
AKT Mediates Adiponectin-Dependent Regulation of VSMC Phenotype

 , , ,
, , , 
Abstract
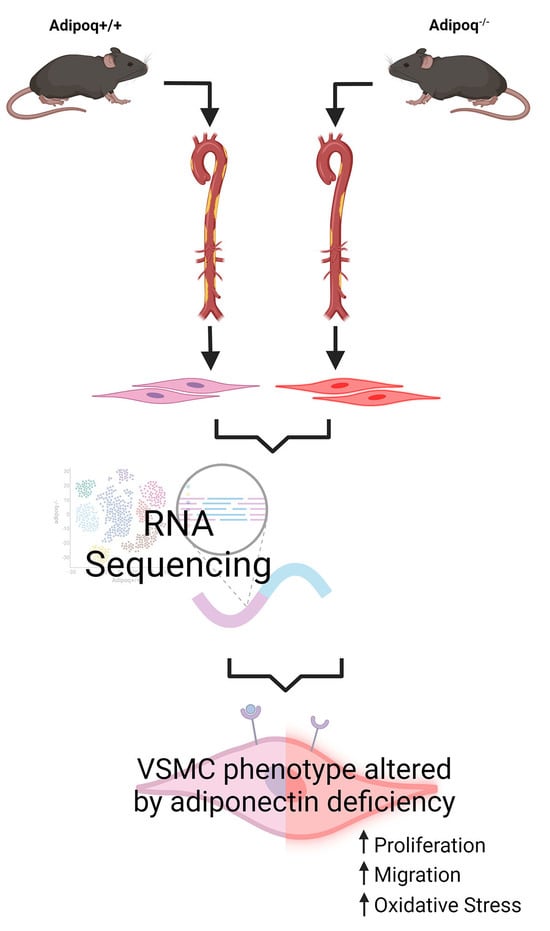
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animal Model
2.2. Preparation of Mouse VSMCs
2.3. Cell Culture
2.4. Cell Lysis and Sample Preparation
2.5. Western Blot
2.6. Proliferation and Cell Migration Assays
2.7. Zymography
2.8. Oroboros Oxygraph System
2.9. Nuclear Fractionation
2.10. RNA Sequence
2.11. Statistics
3. Results
3.1. Adiponectin Deficiency Upregulates the AKT Signaling Pathway and Reduces VSMC Differentiation
3.2. Loss of Adiponectin Promotes a Synthetic Phenotype Altering ECM Composition
3.3. AMPK Inhibits AKT-Dependent Cell Signaling, Proliferation, and Migration but Does Not Reverse the Dedifferentiation of VSMCs
3.4. AKT Mediates the Proliferation and Migration of Adipoq−/− Cells
3.5. Loss of Adiponectin Dysregulates Cellular Signaling in Male and Female Aortas
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Han, S.H.; Sakuma, I.; Shin, E.K.; Koh, K.K. Antiatherosclerotic and anti-insulin resistance effects of adiponectin: Basic and clinical studies. Prog. Cardiovasc. Dis. 2009, 52, 126–140. [Google Scholar] [CrossRef] [PubMed]
- Kitta, Y.; Takano, H.; Nakamura, T.; Kodama, Y.; Umetani, K.; Fujioka, D.; Saito, Y.; Kawabata, K.; Obata, J.E.; Mende, A.; et al. Low adiponectin levels predict late in-stent restenosis after bare metal stenting in native coronary arteries. Int. J. Cardiol. 2008, 131, 78–82. [Google Scholar] [CrossRef] [PubMed]
- Nigro, E.; Scudiero, O.; Monaco, M.L.; Palmieri, A.; Mazzarella, G.; Costagliola, C.; Bianco, A.; Daniele, A. New insight into adiponectin role in obesity and obesity-related diseases. Biomed. Res. Int. 2014, 2014, 658913. [Google Scholar] [CrossRef] [PubMed]
- Achari, A.E.; Jain, S.K. Adiponectin, a Therapeutic Target for Obesity, Diabetes, and Endothelial Dysfunction. Int. J. Mol. Sci. 2017, 18, 1321. [Google Scholar] [CrossRef] [PubMed]
- Ding, M.; Carrao, A.C.; Wagner, R.J.; Xie, Y.; Jin, Y.; Rzucidlo, E.M.; Yu, J.; Li, W.; Tellides, G.; Hwa, J.; et al. Vascular smooth muscle cell-derived adiponectin: A paracrine regulator of contractile phenotype. J. Mol. Cell. Cardiol. 2012, 52, 474–484. [Google Scholar] [CrossRef]
- Parker-Duffen, J.L.; Nakamura, K.; Silver, M.; Kikuchi, R.; Tigges, U.; Yoshida, S.; Denzel, M.S.; Ranscht, B.; Walsh, K. T-cadherin is essential for adiponectin-mediated revascularization. J. Biol. Chem. 2013, 288, 24886–24897. [Google Scholar] [CrossRef]
- Zhan, J.K.; Wang, Y.J.; Wang, Y.; Tang, Z.Y.; Tan, P.; Huang, W.; Liu, Y.S. Adiponectin attenuates the osteoblastic differentiation of vascular smooth muscle cells through the AMPK/mTOR pathway. Exp. Cell Res. 2014, 323, 352–358. [Google Scholar] [CrossRef]
- Cui, X.J.; Lin, X.; Zhong, J.Y.; Li, S.; He, J.Y.; Ni, Y.Q.; Zhan, J.K.; Liu, Y.S. Adiponectin attenuates the premature senescence of vascular smooth muscle cells induced by high glucose through mTOR signaling pathway. Aging Med. 2020, 3, 178–187. [Google Scholar] [CrossRef]
- Ding, M.; Xie, Y.; Wagner, R.J.; Jin, Y.; Carrao, A.C.; Liu, L.S.; Guzman, A.K.; Powell, R.J.; Hwa, J.; Rzucidlo, E.M.; et al. Adiponectin induces vascular smooth muscle cell differentiation via repression of mammalian target of rapamycin complex 1 and FoxO4. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1403–1410. [Google Scholar] [CrossRef]
- Fairaq, A.; Shawky, N.M.; Osman, I.; Pichavaram, P.; Segar, L. AdipoRon, an adiponectin receptor agonist, attenuates PDGF-induced VSMC proliferation through inhibition of mTOR signaling independent of AMPK: Implications toward suppression of neointimal hyperplasia. Pharmacol. Res. 2017, 119, 289–302. [Google Scholar] [CrossRef]
- Nawrocki, A.R.; Hofmann, S.M.; Teupser, D.; Basford, J.E.; Durand, J.L.; Jelicks, L.A.; Woo, C.W.; Kuriakose, G.; Factor, S.M.; Tanowitz, H.B.; et al. Lack of association between adiponectin levels and atherosclerosis in mice. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1159–1165. [Google Scholar] [CrossRef] [PubMed]
- Salazar, G.; Cullen, A.; Huang, J.; Zhao, Y.; Serino, A.; Hilenski, L.; Patrushev, N.; Forouzandeh, F.; Hwang, H.S. SQSTM1/p62 and PPARGC1A/PGC-1alpha at the interface of autophagy and vascular senescence. Autophagy 2020, 16, 1092–1110. [Google Scholar] [CrossRef] [PubMed]
- Fisar, Z.; Hroudova, J.; Singh, N.; Koprivova, A.; Maceckova, D. Effect of Simvastatin, Coenzyme Q10, Resveratrol, Acetylcysteine and Acetylcarnitine on Mitochondrial Respiration. Folia Biol. 2016, 62, 53–66. [Google Scholar]
- Salazar, G.; Huang, J.; Feresin, R.G.; Zhao, Y.; Griendling, K.K. Zinc regulates Nox1 expression through a NF-kappaB and mitochondrial ROS dependent mechanism to induce senescence of vascular smooth muscle cells. Free Radic. Biol. Med. 2017, 108, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Audic, S.; Claverie, J.M. The significance of digital gene expression profiles. Genome Res. 1997, 7, 986–995. [Google Scholar] [CrossRef]
- Yuan, J.; Dong, X.; Yap, J.; Hu, J. The MAPK and AMPK signalings: Interplay and implication in targeted cancer therapy. J. Hematol. Oncol. 2020, 13, 113. [Google Scholar] [CrossRef]
- Kovacic, S.; Soltys, C.L.; Barr, A.J.; Shiojima, I.; Walsh, K.; Dyck, J.R. Akt activity negatively regulates phosphorylation of AMP-activated protein kinase in the heart. J. Biol. Chem. 2003, 278, 39422–39427. [Google Scholar] [CrossRef]
- Xin, X.; Zhou, L.; Reyes, C.M.; Liu, F.; Dong, L.Q. APPL1 mediates adiponectin-stimulated p38 MAPK activation by scaffolding the TAK1-MKK3-p38 MAPK pathway. Am. J. Physiol. Endocrinol. Metab. 2011, 300, E103–E110. [Google Scholar] [CrossRef]
- Chettimada, S.; Joshi, S.R.; Dhagia, V.; Aiezza, A., 2nd; Lincoln, T.M.; Gupte, R.; Miano, J.M.; Gupte, S.A. Vascular smooth muscle cell contractile protein expression is increased through protein kinase G-dependent and -independent pathways by glucose-6-phosphate dehydrogenase inhibition and deficiency. Am. J. Physiol. Heart Circ. Physiol. 2016, 311, H904–H912. [Google Scholar] [CrossRef]
- Lee, J.H.; Liu, R.; Li, J.; Zhang, C.; Wang, Y.; Cai, Q.; Qian, X.; Xia, Y.; Zheng, Y.; Piao, Y.; et al. Stabilization of phosphofructokinase 1 platelet isoform by AKT promotes tumorigenesis. Nat. Commun. 2017, 8, 949. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.X.; Chao, L.; Zhou, G.; Chao, J. Expression and characterization of rat kallikrein-binding protein in Escherichia coli. Biochem. J. 1993, 292 Pt 3, 825–832. [Google Scholar] [CrossRef]
- Kuzuya, M.; Nakamura, K.; Sasaki, T.; Cheng, X.W.; Itohara, S.; Iguchi, A. Effect of MMP-2 deficiency on atherosclerotic lesion formation in apoE-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1120–1125. [Google Scholar] [CrossRef] [PubMed]
- Perrotta, F.; Nigro, E.; Mollica, M.; Costigliola, A.; D’Agnano, V.; Daniele, A.; Bianco, A.; Guerra, G. Pulmonary Hypertension and Obesity: Focus on Adiponectin. Int. J. Mol. Sci. 2019, 20, 912. [Google Scholar] [CrossRef]
- Adiguzel, E.; Hou, G.; Sabatini, P.J.; Bendeck, M.P. Type VIII collagen signals via beta1 integrin and RhoA to regulate MMP-2 expression and smooth muscle cell migration. Matrix Biol. 2013, 32, 332–341. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.B.; McCune, B.K.; Sporn, M.B. TGF-beta: Regulation of extracellular matrix. Kidney Int. 1992, 41, 557–559. [Google Scholar] [CrossRef] [PubMed]
- Pagiatakis, C.; Sun, D.; Tobin, S.W.; Miyake, T.; McDermott, J.C. TGFbeta-TAZ/SRF signalling regulates vascular smooth muscle cell differentiation. FEBS J. 2017, 284, 1644–1656. [Google Scholar] [CrossRef]
- Tatti, O.; Vehvilainen, P.; Lehti, K.; Keski-Oja, J. MT1-MMP releases latent TGF-beta1 from endothelial cell extracellular matrix via proteolytic processing of LTBP-1. Exp. Cell Res. 2008, 314, 2501–2514. [Google Scholar] [CrossRef]
- Sounni, N.E.; Paye, A.; Host, L.; Noel, A. MT-MMPS as Regulators of Vessel Stability Associated with Angiogenesis. Front. Pharmacol. 2011, 2, 111. [Google Scholar] [CrossRef]
- Yasumura, S.; Naito, Y.; Okuno, K.; Sawada, H.; Asakura, M.; Masuyama, T.; Ishihara, M. Effects of Heterozygous TfR1 (Transferrin Receptor 1) Deletion in Pathogenesis of Renal Fibrosis in Mice. Hypertension 2020, 75, 413–421. [Google Scholar] [CrossRef]
- Lei, R.; Zhang, K.; Liu, K.; Shao, X.; Ding, Z.; Wang, F.; Hong, Y.; Zhu, M.; Li, H.; Li, H. Transferrin receptor facilitates TGF-beta and BMP signaling activation to control craniofacial morphogenesis. Cell Death Dis. 2016, 7, e2282. [Google Scholar] [CrossRef] [PubMed]
- Tufveson, G.; Alm, G.V. Effects of mitogens for mouse B lymphocytes on chicken lymphoid cells. Immunology 1975, 29, 697–707. [Google Scholar] [PubMed]
- Hug, C.; Wang, J.; Ahmad, N.S.; Bogan, J.S.; Tsao, T.S.; Lodish, H.F. T-cadherin is a receptor for hexameric and high-molecular-weight forms of Acrp30/adiponectin. Proc. Natl. Acad. Sci. USA 2004, 101, 10308–10313. [Google Scholar] [CrossRef] [PubMed]
- Pierre, P.; Froment, P.; Negre, D.; Rame, C.; Barateau, V.; Chabrolle, C.; Lecomte, P.; Dupont, J. Role of adiponectin receptors, AdipoR1 and AdipoR2, in the steroidogenesis of the human granulosa tumor cell line, KGN. Hum. Reprod. 2009, 24, 2890–2901. [Google Scholar] [CrossRef]
- Das, F.; Ghosh-Choudhury, N.; Lee, D.Y.; Gorin, Y.; Kasinath, B.S.; Choudhury, G.G. Akt2 causes TGFbeta-induced deptor downregulation facilitating mTOR to drive podocyte hypertrophy and matrix protein expression. PLoS ONE 2018, 13, e0207285. [Google Scholar] [CrossRef]
- Zhu, S.B.; Zhu, J.; Zhou, Z.Z.; Xi, E.P.; Wang, R.P.; Zhang, Y. TGF-beta1 induces human aortic vascular smooth muscle cell phenotype switch through PI3K/AKT/ID2 signaling. Am. J. Transl. Res. 2015, 7, 2764–2774. [Google Scholar]
- Yang, X.; Gong, Y.; Tang, Y.; Li, H.; He, Q.; Gower, L.; Liaw, L.; Friesel, R.E. Spry1 and Spry4 Differentially Regulate Human Aortic Smooth Muscle Cell Phenotype via Akt/FoxO/Myocardin Signaling. PLoS ONE 2013, 8, e58746. [Google Scholar] [CrossRef]
- Jin, Y.; Xie, Y.; Ostriker, A.C.; Zhang, X.; Liu, R.; Lee, M.Y.; Leslie, K.L.; Tang, W.; Du, J.; Lee, S.H.; et al. Opposing Actions of AKT (Protein Kinase B) Isoforms in Vascular Smooth Muscle Injury and Therapeutic Response. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 2311–2321. [Google Scholar] [CrossRef]
- Takahashi, M.; Hayashi, K.; Yoshida, K.; Ohkawa, Y.; Komurasaki, T.; Kitabatake, A.; Ogawa, A.; Nishida, W.; Yano, M.; Monden, M.; et al. Epiregulin as a major autocrine/paracrine factor released from ERK- and p38MAPK-activated vascular smooth muscle cells. Circulation 2003, 108, 2524–2529. [Google Scholar] [CrossRef]
- Mack, C.P. Signaling mechanisms that regulate smooth muscle cell differentiation. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1495–1505. [Google Scholar] [CrossRef]
- Martin, K.A.; Merenick, B.L.; Ding, M.; Fetalvero, K.M.; Rzucidlo, E.M.; Kozul, C.D.; Brown, D.J.; Chiu, H.Y.; Shyu, M.; Drapeau, B.L.; et al. Rapamycin promotes vascular smooth muscle cell differentiation through insulin receptor substrate-1/phosphatidylinositol 3-kinase/Akt2 feedback signaling. J. Biol. Chem. 2007, 282, 36112–36120. [Google Scholar] [CrossRef] [PubMed]
- Risinger, G.M., Jr.; Updike, D.L.; Bullen, E.C.; Tomasek, J.J.; Howard, E.W. TGF-beta suppresses the upregulation of MMP-2 by vascular smooth muscle cells in response to PDGF-BB. Am. J. Physiol. Cell Physiol. 2010, 298, C191–C201. [Google Scholar] [CrossRef] [PubMed]
- Saneipour, M.; Ghatreh-Samani, K.; Heydarian, E.; Farrokhi, E.; Abdian, N. Adiponectin inhibits oxidized low density lipoprotein-induced increase in matrix metalloproteinase 9 expression in vascular smooth muscle cells. ARYA Atheroscler. 2015, 11, 191–195. [Google Scholar]
- Wanninger, J.; Walter, R.; Bauer, S.; Eisinger, K.; Schaffler, A.; Dorn, C.; Weiss, T.S.; Hellerbrand, C.; Buechler, C. MMP-9 activity is increased by adiponectin in primary human hepatocytes but even negatively correlates with serum adiponectin in a rodent model of non-alcoholic steatohepatitis. Exp. Mol. Pathol. 2011, 91, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Strickland, D.K.; Bornstein, P. Extracellular Matrix Metalloproteinase 2 Levels Are Regulated by the Low Density Lipoprotein-related Scavenger Receptor and Thrombospondin 2. J. Biol. Chem. 2001, 276, 8403–8408. [Google Scholar] [CrossRef]
- Caldwell, J.T.; Jones, K.M.D.; Park, H.; Pinto, J.R.; Ghosh, P.; Reid-Foley, E.C.; Ulrich, B.; Delp, M.D.; Behnke, B.J.; Muller-Delp, J.M. Aerobic exercise training reduces cardiac function and coronary flow-induced vasodilation in mice lacking adiponectin. Am. J. Physiol. Heart Circ. Physiol. 2021, 321, H1–H14. [Google Scholar] [CrossRef]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cullen, A.E.; Centner, A.M.; Deitado, R.; Ismaeel, A.; Koutakis, P.; Muller-Delp, J.; Salazar, G. AKT Mediates Adiponectin-Dependent Regulation of VSMC Phenotype. Cells 2023, 12, 2493. https://doi.org/10.3390/cells12202493
Cullen AE, Centner AM, Deitado R, Ismaeel A, Koutakis P, Muller-Delp J, Salazar G. AKT Mediates Adiponectin-Dependent Regulation of VSMC Phenotype. Cells. 2023; 12(20):2493. https://doi.org/10.3390/cells12202493
Chicago/Turabian StyleCullen, Abigail E., Ann M. Centner, Riley Deitado, Ahmed Ismaeel, Panagiotis Koutakis, Judy Muller-Delp, and Gloria Salazar. 2023. "AKT Mediates Adiponectin-Dependent Regulation of VSMC Phenotype" Cells 12, no. 20: 2493. https://doi.org/10.3390/cells12202493