

Agomir-331 Suppresses Reactive Gliosis and Neuroinflammation after Traumatic Brain Injury
 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal
2.2. Blade Penetrating Stab Wound
2.3. Intranasal Delivery of Agomir-331
2.4. Cell Transfection
2.5. Primary Astrocyte and Microglia Culture
2.6. Quantitative Real-Time PCR (qRT-PCR)
2.7. Immunostaining
2.8. Cell Viability Assay
2.9. Wound Healing Scratch Assay
2.10. TUNEL Assays
2.11. Behavioral Tests
2.12. Enzyme-Linked Immunosorbent Assay (ELISA)
2.13. Luciferase Reporter Assay
2.14. Statistical Analysis
3. Results
3.1. The Expression of miR-331 Is Downregulated in Brains with Early TBI
3.2. Supplementation with Agomir-331 Protects against Neuronal Apoptosis after HSI
3.3. Agomir-331 Inhibits Glial Scar Formation in HSI Mice
3.4. Administration of Agomir-331 Suppresses Astrocyte Proliferation and Migration
3.5. Agomir-331 Suppresses Inflammatory Response in MCM-Treated Microglia
3.6. Agomir-331 Improves Learning and Memory in HSI Mice
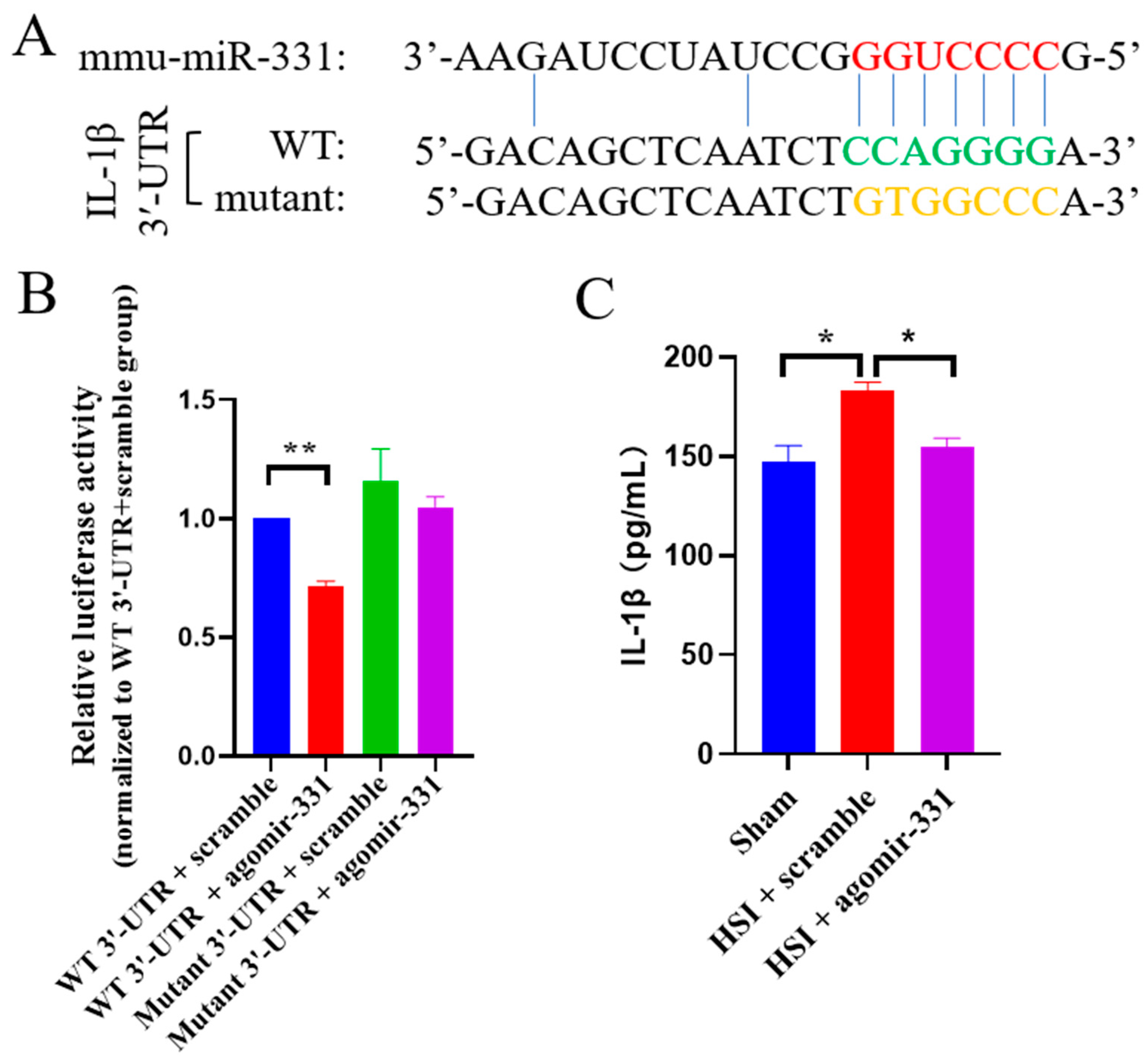
3.7. IL-1β Is a Direct Target Gene of miR-331
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Maas, A.I.R.; Menon, D.K.; Adelson, P.D.; Andelic, N.; Bell, M.J.; Belli, A.; Bragge, P.; Brazinova, A.; Burki, A.; Chesnut, R.M.; et al. Traumatic brain injury: Integrated approaches to improve prevention, clinical care, and research. Lancet Neurol. 2017, 16, 987–1048. [Google Scholar] [CrossRef]
- Galindo, L.T.; Mundim, M.; Pinto, A.S.; Chiarantin, G.M.D.; Almeida, M.E.S.; Lamers, M.L.; Horwitz, A.R.; Santos, M.F.; Porcionatto, M. Chondroitin sulfate impairs neural stem cell migration through rock activation. Mol. Neurobiol. 2018, 55, 3185–3195. [Google Scholar] [CrossRef] [PubMed]
- Adams, K.L.; Gallo, V. The diversity and disparity of the glial scar. Nat. Neurosci. 2018, 21, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Escartin, C.; Galea, E.; Lakatos, A.; O’Callaghan, J.P.; Petzold, G.C.; Serrano-Pozo, A.; Steinhauser, C.; Volterra, A.; Carmignoto, G.; Agarwal, A.; et al. Reactive astrocyte nomenclature, definitions, and future directions. Nat. Neurosci. 2021, 24, 312–325. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Shen, X.; Xu, X.; Liu, H.; Li, F.; Lu, S.; Gao, Z.; Zhang, J.; Wu, Q.; Yang, D.; et al. Astrocytic yap promotes the formation of glia scars and neural regeneration after spinal cord injury. J. Neurosci. 2020, 40, 2644–2662. [Google Scholar] [CrossRef]
- Wen, L.; Wang, Y.D.; Shen, D.F.; Zheng, P.D.; Tu, M.D.; You, W.D.; Zhu, Y.R.; Wang, H.; Feng, J.F.; Yang, X.F. Exosomes derived from bone marrow mesenchymal stem cells inhibit neuroinflammation after traumatic brain injury. Neural Regen. Res. 2022, 17, 2717–2724. [Google Scholar] [CrossRef]
- Guo, M.; Wang, J.; Zhao, Y.; Feng, Y.; Han, S.; Dong, Q.; Cui, M.; Tieu, K. Microglial exosomes facilitate alpha-synuclein transmission in parkinson’s disease. Brain 2020, 143, 1476–1497. [Google Scholar] [CrossRef]
- Zhao, C.; Hou, W.; Lei, H.; Huang, L.; Wang, S.; Cui, D.; Xing, C.; Wang, X.; Peng, Y. Potassium 2-(l-hydroxypentyl)-benzoate attenuates neuroinflammatory responses and upregulates heme oxygenase-1 in systemic lipopolysaccharide-induced inflammation in mice. Acta Pharm. Sin. B 2017, 7, 470–478. [Google Scholar] [CrossRef]
- Liu, W.; Tang, Y.; Feng, J. Cross talk between activation of microglia and astrocytes in pathological conditions in the central nervous system. Life Sci. 2011, 89, 141–146. [Google Scholar] [CrossRef]
- Liu, X.; Li, M.; Peng, Y.; Hu, X.; Xu, J.; Zhu, S.; Yu, Z.; Han, S. Mir-30c regulates proliferation, apoptosis and differentiation via the Shh signaling pathway in P19 cells. Exp. Mol. Med. 2016, 48, e248. [Google Scholar] [CrossRef]
- Bhomia, M.; Balakathiresan, N.S.; Wang, K.K.; Papa, L.; Maheshwari, R.K. A panel of serum mirna biomarkers for the diagnosis of severe to mild traumatic brain injury in humans. Sci. Rep. 2016, 6, 28148. [Google Scholar] [CrossRef] [PubMed]
- Truettner, J.S.; Alonso, O.F.; Bramlett, H.M.; Dietrich, W.D. Therapeutic hypothermia alters microRNA responses to traumatic brain injury in rats. J. Cereb. Blood Flow Metab. 2011, 31, 1897–1907. [Google Scholar] [CrossRef]
- Kheirolomoom, A.; Kim, C.W.; Seo, J.W.; Kumar, S.; Son, D.J.; Gagnon, M.K.; Ingham, E.S.; Ferrara, K.W.; Jo, H. Multifunctional nanoparticles facilitate molecular targeting and mirna delivery to inhibit atherosclerosis in apoE−/− Mice. ACS Nano 2015, 9, 8885–8897. [Google Scholar] [CrossRef] [PubMed]
- Harraz, M.M.; Eacker, S.M.; Wang, X.; Dawson, T.M.; Dawson, V.L. Microrna-223 is neuroprotective by targeting glutamate receptors. Proc. Natl. Acad. Sci. USA 2012, 109, 18962–18967. [Google Scholar] [CrossRef]
- Liu, D.Z.; Jickling, G.C.; Ander, B.P.; Hull, H.; Zhan, X.; Cox, C.; Shroff, N.; Dykstra-Aiello, C.; Stamova, B.; Sharp, F.R. Elevating microrna-122 in blood improves outcomes after temporary middle cerebral artery occlusion in rats. J. Cereb. Blood Flow Metab. 2016, 36, 1374–1383. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Liu, Y.; Wang, X.; Zhang, M.Q.; Hannon, G.J.; Huang, Z.J. Cell-type-based analysis of microrna profiles in the mouse brain. Neuron 2012, 73, 35–48. [Google Scholar] [CrossRef]
- Hoye, M.L.; Koval, E.D.; Wegener, A.J.; Hyman, T.S.; Yang, C.; O’Brien, D.R.; Miller, R.L.; Cole, T.; Schoch, K.M.; Shen, T.; et al. Microrna profiling reveals marker of motor neuron disease in als models. J. Neurosci. 2017, 37, 5574–5586. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, R.; Wang, M. miR-331-3p suppresses cell invasion and migration in colorectal carcinoma by directly targeting nrp2. Oncol. Lett. 2019, 18, 6501–6508. [Google Scholar] [CrossRef]
- Li, R.; Li, X.; Huang, Y.; Qiu, H.; Li, L.; Bi, Z. Lncrna sox2ot knockdown alleviates lipopolysaccharide-induced damage of pc12 cells by regulating mir-331-3p/neurod1 axis. World Neurosurg. 2021, 147, e293–e305. [Google Scholar] [CrossRef]
- Zhao, Q.; Zhao, F.; Liu, C.; Xu, T.; Song, K. Lncrna foxd2-as1 promotes cell proliferation and invasion of fibroblast-like synoviocytes by regulation of mir-331-3p/piaS3 pathway in rheumatoid arthritis. Autoimmunity 2021, 54, 254–263. [Google Scholar] [CrossRef]
- Meissner, L.; Gallozzi, M.; Balbi, M.; Schwarzmaier, S.; Tiedt, S.; Terpolilli, N.A.; Plesnila, N. Temporal profile of microrna expression in contused cortex after traumatic brain injury in mice. J. Neurotrauma 2016, 33, 713–720. [Google Scholar] [CrossRef] [PubMed]
- He, B.D.; Liu, C.M.; Teng, Z.Q. A Mouse model of neurodegeneration induced by blade penetrating stab wound to the hippocampus. Biology 2022, 11, 1365. [Google Scholar] [CrossRef] [PubMed]
- He, X.C.; Wang, J.; Du, H.Z.; Liu, C.M.; Teng, Z.Q. Intranasal administration of agomir-let-7i improves cognitive function in mice with traumatic brain injury. Cells 2022, 11, 1348. [Google Scholar] [CrossRef]
- Mai, H.; Fan, W.; Wang, Y.; Cai, Y.; Li, X.; Chen, F.; Chen, X.; Yang, J.; Tang, P.; Chen, H.; et al. Intranasal administration of mir-146a agomir rescued the pathological process and cognitive impairment in an ad mouse model. Mol. Ther. Nucleic Acids 2019, 18, 681–695. [Google Scholar] [CrossRef]
- Wang, Y.Y.; Deng, Y.S.; Dai, S.K.; Mi, T.W.; Li, R.Y.; Liu, P.P.; Liu, C.; He, B.D.; He, X.C.; Du, H.Z.; et al. Loss of microglial eed impairs synapse density, learning, and memory. Mol. Psychiatry 2022, 27, 2999–3009. [Google Scholar] [CrossRef] [PubMed]
- Saijo, K.; Winner, B.; Carson, C.T.; Collier, J.G.; Boyer, L.; Rosenfeld, M.G.; Gage, F.H.; Glass, C.K. A nurr1/corest pathway in microglia and astrocytes protects dopaminergic neurons from inflammation-induced death. Cell 2009, 137, 47–59. [Google Scholar] [CrossRef]
- Shi, R.X.; Liu, C.; Xu, Y.J.; Wang, Y.Y.; He, B.D.; He, X.C.; Du, H.Z.; Hu, B.; Jiao, J.; Liu, C.M.; et al. The Role and mechanism of transglutaminase 2 in regulating hippocampal neurogenesis after traumatic brain injury. Cells 2023, 12, 558. [Google Scholar] [CrossRef]
- Dai, S.K.; Liu, P.P.; Du, H.Z.; Liu, X.; Xu, Y.J.; Liu, C.; Wang, Y.Y.; Teng, Z.Q.; Liu, C.M. Histone crotonylation regulates neural stem cell fate decisions by activating bivalent promoters. EMBO Rep. 2021, 22, e52023. [Google Scholar] [CrossRef]
- Liu, C.; Dai, S.K.; Shi, R.X.; He, X.C.; Wang, Y.Y.; He, B.D.; Sun, X.W.; Du, H.Z.; Liu, C.M.; Teng, Z.Q. Transcriptional profiling of microglia in the injured brain reveals distinct molecular features underlying neurodegeneration. Glia 2021, 69, 1292–1306. [Google Scholar] [CrossRef]
- Chen, R.; Wen, D.; Fu, W.; Xing, L.; Ma, L.; Liu, Y.; Li, H.; You, C.; Lin, Y. Treatment effect of DNA framework nucleic acids on diffuse microvascular endothelial cell injury after subarachnoid hemorrhage. Cell Prolif. 2022, 55, e13206. [Google Scholar] [CrossRef]
- Viedma-Poyatos, A.; Gonzalez-Jimenez, P.; Pajares, M.A.; Perez-Sala, D. Alexander disease gfap r239c mutant shows increased susceptibility to lipoxidation and elicits mitochondrial dysfunction and oxidative stress. Redox Biol. 2022, 55, 102415. [Google Scholar] [CrossRef] [PubMed]
- Colonna, M.; Butovsky, O. Microglia function in the central nervous system during health and neurodegeneration. Annu. Rev. Immunol. 2017, 35, 441–468. [Google Scholar] [CrossRef] [PubMed]
- Karve, I.P.; Taylor, J.M.; Crack, P.J. The contribution of astrocytes and microglia to traumatic brain injury. Br. J. Pharmacol. 2016, 173, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Bradbury, E.J.; Burnside, E.R. Moving beyond the glial scar for spinal cord repair. Nat. Commun. 2019, 10, 3879. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Liu, J.; Zhao, S.; Zhang, H.; Cai, W.; Cai, M.; Ji, X.; Leak, R.K.; Gao, Y.; Chen, J.; et al. Interleukin-4 is essential for microglia/macrophage m2 polarization and long-Term recovery after cerebral ischemia. Stroke 2016, 47, 498–504. [Google Scholar] [CrossRef]
- Kim, E.C.; Zhang, J.; Tang, A.Y.; Bolton, E.C.; Rhodes, J.S.; Christian-Hinman, C.A.; Chung, H.J. Spontaneous seizure and memory loss in mice expressing an epileptic encephalopathy variant in the calmodulin-binding domain of K(v)7.2. Proc. Natl. Acad. Sci. USA 2021, 118, e2021265118. [Google Scholar] [CrossRef]
- Yuen, T.J.; Johnson, K.R.; Miron, V.E.; Zhao, C.; Quandt, J.; Harrisingh, M.C.; Swire, M.; Williams, A.; McFarland, H.F.; Franklin, R.J.; et al. Identification of endothelin 2 as an inflammatory factor that promotes central nervous system remyelination. Brain 2013, 136, 1035–1047. [Google Scholar] [CrossRef]
- Zhu, J.; Hu, Z.; Han, X.; Wang, D.; Jiang, Q.; Ding, J.; Xiao, M.; Wang, C.; Lu, M.; Hu, G. Dopamine d2 receptor restricts astrocytic nlrp3 inflammasome activation via enhancing the interaction of beta-arrestin2 and nlrp3. Cell Death Differ. 2018, 25, 2037–2049. [Google Scholar] [CrossRef]
- Santos, R.; Vadodaria, K.C.; Jaeger, B.N.; Mei, A.; Lefcochilos-Fogelquist, S.; Mendes, A.P.D.; Erikson, G.; Shokhirev, M.; Randolph-Moore, L.; Fredlender, C.; et al. Differentiation of inflammation-responsive astrocytes from glial progenitors generated from human induced pluripotent stem cells. Stem Cell Rep. 2017, 8, 1757–1769. [Google Scholar] [CrossRef]
- Baxter, P.S.; Dando, O.; Emelianova, K.; He, X.; McKay, S.; Hardingham, G.E.; Qiu, J. Microglial identity and inflammatory responses are controlled by the combined effects of neurons and astrocytes. Cell Rep. 2021, 34, 108882. [Google Scholar] [CrossRef]
- Wu, H.Y.; Chung, M.C.; Wang, C.C.; Huang, C.H.; Liang, H.J.; Jan, T.R. Iron oxide nanoparticles suppress the production of IL-1beta via the secretory lysosomal pathway in murine microglial cells. Part. Fibre Toxicol. 2013, 10, 46. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.M.; Hsu, J.C.; Ko, C.Y.; Chiu, N.E.; Kan, W.M.; Lai, M.D.; Wang, J.M. Astrocytic ccaat/enhancer-binding protein delta contributes to glial scar formation and impairs functional recovery after spinal cord injury. Mol. Neurobiol. 2016, 53, 5912–5927. [Google Scholar] [CrossRef] [PubMed]
- McNamee, E.N.; Griffin, E.W.; Ryan, K.M.; Ryan, K.J.; Heffernan, S.; Harkin, A.; Connor, T.J. Noradrenaline acting at beta-adrenoceptors induces expression of IL-1beta and its negative regulators il-1ra and il-1rii, and drives an overall anti-inflammatory phenotype in rat cortex. Neuropharmacology 2010, 59, 37–48. [Google Scholar] [CrossRef]
- Balosso, S.; Maroso, M.; Sanchez-Alavez, M.; Ravizza, T.; Frasca, A.; Bartfai, T.; Vezzani, A. A novel non-transcriptional pathway mediates the proconvulsive effects of interleukin-1beta. Brain 2008, 131, 3256–3265. [Google Scholar] [CrossRef] [PubMed]
- Ozen, I.; Ruscher, K.; Nilsson, R.; Flygt, J.; Clausen, F.; Marklund, N. Interleukin-1 beta neutralization attenuates traumatic brain injury-induced microglia activation and neuronal changes in the globus pallidus. Int. J. Mol. Sci. 2020, 21, 387. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Cavalli, G.; Dinarello, C.A. Anakinra therapy for non-cancer inflammatory diseases. Front. Pharmacol. 2018, 9, 1157. [Google Scholar] [CrossRef]
- Lu, K.T.; Wang, Y.W.; Yang, J.T.; Yang, Y.L.; Chen, H.I. Effect of interleukin-1 on traumatic brain injury-induced damage to hippocampal neurons. J. Neurotrauma 2005, 22, 885–895. [Google Scholar] [CrossRef]
- Clausen, F.; Hanell, A.; Bjork, M.; Hillered, L.; Mir, A.K.; Gram, H.; Marklund, N. Neutralization of interleukin-1beta modifies the inflammatory response and improves histological and cognitive outcome following traumatic brain injury in mice. Eur. J. Neurosci. 2009, 30, 385–396. [Google Scholar] [CrossRef]
- Clausen, F.; Hanell, A.; Israelsson, C.; Hedin, J.; Ebendal, T.; Mir, A.K.; Gram, H.; Marklund, N. Neutralization of interleukin-1beta reduces cerebral edema and tissue loss and improves late cognitive outcome following traumatic brain injury in mice. Eur. J. Neurosci. 2011, 34, 110–123. [Google Scholar] [CrossRef]
- Van Hoecke, L.; Roose, K. How mRNA therapeutics are entering the monoclonal antibody field. J. Transl. Med. 2019, 17, 54. [Google Scholar] [CrossRef] [PubMed]
- Meziane, H.; Ouagazzal, A.M.; Aubert, L.; Wietrzych, M.; Krezel, W. Estrous cycle effects on behavior of c57bl/6j and balb/cbyj female mice: Implications for phenotyping strategies. Genes Brain Behav. 2007, 6, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Hou, Y.; Zhang, L.; Liu, M.; Zhao, J.; Zhang, Z.; Ma, Y.; Hou, W. Estrogen attenuates traumatic brain injury by inhibiting the activation of microglia and astrocyte-mediated neuroinflammatory responses. Mol. Neurobiol. 2021, 58, 1052–1061. [Google Scholar] [CrossRef] [PubMed]
- Raghava, N.; Das, B.C.; Ray, S.K. Neuroprotective effects of estrogen in cns injuries: Insights from animal models. Neurosci. Neuroeconomics 2017, 6, 15–29. [Google Scholar] [CrossRef] [PubMed]
- Brotfain, E.; Gruenbaum, S.E.; Boyko, M.; Kutz, R.; Zlotnik, A.; Klein, M. Neuroprotection by estrogen and progesterone in traumatic brain injury and spinal cord injury. Curr. Neuropharmacol. 2016, 14, 641–653. [Google Scholar] [CrossRef]
- Martinez, I.; Hayes, K.E.; Barr, J.A.; Harold, A.D.; Xie, M.; Bukhari, S.I.A.; Vasudevan, S.; Steitz, J.A.; DiMaio, D. An exportin-1-dependent microrna biogenesis pathway during human cell quiescence. Proc. Natl. Acad. Sci. USA 2017, 114, E4961–E4970. [Google Scholar] [CrossRef]
- Schneider, R.; McKeever, P.; Kim, T.; Graff, C.; van Swieten, J.C.; Karydas, A.; Boxer, A.; Rosen, H.; Miller, B.L.; Laforce, R., Jr.; et al. Downregulation of exosomal mir-204-5p and mir-632 as a biomarker for ftd: A genfi study. J. Neurol. Neurosurg. Psychiatry 2018, 89, 851–858. [Google Scholar] [CrossRef]
- Dietze, P.; Jauncey, M.; Salmon, A.; Mohebbi, M.; Latimer, J.; van Beek, I.; McGrath, C.; Kerr, D. Effect of intranasal vs intramuscular naloxone on opioid overdose: A randomized clinical trial. JAMA Netw. Open 2019, 2, e1914977. [Google Scholar] [CrossRef]
- Rosenfeld, J.V.; Maas, A.I.; Bragge, P.; Morganti-Kossmann, M.C.; Manley, G.T.; Gruen, R.L. Early management of severe traumatic brain injury. Lancet 2012, 380, 1088–1098. [Google Scholar] [CrossRef]
- Ponsford, J.; Willmott, C.; Rothwell, A.; Cameron, P.; Ayton, G.; Nelms, R.; Curran, C.; Ng, K. Impact of early intervention on outcome after mild traumatic brain injury in children. Pediatrics 2001, 108, 1297–1303. [Google Scholar] [CrossRef]
- Wang, W.X.; Prajapati, P.; Vekaria, H.J.; Spry, M.; Cloud, A.L.; Sullivan, P.G.; Springer, J.E. Temporal changes in inflammatory mitochondria-enriched micrornas following traumatic brain injury and effects of miR-146a nanoparticle delivery. Neural Regen. Res. 2021, 16, 514–522. [Google Scholar] [CrossRef] [PubMed]
- Mendiola, A.S.; Cardona, A.E. The il-1beta phenomena in neuroinflammatory diseases. J. Neural Transm. 2018, 125, 781–795. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer Sequence (5’-3’) | |
|---|---|---|
| Actin | Forward | TGCACCACCAACTGCTTAG |
| Reverse | GGATGCAGGGATGATGTTC | |
| TNF-α | Forward | ACGGCATGGATCTCAAAGAC |
| Reverse | GTGGGTGAGGAGCACGTAGT | |
| IL-1β | Forward | CAGGCAGGCAGTATCACTCA |
| Reverse | TGTCCTCATCCTGGAAGGTC | |
| iNOS | Forward | GGCAGCCTGTGAGACCTTTG |
| Reverse | GCATTGGAAGTGAAGCGTTTC | |
| Arg1 | Forward | ACATTGGCTTGCGAGACGTA |
| Reverse | TCCATCACCTTGCCAATCCC | |
| IL-6 | Forward | ATGGATGCTACCAAACTGGAT |
| Reverse | TGAAGGACTCTGGCTTTGTCT | |
| IL-10 | Forward | CCCTGGCTCGTGTGGATTT |
| Reverse | GACCGATACCACTCCTCTGTC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.-X.; Xiao, X.; He, X.-C.; He, B.-D.; Liu, C.-M.; Teng, Z.-Q. Agomir-331 Suppresses Reactive Gliosis and Neuroinflammation after Traumatic Brain Injury. Cells 2023, 12, 2429. https://doi.org/10.3390/cells12202429
Wang J-X, Xiao X, He X-C, He B-D, Liu C-M, Teng Z-Q. Agomir-331 Suppresses Reactive Gliosis and Neuroinflammation after Traumatic Brain Injury. Cells. 2023; 12(20):2429. https://doi.org/10.3390/cells12202429
Chicago/Turabian StyleWang, Jin-Xing, Xiao Xiao, Xuan-Cheng He, Bao-Dong He, Chang-Mei Liu, and Zhao-Qian Teng. 2023. "Agomir-331 Suppresses Reactive Gliosis and Neuroinflammation after Traumatic Brain Injury" Cells 12, no. 20: 2429. https://doi.org/10.3390/cells12202429