
CEP55 as a Promising Immune Intervention Marker to Regulate Tumor Progression: A Pan-Cancer Analysis with Experimental Verification

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Collection
2.2. Data Processing and Analysis
2.3. Cell Culture and Transfection
2.4. RNA Extraction and qRT-PCR
2.5. Western Blot Analysis
2.6. Cell Viability
2.7. Cell Proliferation and Migration Assays
2.8. Statistical Analysis
3. Results
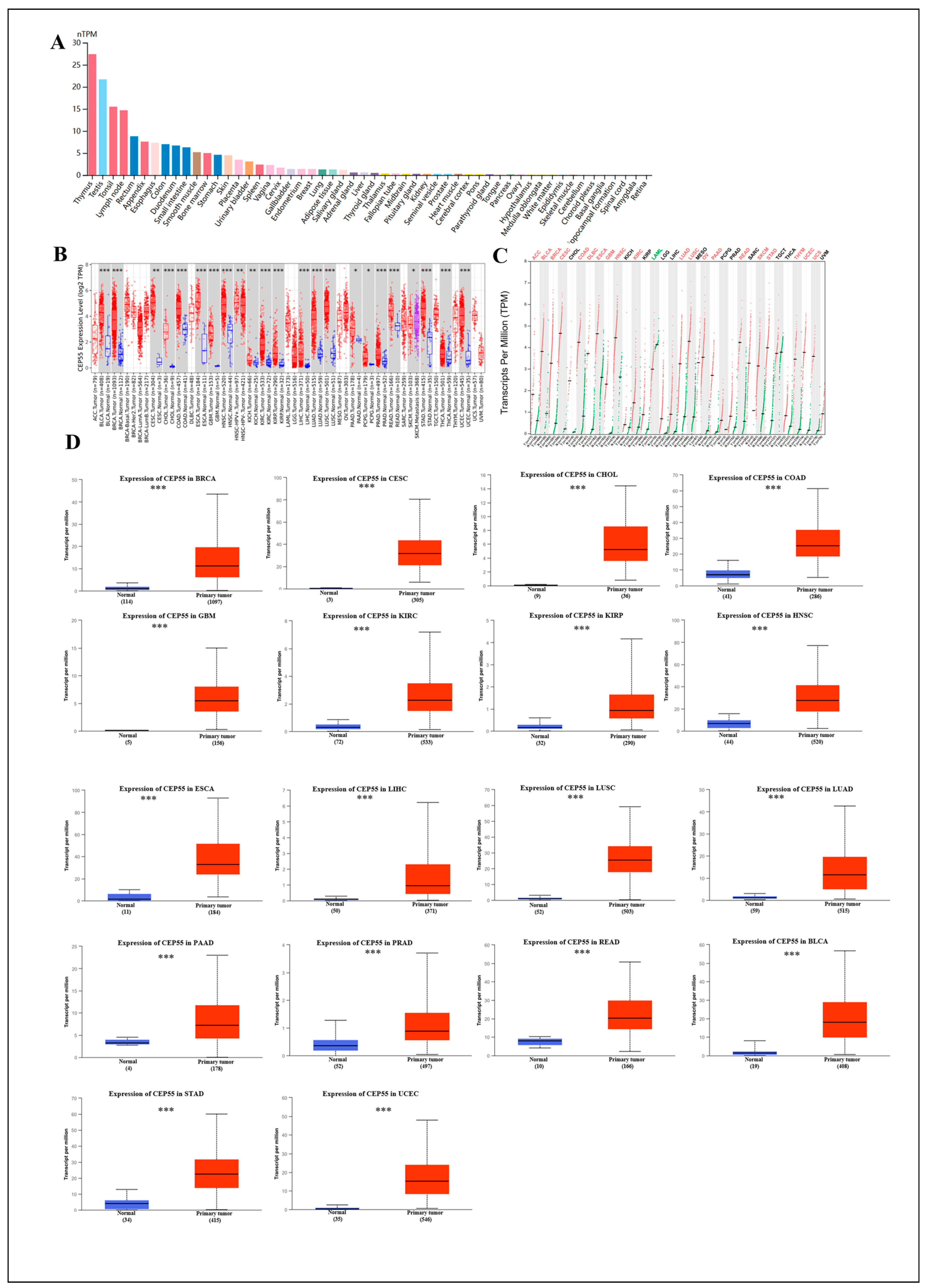
3.1. Expression of CEP55 mRNA and Protein in Normal and Tumor Tissues
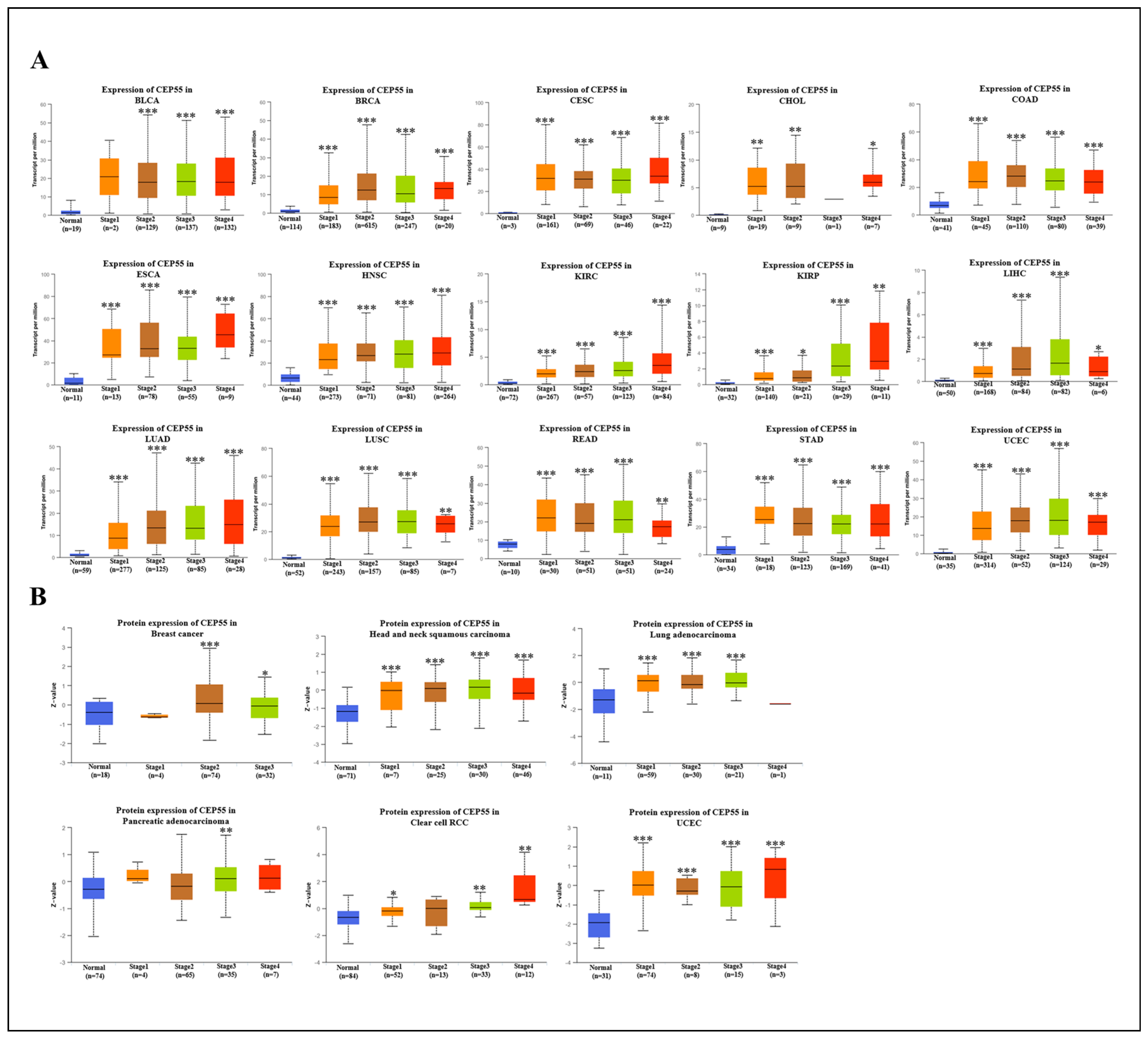
3.2. Correlation between CEP55 Levels and Clinicopathology in Various Tumors


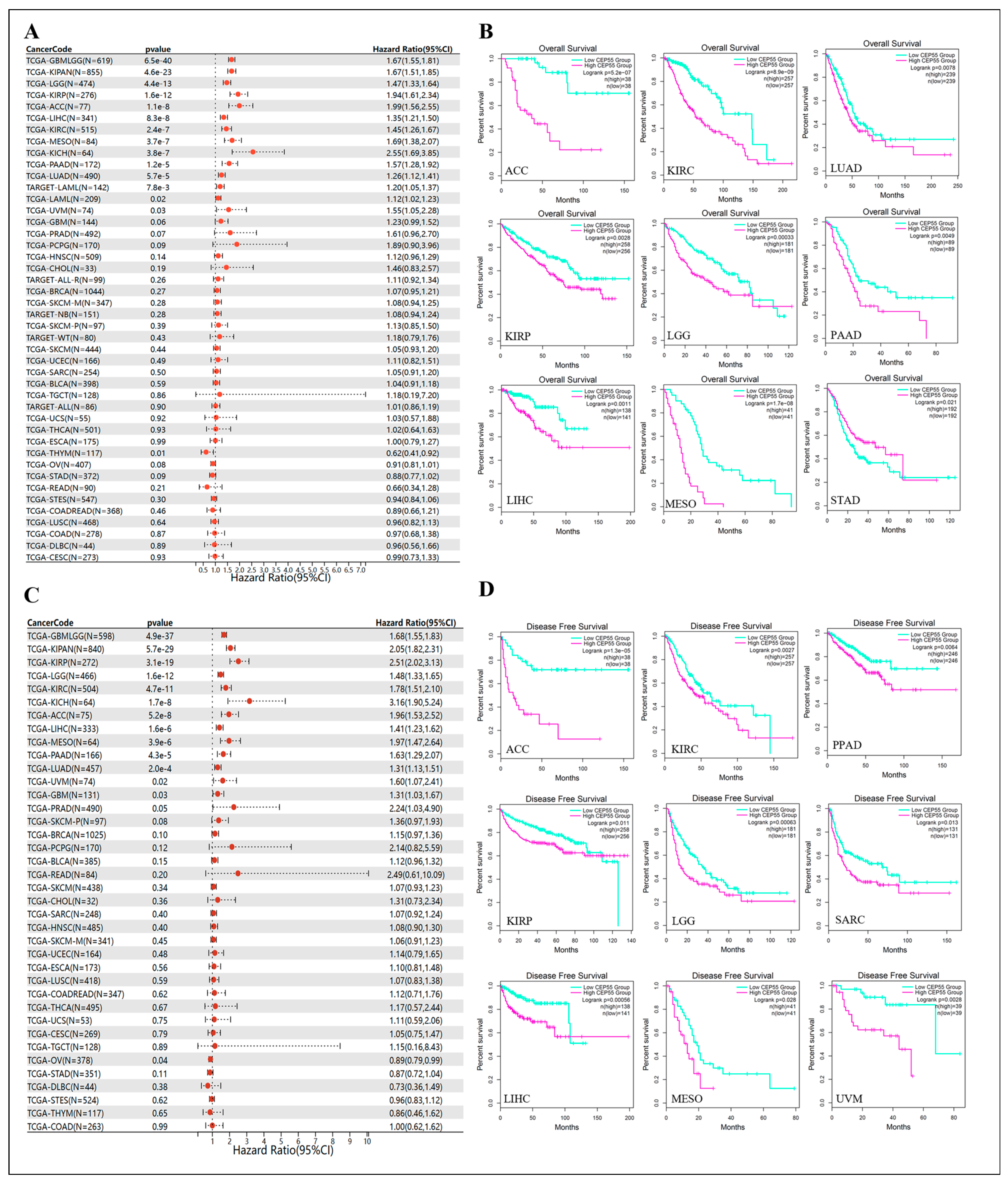
3.3. Prognostic Value of CEP55 across Cancers
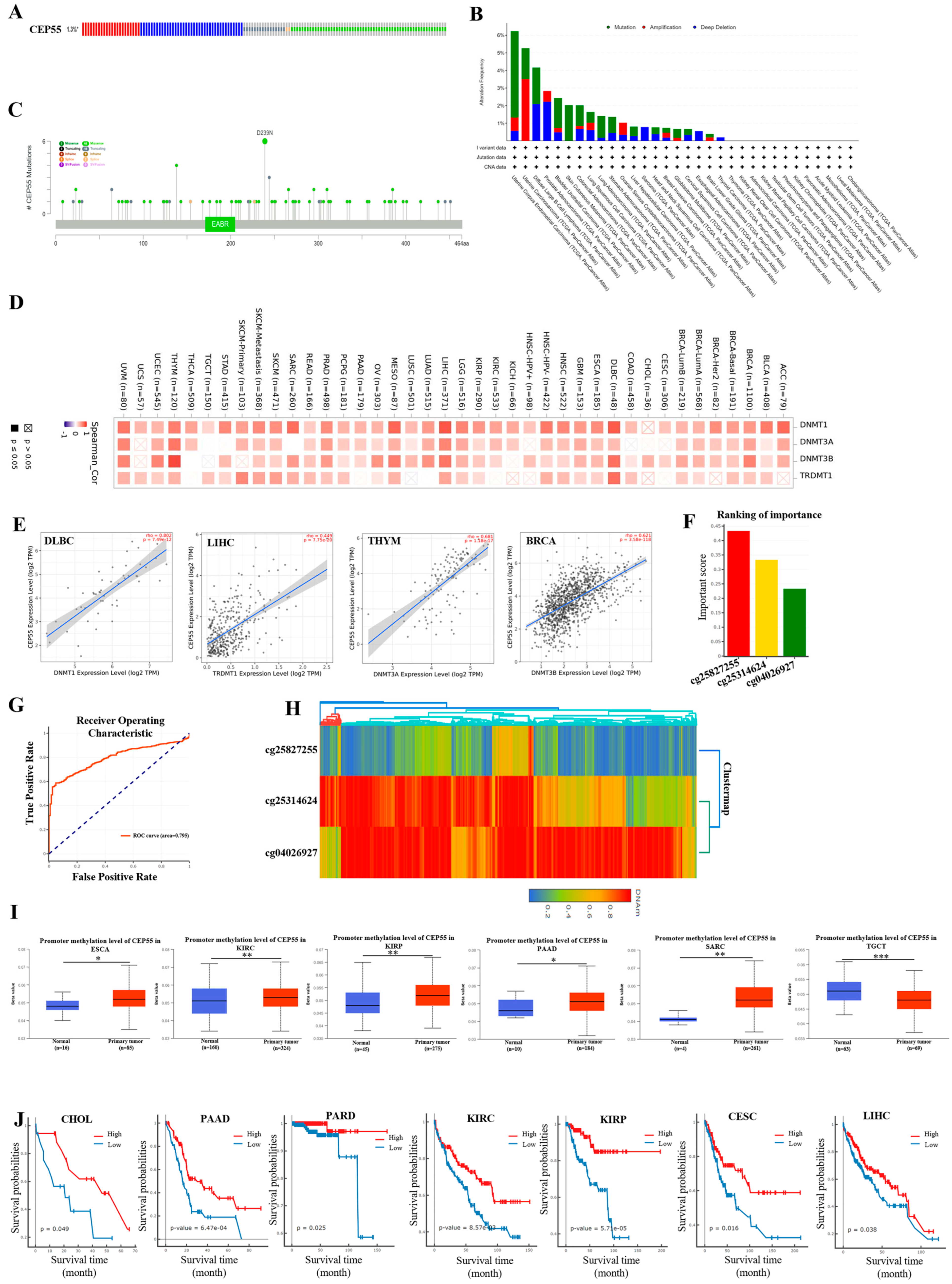
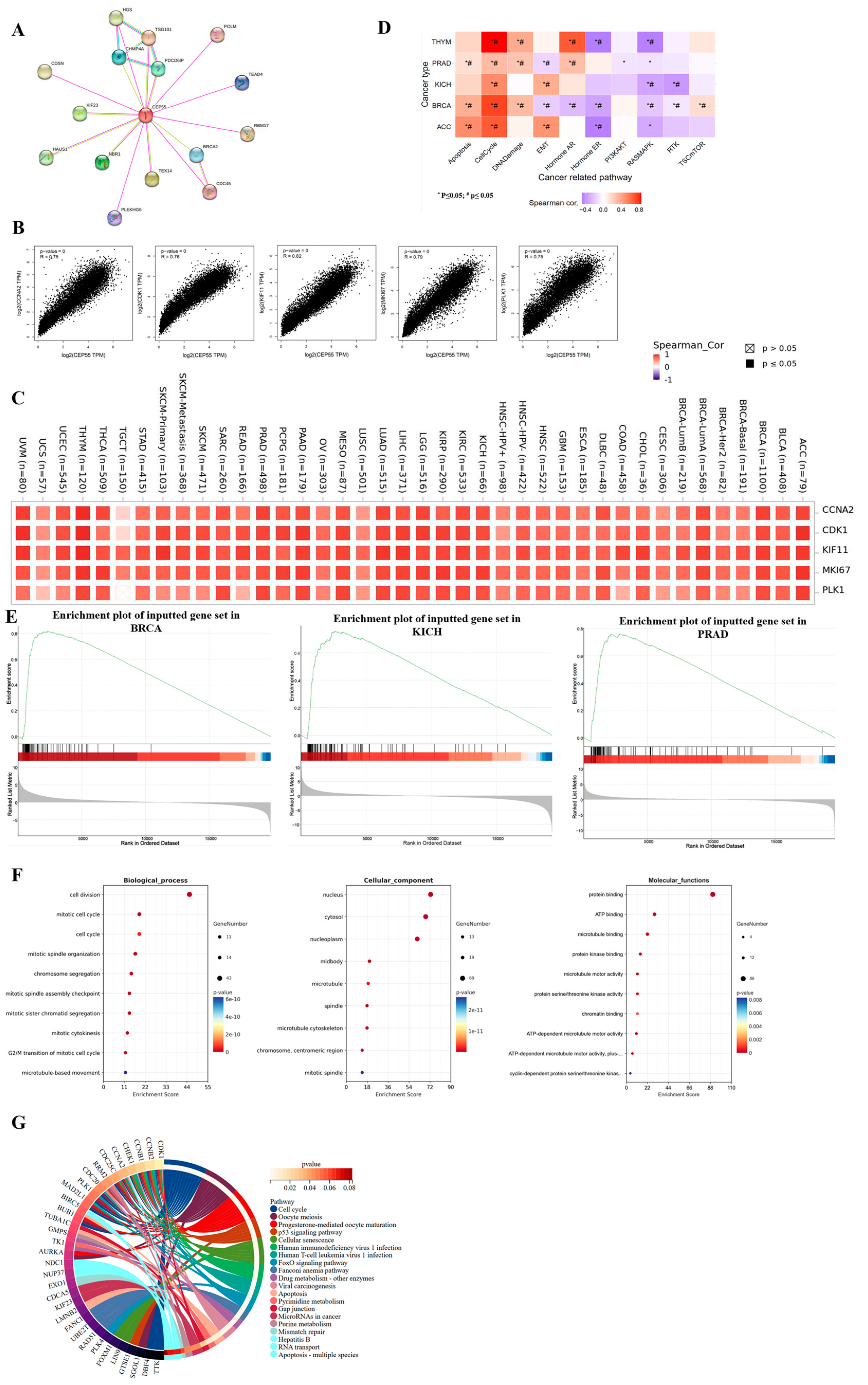
3.4. Gene Stability Analysis of CEP55 in Pan-Cancer
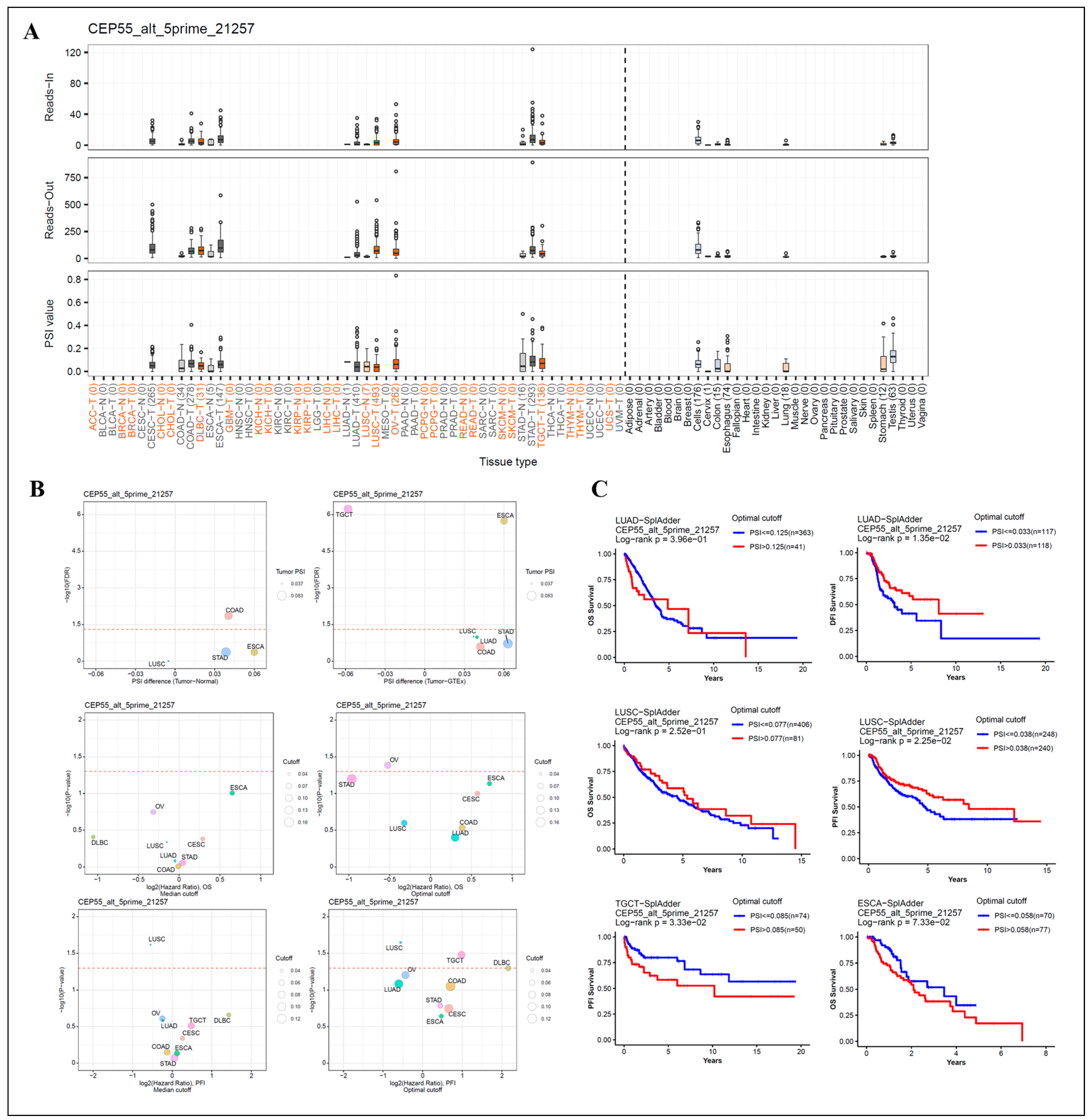
3.5. Analysis of Patient Survival by Alternative Splicing of Differentially Expressed CEP55
3.6. Gene Enrichment and Functional Analysis of CEP55 in Pan-Cancer


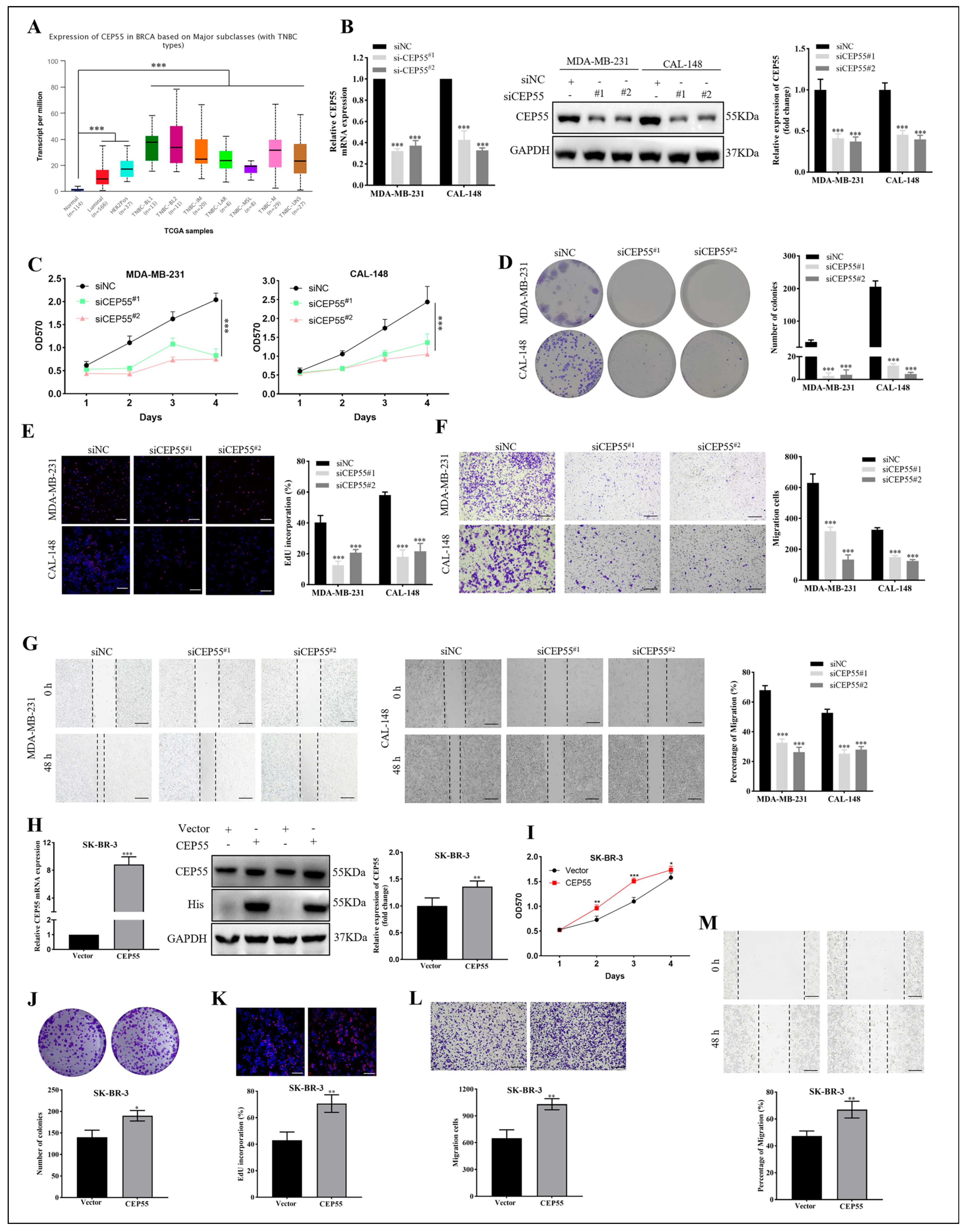
3.7. The Effects of CEP55 on Proliferation and Migration of Breast Cancer
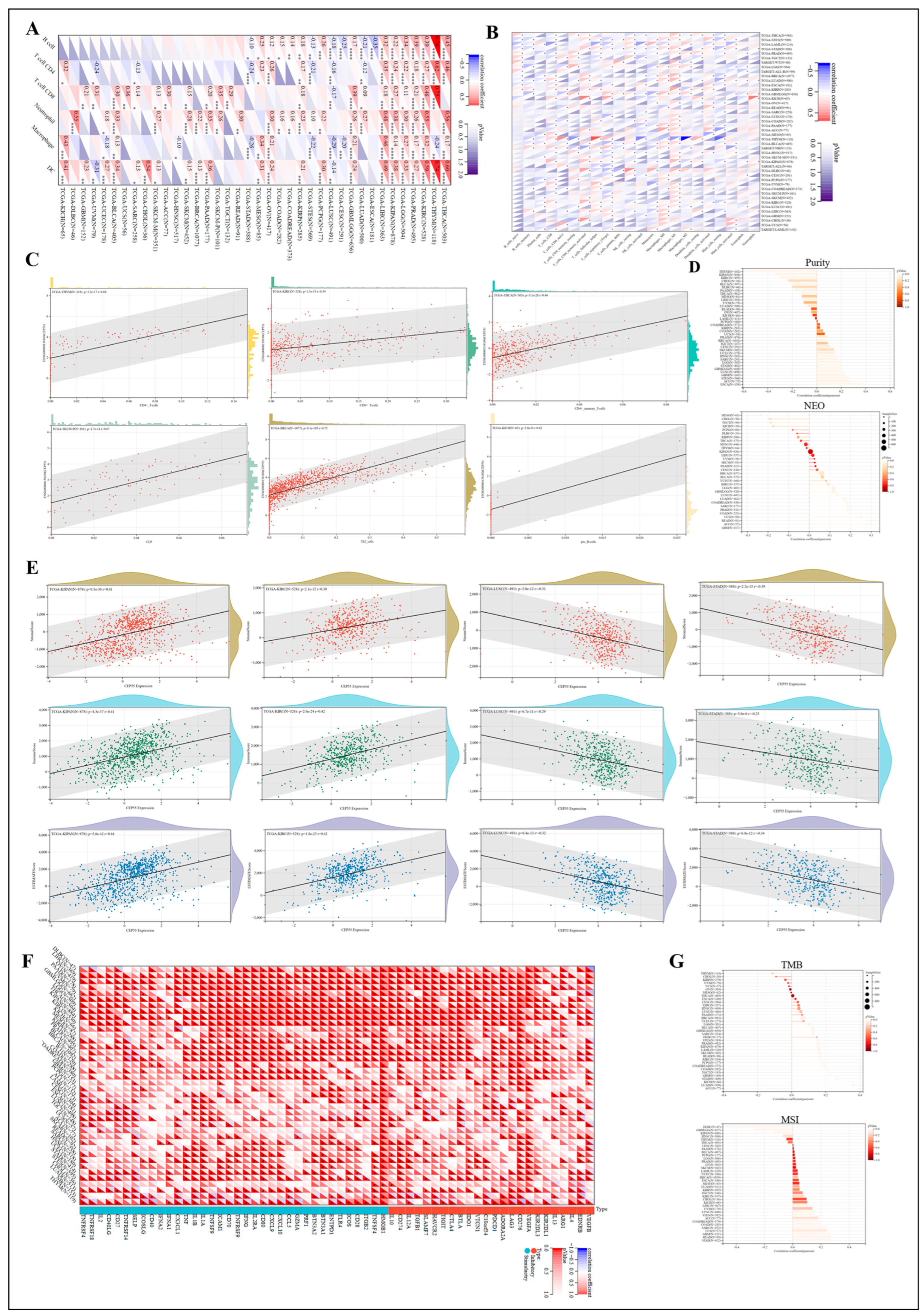
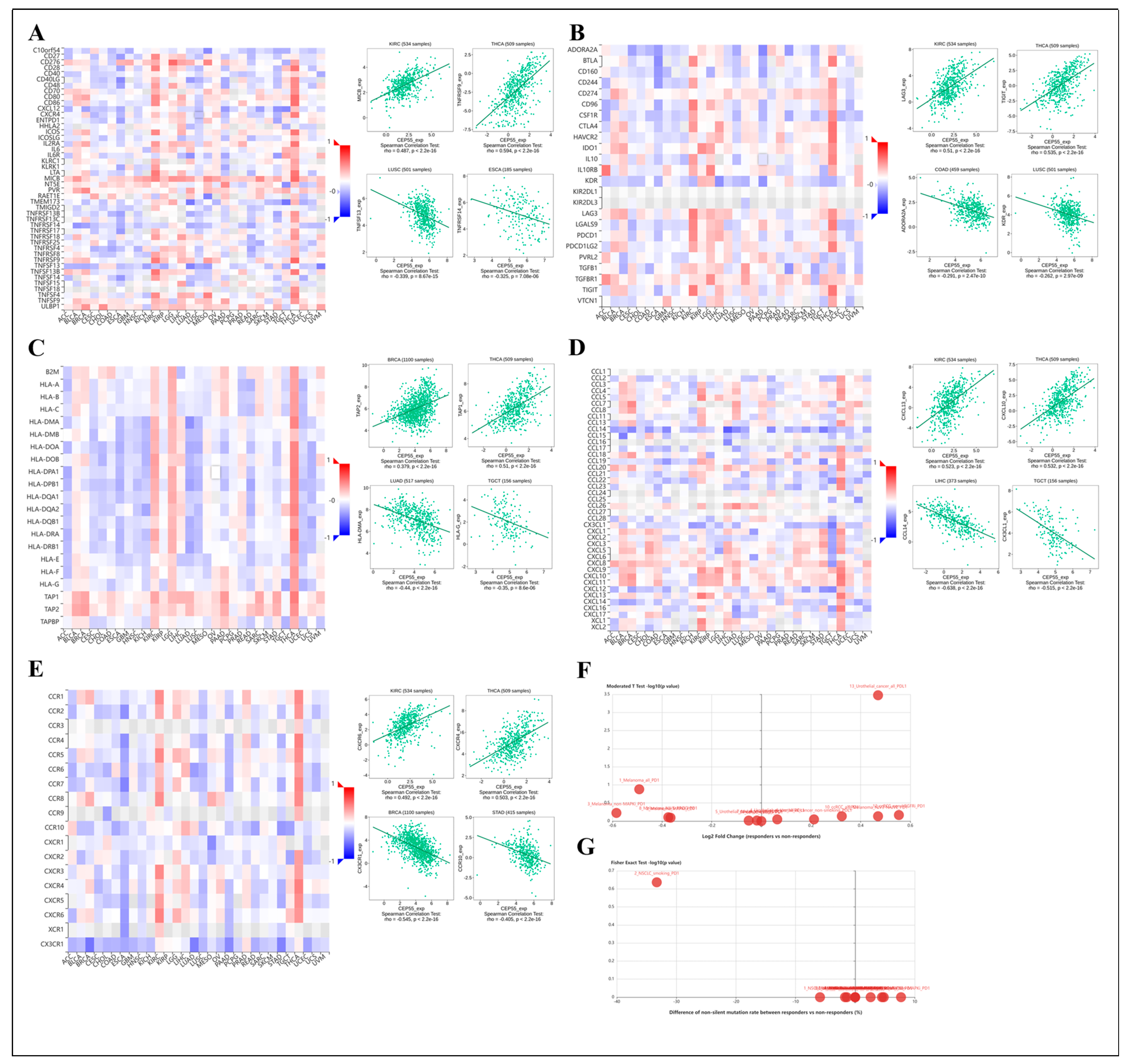
3.8. Regulation of Immune Cell Infiltration by CEP55 in Human Tumors

3.9. Association of CEP55 with Immune-Related Genes
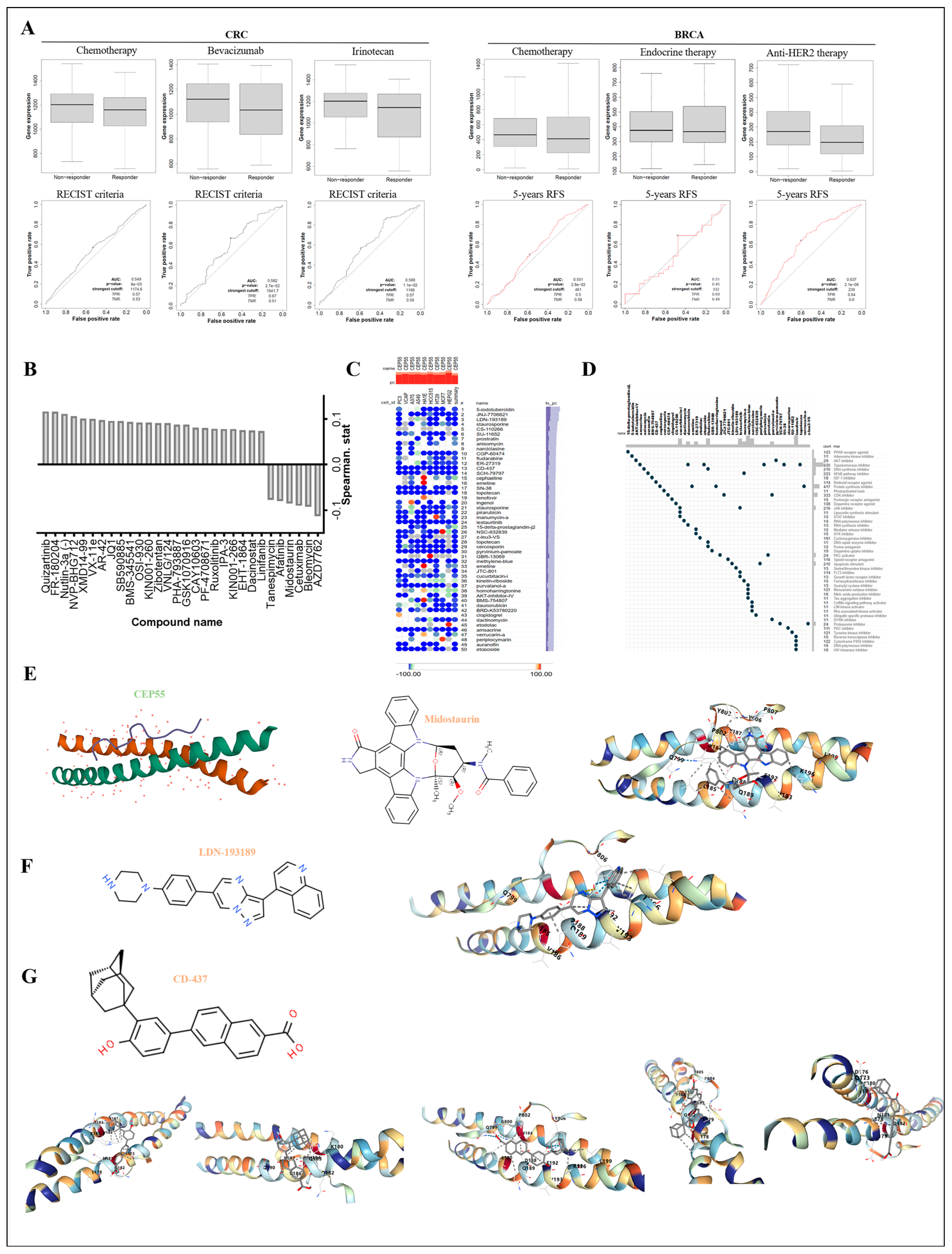
3.10. Exploring the Responsiveness of CEP55 in Oncology Therapy and Potential Drugs Targeting CEP55


4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lin, L.; Yan, L.; Liu, Y.; Yuan, F.; Li, H.; Ni, J. Incidence and death in 29 cancer groups in 2017 and trend analysis from 1990 to 2017 from the Global Burden of Disease Study. J. Hematol. Oncol. 2019, 12, 96. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Chandra, R.A.; Keane, F.K.; Voncken, F.E.M.; Thomas, C.R. Contemporary radiotherapy: Present and future. Lancet 2021, 398, 171–184. [Google Scholar] [CrossRef] [PubMed]
- Tsimberidou, A.M.; Fountzilas, E.; Nikanjam, M.; Kurzrock, R. Review of precision cancer medicine: Evolution of the treatment paradigm. Cancer Treat. Rev. 2020, 86, 102019. [Google Scholar] [CrossRef] [PubMed]
- Mun, E.J.; Babiker, H.M.; Weinberg, U.; Kirson, E.D.; Von Hoff, D.D. Tumor-Treating Fields: A Fourth Modality in Cancer Treatment. Clin. Cancer Res. 2018, 24, 266–275. [Google Scholar] [CrossRef]
- Kennedy, L.B.; Salama, A.K.S. A review of cancer immunotherapy toxicity. CA Cancer J. Clin. 2020, 70, 86–104. [Google Scholar] [CrossRef]
- Wu, Q.; Li, B.; Liu, L.; Sun, S.; Sun, S. Centrosome dysfunction: A link between senescence and tumor immunity. Signal Transduct. Target. Ther. 2020, 5, 107. [Google Scholar] [CrossRef]
- Sinha, D.; Nag, P.; Nanayakkara, D.; Duijf, P.H.G.; Burgess, A.; Raninga, P.; Smits, V.A.J.; Bain, A.L.; Subramanian, G.; Wall, M.; et al. Cep55 overexpression promotes genomic instability and tumorigenesis in mice. Commun. Biol. 2020, 3, 593. [Google Scholar] [CrossRef] [PubMed]
- Phan, T.P.; Holland, A.J. Time is of the essence: The molecular mechanisms of primary microcephaly. Genes Dev. 2021, 35, 1551–1578. [Google Scholar] [CrossRef]
- Kalimutho, M.; Sinha, D.; Jeffery, J.; Nones, K.; Srihari, S.; Fernando, W.C.; Duijf, P.H.; Vennin, C.; Raninga, P.; Nanayakkara, D.; et al. CEP 55 is a determinant of cell fate during perturbed mitosis in breast cancer. EMBO Mol. Med. 2018, 10, e8566. [Google Scholar] [CrossRef]
- Chen, C.-H.; Lu, P.-J.; Chen, Y.-C.; Fu, S.-L.; Wu, K.-J.; Tsou, A.-P.; Lee, Y.-C.G.; Lin, T.-C.E.; Hsu, S.-L.; Lin, W.-J.; et al. FLJ10540-elicited cell transformation is through the activation of PI3-kinase/AKT pathway. Oncogene 2007, 26, 4272–4283. [Google Scholar] [CrossRef]
- Yang, C.; Yang, Y.; Wang, W.; Zhou, W.; Zhang, X.; Xiao, Y.; Zhang, H. CEP55 3′-UTR promotes epithelial–mesenchymal transition and enhances tumorigenicity of bladder cancer cells by acting as a ceRNA regulating miR-497-5p. Cell Oncol. 2022, 45, 1217–1236. [Google Scholar] [CrossRef]
- Zhang, X.; Xu, Q.; Li, E.; Shi, T.; Chen, H. CEP55 predicts the poor prognosis and promotes tumorigenesis in endometrial cancer by regulating the Foxo1 signaling. Mol. Cell Biochem. 2022, 478, 1561–1571. [Google Scholar] [CrossRef]
- Fekete, J.T.; Győrffy, B. ROCplot.org: Validating predictive biomarkers of chemotherapy/hormonal therapy/anti-HER2 therapy using transcriptomic data of 3,104 breast cancer patients. Int. J. Cancer 2019, 145, 3140–3151. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Liang, X.; Zhang, H.; Wang, Z.; Zhang, X.; Dai, Z.; Zhang, J.; Luo, P.; Zhang, L.; Hu, J.; Liu, Z.; et al. JMJD8 Is an M2 Macrophage Biomarker, and It Associates with DNA Damage Repair to Facilitate Stemness Maintenance, Chemoresistance, and Immunosuppression in Pan-Cancer. Front. Immunol. 2022, 13, 875786. [Google Scholar] [CrossRef] [PubMed]
- Tandon, D.; Banerjee, M. Centrosomal protein 55: A new paradigm in tumorigenesis. Eur. J. Cell Biol. 2020, 99, 151086. [Google Scholar] [CrossRef]
- Baralle, F.E.; Giudice, J. Alternative splicing as a regulator of development and tissue identity. Nat. Rev. Mol. Cell Biol. 2017, 18, 437–451. [Google Scholar] [CrossRef]
- Zhao, S. Alternative splicing, RNA-seq and drug discovery. Drug Discov. Today 2019, 24, 1258–1267. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.F.; Dannenberg, A.J. The obese adipose tissue microenvironment in cancer development and progression. Nat. Rev. Endocrinol. 2019, 15, 139–154. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Li, F.; Ye, T.; Wang, J.; Lyu, C.; Qing, S.; Ding, Z.; Gao, X.; Jia, R.; Yu, D.; et al. Macrophage-tumor chimeric exosomes accumulate in lymph node and tumor to activate the immune response and the tumor microenvironment. Sci. Transl. Med. 2021, 13, eabb6981. [Google Scholar] [CrossRef]
- Liu, K.; Cui, J.-J.; Zhan, Y.; Ouyang, Q.-Y.; Lu, Q.-S.; Yang, D.-H.; Li, X.-P.; Yin, J.-Y. Reprogramming the tumor microenvironment by genome editing for precision cancer therapy. Mol. Cancer 2022, 21, 98. [Google Scholar] [CrossRef]
- Yoshihara, K.; Shahmoradgoli, M.; Martínez, E.; Vegesna, R.; Kim, H.; Torres-Garcia, W.; Treviño, V.; Shen, H.; Laird, P.W.; Levine, D.A.; et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 2013, 4, 2612. [Google Scholar] [CrossRef] [PubMed]
- Lv, M.; Chen, M.; Zhang, R.; Zhang, W.; Wang, C.; Zhang, Y.; Wei, X.; Guan, Y.; Liu, J.; Feng, K.; et al. Manganese is critical for antitumor immune responses via cGAS-STING and improves the efficacy of clinical immunotherapy. Cell Res. 2020, 30, 966–979. [Google Scholar] [CrossRef] [PubMed]
- Jardim, D.L.; Goodman, A.; de Melo Gagliato, D.; Kurzrock, R. The Challenges of Tumor Mutational Burden as an Immunotherapy Biomarker. Cancer Cell 2021, 39, 154–173. [Google Scholar] [CrossRef] [PubMed]
- Huse, M. Mechanical forces in the immune system. Nat. Rev. Immunol. 2017, 17, 679–690. [Google Scholar] [CrossRef]
- Brodin, P. Immune-microbe interactions early in life: A determinant of health and disease long term. Science 2022, 376, 945–950. [Google Scholar] [CrossRef] [PubMed]
- Axelrod, M.L.; Cook, R.S.; Johnson, D.B.; Balko, J.M. Biological Consequences of MHC-II Expression by Tumor Cells in Cancer. Clin. Cancer Res. 2019, 25, 2392–2402. [Google Scholar] [CrossRef]
- Charo, I.F.; Ransohoff, R.M. The many roles of chemokines and chemokine receptors in inflammation. N. Engl. J. Med. 2006, 354, 610–621. [Google Scholar] [CrossRef]
- Sharma, R.; Abbasi-Kangevari, M.; Abd-Rabu, R.; Abidi, H.; Abu-Gharbieh, E.; Acuna, J.M.; Adhikari, S.; Advani, S.M.; Afzal, M.S.; Meybodi, M.A.; et al. Global, regional, and national burden of colorectal cancer and its risk factors, 1990–2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet Gastroenterol. Hepatol. 2022, 7, 627–647. [Google Scholar] [CrossRef]
- Qiu, H.; Cao, S.; Xu, R. Cancer incidence, mortality, and burden in China: A time-trend analysis and comparison with the United States and United Kingdom based on the global epidemiological data released in 2020. Cancer Commun. 2021, 41, 1037–1048. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Garay, I.; Rustom, A.; Gerdes, H.-H.; Kutsche, K. The novel centrosomal associated protein CEP55 is present in the spindle midzone and the midbody. Genomics 2006, 87, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Zhang, Y.; Li, Y.; Lu, J.; Huang, Q.; Xu, R.; Feng, Y.; Yan, S. High CEP55 expression is associated with poor prognosis in non-small-cell lung cancer. OncoTargets Ther. 2018, 11, 4979–4990. [Google Scholar] [CrossRef]
- Xu, L.; Xia, C.; Sheng, F.; Sun, Q.; Xiong, J.; Wang, S. CEP55 promotes the proliferation and invasion of tumour cells via the AKT signalling pathway in osteosarcoma. Carcinogenesis 2018, 39, 623–631. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.-M.; Liu, L.; Gu, W.-Y.; Huang, L.-Y.; Yang, Y.; Huang, Y.-H.; Luo, R.-Z. CEP55 Positively Affects Tumorigenesis of Esophageal Squamous Cell Carcinoma and Is Correlated with Poor Prognosis. J. Oncol. 2021, 2021, 8890715. [Google Scholar] [CrossRef]
- Wu, S.; Wu, D.; Pan, Y.; Liu, H.; Shao, Z.; Wang, M. Correlation between EZH2 and CEP55 and lung adenocarcinoma prognosis. Pathol.-Res. Pract. 2019, 215, 292–301. [Google Scholar] [CrossRef]
- Thandapani, P. Super-enhancers in cancer. Pharmacol. Ther. 2019, 199, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Jiménez, F.; Muiños, F.; Sentís, I.; Deu-Pons, J.; Reyes-Salazar, I.; Arnedo-Pac, C.; Mularoni, L.; Pich, O.; Bonet, J.; Kranas, H.; et al. A compendium of mutational cancer driver genes. Nat. Rev. Cancer 2020, 20, 555–572. [Google Scholar] [CrossRef]
- Pan, S.; Chen, R. Pathological implication of protein post-translational modifications in cancer. Mol. Asp. Med. 2022, 86, 101097. [Google Scholar] [CrossRef]
- Deribe, Y.L.; Pawson, T.; Dikic, I. Post-translational modifications in signal integration. Nat. Struct. Mol. Biol. 2010, 17, 666–672. [Google Scholar] [CrossRef]
- Yang, L.; He, Y.; Zhang, Z.; Wang, W. Upregulation of CEP55 Predicts Dismal Prognosis in Patients with Liver Cancer. BioMed Res. Int. 2020, 2020, 4139320. [Google Scholar] [CrossRef] [PubMed]
- Fabbro, M.; Zhou, B.B.; Takahashi, M.; Sarcevic, B.; Lal, P.; Graham, M.E.; Gabrielli, B.G.; Robinson, P.J.; Nigg, E.A.; Ono, Y.; et al. Cdk1/Erk2-and Plk1-dependent phosphorylation of a centrosome protein, Cep55, is required for its recruitment to midbody and cytokinesis. Dev. Cell 2005, 9, 477–488. [Google Scholar] [CrossRef]
- Frosk, P.; Arts, H.H.; Philippe, J.; Gunn, C.S.; Brown, E.L.; Chodirker, B.; Simard, L.; Majewski, J.; Fahiminiya, S.; Russell, C.; et al. A truncating mutation in CEP55 is the likely cause of MARCH, a novel syndrome affecting neuronal mitosis. J. Med. Genet. 2017, 54, 490–501. [Google Scholar] [CrossRef]
- Bondeson, M.L.; Ericson, K.; Gudmundsson, S.; Ameur, A.; Pontén, F.; Wesström, J.; Frykholm, C.; Wilbe, M. A nonsense mutation in CEP55 defines a new locus for a Meckel-like syndrome, an autosomal recessive lethal fetal ciliopathy. Clin. Genet. 2017, 92, 510–516. [Google Scholar] [CrossRef]
- Cuzick, J.; Swanson, G.P.; Fisher, G.; Brothman, A.R.; Berney, D.M.; Reid, J.E.; Mesher, D.; Speights, V.O.; Stankiewicz, E.; Foster, C.S.; et al. Prognostic value of an RNA expression signature derived from cell cycle proliferation genes in patients with prostate cancer: A retrospective study. Lancet Oncol. 2011, 12, 245–255. [Google Scholar] [CrossRef]
- Al-Ejeh, F.; Simpson, P.T.; Sanus, J.M.; Klein, K.; Kalimutho, M.; Shi, W.; Miranda, M.; Kutasovic, J.; Raghavendra, A.; Madore, J.; et al. Meta-analysis of the global gene expression profile of triple-negative breast cancer identifies genes for the prognostication and treatment of aggressive breast cancer. Oncogenesis 2014, 3, e100. [Google Scholar] [CrossRef]
- Chen, C.-H.; Lai, J.-M.; Chou, T.-Y.; Chen, C.-Y.; Su, L.-J.; Lee, Y.-C.; Cheng, T.-S.; Hong, Y.-R.; Chou, C.-K.; Whang-Peng, J.; et al. VEGFA Upregulates FLJ10540 and Modulates Migration and Invasion of Lung Cancer via PI3K/AKT Pathway. PLoS ONE 2009, 4, e5052. [Google Scholar] [CrossRef] [PubMed]
- de Visser, K.E.; Joyce, J.A. The evolving tumor microenvironment: From cancer initiation to metastatic outgrowth. Cancer Cell 2023, 41, 374–403. [Google Scholar] [CrossRef]
- Hinshaw, D.C.; Shevde, L.A. The tumor microenvironment innately modulates cancer progression. Cancer Res. 2019, 79, 4557–4566. [Google Scholar] [CrossRef] [PubMed]
- Bejarano, L.; Jordāo, M.J.; Joyce, J.A. Therapeutic Targeting of the Tumor Microenvironment. Cancer Discov. 2021, 11, 933–959. [Google Scholar] [CrossRef]
- Qadir, F.; Aziz, M.A.; Sari, C.P.; Ma, H.; Dai, H.; Wang, X.; Raithatha, D.; Da Silva, L.G.L.; Hussain, M.; Poorkasreiy, S.P.; et al. Transcriptome reprogramming by cancer exosomes: Identification of novel molecular targets in matrix and immune modulation. Mol. Cancer 2018, 17, 97. [Google Scholar] [CrossRef] [PubMed]
- Aran, D.; Sirota, M.; Butte, A.J. Systematic pan-cancer analysis of tumour purity. Nat. Commun. 2015, 6, 8971. [Google Scholar] [CrossRef]
- Kalbasi, A.; Ribas, A. Tumour-intrinsic resistance to immune checkpoint blockade. Nat. Rev. Immunol. 2020, 20, 25–39. [Google Scholar] [CrossRef] [PubMed]
- Xia, L.; Oyang, L.; Lin, J.; Tan, S.; Han, Y.; Wu, N.; Yi, P.; Tang, L.; Pan, Q.; Rao, S.; et al. The cancer metabolic reprogramming and immune response. Mol. Cancer 2021, 20, 28. [Google Scholar] [CrossRef] [PubMed]
- Rock, K.L.; Reits, E.; Neefjes, J. Present Yourself! By MHC Class I and MHC Class II Molecules. Trends Immunol. 2016, 37, 724–737. [Google Scholar] [CrossRef] [PubMed]
- Propper, D.J.; Balkwill, F.R. Harnessing cytokines and chemokines for cancer therapy. Nat. Rev. Clin. Oncol. 2022, 19, 237–253. [Google Scholar] [CrossRef]
- Carter, P.J.; Lazar, G.A. Next generation antibody drugs: Pursuit of the ‘high-hanging fruit’. Nat. Rev. Drug Discov. 2018, 17, 197–223. [Google Scholar] [CrossRef] [PubMed]
- Palve, V.; Liao, Y.; Rix, L.L.R.; Rix, U. Turning liabilities into opportunities: Off-target based drug repurposing in cancer. Semin. Cancer Biol. 2021, 68, 209–229. [Google Scholar] [CrossRef] [PubMed]
- Brown, D. Unfinished business: Target-based drug discovery. Drug Discov. Today 2007, 12, 1007–1012. [Google Scholar] [CrossRef] [PubMed]
- de Siqueira, L.R.; de Moraes Gomes, P.A.; de Lima Ferreira, L.P.; de Melo Rêgo, M.J.; Leite, A.C. Multi-target compounds acting in cancer progression: Focus on thiosemicarbazone, thiazole and thiazolidinone analogues. Eur. J. Med. Chem. 2019, 170, 237–260. [Google Scholar] [CrossRef]
- Li, Y.; Chen, J.; Bolinger, A.A.; Chen, H.; Liu, Z.; Cong, Y.; Brasier, A.R.; Pinchuk, I.V.; Tian, B.; Zhou, J. Target-based small molecule drug discovery towards novel therapeutics for inflammatory bowel diseases. Inflamm. Bowel Dis. 2021, 27, S38–S62. [Google Scholar] [CrossRef] [PubMed]
- Shaik, B.; Zafar, T.; Balasubramanian, K.; Gupta, S.P. An Overview of Ovarian Cancer: Molecular Processes Involved and Development of Target-based Chemotherapeutics. Curr. Top. Med. Chem. 2021, 21, 329–346. [Google Scholar] [CrossRef] [PubMed]
- Lestari, B.; Utomo, R.Y. CEP55 Inhibitor: Extensive Computational Approach Defining a New Target of Cell Cycle Machinery Agent. Adv. Pharm. Bull. 2022, 12, 191–199. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, G.; Chen, B.; Su, Y.; Qu, N.; Zhou, D.; Zhou, W. CEP55 as a Promising Immune Intervention Marker to Regulate Tumor Progression: A Pan-Cancer Analysis with Experimental Verification. Cells 2023, 12, 2457. https://doi.org/10.3390/cells12202457
Wang G, Chen B, Su Y, Qu N, Zhou D, Zhou W. CEP55 as a Promising Immune Intervention Marker to Regulate Tumor Progression: A Pan-Cancer Analysis with Experimental Verification. Cells. 2023; 12(20):2457. https://doi.org/10.3390/cells12202457
Chicago/Turabian StyleWang, Gang, Bo Chen, Yue Su, Na Qu, Duanfang Zhou, and Weiying Zhou. 2023. "CEP55 as a Promising Immune Intervention Marker to Regulate Tumor Progression: A Pan-Cancer Analysis with Experimental Verification" Cells 12, no. 20: 2457. https://doi.org/10.3390/cells12202457