

Mammalian Models in Alzheimer’s Research: An Update



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
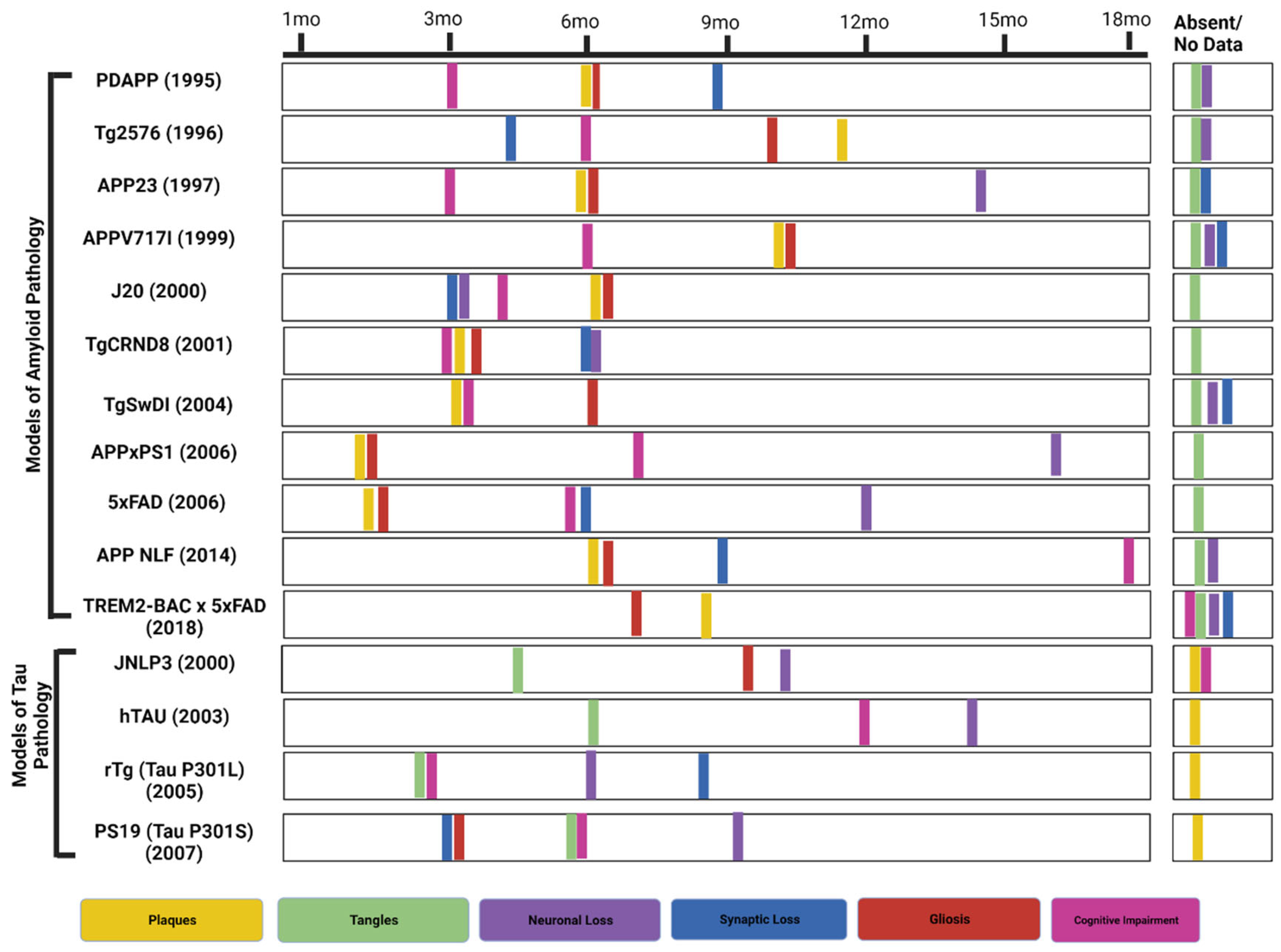
2. Murine Models of Amyloid Pathology
2.1. PDAPP
2.2. Tg2576
2.3. APP23
2.4. APP(V717I) & J20 Models
2.5. TgCRND8 Model
2.6. TgSwDI Model
2.7. 5xFAD-
2.8. APPxPS1-
2.9. APP NL-F KI-
2.10. TREM2- BAC × 5XFAD-
3. Prominent Tau Mice Models
3.1. JNPL3 (P301L)-
3.2. hTau-
3.3. rTg (tauP301L)-
3.4. PS19 (tau P301S)-
3.5. 3xTg A Composite Model-
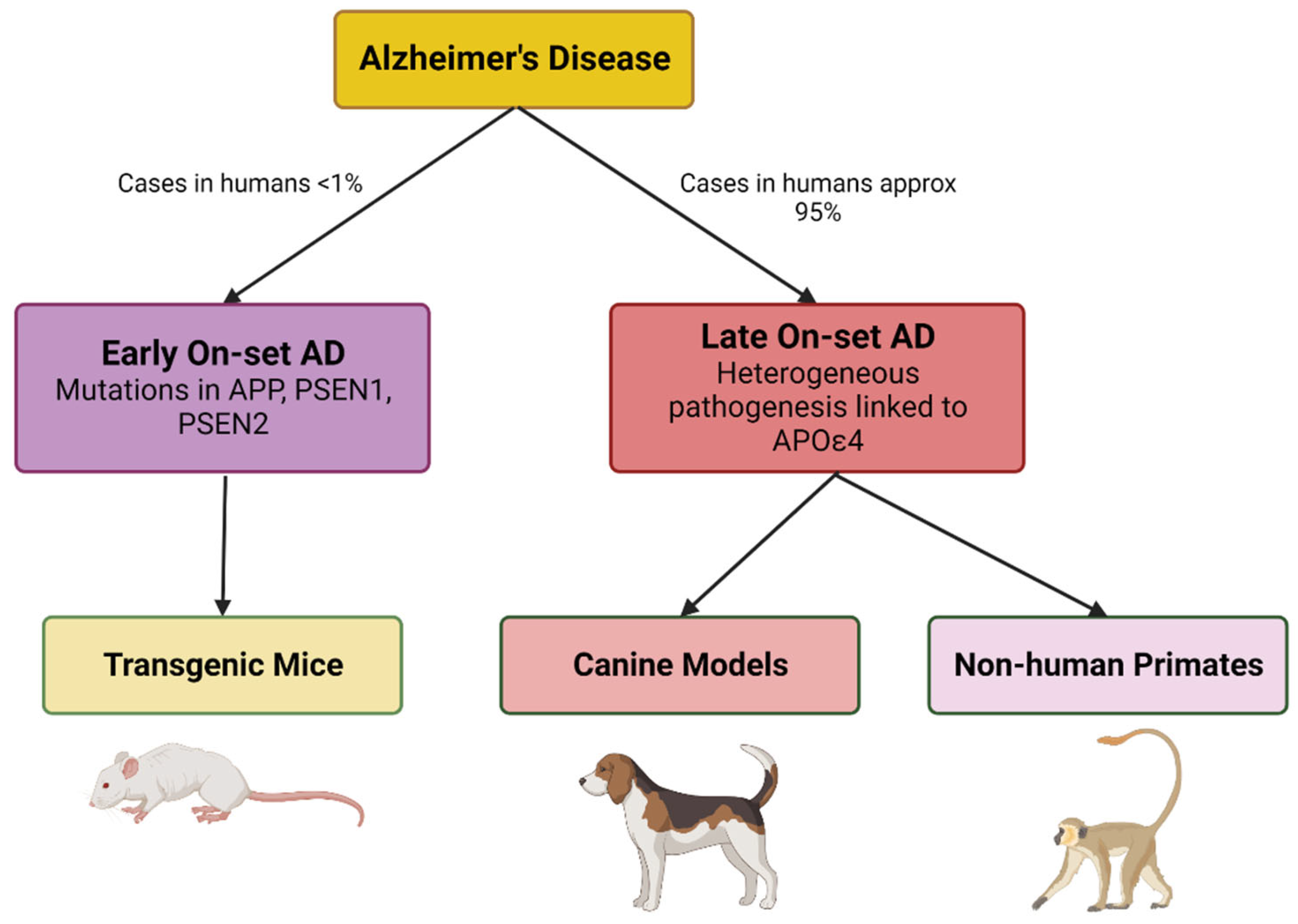
4. LOAD Murine Models
5. Alternative Murine Models
6. Emerging Models
7. Behavioral Studies
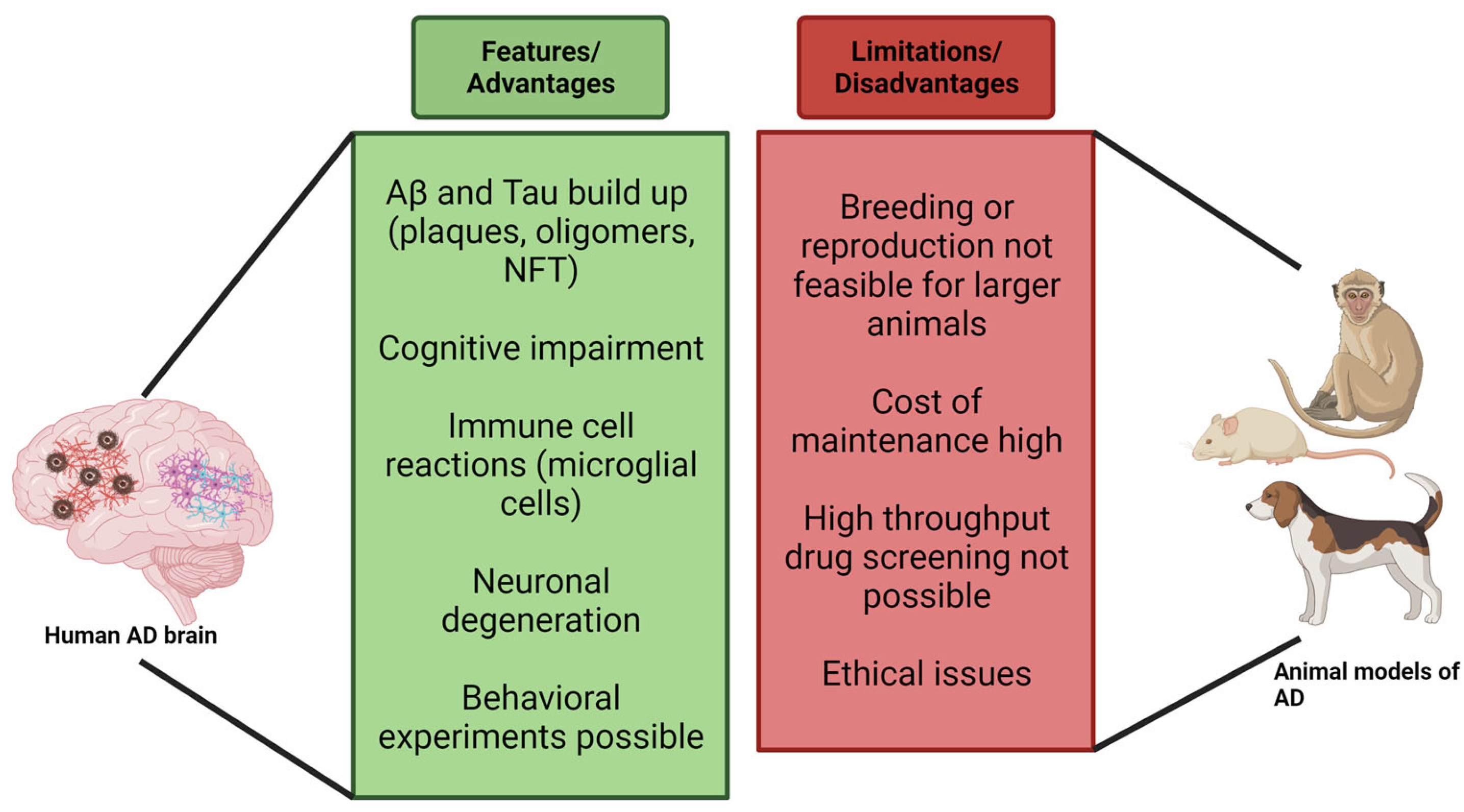
8. Limitations of Murine Models
9. Neuro Imaging in Murine Models
10. Evolutionarily Closer Models
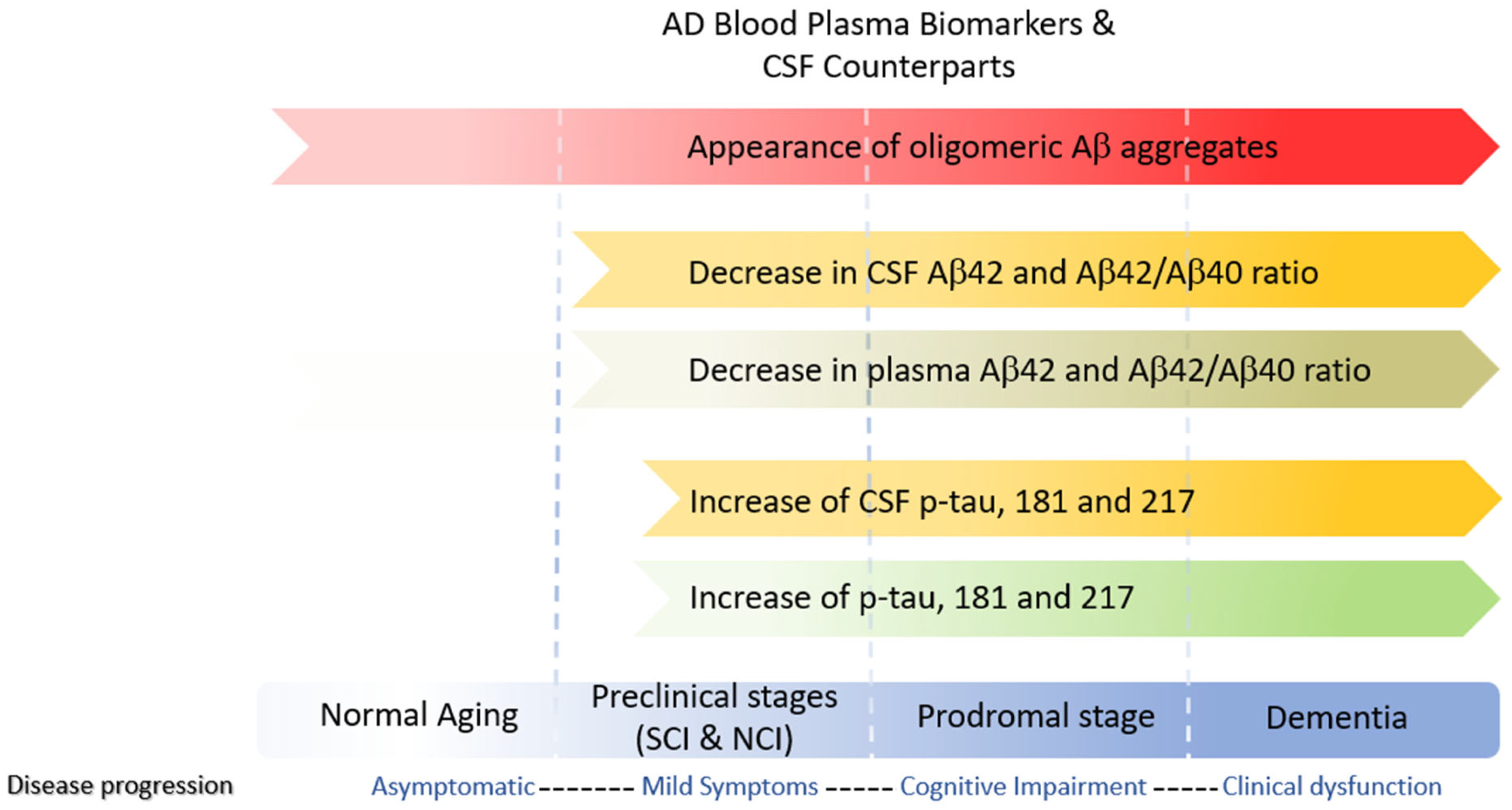
11. Fluid Biomarkers
12. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Cummings, J.L.; Goldman, D.P.; Simmons-Stern, N.R.; Ponton, E. The costs of developing treatments for Alzheimer’s disease: A retrospective exploration. Alzheimer’s Dement. 2022, 18, 469–477. [Google Scholar] [CrossRef]
- Poon, C.H.; Wang, Y.; Fung, M.-L.; Zhang, C.; Lim, L.W. Rodent models of amyloid-beta feature of alzheimer’s disease: Development and potential treatment implications. Aging Dis. 2020, 11, 1235. [Google Scholar] [CrossRef]
- Therriault, J.; Pascoal, T.A.; Lussier, F.Z.; Tissot, C.; Chamoun, M.; leb Bezgin, G.; Servaes, S.; Benedet, A.L.; Ashton, N.J.; Karikari, T.K.; et al. Biomarker modeling of Alzheimer’s disease using PET-based Braak staging. Nat. Aging 2022, 2, 526–535. [Google Scholar] [CrossRef]
- De Ture, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef]
- Thal, D.R.; Capetillo-Zarate, E.; Tredici, K.D.; Braak, H. The development of amyloid β protein deposits in the aged brain. Sci. Aging Knowl. Environ. 2006, 2006, re1. [Google Scholar] [CrossRef]
- Jankowsky, J.L.; Zheng, H. Practical considerations for choosing a mouse model of Alzheimer’s disease. Mol. Neurodegener. 2017, 12, 89. [Google Scholar] [CrossRef]
- Wattmo, C.; Wallin, A.K. Early- versus late-onset Alzheimer’s disease in clinical practice: Cognitive and global outcomes over 3 years. Alzheimer’s Res. Ther. 2017, 9, 70. [Google Scholar] [CrossRef]
- Verghese, P.B.; Castellano, J.M.; Holtzman, D.M. Apolipoprotein E in Alzheimer’s disease and other neurological disorders. Lancet Neurol. 2011, 10, 241–252. [Google Scholar] [CrossRef]
- Kim, J.; Basak, J.M.; Holtzman, D.M. The role of apolipoprotein E in Alzheimer’s disease. Neuron 2009, 63, 287–303. [Google Scholar] [CrossRef]
- Vassar, R. BACE1: The beta-secretase enzyme in Alzheimer’s disease. J. Mol. Neurosci. 2004, 23, 105–114. [Google Scholar] [CrossRef]
- Drummond, E.; Wisniewski, T. Alzheimer’s disease: Experimental models and reality. Acta Neuropathol. 2017, 133, 155–175. [Google Scholar] [CrossRef]
- Cole, S.L.; Vassar, R. The Alzheimer’s disease beta-secretase enzyme, BACE1. Mol. Neurodegener. 2007, 2, 22. [Google Scholar] [CrossRef]
- Hampel, H.; Vassar, R.; De Strooper, B.; Hardy, J.; Willem, M.; Singh, N.; Zhou, J.; Yan, R.; Vanmechelen, E.; De Vos, A.; et al. The beta-Secretase BACE1 in Alzheimer’s Disease. Biol. Psychiatry 2021, 89, 745–756. [Google Scholar] [CrossRef]
- Hussain, I.; Powell, D.; Howlett, D.R.; Tew, D.G.; Meek, T.D.; Chapman, C.; Gloger, I.S.; Murphy, K.E.; Southan, C.D.; Ryan, D.M.; et al. Identification of a novel aspartic protease (Asp 2) as beta-secretase. Mol. Cell. Neurosci. 1999, 14, 419–427. [Google Scholar] [CrossRef]
- Sinha, S.; Anderson, J.P.; Barbour, R.; Basi, G.S.; Caccavello, R.; Davis, D.; Doan, M.; Dovey, H.F.; Frigon, N.; Hong, J.; et al. Purification and cloning of amyloid precursor protein beta-secretase from human brain. Nature 1999, 402, 537–540. [Google Scholar] [CrossRef]
- Vassar, R.; Bennett, B.D.; Babu-Khan, S.; Kahn, S.; Mendiaz, E.A.; Denis, P.; Teplow, D.B.; Ross, S.; Amarante, P.; Loeloff, R.; et al. Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science 1999, 286, 735–741. [Google Scholar] [CrossRef]
- Lin, X.; Koelsch, G.; Wu, S.; Downs, D.; Dashti, A.; Tang, J. Human aspartic protease memapsin 2 cleaves the beta-secretase site of beta-amyloid precursor protein. Proc. Natl. Acad. Sci. USA 2000, 97, 1456–1460. [Google Scholar] [CrossRef]
- Goate, A.; Chartier-Harlin, M.C.; Mullan, M.; Brown, J.; Crawford, F.; Fidani, L.; Giuffra, L.; Haynes, A.; Irving, N.; James, L.; et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature 1991, 349, 704–706. [Google Scholar] [CrossRef]
- Petit, D.; Fernández, S.G.; Zoltowska, K.M.; Enzlein, T.; Ryan, N.S.; O’Connor, A.; Szaruga, M.; Hill, E.; Vandenberghe, R.; Fox, N.C.; et al. Aβ profiles generated by Alzheimer’s disease causing PSEN1 variants determine the pathogenicity of the mutation and predict age at disease onset. Mol. Psychiatry 2022, 27, 2821–2832. [Google Scholar] [CrossRef]
- Hardy, J.; Allsop, D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol. Sci. 1991, 12, 383–388. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Mandelkow, E.-M.; Mandelkow, E. Biochemistry and cell biology of tau protein in neurofibrillary degeneration. Cold Spring Harb. Perspect. Med. 2012, 2, a006247. [Google Scholar] [CrossRef]
- Gendron, T.F.; Petrucelli, L. The role of tau in neurodegeneration. Mol. Neurodegener. 2009, 4, 13. [Google Scholar] [CrossRef]
- Ribeiro, F.M.; Camargos, E.R.d.S.; de Souza, L.C.; Teixeira, A.L. Animal models of neurodegenerative diseases. Rev. Bras. De Psiquiatr. 2013, 35 (Suppl. S2), S82–S91. [Google Scholar] [CrossRef]
- KoSIK, K.S.; Joachim, C.L.; Selkoe, D.J. Microtubule-associated protein tau (tau) is a major antigenic component of paired helical filaments in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1986, 83, 4044–4048. [Google Scholar] [CrossRef]
- Research Models Alzheimer’s Disease. Available online: https://www.alzforum.org/research-models/alzheimers-disease (accessed on 3 September 2023).
- Puzzo, D.; Gulisano, W.; Palmeri, A.; Arancio, O. Rodent models for Alzheimer’s disease drug discovery. Expert. Opin. Drug Discov. 2015, 10, 703–711. [Google Scholar] [CrossRef]
- Games, D.; Adams, D.; Alessandrini, R.; Barbour, R.; Berthelette, P.; Blackwell, C.; Carr, T.; Clemens, J.; Donaldson, T.; Gillespie, F. Alzheimer-type neuropathology in transgenic mice overexpressing V717F beta-amyloid precursor protein. Nature 1995, 373, 523–527. [Google Scholar] [CrossRef]
- Masliah, E.; Sisk, A.; Mallory, M.; Mucke, L.; Schenk, D.; Games, D. Comparison of Neurodegenerative Pathology in Transgenic Mice Overexpressing V717F β-Amyloid Precursor Protein and Alzheimer’s Disease. J. Neurosci. 1996, 16, 5795–5811. [Google Scholar] [CrossRef]
- Chen, G.; Chen, K.S.; Knox, J.; Inglis, J.; Bernard, A.; Martin, S.J.; Justice, A.; McConlogue, L.; Games, D.; Freedman, S.B.; et al. A learning deficit related to age and β-amyloid plaques in a mouse model of Alzheimer’s disease. Nature 2000, 408, 975–979. [Google Scholar] [CrossRef]
- Dodart, J.-C.; Mathis, C.; Saura, J.; Bales, K.R.; Paul, S.M.; Ungerer, A. Neuroanatomical Abnormalities in Behaviorally Characterized APPV717F Transgenic Mice. Neurobiol. Dis. 2000, 7, 71–85. [Google Scholar] [CrossRef]
- Reilly, J.F.; Games, D.; Rydel, R.E.; Freedman, S.; Schenk, D.; Young, W.G.; Morrison, J.H.; Bloom, F.E. Amyloid deposition in the hippocampus and entorhinal cortex: Quantitative analysis of a transgenic mouse model. Proc. Natl. Acad. Sci. USA 2003, 100, 4837–4842. [Google Scholar] [CrossRef]
- Hsiao, K.; Chapman, P.; Nilsen, S.; Eckman, C.; Harigaya, Y.; Younkin, S.; Yang, F.; Cole, G. Correlative Memory Deficits, Aβ Elevation, and Amyloid Plaques in Transgenic Mice. Science 1996, 274, 99–103. [Google Scholar] [CrossRef]
- Spires-Jones, T.L.; Meyer-Luehmann, M.; Osetek, J.D.; Jones, P.B.; Stern, E.A.; Bacskai, B.J.; Hyman, B.T. Impaired Spine Stability Underlies Plaque-Related Spine Loss in an Alzheimer’s Disease Mouse Model. Am. J. Pathol. 2007, 171, 1304–1311. [Google Scholar] [CrossRef]
- Irizarry, M.C.; Mcnamara, M.; Fedorchak, K.; Hsiao, K.; Hyman, B.T. APPSW Transgenic Mice Develop Age-related Aβ Deposits and Neuropil Abnormalities, but no Neuronal Loss in CA1. J. Neuropathol. Exp. Neurol. 1997, 56, 965–973. [Google Scholar] [CrossRef]
- King, D.L.; Arendash, G.W. Behavioral characterization of the Tg2576 transgenic model of Alzheimer’s disease through 19 months. Physiol. Behav. 2002, 75, 627–642. [Google Scholar] [CrossRef]
- Sturchler-Pierrat, C.; Abramowski, D.; Duke, M.; Wiederhold, K.H.; Mistl, C.; Rothacher, S.; Ledermann, B.; Burki, K.; Frey, P.; Paganetti, P.A.; et al. Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc. Natl. Acad. Sci. USA 1997, 94, 13287–13292. [Google Scholar] [CrossRef]
- Thal, D.R.; Griffin, W.S.; Braak, H. Parenchymal and vascular Abeta-deposition and its effects on the degeneration of neurons and cognition in Alzheimer’s disease. J. Cell Mol. Med. 2008, 12, 1848–1862. [Google Scholar] [CrossRef]
- Reuter, B.; Venus, A.; Grudzenski, S.; Heiler, P.; Schad, L.; Staufenbiel, M.; Hennerici, M.G.; Fatar, M. Statin Therapy and the Development of Cerebral Amyloid Angiopathy—A Rodent in Vivo Approach. Int. J. Mol. Sci. 2016, 17, 126. [Google Scholar] [CrossRef]
- Fryer, J.D.; Simmons, K.; Parsadanian, M.; Bales, K.R.; Paul, S.M.; Sullivan, P.M.; Holtzman, D.M. Human apolipoprotein E4 alters the amyloid-beta 40:42 ratio and promotes the formation of cerebral amyloid angiopathy in an amyloid precursor protein transgenic model. J. Neurosci. 2005, 25, 2803–2810. [Google Scholar] [CrossRef]
- Meyer, E.P.; Ulmann-Schuler, A.; Staufenbiel, M.; Krucker, T. Altered morphology and 3D architecture of brain vasculature in a mouse model for Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2008, 105, 3587–3592. [Google Scholar] [CrossRef]
- Nehra, G.; Bauer, B.; Hartz, A.M. Blood-brain barrier leakage in Alzheimer’s disease: From discovery to clinical relevance. Pharmacol. Ther. 2022, 234, 108119. [Google Scholar] [CrossRef]
- Caroni, P. Overexpression of growth-associated proteins in the neurons of adult transgenic mice. J. Neurosci. Methods 1997, 71, 3–9. [Google Scholar] [CrossRef]
- Lloyd, G.M.; Trejo-Lopez, J.A.; Xia, Y.; McFarland, K.N.; Lincoln, S.J.; Ertekin-Taner, N.; Giasson, B.I.; Yachnis, A.T.; Prokop, S. Prominent amyloid plaque pathology and cerebral amyloid angiopathy in APP V717I (London) carrier—Phenotypic variability in autosomal dominant Alzheimer’s disease. Acta Neuropathol. Commun. 2020, 8, 31. [Google Scholar] [CrossRef]
- Mucke, L.; Masliah, E.; Yu, G.Q.; Mallory, M.; Rockenstein, E.M.; Tatsuno, G.; Hu, K.; Kholodenko, D.; Johnson-Wood, K.; McConlogue, L. High-level neuronal expression of abeta 1-42 in wild-type human amyloid protein precursor transgenic mice: Synaptotoxicity without plaque formation. J. Neurosci. 2000, 20, 4050–4058. [Google Scholar] [CrossRef]
- Hong, J.H.; Kang, J.W.; Kim, D.K.; Baik, S.H.; Kim, K.H.; Shanta, S.R.; Jung, J.H.; Mook-Jung, I.; Kim, K.P. Global changes of phospholipids identified by MALDI imaging mass spectrometry in a mouse model of Alzheimer’s disease. J. Lipid Res. 2016, 57, 36–45. [Google Scholar] [CrossRef]
- Ameen-Ali, K.E.; Simpson, J.E.; Wharton, S.B.; Heath, P.R.; Sharp, P.S.; Brezzo, G.; Berwick, J. The Time Course of Recognition Memory Impairment and Glial Pathology in the hAPP-J20 Mouse Model of Alzheimer’s Disease. J. Alzheimer’s Dis. 2019, 68, 609–624. [Google Scholar] [CrossRef]
- Shabir, O.; Sharp, P.; Rebollar, M.A.; Boorman, L.; Howarth, C.; Wharton, S.B.; Francis, S.E.; Berwick, J. Enhanced Cerebral Blood Volume under Normobaric Hyperoxia in the J20-hAPP Mouse Model of Alzheimer’s Disease. Sci. Rep. 2020, 10, 7518. [Google Scholar] [CrossRef]
- Chishti, M.A.; Yang, D.S.; Janus, C.; Phinney, A.L.; Horne, P.; Pearson, J.; Strome, R.; Zuker, N.; Loukides, J.; French, J.; et al. Early-onset amyloid deposition and cognitive deficits in transgenic mice expressing a double mutant form of amyloid precursor protein 695. J. Biol. Chem. 2001, 276, 21562–21570. [Google Scholar] [CrossRef]
- Boy, J.; Leergaard, T.B.; Schmidt, T.; Odeh, F.; Bichelmeier, U.; Nuber, S.; Holzmann, C.; Wree, A.; Prusiner, S.B.; Bujard, H.; et al. Expression mapping of tetracycline-responsive prion protein promoter: Digital atlasing for generating cell-specific disease models. Neuroimage 2006, 33, 449–462. [Google Scholar] [CrossRef]
- Cortes-Canteli, M.; Mattei, L.; Richards, A.T.; Norris, E.H.; Strickland, S. Fibrin deposited in the Alzheimer’s disease brain promotes neuronal degeneration. Neurobiol. Aging 2015, 36, 608–617. [Google Scholar] [CrossRef]
- Brautigam, H.; Steele, J.W.; Westaway, D.; Fraser, P.E.; St George-Hyslop, P.H.; Gandy, S.; Hof, P.R.; Dickstein, D.L. The isotropic fractionator provides evidence for differential loss of hippocampal neurons in two mouse models of Alzheimer’s disease. Mol. Neurodegener. 2012, 7, 58. [Google Scholar] [CrossRef]
- Davis, J.; Xu, F.; Deane, R.; Romanov, G.; Previti, M.L.; Zeigler, K.; Zlokovic, B.V.; Van Nostrand, W.E. Early-onset and robust cerebral microvascular accumulation of amyloid beta-protein in transgenic mice expressing low levels of a vasculotropic Dutch/Iowa mutant form of amyloid beta-protein precursor. J. Biol. Chem. 2004, 279, 20296–20306. [Google Scholar] [CrossRef]
- Davis, J.; Xu, F.; Miao, J.; Previti, M.L.; Romanov, G.; Ziegler, K.; Van Nostrand, W.E. Deficient cerebral clearance of vasculotropic mutant Dutch/Iowa Double A beta in human A betaPP transgenic mice. Neurobiol. Aging 2006, 27, 946–954. [Google Scholar] [CrossRef]
- Choi, S.; Singh, I.; Singh, A.K.; Khan, M.; Won, J. Asymmetric dimethylarginine exacerbates cognitive dysfunction associated with cerebrovascular pathology. FASEB J. 2020, 34, 6808–6823. [Google Scholar] [CrossRef]
- Searcy, J.L.; Le Bihan, T.; Salvadores, N.; McCulloch, J.; Horsburgh, K. Impact of age on the cerebrovascular proteomes of wild-type and Tg-SwDI mice. PLoS ONE 2014, 9, e89970. [Google Scholar] [CrossRef]
- Oakley, H.; Cole, S.L.; Logan, S.; Maus, E.; Shao, P.; Craft, J.; Guillozet-Bongaarts, A.; Ohno, M.; Disterhoft, J.; Van Eldik, L. Intraneuronal β-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: Potential factors in amyloid plaque formation. J. Neurosci. 2006, 26, 10129–10140. [Google Scholar] [CrossRef]
- Forner, S.; Kawauchi, S.; Balderrama-Gutierrez, G.; Kramár, E.A.; Matheos, D.P.; Phan, J.; Javonillo, D.I.; Tran, K.M.; Hingco, E.; da Cunha, C. Systematic phenotyping and characterization of the 5xFAD mouse model of Alzheimer’s disease. Sci. Data 2021, 8, 270. [Google Scholar] [CrossRef]
- Jawhar, S.; Trawicka, A.; Jenneckens, C.; Bayer, T.A.; Wirths, O. Motor deficits, neuron loss, and reduced anxiety coinciding with axonal degeneration and intraneuronal Aβ aggregation in the 5XFAD mouse model of Alzheimer’s disease. Neurobiol. Aging 2012, 33, 196.e129–196.e140. [Google Scholar] [CrossRef]
- Oblak, A.L.; Lin, P.B.; Kotredes, K.P.; Pandey, R.S.; Garceau, D.; Williams, H.M.; Uyar, A.; O’Rourke, R.; O’Rourke, S.; Ingraham, C. Comprehensive evaluation of the 5XFAD mouse model for preclinical testing applications: A MODEL-AD study. Front. Aging Neurosci. 2021, 13, 713726. [Google Scholar] [CrossRef]
- Chu, T.-H.; Cummins, K.; Sparling, J.S.; Tsutsui, S.; Brideau, C.; Nilsson, K.P.R.; Joseph, J.T.; Stys, P.K. Axonal and myelinic pathology in 5xFAD Alzheimer’s mouse spinal cord. PLoS ONE 2017, 12, e0188218. [Google Scholar] [CrossRef]
- Radde, R.; Bolmont, T.; Kaeser, S.A.; Coomaraswamy, J.; Lindau, D.; Stoltze, L.; Calhoun, M.E.; Jaggi, F.; Wolburg, H.; Gengler, S.; et al. Aβ42-driven cerebral amyloidosis in transgenic mice reveals early and robust pathology. EMBO Rep. 2006, 7, 940–946. [Google Scholar] [CrossRef]
- Bittner, T.; Burgold, S.; Dorostkar, M.M.; Fuhrmann, M.; Wegenast-Braun, B.M.; Schmidt, B.; Kretzschmar, H.; Herms, J. Amyloid plaque formation precedes dendritic spine loss. Acta Neuropathol. 2012, 124, 797–807. [Google Scholar] [CrossRef]
- Serneels, L.; Van Biervliet, J.; Craessaerts, K.; Dejaegere, T.; Horre, K.; Van Houtvin, T.; Esselmann, H.; Paul, S.; Schafer, M.K.; Berezovska, O.; et al. gamma-Secretase heterogeneity in the Aph1 subunit: Relevance for Alzheimer’s disease. Science 2009, 324, 639–642. [Google Scholar] [CrossRef]
- Gengler, S.; Hamilton, A.; Holscher, C. Synaptic plasticity in the hippocampus of a APP/PS1 mouse model of Alzheimer’s disease is impaired in old but not young mice. PLoS ONE 2010, 5, e9764. [Google Scholar] [CrossRef]
- Rupp, N.J.; Wegenast-Braun, B.M.; Radde, R.; Calhoun, M.E.; Jucker, M. Early onset amyloid lesions lead to severe neuritic abnormalities and local, but not global neuron loss in APPPS1 transgenic mice. Neurobiol. Aging 2011, 32, 2324.e1–2324.e6. [Google Scholar] [CrossRef]
- Saito, T.; Matsuba, Y.; Mihira, N.; Takano, J.; Nilsson, P.; Itohara, S.; Iwata, N.; Saido, T.C. Single App knock-in mouse models of Alzheimer’s disease. Nat. Neurosci. 2014, 17, 661–663. [Google Scholar] [CrossRef]
- Udeochu, J.; Sayed, F.A.; Gan, L. TREM2 and Amyloid Beta: A Love-Hate Relationship. Neuron 2018, 97, 991–993. [Google Scholar] [CrossRef]
- Lee, C.Y.D.; Daggett, A.; Gu, X.; Jiang, L.-L.; Langfelder, P.; Li, X.; Wang, N.; Zhao, Y.; Park, C.S.; Cooper, Y.; et al. Elevated TREM2 Gene Dosage Reprograms Microglia Responsivity and Ameliorates Pathological Phenotypes in Alzheimer’s Disease Models. Neuron 2018, 97, 1032–1048.e5. [Google Scholar] [CrossRef]
- Song, W.M.; Joshita, S.; Zhou, Y.; Ulland, T.K.; Gilfillan, S.; Colonna, M. Humanized TREM2 mice reveal microglia-intrinsic and -extrinsic effects of R47H polymorphism. J. Exp. Med. 2018, 215, 745–760. [Google Scholar] [CrossRef]
- Barthelemy, N.R.; Horie, K.; Sato, C.; Bateman, R.J. Blood plasma phosphorylated-tau isoforms track CNS change in Alzheimer’s disease. J. Exp. Med. 2020, 217, e20200861. [Google Scholar] [CrossRef]
- Hall, A.M.; Roberson, E.D. Mouse models of Alzheimer’s disease. Brain Res. Bull. 2012, 88, 3–12. [Google Scholar] [CrossRef]
- Banerjee, R.; Rai, A.; Iyer, S.M.; Narwal, S.; Tare, M. Animal models in the study of Alzheimer’s disease and Parkinson’s disease: A historical perspective. Anim. Models Exp. Med. 2022, 5, 27–37. [Google Scholar] [CrossRef]
- Probst, A.; Götz, J.; Wiederhold, K.H.; Tolnay, M.; Mistl, C.; Jaton, A.L.; Hong, M.; Ishihara, T.; Lee, V.M.-Y.; Trojanowski, J.Q.; et al. Axonopathy and amyotrophy in mice transgenic for human four-repeat tau protein. Acta Neuropathol. 2000, 99, 469–481. [Google Scholar] [CrossRef]
- LaFerla, F.M.; Green, K.N. Animal Models of Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006320. [Google Scholar] [CrossRef]
- Goedert, M.; Eisenberg, D.S.; Crowther, R.A. Propagation of Tau Aggregates and Neurodegeneration. Annu. Rev. Neurosci. 2017, 40, 189–210. [Google Scholar] [CrossRef]
- Dawson, T.M.; Golde, T.E.; Lagier-Tourenne, C. Animal models of neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1370–1379. [Google Scholar] [CrossRef]
- Lewis, J.; McGowan, E.; Rockwood, J.; Melrose, H.; Nacharaju, P.; Van Slegtenhorst, M.; Gwinn-Hardy, K.; Paul Murphy, M.; Baker, M.; Yu, X.; et al. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat. Genet. 2000, 25, 402–405. [Google Scholar] [CrossRef]
- Hutton, M. Missense and splice site mutations in tau associated with FTDP-17: Multiple pathogenic mechanisms. Neurology 2001, 56, S21–S25. [Google Scholar] [CrossRef]
- Duff, K.; Knight, H.; Refolo, L.M.; Sanders, S.; Yu, X.; Picciano, M.; Malester, B.; Hutton, M.; Adamson, J.; Goedert, M.; et al. Characterization of pathology in transgenic mice over-expressing human genomic and cDNA tau transgenes. Neurobiol. Dis. 2000, 7, 87–98. [Google Scholar] [CrossRef]
- Hutton, M.; Lewis, J.; Dickson, D.; Yen, S.H.; McGowan, E. Analysis of tauopathies with transgenic mice. Trends Mol. Med. 2001, 7, 467–470. [Google Scholar] [CrossRef]
- Andorfer, C.; Kress, Y.; Espinoza, M.; Silva, R.D.; Tucker, K.L.; Barde, Y.-A.; Duff, K.; Davies, P. Hyperphosphorylation and aggregation of tau in mice expressing normal human tau isoforms. J. Neurochem. 2003, 86, 582–590. [Google Scholar] [CrossRef]
- Andorfer, C.; Acker, C.M.; Kress, Y.; Hof, P.R.; Duff, K.; Davies, P. Cell-cycle reentry and cell death in transgenic mice expressing nonmutant human tau isoforms. J. Neurosci. Off. J. Soc. Neurosci. 2005, 25, 5446–5454. [Google Scholar] [CrossRef]
- Ramsden, M.; Kotilinek, L.; Forster, C.; Paulson, J.; McGowan, E.; SantaCruz, K.; Guimaraes, A.; Yue, M.; Lewis, J.; Carlson, G.; et al. Age-dependent neurofibrillary tangle formation, neuron loss, and memory impairment in a mouse model of human tauopathy (P301L). J. Neurosci. Off. J. Soc. Neurosci. 2005, 25, 10637–10647. [Google Scholar] [CrossRef]
- Santacruz, K.; Lewis, J.; Spires, T.; Paulson, J.; Kotilinek, L.; Ingelsson, M.; Guimaraes, A.; DeTure, M.; Ramsden, M.; McGowan, E.; et al. Tau suppression in a neurodegenerative mouse model improves memory function. Science 2005, 309, 476–481. [Google Scholar] [CrossRef]
- Tucker, K.L.; Meyer, M.; Barde, Y.A. Neurotrophins are required for nerve growth during development. Nat. Neurosci. 2001, 4, 29–37. [Google Scholar] [CrossRef]
- Neddens, J.; Daurer, M.; Loeffler, T.; Alzola Aldamizetxebarria, S.; Flunkert, S.; Hutter-Paier, B. Constant Levels of Tau Phosphorylation in the Brain of htau Mice. Front. Mol. Neurosci. 2020, 13, 136. [Google Scholar] [CrossRef]
- Wenger, K.; Viode, A.; Schlaffner, C.N.; van Zalm, P.; Cheng, L.; Dellovade, T.; Langlois, X.; Bannon, A.; Chang, R.; Connors, T.R.; et al. Common mouse models of tauopathy reflect early but not late human disease. Mol. Neurodegener. 2023, 18, 10. [Google Scholar] [CrossRef]
- Billings, L.M.; Oddo, S.; Green, K.N.; McGaugh, J.L.; LaFerla, F.M. Intraneuronal Aβ causes the onset of early Alzheimer’s disease-related cognitive deficits in transgenic mice. Neuron 2005, 45, 675–688. [Google Scholar] [CrossRef]
- Yue, M.; Hanna, A.; Wilson, J.; Roder, H.; Janus, C. Sex difference in pathology and memory decline in rTg4510 mouse model of tauopathy. Neurobiol. Aging 2011, 32, 590–603. [Google Scholar] [CrossRef]
- Song, L.; Lu, S.X.; Ouyang, X.; Melchor, J.; Lee, J.; Terracina, G.; Wang, X.; Hyde, L.; Hess, J.F.; Parker, E.M.; et al. Analysis of tau post-translational modifications in rTg4510 mice, a model of tau pathology. Mol. Neurodegener. 2015, 10, 14. [Google Scholar] [CrossRef]
- Helboe, L.; Egebjerg, J.; Barkholt, P.; Volbracht, C. Early depletion of CA1 neurons and late neurodegeneration in a mouse tauopathy model. Brain Res. 2017, 1665, 22–35. [Google Scholar] [CrossRef]
- Hoover, B.R.; Reed, M.N.; Su, J.; Penrod, R.D.; Kotilinek, L.A.; Grant, M.K.; Pitstick, R.; Carlson, G.A.; Lanier, L.M.; Yuan, L.L.; et al. Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron 2010, 68, 1067–1081. [Google Scholar] [CrossRef]
- Gamache, J.; Benzow, K.; Forster, C.; Kemper, L.; Hlynialuk, C.; Furrow, E.; Ashe, K.H.; Koob, M.D. Factors other than hTau overexpression that contribute to tauopathy-like phenotype in rTg4510 mice. Nat. Commun. 2019, 10, 2479. [Google Scholar] [CrossRef]
- Yoshiyama, Y.; Higuchi, M.; Zhang, B.; Huang, S.M.; Iwata, N.; Saido, T.C.; Maeda, J.; Suhara, T.; Trojanowski, J.Q.; Lee, V.M. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 2007, 53, 337–351. [Google Scholar] [CrossRef]
- Oddo, S.; Caccamo, A.; Kitazawa, M.; Tseng, B.P.; LaFerla, F.M. Amyloid deposition precedes tangle formation in a triple transgenic model of Alzheimer’s disease. Neurobiol. Aging 2003, 24, 1063–1070. [Google Scholar] [CrossRef]
- Rodriguez, J.J.; Jones, V.C.; Tabuchi, M.; Allan, S.M.; Knight, E.M.; LaFerla, F.M.; Oddo, S.; Verkhratsky, A. Impaired adult neurogenesis in the dentate gyrus of a triple transgenic mouse model of Alzheimer’s disease. PLoS ONE 2008, 3, e2935. [Google Scholar] [CrossRef]
- Do, T.M.; Dodacki, A.; Alata, W.; Calon, F.; Nicolic, S.; Scherrmann, J.M.; Farinotti, R.; Bourasset, F. Age-Dependent Regulation of the Blood-Brain Barrier Influx/Efflux Equilibrium of Amyloid-beta Peptide in a Mouse Model of Alzheimer’s Disease (3xTg-AD). J. Alzheimer’s Dis. 2016, 49, 287–300. [Google Scholar] [CrossRef]
- Origlia, N.; Arancio, O.; Domenici, L.; Yan, S.S. MAPK, beta-amyloid and synaptic dysfunction: The role of RAGE. Expert. Rev. Neurother. 2009, 9, 1635–1645. [Google Scholar] [CrossRef]
- Padmanabhan, P.; Götz, J. Clinical relevance of animal models in aging-related dementia research. Nat. Aging 2023, 3, 481–493. [Google Scholar] [CrossRef]
- Badea, A.; Wu, W.; Shuff, J.; Wang, M.; Anderson, R.J.; Qi, Y.; Johnson, G.A.; Wilson, J.G.; Koudoro, S.; Garyfallidis, E.; et al. Identifying Vulnerable Brain Networks in Mouse Models of Genetic Risk Factors for Late Onset Alzheimer’s Disease. Front. Neuroinform 2019, 13, 72. [Google Scholar] [CrossRef]
- Bagyinszky, E.; Giau, V.V.; Shim, K.; Suk, K.; An, S.S.A.; Kim, S. Role of inflammatory molecules in the Alzheimer’s disease progression and diagnosis. J. Neurol. Sci. 2017, 376, 242–254. [Google Scholar] [CrossRef]
- Reiman, E.M.; Arboleda-Velasquez, J.F.; Quiroz, Y.T.; Huentelman, M.J.; Beach, T.G.; Caselli, R.J.; Chen, Y.; Su, Y.; Myers, A.J.; Hardy, J.; et al. Exceptionally low likelihood of Alzheimer’s dementia in APOE2 homozygotes from a 5000-person neuropathological study. Nat. Commun. 2020, 11, 667. [Google Scholar] [CrossRef]
- Chambers, J.K.; Uchida, K.; Nakayama, H. White matter myelin loss in the brains of aged dogs. Exp. Gerontol. 2012, 47, 263–269. [Google Scholar] [CrossRef]
- Huang, Y.; Mahley, R.W. Apolipoprotein E: Structure and function in lipid metabolism, neurobiology, and Alzheimer’s diseases. Neurobiol. Dis. 2014, 72 Pt A, 3–12. [Google Scholar] [CrossRef]
- Liu, C.C.; Liu, C.C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein E and Alzheimer disease: Risk, mechanisms and therapy. Nat. Rev. Neurol. 2013, 9, 106–118. [Google Scholar] [CrossRef]
- Bales, K.R. The value and limitations of transgenic mouse models used in drug discovery for Alzheimer’s disease: An update. Expert. Opin. Drug Discov. 2012, 7, 281–297. [Google Scholar] [CrossRef]
- Shineman, D.W.; Basi, G.S.; Bizon, J.L.; Colton, C.A.; Greenberg, B.D.; Hollister, B.A.; Lincecum, J.; Leblanc, G.G.; Lee, L.B.; Luo, F.; et al. Accelerating drug discovery for Alzheimer’s disease: Best practices for preclinical animal studies. Alzheimer’s Res. Ther. 2011, 3, 28. [Google Scholar] [CrossRef]
- Oblak, A.L.; Forner, S.; Territo, P.R.; Sasner, M.; Carter, G.W.; Howell, G.R.; Sukoff-Rizzo, S.J.; Logsdon, B.A.; Mangravite, L.M.; Mortazavi, A.; et al. Model organism development and evaluation for late-onset Alzheimer’s disease: MODEL-AD. Alzheimer’s Dement. 2020, 6, e12110. [Google Scholar] [CrossRef]
- Scheiber, I.F.; Mercer, J.F.; Dringen, R. Metabolism and functions of copper in brain. Prog. Neurobiol. 2014, 116, 33–57. [Google Scholar] [CrossRef]
- Pal, A.; Prasad, R. An overview of various mammalian models to study chronic copper intoxication associated Alzheimer’s disease like pathology. BioMetals 2015, 28, 1–9. [Google Scholar] [CrossRef]
- Singh, I.; Sagare, A.P.; Coma, M.; Perlmutter, D.; Gelein, R.; Bell, R.D.; Deane, R.J.; Zhong, E.; Parisi, M.; Ciszewski, J.; et al. Low levels of copper disrupt brain amyloid-beta homeostasis by altering its production and clearance. Proc. Natl. Acad. Sci. USA 2013, 110, 14771–14776. [Google Scholar] [CrossRef]
- Kong, G.K.-W.; Miles, L.A.; Crespi, G.A.; Morton, C.J.; Ng, H.L.; Barnham, K.J.; McKinstry, W.J.; Cappai, R.; Parker, M.W. Copper binding to the Alzheimer’s disease amyloid precursor protein. Eur. Biophys. J. 2008, 37, 269–279. [Google Scholar] [CrossRef]
- Huang, X.; Cuajungco, M.P.; Atwood, C.S.; Hartshorn, M.A.; Tyndall, J.D.; Hanson, G.R.; Stokes, K.C.; Leopold, M.; Multhaup, G.; Goldstein, L.E. Cu (II) potentiation of Alzheimer Aβ neurotoxicity: Correlation with cell-free hydrogen peroxide production and metal reduction. J. Biol. Chem. 1999, 274, 37111–37116. [Google Scholar] [CrossRef]
- White, A.R.; Reyes, R.; Mercer, J.F.; Camakaris, J.; Zheng, H.; Bush, A.I.; Multhaup, G.; Beyreuther, K.; Masters, C.L.; Cappai, R. Copper levels are increased in the cerebral cortex and liver of APP and APLP2 knockout mice. Brain Res. 1999, 842, 439–444. [Google Scholar] [CrossRef]
- Sparks, D.L.; Friedland, R.; Petanceska, S.; Schreurs, B.G.; Shi, J.; Perry, G.; Smith, M.A.; Sharma, A.; Derosa, S.; Ziolkowski, C.; et al. Trace copper levels in the drinking water, but not zinc or aluminum influence CNS Alzheimer-like pathology. J. Nutr. Health Aging 2006, 10, 247–254. [Google Scholar]
- Lu, J.; Zheng, Y.; Wu, D.; Sun, D.; Shan, Q.; Fan, S. Trace amounts of copper induce neurotoxicity in the cholesterol-fed mice through apoptosis. FEBS Lett. 2006, 580, 6730–6740. [Google Scholar] [CrossRef]
- Arnal, N.; Morel, G.R.; de Alaniz, M.J.T.; Castillo, O.; Marra, C.A. Role of Copper and Cholesterol Association in the Neurodegenerative Process. Int. J. Alzheimer’s Dis. 2013, 2013, 414817. [Google Scholar] [CrossRef]
- Arnal, N.; Dominici, L.; Tacconi, M.J.T.d.; Marra, C.A. Copper-induced alterations in rat brain depends on route of overload and basal copper levels. Nutrition 2014, 30, 96–106. [Google Scholar] [CrossRef]
- Chen, Z.Y.; Zhang, Y. Animal models of Alzheimer’s disease: Applications, evaluation, and perspectives. Zool. Res. 2022, 43, 1026–1040. [Google Scholar] [CrossRef]
- Krstic, D.; Madhusudan, A.; Doehner, J.; Vogel, P.; Notter, T.; Imhof, C.; Manalastas, A.; Hilfiker, M.; Pfister, S.; Schwerdel, C. Systemic immune challenges trigger and drive Alzheimer-like neuropathology in mice. J. Neuroinflammation 2012, 9, 151. [Google Scholar] [CrossRef]
- Ahlijanian, M.K.; Barrezueta, N.X.; Williams, R.D.; Jakowski, A.; Kowsz, K.P.; McCarthy, S.; Coskran, T.; Carlo, A.; Seymour, P.A.; Burkhardt, J.E. Hyperphosphorylated tau and neurofilament and cytoskeletal disruptions in mice overexpressing human p25, an activator of cdk5. Proc. Natl. Acad. Sci. USA 2000, 97, 2910–2915. [Google Scholar] [CrossRef]
- Sundaram, J.R.; Chan, E.S.; Poore, C.P.; Pareek, T.K.; Cheong, W.F.; Shui, G.; Tang, N.; Low, C.-M.; Wenk, M.R.; Kesavapany, S. Cdk5/p25-induced cytosolic PLA2-mediated lysophosphatidylcholine production regulates neuroinflammation and triggers neurodegeneration. J. Neurosci. 2012, 32, 1020–1034. [Google Scholar] [CrossRef]
- Fischer, A.; Sananbenesi, F.; Pang, P.T.; Lu, B.; Tsai, L.-H. Opposing roles of transient and prolonged expression of p25 in synaptic plasticity and hippocampus-dependent memory. Neuron 2005, 48, 825–838. [Google Scholar] [CrossRef]
- Muyllaert, D.; Terwel, D.; Kremer, A.; Sennvik, K.; Borghgraef, P.; Devijver, H.; Dewachter, I.; Van Leuven, F. Neurodegeneration and neuroinflammation in cdk5/p25-inducible mice: A model for hippocampal sclerosis and neocortical degeneration. Am. J. Pathol. 2008, 172, 470–485. [Google Scholar] [CrossRef]
- Tapia, R.; Peña, F.; Arias, C. Neurotoxic and synaptic effects of okadaic acid, an inhibitor of protein phosphatases. Neurochem. Res. 1999, 24, 1423–1430. [Google Scholar] [CrossRef]
- Cohen, P.T.; Brewis, N.D.; Hughes, V.; Mann, D.J. Protein serine/threonine phosphatases; an expanding family. FEBS Lett. 1990, 268, 355–359. [Google Scholar] [CrossRef]
- Sontag, J.-M.; Sontag, E. Protein phosphatase 2A dysfunction in Alzheimer’s disease. Front. Mol. Neurosci. 2014, 7, 16. [Google Scholar] [CrossRef]
- Costa, A.P.; Tramontina, A.C.; Biasibetti, R.; Batassini, C.; Lopes, M.W.; Wartchow, K.M.; Bernardi, C.; Tortorelli, L.S.; Leal, R.B.; Gonçalves, C.-A. Neuroglial alterations in rats submitted to the okadaic acid-induced model of dementia. Behav. Brain Res. 2012, 226, 420–427. [Google Scholar] [CrossRef]
- Kamat, P.K.; Tota, S.; Saxena, G.; Shukla, R.; Nath, C. Okadaic acid (ICV) induced memory impairment in rats: A suitable experimental model to test anti-dementia activity. Brain Res. 2010, 1309, 66–74. [Google Scholar] [CrossRef]
- Arendt, T.; Holzer, M.; Fruth, R.; Brückner, M.; Gärtner, U. Phosphorylation of tau, Aβ-formation, and apoptosis after in vivo inhibition of PP-1 and PP-2A. Neurobiol. Aging 1998, 19, 3–13. [Google Scholar] [CrossRef]
- Yagi, H.; Katoh, S.; Akiguchi, I.; Takeda, T. Age-related deterioration of ability of acquisition in memory and learning in senescence accelerated mouse: SAM-P/8 as an animal model of disturbances in recent memory. Brain Res. 1988, 474, 86–93. [Google Scholar] [CrossRef]
- Tanisawa, K.; Mikami, E.; Fuku, N.; Honda, Y.; Honda, S.; Ohsawa, I.; Ito, M.; Endo, S.; Ihara, K.; Ohno, K. Exome sequencing of senescence-accelerated mice (SAM) reveals deleterious mutations in degenerative disease-causing genes. BMC Genom. 2013, 14, 248. [Google Scholar] [CrossRef]
- Stefanova, N.A.; Maksimova, K.Y.; Rudnitskaya, E.A.; Muraleva, N.A.; Kolosova, N.G. Association of cerebrovascular dysfunction with the development of Alzheimer’s disease-like pathology in OXYS rats. BMC Genom. 2018, 19, 51–63. [Google Scholar] [CrossRef]
- Kolosova, N.; Stefanova, N.; Korbolina, E.; Fursova, A.Z.; Kozhevnikova, O. Senescence-accelerated OXYS rats: A genetic model of premature aging and age-related diseases. Adv. Gerontol. 2014, 4, 294–298. [Google Scholar] [CrossRef]
- Liu, B.; Liu, J.; Shi, J.S. SAMP8 Mice as a Model of Age-Related Cognition Decline with Underlying Mechanisms in Alzheimer’s Disease. J. Alzheimer’s Dis. 2020, 75, 385–395. [Google Scholar] [CrossRef]
- Kepchia, D.; Huang, L.; Currais, A.; Liang, Z.; Fischer, W.; Maher, P. The Alzheimer’s disease drug candidate J147 decreases blood plasma fatty acid levels via modulation of AMPK/ACC1 signaling in the liver. Biomed. Pharmacother. 2022, 147, 112648. [Google Scholar] [CrossRef]
- Codony, S.; Pont, C.; Grinan-Ferre, C.; Di Pede-Mattatelli, A.; Calvo-Tusell, C.; Feixas, F.; Osuna, S.; Jarne-Ferrer, J.; Naldi, M.; Bartolini, M.; et al. Discovery and In Vivo Proof of Concept of a Highly Potent Dual Inhibitor of Soluble Epoxide Hydrolase and Acetylcholinesterase for the Treatment of Alzheimer’s Disease. J. Med. Chem. 2022, 65, 4909–4925. [Google Scholar] [CrossRef]
- Kolosova, N.G.; Stefanova, N.A.; Korbolina, E.E.; Fursova, A.; Kozhevnikova, O.S. The senescence-accelerated oxys rats—A genetic model of premature aging and age-dependent degenerative diseases. Adv. Gerontol. 2014, 27, 336–340. [Google Scholar]
- Hurley, M.J.; Deacon, R.M.J.; Beyer, K.; Ioannou, E.; Ibáñez, A.; Teeling, J.L.; Cogram, P. The long-lived Octodon degus as a rodent drug discovery model for Alzheimer’s and other age-related diseases. Pharmacol. Ther. 2018, 188, 36–44. [Google Scholar] [CrossRef]
- Inestrosa, N.C.; Reyes, A.E.; Chacón, M.A.; Cerpa, W.; Villalón, A.; Montiel, J.; Merabachvili, G.; Aldunate, R.; Bozinovic, F.; Aboitiz, F. Human-like rodent amyloid-beta-peptide determines Alzheimer pathology in aged wild-type Octodon degu. Neurobiol. Aging 2005, 26, 1023–1028. [Google Scholar] [CrossRef]
- Braidy, N.; Poljak, A.; Jayasena, T.; Mansour, H.; Inestrosa, N.C.; Sachdev, P.S. Accelerating Alzheimer’s research through ‘natural’ animal models. Curr. Opin. Psychiatry 2015, 28, 155–164. [Google Scholar] [CrossRef]
- Steffen, J.; Krohn, M.; Paarmann, K.; Schwitlick, C.; Brüning, T.; Marreiros, R.; Müller-Schiffmann, A.; Korth, C.; Braun, K.; Pahnke, J. Revisiting rodent models: Octodon degus as Alzheimer’s disease model? Acta Neuropathol. Commun. 2016, 4, 91. [Google Scholar] [CrossRef]
- hAβ-KI Alzforum. Available online: https://www.alzforum.org/research-models/hav-ki (accessed on 3 September 2023).
- Baglietto-Vargas, D.; Forner, S.; Cai, L.; Martini, A.C.; Trujillo-Estrada, L.; Swarup, V.; Nguyen, M.M.T.; Do Huynh, K.; Javonillo, D.I.; Tran, K.M.; et al. Generation of a humanized Aβ expressing mouse demonstrating aspects of Alzheimer’s disease-like pathology. Nat. Commun. 2021, 12, 2421. [Google Scholar] [CrossRef]
- Xia, D.; Lianoglou, S.; Sandmann, T.; Calvert, M.; Suh, J.H.; Thomsen, E.; Dugas, J.; Pizzo, M.E.; DeVos, S.L.; Earr, T.K. Novel App knock-in mouse model shows key features of amyloid pathology and reveals profound metabolic dysregulation of microglia. Mol. Neurodegener. 2022, 17, 41. [Google Scholar] [CrossRef]
- Jankowsky, J.L.; Younkin, L.H.; Gonzales, V.; Fadale, D.J.; Slunt, H.H.; Lester, H.A.; Younkin, S.G.; Borchelt, D.R. Rodent Aβ modulates the solubility and distribution of amyloid deposits in transgenic mice. J. Biol. Chem. 2007, 282, 22707–22720. [Google Scholar] [CrossRef]
- Newcombe, E.A.; Camats-Perna, J.; Silva, M.L.; Valmas, N.; Huat, T.J.; Medeiros, R. Inflammation: The link between comorbidities, genetics, and Alzheimer’s disease. J. Neuroinflammation 2018, 15, 276. [Google Scholar] [CrossRef]
- Le Bras, A. A new mouse model to study late-onset Alzheimer’s disease. Lab. Anim. 2021, 50, 151. [Google Scholar] [CrossRef]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef]
- Squire, L.R.; Knowlton, B.; Musen, G. The structure and organization of memory. Annu. Rev. Psychol. 1993, 44, 453–495. [Google Scholar] [CrossRef]
- Bussière, T.; Friend, P.; Sadeghi, N.a.; Wicinski, B.; Lin, G.; Bouras, C.; Giannakopoulos, P.; Robakis, N.; Morrison, J.; Perl, D. Stereologic assessment of the total cortical volume occupied by amyloid deposits and its relationship with cognitive status in aging and Alzheimer’s disease. Neuroscience 2002, 112, 75–91. [Google Scholar] [CrossRef]
- Wang, L.; Miller, J.P.; Gado, M.H.; McKeel, D.W.; Rothermich, M.; Miller, M.I.; Morris, J.C.; Csernansky, J.G. Abnormalities of hippocampal surface structure in very mild dementia of the Alzheimer type. Neuroimage 2006, 30, 52–60. [Google Scholar] [CrossRef]
- Puzzo, D.; Lee, L.; Palmeri, A.; Calabrese, G.; Arancio, O. Behavioral assays with mouse models of Alzheimer’s disease: Practical considerations and guidelines. Biochem. Pharmacol. 2014, 88, 450–467. [Google Scholar] [CrossRef]
- Clark, R.E.; Squire, L.R. Similarity in form and function of the hippocampus in rodents, monkeys, and humans. Proc. Natl. Acad. Sci. USA 2013, 110, 10365–10370. [Google Scholar] [CrossRef]
- Watrous, A.J.; Lee, D.J.; Izadi, A.; Gurkoff, G.G.; Shahlaie, K.; Ekstrom, A.D. A comparative study of human and rat hippocampal low-frequency oscillations during spatial navigation. Hippocampus 2013, 23, 656–661. [Google Scholar] [CrossRef]
- Vitek, M.P.; Araujo, J.A.; Fossel, M.; Greenberg, B.D.; Howell, G.R.; Rizzo, S.J.S.; Seyfried, N.T.; Tenner, A.J.; Territo, P.R.; Windisch, M.; et al. Translational animal models for Alzheimer’s disease: An Alzheimer’s Association Business Consortium Think Tank. Alzheimer’s Dement. Transl. Res. Clin. 2020, 6, e12114. [Google Scholar] [CrossRef]
- Bruce-Keller, A.J.; Gupta, S.; Knight, A.G.; Beckett, T.L.; McMullen, J.M.; Davis, P.R.; Murphy, M.P.; Eldik, L.J.V.; Clair, D.S.; Keller, J.N. Cognitive impairment in humanized APP × PS1 mice is linked to Aβ1–42 and NOX activation. Neurobiol. Dis. 2011, 44, 317–326. [Google Scholar] [CrossRef]
- Murphy, M.P.; Beckett, T.L.; Ding, Q.; Patel, E.; Markesbery, W.R.; St Clair, D.K.; LeVine, H.r.; Keller, J.N. Abeta solubility and deposition during AD progression and in APPxPS-1 knock-in mice. Neurobiol. Dis. 2007, 27, 301–311. [Google Scholar] [CrossRef]
- Xu, W.; Xu, F.; Anderson, M.E.; Kotarba, A.E.; Davis, J.; Robinson, J.K.; Van Nostrand, W.E. Cerebral microvascular rather than parenchymal amyloid-β protein pathology promotes early cognitive impairment in transgenic mice. J. Alzheimer’s Dis. 2014, 38, 621–632. [Google Scholar] [CrossRef]
- Fisher, E.M.C.; Bannerman, D.M. Mouse models of neurodegeneration: Know your question, know your mouse. Sci. Transl. Med. 2019, 11, eaaq1818. [Google Scholar] [CrossRef]
- Sabbagh, J.J.; Kinney, J.W.; Cummings, J.L. Animal systems in the development of treatments for Alzheimer’s disease: Challenges, methods, and implications. Neurobiol. Aging 2013, 34, 169–183. [Google Scholar] [CrossRef]
- Sasaguri, H.; Nilsson, P.; Hashimoto, S.; Nagata, K.; Saito, T.; De Strooper, B.; Hardy, J.; Vassar, R.; Winblad, B.; Saido, T.C. APP mouse models for Alzheimer’s disease preclinical studies. EMBO J. 2017, 36, 2473–2487. [Google Scholar] [CrossRef]
- Webster, S.J.; Bachstetter, A.D.; Nelson, P.T.; Schmitt, F.A.; Van Eldik, L.J. Using mice to model Alzheimer’s dementia: An overview of the clinical disease and the preclinical behavioral changes in 10 mouse models. Front. Genet. 2014, 5, 88. [Google Scholar] [CrossRef]
- Foley, A.M.; Ammar, Z.M.; Lee, R.H.; Mitchell, C.S. Systematic review of the relationship between amyloid-β levels and measures of transgenic mouse cognitive deficit in Alzheimer’s disease. J. Alzheimer’s Dis. 2015, 44, 787–795. [Google Scholar] [CrossRef]
- Higuchi, M.; Iwata, N.; Matsuba, Y.; Takano, J.; Suemoto, T.; Maeda, J.; Ji, B.; Ono, M.; Staufenbiel, M.; Suhara, T. Mechanistic involvement of the calpain-calpastatin system in Alzheimer neuropathology. FASEB J. 2012, 26, 1204–1217. [Google Scholar] [CrossRef]
- Jack, C.R., Jr.; Marjanska, M.; Wengenack, T.M.; Reyes, D.A.; Curran, G.L.; Lin, J.; Preboske, G.M.; Poduslo, J.F.; Garwood, M. Magnetic resonance imaging of Alzheimer’s pathology in the brains of living transgenic mice: A new tool in Alzheimer’s disease research. Neuroscientist 2007, 13, 38–48. [Google Scholar] [CrossRef]
- Oh, S.J.; Lee, H.-J.; Jeong, Y.J.; Nam, K.R.; Kang, K.J.; Han, S.J.; Lee, K.C.; Lee, Y.J.; Choi, J.Y. Evaluation of the neuroprotective effect of taurine in Alzheimer’s disease using functional molecular imaging. Sci. Rep. 2020, 10, 15551. [Google Scholar] [CrossRef]
- Chiquita, S.; Ribeiro, M.; Castelhano, J.; Oliveira, F.; Sereno, J.; Batista, M.; Abrunhosa, A.; Rodrigues-Neves, A.C.; Carecho, R.; Baptista, F.; et al. A longitudinal multimodal in vivo molecular imaging study of the 3xTg-AD mouse model shows progressive early hippocampal and taurine loss. Hum. Mol. Genet. 2019, 28, 2174–2188. [Google Scholar] [CrossRef]
- Guell-Bosch, J.; Lope-Piedrafita, S.; Esquerda-Canals, G.; Montoliu-Gaya, L.; Villegas, S. Progression of Alzheimer’s disease and effect of scFv-h3D6 immunotherapy in the 3xTg-AD mouse model: An in vivo longitudinal study using Magnetic Resonance Imaging and Spectroscopy. NMR Biomed. 2020, 33, e4263. [Google Scholar] [CrossRef]
- Mlynarik, V.; Cacquevel, M.; Sun-Reimer, L.; Janssens, S.; Cudalbu, C.; Lei, H.; Schneider, B.L.; Aebischer, P.; Gruetter, R. Proton and phosphorus magnetic resonance spectroscopy of a mouse model of Alzheimer’s disease. J. Alzheimer’s Dis. 2012, 31 (Suppl. S3), S87–S99. [Google Scholar] [CrossRef]
- Cotman, C.W.; Head, E. The canine (dog) model of human aging and disease: Dietary, environmental and immunotherapy approaches. J. Alzheimer’s Dis. JAD 2008, 15, 685–707. [Google Scholar] [CrossRef]
- Head, E. A canine model of human aging and Alzheimer’s disease. Biochim. Biophys. Acta 2013, 1832, 1384–1389. [Google Scholar] [CrossRef]
- Wallace, T.L.; Ballard, T.M.; Glavis-Bloom, C. Animal paradigms to assess cognition with translation to humans. Handb. Exp. Pharmacol. 2015, 228, 27–57. [Google Scholar]
- Perez, S.E.; Raghanti, M.A.; Hof, P.R.; Kramer, L.; Ikonomovic, M.D.; Lacor, P.N.; Erwin, J.M.; Sherwood, C.C.; Mufson, E.J. Alzheimer’s disease pathology in the neocortex and hippocampus of the western lowland gorilla (Gorilla gorilla gorilla). J. Comp. Neurol. 2013, 521, 4318–4338. [Google Scholar] [CrossRef]
- Perez, S.E.; Sherwood, C.C.; Cranfield, M.R.; Erwin, J.M.; Mudakikwa, A.; Hof, P.R.; Mufson, E.J. Early Alzheimer’s disease-type pathology in the frontal cortex of wild mountain gorillas (Gorilla beringei beringei). Neurobiol. Aging 2016, 39, 195–201. [Google Scholar] [CrossRef]
- Edler, M.K.; Sherwood, C.C.; Meindl, R.S.; Hopkins, W.D.; Ely, J.J.; Erwin, J.M.; Mufson, E.J.; Hof, P.R.; Raghanti, M.A. Aged chimpanzees exhibit pathologic hallmarks of Alzheimer’s disease. Neurobiol. Aging 2017, 59, 107–120. [Google Scholar] [CrossRef]
- Oikawa, N.; Kimura, N.; Yanagisawa, K. Alzheimer-type tau pathology in advanced aged nonhuman primate brains harboring substantial amyloid deposition. Brain Res. 2010, 1315, 137–149. [Google Scholar] [CrossRef]
- Gearing, M.; Rebeck, G.W.; Hyman, B.T.; Tigges, J.; Mirra, S.S. Neuropathology and apolipoprotein E profile of aged chimpanzees: Implications for Alzheimer disease. Proc. Natl. Acad. Sci. USA 1994, 91, 9382–9386. [Google Scholar] [CrossRef]
- Gearing, M.; Tigges, J.; Mori, H.; Mirra, S.S. Beta-Amyloid (A beta) deposition in the brains of aged orangutans. Neurobiol. Aging 1997, 18, 139–146. [Google Scholar] [CrossRef]
- Heuer, E.; Rosen, R.F.; Cintron, A.; Walker, L.C. Nonhuman primate models of Alzheimer-like cerebral proteopathy. Curr. Pharm. Des. 2012, 18, 1159–1169. [Google Scholar] [CrossRef]
- Kitt Cheryl, A.; Price Donald, L.; Struble Robert, G.; Cork Linda, C.; Wainer Bruce, H.; Becher Mark, W.; Mobley William, C. Evidence for Cholinergic Neurites in Senile Plaques. Science 1984, 226, 1443–1445. [Google Scholar] [CrossRef]
- Mufson, E.J.; Benzing, W.C.; Cole, G.M.; Wang, H.; Emerich, D.F.; Sladek, J.R.; Morrison, J.H.; Kordower, J.H. Apolipoprotein E-immunoreactivity in aged rhesus monkey cortex: Colocalization with amyloid plaques. Neurobiol. Aging 1994, 15, 621–627. [Google Scholar] [CrossRef]
- Didier, E.S.; MacLean, A.G.; Mohan, M.; Didier, P.J.; Lackner, A.A.; Kuroda, M.J. Contributions of Nonhuman Primates to Research on Aging. Vet. Pathol. 2016, 53, 277–290. [Google Scholar] [CrossRef]
- Rodriguez-Callejas, J.D.; Fuchs, E.; Perez-Cruz, C. Evidence of Tau Hyperphosphorylation and Dystrophic Microglia in the Common Marmoset. Front. Aging Neurosci. 2016, 8, 315. [Google Scholar] [CrossRef]
- Peters, A.; Rosene, D.L.; Moss, M.B.; Kemper, T.L.; Abraham, C.R.; Tigges, J.; Albert, M.S. Neurobiological bases of age-related cognitive decline in the rhesus monkey. J. Neuropathol. Exp. Neurol. 1996, 55, 861–874. [Google Scholar] [CrossRef]
- Price, D.L.; Martin, L.J.; Sisodia, S.S.; Wagster, M.V.; Koo, E.H.; Walker, L.C.; Koliatsos, V.E.; Cork, L.C. Aged non-human primates: An animal model of age-associated neurodegenerative disease. Brain Pathol. 1991, 1, 287–296. [Google Scholar] [CrossRef]
- Emborg, M.E. Nonhuman Primate Models of Neurodegenerative Disorders. ILAR J. 2017, 58, 190–201. [Google Scholar] [CrossRef]
- Geula, C.; Nagykery, N.; Wu, C.-K. Amyloid-β deposits in the cerebral cortex of the aged common marmoset (Callithrix jacchus): Incidence and chemical composition. Acta Neuropathol. 2002, 103, 48–58. [Google Scholar] [CrossRef]
- Zhao, Q.; Lu, J.; Yao, Z.; Wang, S.; Zhu, L.; Wang, J.; Chen, B. Upregulation of Aβ42 in the brain and bodily fluids of rhesus monkeys with aging. J. Mol. Neurosci. 2017, 61, 79–87. [Google Scholar] [CrossRef]
- Baker, H.; Ridley, R.; Duchen, L.; Crow, T.; Bruton, C. Evidence for the experimental transmission of cerebral beta-amyloidosis to primates. Int. J. Exp. Pathol. 1993, 74, 441. [Google Scholar]
- Kastman, E.K.; Willette, A.A.; Coe, C.L.; Bendlin, B.B.; Kosmatka, K.J.; McLaren, D.G.; Xu, G.; Canu, E.; Field, A.S.; Alexander, A.L. A calorie-restricted diet decreases brain iron accumulation and preserves motor performance in old rhesus monkeys. J. Neurosci. 2012, 32, 11897–11904. [Google Scholar] [CrossRef]
- Sridharan, A.; Bendlin, B.B.; Gallagher, C.L.; Oh, J.M.; Willette, A.A.; Alexander, A.L.; Kemnitz, J.W.; Colman, R.J.; Weindruch, R.H.; Johnson, S.C. Effect of age and calorie restriction on corpus callosal integrity in rhesus macaques: A fiber tractography study. Neurosci. Lett. 2014, 569, 38–42. [Google Scholar] [CrossRef]
- Willette, A.; Coe, C.; Birdsill, A.; Bendlin, B.; Colman, R.; Alexander, A.; Allison, D.; Weindruch, R.; Johnson, S. Interleukin-8 and interleukin-10, brain volume and microstructure, and the influence of calorie restriction in old rhesus macaques. Age 2013, 35, 2215–2227. [Google Scholar] [CrossRef]
- Fang, R.; Xia, C.; Close, J.L.; Zhang, M.; He, J.; Huang, Z.; Halpern, A.R.; Long, B.; Miller, J.A.; Lein, E.S.; et al. Conservation and divergence of cortical cell organization in human and mouse revealed by MERFISH. Science 2022, 377, 56–62. [Google Scholar] [CrossRef]
- Li, B.; He, D.-J.; Li, X.-J.; Guo, X.-Y. Modeling neurodegenerative diseases using non-human primates: Advances and challenges. Ageing Neurodegener. Dis. 2022, 2, 12. [Google Scholar] [CrossRef]
- Frye, B.M.; Craft, S.; Register, T.C.; Kim, J.; Whitlow, C.T.; Barcus, R.A.; Lockhart, S.N.; Sai, K.K.S.; Shively, C.A. Early Alzheimer’s disease-like reductions in gray matter and cognitive function with aging in nonhuman primates. Alzheimer’s Dement. 2022, 8, e12284. [Google Scholar] [CrossRef]
- Visser, P.J.; Reus, L.M.; Gobom, J.; Jansen, I.; Dicks, E.; van der Lee, S.J.; Tsolaki, M.; Verhey, F.R.J.; Popp, J.; Martinez-Lage, P.; et al. Cerebrospinal fluid tau levels are associated with abnormal neuronal plasticity markers in Alzheimer’s disease. Mol. Neurodegener. 2022, 17, 27. [Google Scholar] [CrossRef]
- An, S.S.A.; Hulme, J.P. Plasma amyloid-beta oligomer and phosphorylated tau: Diagnostic tools for progressive Alzheimer’s disease. Neural Regen. Res. 2023, 18, 2391–2392. [Google Scholar] [CrossRef]
- Li, Y.; Xu, D.; Chan, H.N.; Poon, C.Y.; Ho, S.L.; Li, H.W.; Wong, M.S. Dual-Modal NIR-Fluorophore Conjugated Magnetic Nanoparticle for Imaging Amyloid-beta Species In Vivo. Small 2018, 14, e1800901. [Google Scholar] [CrossRef]
- Peng, C.; Wang, X.; Li, Y.; Li, H.-W.; Wong, M.S. Versatile fluorescent probes for near-infrared imaging of amyloid-β species in Alzheimer’s disease mouse model. J. Mater. Chem. B 2019, 7, 1986–1995. [Google Scholar] [CrossRef]
- Kawarabayashi, T.; Younkin, L.H.; Saido, T.C.; Shoji, M.; Ashe, K.H.; Younkin, S.G. Age-dependent changes in brain, CSF, and plasma amyloid β protein in the Tg2576 transgenic mouse model of Alzheimer’s disease. J. Neurosci. 2001, 21, 372–381. [Google Scholar] [CrossRef]
- Cho, S.M.; Lee, S.; Yang, S.-H.; Kim, H.Y.; Lee, M.J.; Kim, H.V.; Kim, J.; Baek, S.; Yun, J.; Kim, D. Age-dependent inverse correlations in CSF and plasma amyloid-β (1–42) concentrations prior to amyloid plaque deposition in the brain of 3xTg-AD mice. Sci. Rep. 2016, 6, 20185. [Google Scholar] [CrossRef]
- Qin, T.; Prins, S.; Groeneveld, G.J.; Van Westen, G.; de Vries, H.E.; Wong, Y.C.; Bischoff, L.J.M.; de Lange, E.C.M. Utility of Animal Models to Understand Human Alzheimer’s Disease, Using the Mastermind Research Approach to Avoid Unnecessary Further Sacrifices of Animals. Int. J. Mol. Sci. 2020, 21, 3158. [Google Scholar] [CrossRef]
- DeMattos, R.B.; Bales, K.R.; Parsadanian, M.; O’Dell, M.A.; Foss, E.M.; Paul, S.M.; Holtzman, D.M. Plaque-associated disruption of CSF and plasma amyloid-β (Aβ) equilibrium in a mouse model of Alzheimer’s disease. J. Neurochem. 2002, 81, 229–236. [Google Scholar] [CrossRef]
- Fagan, A.M.; Mintun, M.A.; Mach, R.H.; Lee, S.Y.; Dence, C.S.; Shah, A.R.; LaRossa, G.N.; Spinner, M.L.; Klunk, W.E.; Mathis, C.A. Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid Aβ42 in humans. Ann. Neurol. 2006, 59, 512–519. [Google Scholar] [CrossRef]
- Dubois, B. The emergence of a new conceptual framework for Alzheimer’s disease. J. Alzheimer’s Dis. 2018, 62, 1059–1066. [Google Scholar] [CrossRef]
- Maia, L.F.; Kaeser, S.A.; Reichwald, J.; Hruscha, M.; Martus, P.; Staufenbiel, M.; Jucker, M. Changes in amyloid-β and Tau in the cerebrospinal fluid of transgenic mice overexpressing amyloid precursor protein. Sci. Transl. Med. 2013, 5, re192–re194. [Google Scholar] [CrossRef]
- Li, L.; Jiang, Y.; Hu, W.; Tung, Y.C.; Dai, C.; Chu, D.; Gong, C.-X.; Iqbal, K.; Liu, F. Pathological alterations of tau in Alzheimer’s disease and 3xTg-AD mouse brains. Mol. Neurobiol. 2019, 56, 6168–6183. [Google Scholar] [CrossRef]
- Park, J.-C.; Han, S.-H.; Yi, D.; Byun, M.S.; Lee, J.H.; Jang, S.; Ko, K.; Jeon, S.Y.; Lee, Y.-S.; Kim, Y.K. Plasma tau/amyloid-β1–42 ratio predicts brain tau deposition and neurodegeneration in Alzheimer’s disease. Brain 2019, 142, 771–786. [Google Scholar] [CrossRef]
- Antonell, A.; Mansilla, A.; Rami, L.; Lladó, A.; Iranzo, A.; Olives, J.; Balasa, M.; Sánchez-Valle, R.; Molinuevo, J.L. Cerebrospinal fluid level of YKL-40 protein in preclinical and prodromal Alzheimer’s disease. J. Alzheimer’s Dis. 2014, 42, 901–908. [Google Scholar] [CrossRef]
- Zetterberg, H.; Skillbäck, T.; Mattsson, N.; Trojanowski, J.Q.; Portelius, E.; Shaw, L.M.; Weiner, M.W.; Blennow, K.; Initiative, A.s.D.N. Association of cerebrospinal fluid neurofilament light concentration with Alzheimer disease progression. JAMA Neurol. 2016, 73, 60–67. [Google Scholar] [CrossRef]
- Palmqvist, S.; Janelidze, S.; Stomrud, E.; Zetterberg, H.; Karl, J.; Zink, K.; Bittner, T.; Mattsson, N.; Eichenlaub, U.; Blennow, K. Performance of fully automated plasma assays as screening tests for Alzheimer disease–related β-amyloid status. JAMA Neurol. 2019, 76, 1060–1069. [Google Scholar] [CrossRef]
- Lewczuk, P.; Ermann, N.; Andreasson, U.; Schultheis, C.; Podhorna, J.; Spitzer, P.; Maler, J.M.; Kornhuber, J.; Blennow, K.; Zetterberg, H. Plasma neurofilament light as a potential biomarker of neurodegeneration in Alzheimer’s disease. Alzheimer’s Res. Ther. 2018, 10, 71. [Google Scholar] [CrossRef]
- Abu-Rumeileh, S.; Steinacker, P.; Polischi, B.; Mammana, A.; Bartoletti-Stella, A.; Oeckl, P.; Baiardi, S.; Zenesini, C.; Huss, A.; Cortelli, P. CSF biomarkers of neuroinflammation in distinct forms and subtypes of neurodegenerative dementia. Alzheimer’s Res. Ther. 2020, 12, 2. [Google Scholar] [CrossRef]
- Bacioglu, M.; Maia, L.F.; Preische, O.; Schelle, J.; Apel, A.; Kaeser, S.A.; Schweighauser, M.; Eninger, T.; Lambert, M.; Pilotto, A. Neurofilament light chain in blood and CSF as marker of disease progression in mouse models and in neurodegenerative diseases. Neuron 2016, 91, 56–66. [Google Scholar] [CrossRef]
- Öhrfelt, A.; Brinkmalm, A.; Dumurgier, J.; Zetterberg, H.; Bouaziz-Amar, E.; Hugon, J.; Paquet, C.; Blennow, K. A novel ELISA for the measurement of cerebrospinal fluid SNAP-25 in patients with Alzheimer’s disease. Neuroscience 2019, 420, 136–144. [Google Scholar] [CrossRef]
- Brinkmalm, A.; Brinkmalm, G.; Honer, W.G.; Frölich, L.; Hausner, L.; Minthon, L.; Hansson, O.; Wallin, A.; Zetterberg, H.; Blennow, K. SNAP-25 is a promising novel cerebrospinal fluid biomarker for synapse degeneration in Alzheimer’s disease. Mol. Neurodegener. 2014, 9, 53. [Google Scholar] [CrossRef]
- Hong, S.; Beja-Glasser, V.F.; Nfonoyim, B.M.; Frouin, A.; Li, S.; Ramakrishnan, S.; Merry, K.M.; Shi, Q.; Rosenthal, A.; Barres, B.A. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 2016, 352, 712–716. [Google Scholar] [CrossRef]
- Caruso, D.; Barron, A.M.; Brown, M.A.; Abbiati, F.; Carrero, P.; Pike, C.J.; Garcia-Segura, L.M.; Melcangi, R.C. Age-related changes in neuroactive steroid levels in 3xTg-AD mice. Neurobiol. Aging 2013, 34, 1080–1089. [Google Scholar] [CrossRef]
- Minkeviciene, R.; Ihalainen, J.; Malm, T.; Matilainen, O.; Keksa-Goldsteine, V.; Goldsteins, G.; Iivonen, H.; Leguit, N.; Glennon, J.; Koistinaho, J. Age-related decrease in stimulated glutamate release and vesicular glutamate transporters in APP/PS1 transgenic and wild-type mice. J. Neurochem. 2008, 105, 584–594. [Google Scholar] [CrossRef]
- Head, E.; Pop, V.; Sarsoza, F.; Kayed, R.; Beckett, T.L.; Studzinski, C.M.; Tomic, J.L.; Glabe, C.G.; Murphy, M.P. Amyloid-β peptide and oligomers in the brain and cerebrospinal fluid of aged canines. J. Alzheimer’s Dis. 2010, 20, 637–646. [Google Scholar] [CrossRef]
- Wu, G.; Sankaranarayanan, S.; Wong, J.; Tugusheva, K.; Michener, M.S.; Shi, X.; Cook, J.J.; Simon, A.J.; Savage, M.J. Characterization of plasma β-secretase (BACE1) activity and soluble amyloid precursor proteins as potential biomarkers for Alzheimer’s disease. J. Neurosci. Res. 2012, 90, 2247–2258. [Google Scholar] [CrossRef]
- Sabbagh, J.J.; Kinney, J.W.; Cummings, J.L. Alzheimer’s disease biomarkers in animal models: Closing the translational gap. Am. J. Neurodegener. Dis. 2013, 2, 108–120. [Google Scholar]
- Xu, Q.-Q.; Yang, W.; Zhong, M.; Lin, Z.-X.; Gray, N.E.; Xian, Y.-F. Animal models of Alzheimer’s disease: Preclinical insights and challenges. Acta Mater. Medica 2023, 2, 192–215. [Google Scholar] [CrossRef]
- Miller, J.A.; Horvath, S.; Geschwind, D.H. Divergence of human and mouse brain transcriptome highlights Alzheimer disease pathways. Proc. Natl. Acad. Sci. USA 2010, 107, 12698–12703. [Google Scholar] [CrossRef]
- Benavides, F.; Rulicke, T.; Prins, J.B.; Bussell, J.; Scavizzi, F.; Cinelli, P.; Herault, Y.; Wedekind, D. Genetic quality assurance and genetic monitoring of laboratory mice and rats: FELASA Working Group Report. Lab. Anim. 2020, 54, 135–148. [Google Scholar] [CrossRef]
- Mekada, K.; Hirose, M.; Murakami, A.; Yoshiki, A. Development of SNP markers for C57BL/6N-derived mouse inbred strains. Exp. Anim. 2015, 64, 91–100. [Google Scholar] [CrossRef]
- Shinohara, M.; Fujioka, S.; Murray, M.E.; Wojtas, A.; Baker, M.; Rovelet-Lecrux, A.; Rademakers, R.; Das, P.; Parisi, J.E.; Graff-Radford, N.R.; et al. Regional distribution of synaptic markers and APP correlate with distinct clinicopathological features in sporadic and familial Alzheimer’s disease. Brain 2014, 137, 1533–1549. [Google Scholar] [CrossRef]
- Nisbet, R.M.; Polanco, J.C.; Ittner, L.M.; Gotz, J. Tau aggregation and its interplay with amyloid-beta. Acta Neuropathol. 2015, 129, 207–220. [Google Scholar] [CrossRef]
- Hyde, L.A.; McHugh, N.A.; Chen, J.; Zhang, Q.; Manfra, D.; Nomeir, A.A.; Josien, H.; Bara, T.; Clader, J.W.; Zhang, L. Studies to investigate the in vivo therapeutic window of the γ-secretase inhibitor N2-[(2S)-2-(3, 5-difluorophenyl)-2-hydroxyethanoyl]-N1-[(7S)-5-methyl-6-oxo-6, 7-dihydro-5H-dibenzo [b, d] azepin-7-yl]-L-alaninamide (LY411, 575) in the CRND8 mouse. J. Pharmacol. Exp. Ther. 2006, 319, 1133–1143. [Google Scholar] [CrossRef]
- Head, E.; Pop, V.; Vasilevko, V.; Hill, M.; Saing, T.; Sarsoza, F.; Nistor, M.; Christie, L.-A.; Milton, S.; Glabe, C.; et al. A Two-Year Study with Fibrillar β-Amyloid (Aβ) Immunization in Aged Canines: Effects on Cognitive Function and Brain Aβ. J. Neurosci. 2008, 28, 3555. [Google Scholar] [CrossRef]
- Davis, P.R.; Giannini, G.; Rudolph, K.; Calloway, N.; Royer, C.M.; Beckett, T.L.; Murphy, M.P.; Bresch, F.; Pagani, D.; Platt, T.; et al. Aβ vaccination in combination with behavioral enrichment in aged beagles: Effects on cognition, Aβ, and microhemorrhages. Neurobiol. Aging 2017, 49, 86–99. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharma, H.; Chang, K.-A.; Hulme, J.; An, S.S.A. Mammalian Models in Alzheimer’s Research: An Update. Cells 2023, 12, 2459. https://doi.org/10.3390/cells12202459
Sharma H, Chang K-A, Hulme J, An SSA. Mammalian Models in Alzheimer’s Research: An Update. Cells. 2023; 12(20):2459. https://doi.org/10.3390/cells12202459
Chicago/Turabian StyleSharma, Himadri, Keun-A Chang, John Hulme, and Seong Soo A. An. 2023. "Mammalian Models in Alzheimer’s Research: An Update" Cells 12, no. 20: 2459. https://doi.org/10.3390/cells12202459