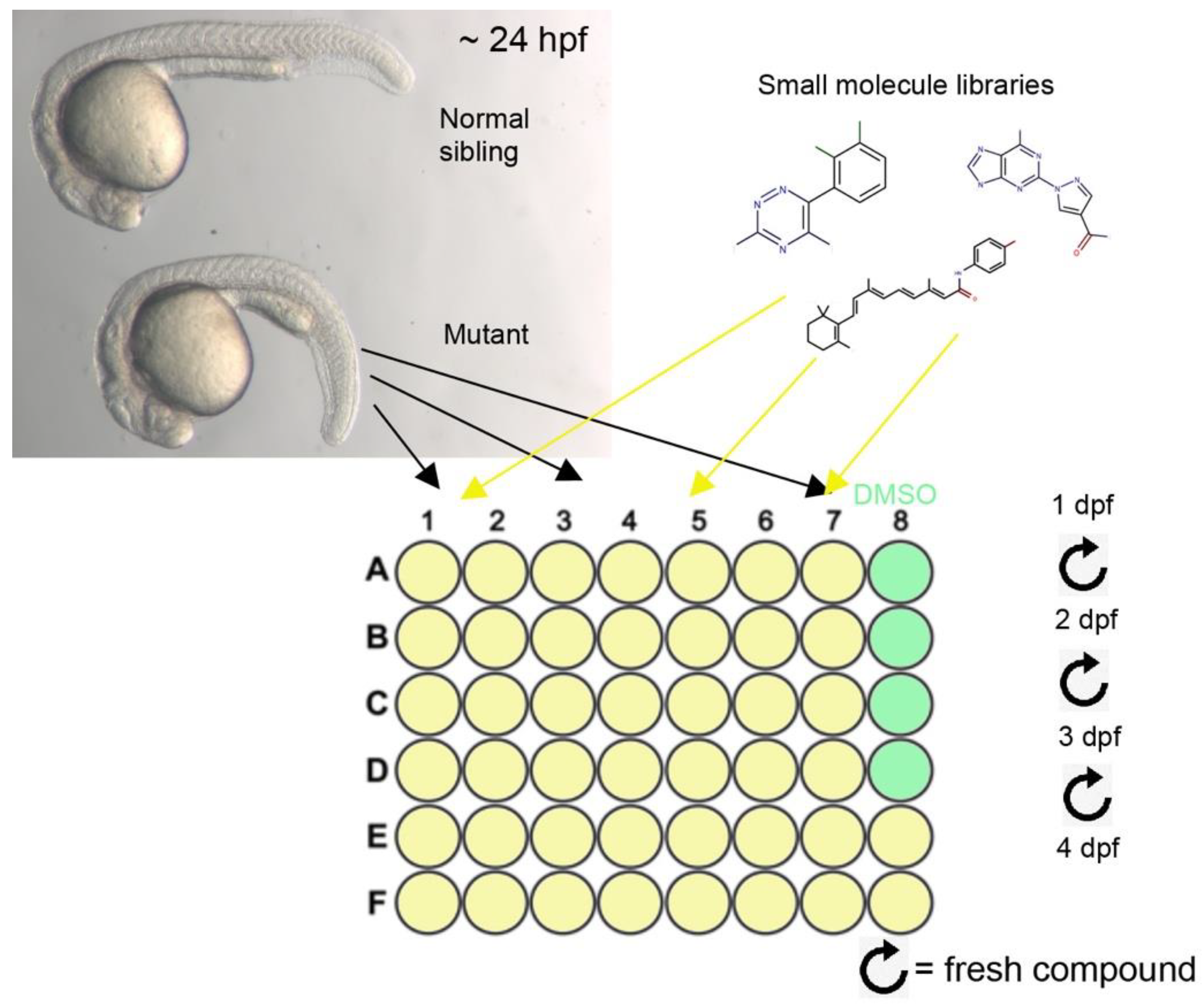
3.3. A Small Molecule Screen for Compounds That Can Rescue the Lens Phenotype of Lh3 Deficiency
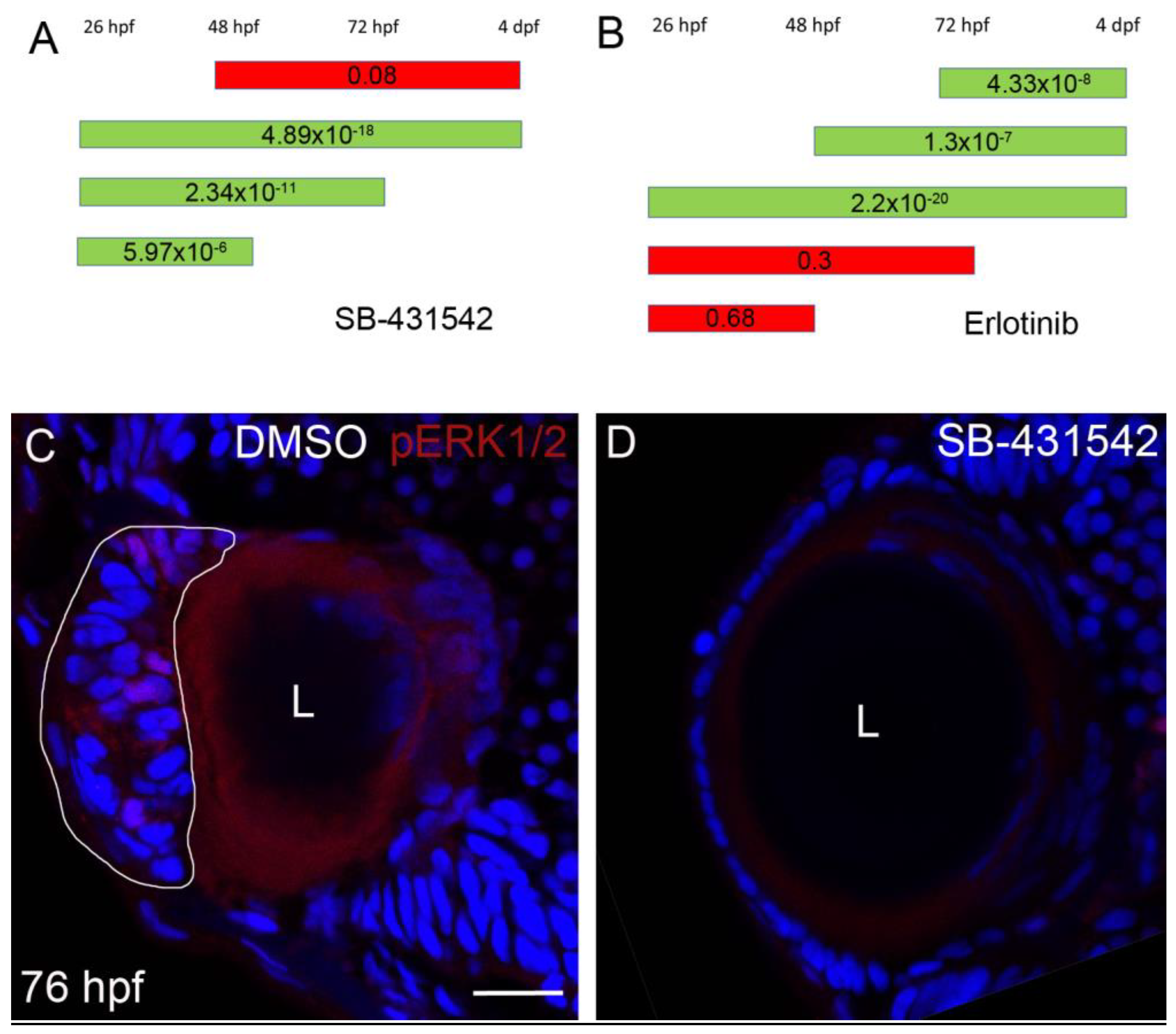
3.4. Suppression of plod3 Mutant Lens Phenotype by Erlotinib
3.5. TGFβ Signaling Functions Upstream of EGFR Signaling in Cell Mass Formation
3.6. 4-PBA Inhibits Only Late TGFβ Signaling and Does Not Inhibit EGFR Signaling
3.7. Inhibition of mTOR Signaling Only Partially Limits Cell Mass Formation
4. Discussion
4.1. Inhibition of Cell Mass Formation by 4-PBA
4.2. Inhibition of Cell Mass Formation by Blocking TGFβ and EGFR Signaling
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- GBD 2019 Blindness and Vision Impairment Collaborators. Causes of Blindness and Vision Impairment in 2020 and Trends over 30 Years, and Prevalence of Avoidable Blindness in Relation to VISION 2020: The Right to Sight: An Analysis for the Global Burden of Disease Study. Lancet Glob. Health 2021, 9, e144–e160. [Google Scholar] [CrossRef]
- Liu, Y.C.; Wilkins, M.; Kim, T.; Malyugin, B.; Mehta, J.S. Cataracts. Lancet 2017, 390, 600–612. [Google Scholar] [CrossRef] [PubMed]
- Wormstone, I.M.; Wang, L.; Liu, C.S. Posterior Capsule Opacification. Exp. Eye Res. 2009, 88, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Saika, S. Relationship between Posterior Capsule Opacification and Intraocular Lens Biocompatibility. Prog. Retin. Eye Res. 2004, 23, 283–305. [Google Scholar] [CrossRef] [PubMed]
- Findl, O.; Buehl, W.; Bauer, P.; Sycha, T. Interventions for Preventing Posterior Capsule Opacification. Cochrane Database Syst. Rev. 2010, CD003738. [Google Scholar] [CrossRef]
- Cheng, C.-Y.; Yen, M.-Y.; Chen, S.-J.; Kao, S.-C.; Hsu, W.-M.; Liu, J.-H. Visual Acuity and Contrast Sensitivity in Different Types of Posterior Capsule Opacification. J. Cataract. Refract. Surg. 2001, 27, 1055–1060. [Google Scholar] [CrossRef]
- Van Bree, M.C.J.; Van Der Meulen, I.J.E.; Franssen, L.; Coppens, J.E.; Reus, N.J.; Zijlmans, B.L.M.; Van Den Berg, T.J.T.P. Imaging of Forward Light-Scatter by Opacified Posterior Capsules Isolated from Pseudophakic Donor Eyes. Investig. Opthalmol. Vis. Sci. 2011, 52, 5587. [Google Scholar] [CrossRef]
- Javitt, J.C.; Tielsch, J.M.; Canner, J.K.; Kolb, M.M.; Sommer, A.; Steinberg, E.P. National Outcomes of Cataract Extraction. Increased Risk of Retinal Complications Associated with Nd:YAG Laser Capsulotomy. The Cataract Patient Outcomes Research Team. Ophthalmology 1992, 99, 1487–1497. [Google Scholar] [CrossRef]
- Pandey, S.K.; Apple, D.J.; Werner, L.; Maloof, A.J.; Milverton, E.J. Posterior Capsule Opacification: A Review of the Aetiopathogenesis, Experimental and Clinical Studies and Factors for Prevention. Indian J. Ophthalmol. 2004, 52, 99–112. [Google Scholar]
- West-Mays, J.A.; Pino, G.; Lovicu, F.J. Development and Use of the Lens Epithelial Explant System to Study Lens Differentiation and Cataractogenesis. Prog. Retin. Eye Res. 2010, 29, 135–143. [Google Scholar] [CrossRef]
- Wormstone, I.M.; Wormstone, Y.M.; Smith, A.J.O.; Eldred, J.A. Posterior Capsule Opacification: What’s in the Bag? Prog. Retin. Eye Res. 2021, 82, 100905. [Google Scholar] [CrossRef]
- Srinivasan, Y.; Lovicu, F.J.; Overbeek, P.A. Lens-Specific Expression of Transforming Growth Factor Beta1 in Transgenic Mice Causes Anterior Subcapsular Cataracts. J. Clin. Invest. 1998, 101, 625–634. [Google Scholar] [CrossRef]
- Taler, K.; Weiss, O.; Rotem-Bamberger, S.; Rubinstein, A.M.; Seritrakul, P.; Gross, J.M.; Inbal, A. Lysyl Hydroxylase 3 Is Required for Normal Lens Capsule Formation and Maintenance of Lens Epithelium Integrity and Fate. Dev. Biol. 2020, 458, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Salo, A.M.; Cox, H.; Farndon, P.; Moss, C.; Grindulis, H.; Risteli, M.; Robins, S.P.; Myllyla, R. A Connective Tissue Disorder Caused by Mutations of the Lysyl Hydroxylase 3 Gene. Am. J. Hum. Genet. 2008, 83, 495–503. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Hales, A.M.; Chamberlain, C.G.; McAvoy, J.W. Induction of Cataract-like Changes in Rat Lens Epithelial Explants by Transforming Growth Factor Beta. Invest. Ophthalmol. Vis. Sci. 1994, 35, 388–401. [Google Scholar] [PubMed]
- Hales, A.M.; Chamberlain, C.G.; McAvoy, J.W. Cataract Induction in Lenses Cultured with Transforming Growth Factor-Beta. Invest. Ophthalmol. Vis. Sci. 1995, 36, 1709–1713. [Google Scholar]
- Lovicu, F.J.; Steven, P.; Saika, S.; McAvoy, J.W. Aberrant Lens Fiber Differentiation in Anterior Subcapsular Cataract Formation: A Process Dependent on Reduced Levels of Pax6. Invest. Ophthalmol. Vis. Sci. 2004, 45, 1946–1953. [Google Scholar] [CrossRef]
- Schneider, V.A.; Granato, M. The Myotomal Diwanka (Lh3) Glycosyltransferase and Type XVIII Collagen Are Critical for Motor Growth Cone Migration. Neuron 2006, 50, 683–695. [Google Scholar] [CrossRef]
- Westerfield, M. The Zebrafish Book. A Guide for the Laboratory Use of Zebrafish (Danio Rerio), 4th ed.; University of Oregon Press: Eugene, OR, USA, 2000. [Google Scholar]
- Humphrey, C.D.; Pittman, F.E. A Simple Methylene Blue-Azure II-Basic Fuchsin Stain for Epoxy-Embedded Tissue Sections. Stain. Technol. 1974, 49, 9–14. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 Years of Image Analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Rautavuoma, K.; Takaluoma, K.; Sormunen, R.; Myllyharju, J.; Kivirikko, K.I.; Soininen, R. Premature Aggregation of Type IV Collagen and Early Lethality in Lysyl Hydroxylase 3 Null Mice. Proc. Natl. Acad. Sci. USA 2004, 101, 14120–14125. [Google Scholar] [CrossRef] [PubMed]
- Kropski, J.A.; Blackwell, T.S. Endoplasmic Reticulum Stress in the Pathogenesis of Fibrotic Disease. J. Clin. Invest. 2018, 128, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Morishita, H.; Eguchi, T.; Tsukamoto, S.; Sakamaki, Y.; Takahashi, S.; Saito, C.; Koyama-Honda, I.; Mizushima, N. Organelle Degradation in the Lens by PLAAT Phospholipases. Nature 2021, 592, 634–638. [Google Scholar] [CrossRef]
- Greiling, T.M.; Clark, J.I. Early Lens Development in the Zebrafish: A Three-Dimensional Time-Lapse Analysis. Dev. Dyn. 2009, 238, 2254–2265. [Google Scholar] [CrossRef] [PubMed]
- Özcan, U.; Yilmaz, E.; Özcan, L.; Furuhashi, M.; Vaillancourt, E.; Smith, R.O.; Görgün, C.Z.; Hotamisligil, G.S. Chemical Chaperones Reduce ER Stress and Restore Glucose Homeostasis in a Mouse Model of Type 2 Diabetes. Science 2006, 313, 1137–1140. [Google Scholar] [CrossRef]
- Iannitti, T.; Palmieri, B. Clinical and Experimental Applications of Sodium Phenylbutyrate. Drugs RD 2011, 11, 227–249. [Google Scholar] [CrossRef] [PubMed]
- Inman, G.J.; Nicolás, F.J.; Callahan, J.F.; Harling, J.D.; Gaster, L.M.; Reith, A.D.; Laping, N.J.; Hill, C.S. SB-431542 Is a Potent and Specific Inhibitor of Transforming Growth Factor-Beta Superfamily Type I Activin Receptor-like Kinase (ALK) Receptors ALK4, ALK5, and ALK7. Mol. Pharmacol. 2002, 62, 65–74. [Google Scholar] [CrossRef]
- Moyer, J.D.; Barbacci, E.G.; Iwata, K.K.; Arnold, L.; Boman, B.; Cunningham, A.; DiOrio, C.; Doty, J.; Morin, M.J.; Moyer, M.P.; et al. Induction of Apoptosis and Cell Cycle Arrest by CP-358,774, an Inhibitor of Epidermal Growth Factor Receptor Tyrosine Kinase. Cancer Res. 1997, 57, 4838–4848. [Google Scholar]
- Wee, P.; Wang, Z. Epidermal Growth Factor Receptor Cell Proliferation Signaling Pathways. Cancers 2017, 9, 52. [Google Scholar] [CrossRef]
- Shu, D.Y.; Lovicu, F.J. Enhanced EGF Receptor-Signaling Potentiates TGFbeta-Induced Lens Epithelial-Mesenchymal Transition. Exp. Eye Res. 2019, 185, 107693. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. MTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [PubMed]
- Brown, E.J.; Beal, P.A.; Keith, C.T.; Chen, J.; Bum Shin, T.; Schreiber, S.L. Control of P70 S6 Kinase by Kinase Activity of FRAP in Vivo. Nature 1995, 377, 441–446. [Google Scholar] [CrossRef] [PubMed]
- Hay, N.; Sonenberg, N. Upstream and Downstream of MTOR. Genes. Dev. 2004, 18, 1926–1945. [Google Scholar] [CrossRef]
- Li, J.; Kim, S.G.; Blenis, J. Rapamycin: One Drug, Many Effects. Cell Metab. 2014, 19, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Heikkinen, J.; Risteli, M.; Wang, C.; Latvala, J.; Rossi, M.; Valtavaara, M.; Myllyla, R. Lysyl Hydroxylase 3 Is a Multifunctional Protein Possessing Collagen Glucosyltransferase Activity. J. Biol. Chem. 2000, 275, 36158–36163. [Google Scholar] [CrossRef]
- Ruotsalainen, H.; Sipila, L.; Vapola, M.; Sormunen, R.; Salo, A.M.; Uitto, L.; Mercer, D.K.; Robins, S.P.; Risteli, M.; Aszodi, A.; et al. Glycosylation Catalyzed by Lysyl Hydroxylase 3 Is Essential for Basement Membranes. J. Cell Sci. 2006, 119, 625–635. [Google Scholar] [CrossRef]
- Bonifacino, J.S.; Weissman, A.M. Ubiquitin and the Control of Protein Fate in the Secretory and Endocytic Pathways. Annu. Rev. Cell Dev. Biol. 1998, 14, 19–57. [Google Scholar] [CrossRef] [PubMed]
- Schuck, S.; Prinz, W.A.; Thorn, K.S.; Voss, C.; Walter, P. Membrane Expansion Alleviates Endoplasmic Reticulum Stress Independently of the Unfolded Protein Response. J. Cell Biol. 2009, 187, 525–536. [Google Scholar] [CrossRef]
- Maidment, J.M. Regional Differences in Tyrosine Kinase Receptor Signaling Components Determine Differential Growth Patterns in the Human Lens. Invest. Ophthalmol. Vis. Sci. 2004, 45, 1427–1435. [Google Scholar] [CrossRef]
- Huang, W.-R.; Fan, X.-X.; Tang, X. SiRNA Targeting EGFR Effectively Prevents Posterior Capsular Opacification after Cataract Surgery. Mol. Vis. 2011, 17, 2349–2355. [Google Scholar]
- Wertheimer, C.; Liegl, R.; Kernt, M.; Mayer, W.; Docheva, D.; Kampik, A.; Eibl-Lindner, K.H. EGF Receptor Inhibitor Erlotinib as a Potential Pharmacological Prophylaxis for Posterior Capsule Opacification. Graefes Arch. Clin. Exp. Ophthalmol. 2013, 251, 1529–1540. [Google Scholar] [CrossRef]
- Wertheimer, C.; Liegl, R.; Kernt, M.; Docheva, D.; Kampik, A.; Eibl-Lindner, K.H. EGFR-Blockade with Erlotinib Reduces EGF and TGF-Β2 Expression and the Actin-Cytoskeleton Which Influences Different Aspects of Cellular Migration in Lens Epithelial Cells. Curr. Eye Res. 2014, 39, 1000–1012. [Google Scholar] [CrossRef]
- Wertheimer, C.; Siedlecki, J.; Kook, D.; Mayer, W.J.; Wolf, A.; Klingenstein, A.; Kampik, A.; Eibl-Lindner, K. EGFR Inhibitor Gefitinib Attenuates Posterior Capsule Opacification in Vitro and in the Ex Vivo Human Capsular Bag Model. Graefes Arch. Clin. Exp. Ophthalmol. 2015, 253, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Shu, D.Y.; Wojciechowski, M.; Lovicu, F.J. ERK1/2-Mediated EGFR-Signaling Is Required for TGFβ-Induced Lens Epithelial-Mesenchymal Transition. Exp. Eye Res. 2019, 178, 108–121. [Google Scholar] [CrossRef] [PubMed]
- VanSlyke, J.K.; Boswell, B.A.; Musil, L.S. ErbBs in Lens Cell Fibrosis and Secondary Cataract. Investig. Opthalmol. Vis. Sci. 2023, 64, 6. [Google Scholar] [CrossRef] [PubMed]
- Wormstone, I.M.; Eldred, J.A.; Dawes, L.J. Fibrotic Modifications of the Lens Epithelium. In Lens Epithelium and Posterior Capsular Opacification; Saika, S., Werner, L., Lovicu, F.J., Eds.; Springer Japan: Tokyo, Japan, 2014; pp. 143–158. ISBN 978-4-431-54299-5. [Google Scholar]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Taler, K.; Zatari, N.; Lone, M.I.; Rotem-Bamberger, S.; Inbal, A. Identification of Small Molecules for Prevention of Lens Epithelium-Derived Cataract Using Zebrafish. Cells 2023, 12, 2540. https://doi.org/10.3390/cells12212540
Taler K, Zatari N, Lone MI, Rotem-Bamberger S, Inbal A. Identification of Small Molecules for Prevention of Lens Epithelium-Derived Cataract Using Zebrafish. Cells. 2023; 12(21):2540. https://doi.org/10.3390/cells12212540
Chicago/Turabian StyleTaler, Kineret, Nour Zatari, Mohammad Iqbal Lone, Shahar Rotem-Bamberger, and Adi Inbal. 2023. "Identification of Small Molecules for Prevention of Lens Epithelium-Derived Cataract Using Zebrafish" Cells 12, no. 21: 2540. https://doi.org/10.3390/cells12212540