
Biochemical and Molecular Pathways in Neurodegenerative Diseases: An Integrated View

Abstract
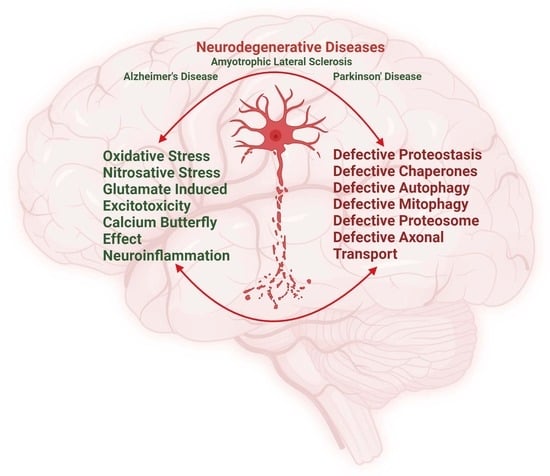
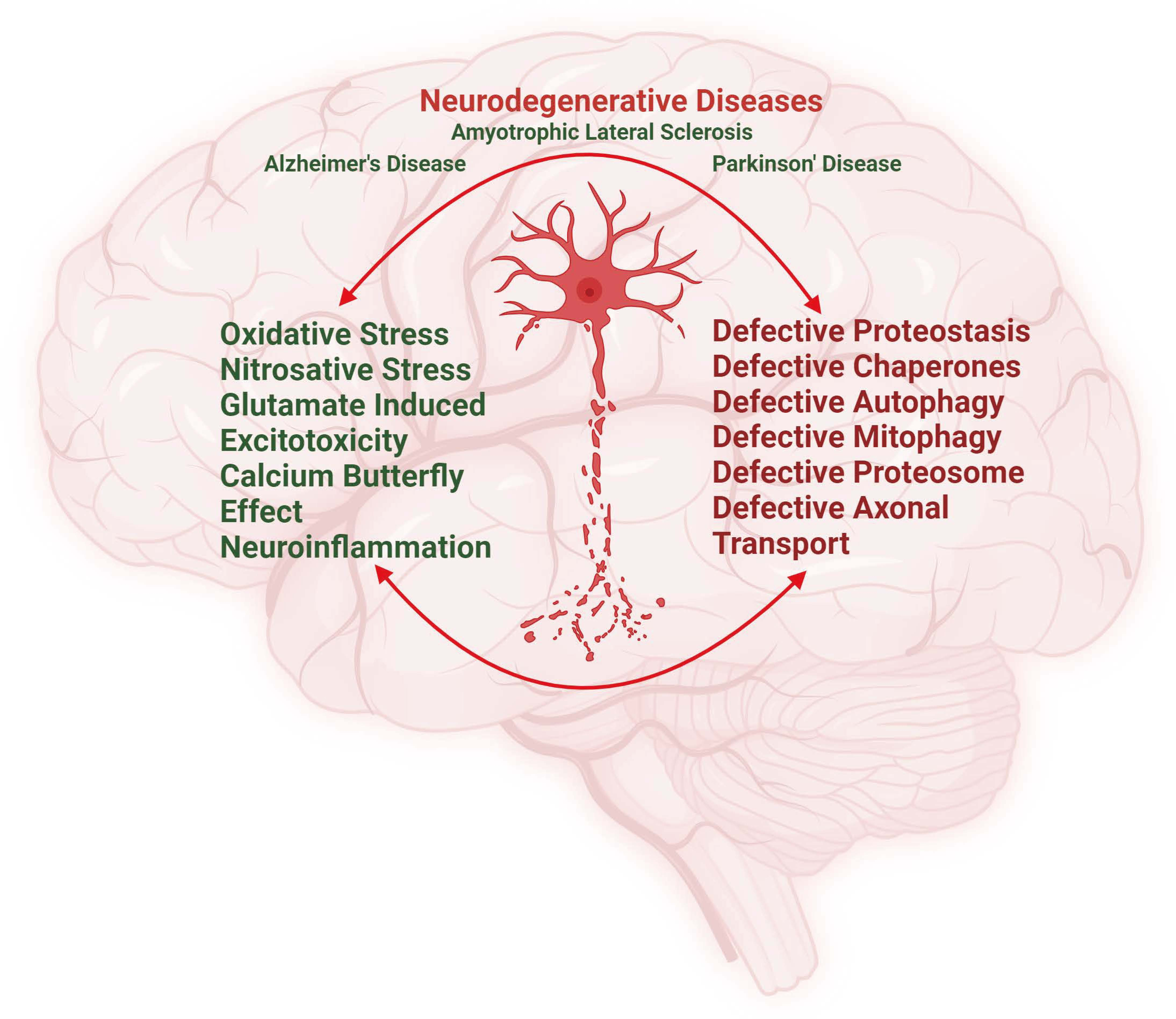
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
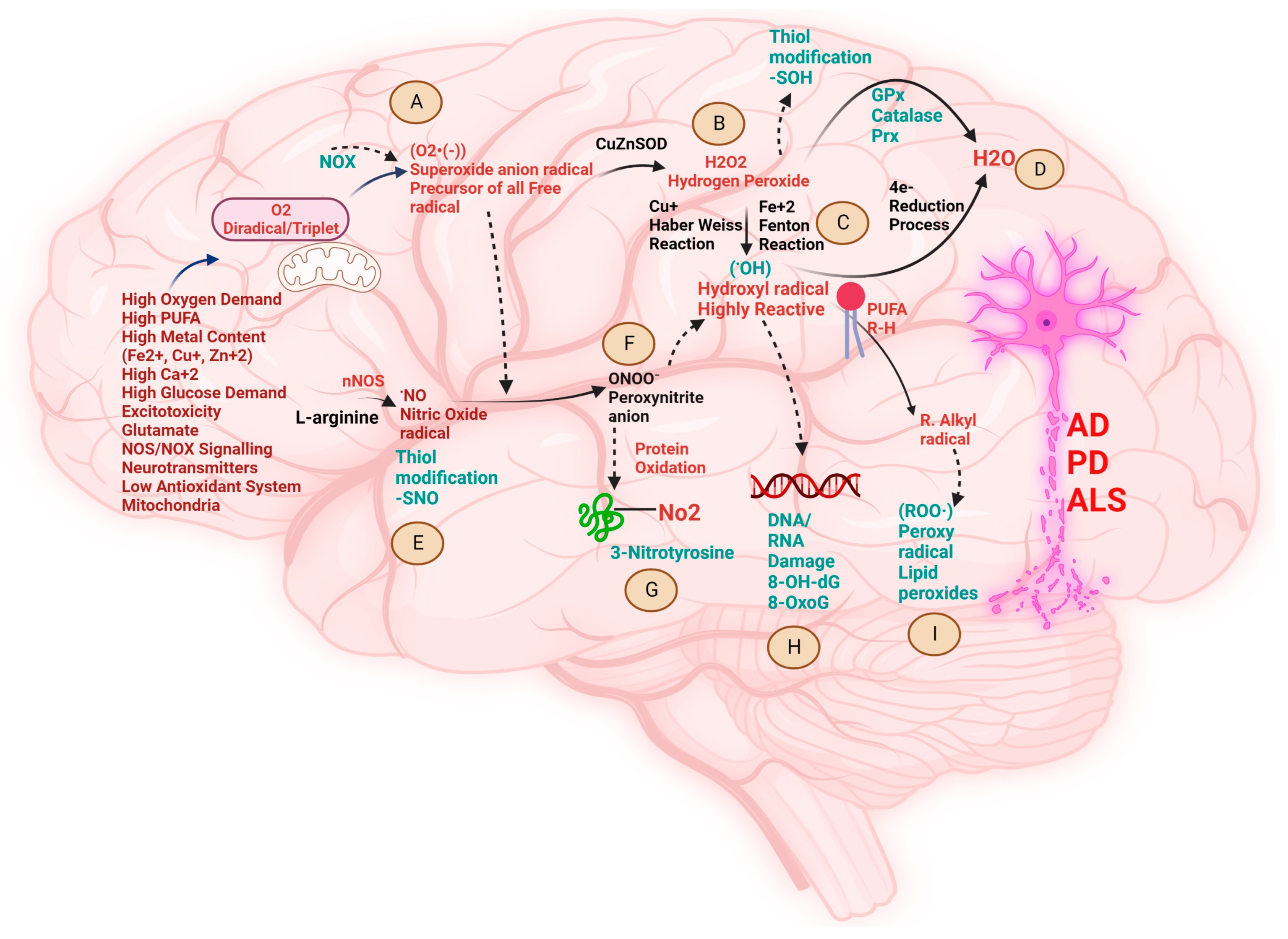
2. Biochemical Pathways Perturbed by Different Metal Ions and the Pathological Role of Free Radicals in NDDs

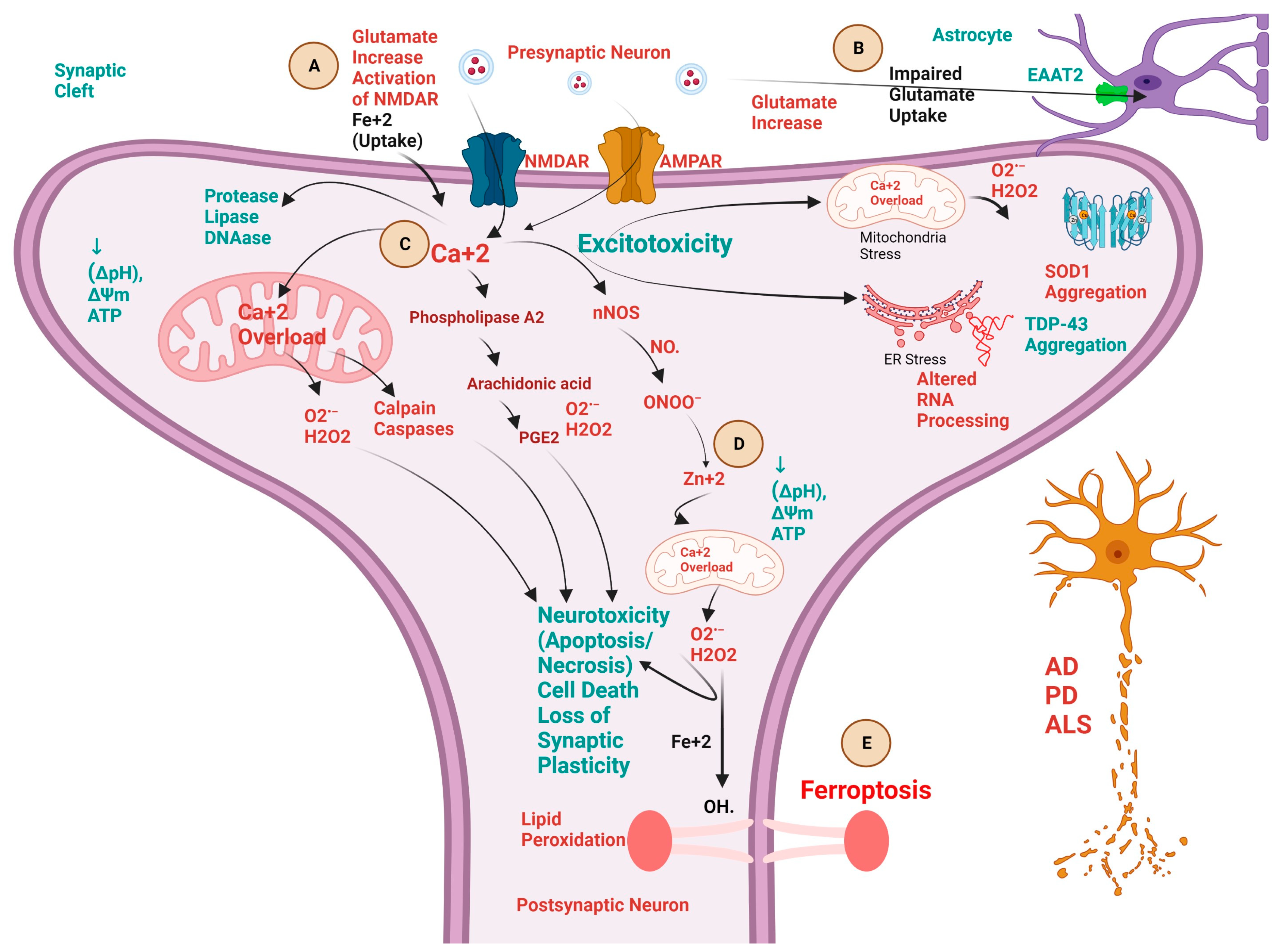
3. Biochemical Pathways and Cross-Talk between Excitotoxicity, Calcium ion (Ca+2), Fe+2, and Zn+2 in NDDs

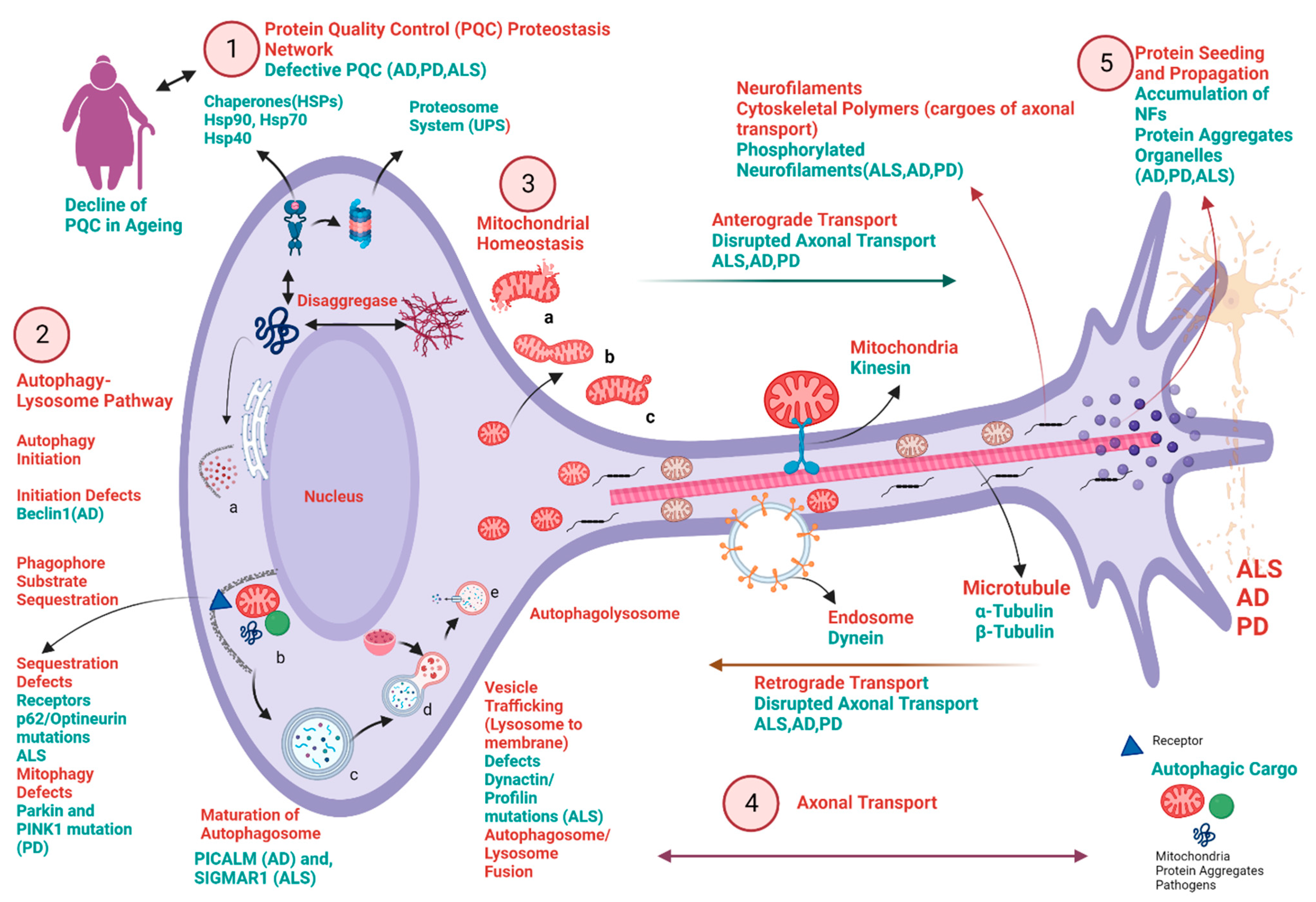
4. Biochemical Pathways Involving Protein Homeostasis, Autophagy, Mitochondrial Homeostasis, Axonal Transport, Protein Seeding, and Propagation and Their Implication in the Pathophysiology of NDDs

5. Biochemical Pathways Altered in NDDs Due to Neuroinflammation
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Kovacs, G.G. Molecular pathology of neurodegenerative diseases: Principles and practice. J. Clin. Pathol. 2019, 72, 725–735. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.M., 3rd; Cookson, M.R.; Van Den Bosch, L.; Zetterberg, H.; Holtzman, D.M.; Dewachter, I. Hallmarks of neurodegenerative diseases. Cell 2023, 186, 693–714. [Google Scholar] [CrossRef] [PubMed]
- Gitler, A.D.; Dhillon, P.; Shorter, J. Neurodegenerative disease: Models, mechanisms, and a new hope. Dis. Model. Mech. 2017, 10, 499–502. [Google Scholar] [CrossRef] [PubMed]
- Gan, L.; Cookson, M.R.; Petrucelli, L.; La Spada, A.R. Converging pathways in neurodegeneration, from genetics to mechanisms. Nat. Neurosci. 2018, 21, 1300–1309. [Google Scholar] [CrossRef] [PubMed]
- Cicero, C.E.; Mostile, G.; Vasta, R.; Rapisarda, V.; Signorelli, S.S.; Ferrante, M.; Zappia, M.; Nicoletti, A. Metals and neurodegenerative diseases. A systematic review. Environ. Res. 2017, 159, 82–94. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.R. Metals in neurodegenerative disease. Metallomics 2011, 3, 226–228. [Google Scholar] [CrossRef] [PubMed]
- Hayflick, S.J.; Kurian, M.A.; Hogarth, P. Neurodegeneration with brain iron accumulation. Handb. Clin. Neurol. 2018, 147, 293–305. [Google Scholar] [CrossRef]
- Ward, R.J.; Zucca, F.A.; Duyn, J.H.; Crichton, R.R.; Zecca, L. The role of iron in brain ageing and neurodegenerative disorders. Lancet Neurol. 2014, 13, 1045–1060. [Google Scholar] [CrossRef]
- Barnham, K.J.; Masters, C.L.; Bush, A.I. Neurodegenerative diseases and oxidative stress. Nat. Rev. Drug Discov. 2004, 3, 205–214. [Google Scholar] [CrossRef]
- Sbodio, J.I.; Snyder, S.H.; Paul, B.D. Redox Mechanisms in Neurodegeneration: From Disease Outcomes to Therapeutic Opportunities. Antioxid. Redox Signal. 2019, 30, 1450–1499. [Google Scholar] [CrossRef]
- Iskusnykh, I.Y.; Zakharova, A.A.; Pathak, D. Glutathione in Brain Disorders and Aging. Molecules 2022, 27, 324. [Google Scholar] [CrossRef] [PubMed]
- Armada-Moreira, A.; Gomes, J.I.; Pina, C.C.; Savchak, O.K.; Gonçalves-Ribeiro, J.; Rei, N.; Pinto, S.; Morais, T.P.; Martins, R.S.; Ribeiro, F.F.; et al. Going the Extra (Synaptic) Mile: Excitotoxicity as the Road Toward Neurodegenerative Diseases. Front. Cell. Neurosci. 2020, 14, 90. [Google Scholar] [CrossRef] [PubMed]
- Schrank, S.; Barrington, N.; Stutzmann, G.E. Calcium-Handling Defects and Neurodegenerative Disease. Cold Spring Harb. Perspect. Biol. 2020, 12, a035212. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.; Kim, S.Y.; Canbakis Cecen, F.S.; Cho, Y.; Kwon, S.K. Dysfunction of Mitochondrial Ca(2+) Regulatory Machineries in Brain Aging and Neurodegenerative Diseases. Front. Cell Dev. Biol. 2020, 8, 599792. [Google Scholar] [CrossRef]
- Pchitskaya, E.; Popugaeva, E.; Bezprozvanny, I. Calcium signaling and molecular mechanisms underlying neurodegenerative diseases. Cell Calcium 2018, 70, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Hipp, M.S.; Kasturi, P.; Hartl, F.U. The proteostasis network and its decline in ageing. Nat. Rev. Mol. Cell Biol. 2019, 20, 421–435. [Google Scholar] [CrossRef] [PubMed]
- Sontag, E.M.; Samant, R.S.; Frydman, J. Mechanisms and Functions of Spatial Protein Quality Control. Annu. Rev. Biochem. 2017, 86, 97–122. [Google Scholar] [CrossRef]
- Soto, C.; Pritzkow, S. Protein misfolding, aggregation, and conformational strains in neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1332–1340. [Google Scholar] [CrossRef]
- Koopman, M.B.; Ferrari, L.; Rüdiger, S.G.D. How do protein aggregates escape quality control in neurodegeneration? Trends Neurosci. 2022, 45, 257–271. [Google Scholar] [CrossRef]
- Ciechanover, A.; Kwon, Y.T. Protein Quality Control by Molecular Chaperones in Neurodegeneration. Front. Neurosci. 2017, 11, 185. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, Y.; Zhou, J. Neuroinflammation in Parkinson’s disease and its potential as therapeutic target. Transl. Neurodegener. 2015, 4, 19. [Google Scholar] [CrossRef] [PubMed]
- Leng, F.; Edison, P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat. Rev. Neurol. 2021, 17, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Philips, T.; Robberecht, W. Neuroinflammation in amyotrophic lateral sclerosis: Role of glial activation in motor neuron disease. Lancet Neurol. 2011, 10, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P.; Magnus, T. Ageing and neuronal vulnerability. Nat. Rev. Neurosci. 2006, 7, 278–294. [Google Scholar] [CrossRef]
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef] [PubMed]
- Wyss-Coray, T. Ageing, neurodegeneration and brain rejuvenation. Nature 2016, 539, 180–186. [Google Scholar] [CrossRef]
- Cobley, J.N.; Fiorello, M.L.; Bailey, D.M. 13 reasons why the brain is susceptible to oxidative stress. Redox Biol. 2018, 15, 490–503. [Google Scholar] [CrossRef]
- Imlay, J.A. Iron-sulphur clusters and the problem with oxygen. Mol. Microbiol. 2006, 59, 1073–1082. [Google Scholar] [CrossRef]
- Connor, J.R.; Menzies, S.L. Relationship of iron to oligodendrocytes and myelination. Glia 1996, 17, 83–93. [Google Scholar] [CrossRef]
- Todorich, B.; Pasquini, J.M.; Garcia, C.I.; Paez, P.M.; Connor, J.R. Oligodendrocytes and myelination: The role of iron. Glia 2009, 57, 467–478. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Weiland, A.; Wang, Y.; Wu, W.; Lan, X.; Han, X.; Li, Q.; Wang, J. Ferroptosis and Its Role in Diverse Brain Diseases. Mol. Neurobiol. 2019, 56, 4880–4893. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.F.; Zou, T.; Tuo, Q.Z.; Xu, S.; Li, H.; Belaidi, A.A.; Lei, P. Ferroptosis: Mechanisms and links with diseases. Signal Transduct. Target. Ther. 2021, 6, 49. [Google Scholar] [CrossRef] [PubMed]
- Banci, L.; Bertini, I.; Ciofi-Baffoni, S.; Kozyreva, T.; Zovo, K.; Palumaa, P. Affinity gradients drive copper to cellular destinations. Nature 2010, 465, 645–648. [Google Scholar] [CrossRef] [PubMed]
- Swaminathan, A.B.; Gohil, V.M. The Role of COA6 in the Mitochondrial Copper Delivery Pathway to Cytochrome c Oxidase. Biomolecules 2022, 12, 125. [Google Scholar] [CrossRef] [PubMed]
- Garcia, L.; Mansilla, N.; Ocampos, N.; Pagani, M.A.; Welchen, E.; Gonzalez, D.H. The mitochondrial copper chaperone COX19 influences copper and iron homeostasis in arabidopsis. Plant Mol. Biol. 2019, 99, 621–638. [Google Scholar] [CrossRef] [PubMed]
- Kawamata, H.; Manfredi, G. Import, maturation, and function of SOD1 and its copper chaperone CCS in the mitochondrial intermembrane space. Antioxid. Redox Signal. 2010, 13, 1375–1384. [Google Scholar] [CrossRef]
- Luchinat, E.; Barbieri, L.; Banci, L. A molecular chaperone activity of CCS restores the maturation of SOD1 fALS mutants. Sci. Rep. 2017, 7, 17433. [Google Scholar] [CrossRef]
- Tokuda, E.; Furukawa, Y. Copper Homeostasis as a Therapeutic Target in Amyotrophic Lateral Sclerosis with SOD1 Mutations. Int. J. Mol. Sci. 2016, 17, 636. [Google Scholar] [CrossRef]
- Opazo, C.M.; Greenough, M.A.; Bush, A.I. Copper: From neurotransmission to neuroproteostasis. Front. Aging Neurosci. 2014, 6, 143. [Google Scholar] [CrossRef]
- Kesidou, E.; Theotokis, P.; Damianidou, O.; Boziki, M.; Konstantinidou, N.; Taloumtzis, C.; Sintila, S.-A.; Grigoriadis, P.; Evangelopoulos, M.E.; Bakirtzis, C. CNS Ageing in Health and Neurodegenerative Disorders. J. Clin. Med. 2023, 12, 2255. [Google Scholar] [CrossRef] [PubMed]
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, F.; Bonaduce, D.; et al. Oxidative stress, aging, and diseases. Clin. Interv. Aging 2018, 13, 757–772. [Google Scholar] [CrossRef] [PubMed]
- Tan, B.L.; Norhaizan, M.E.; Liew, W.P.; Sulaiman Rahman, H. Antioxidant and Oxidative Stress: A Mutual Interplay in Age-Related Diseases. Front. Pharmacol. 2018, 9, 1162. [Google Scholar] [CrossRef] [PubMed]
- Olanow, C.W.; Arendash, G.W. Metals and free radicals in neurodegeneration. Curr. Opin. Neurol. 1994, 7, 548–558. [Google Scholar] [CrossRef] [PubMed]
- Shafiq, K.; Sanghai, N.; Guo, Y.; Kong, J. Implication of post-translationally modified SOD1 in pathological aging. Geroscience 2021, 43, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Petillon, C.; Hergesheimer, R.; Puy, H.; Corcia, P.; Vourc’h, P.; Andres, C.; Karim, Z.; Blasco, H. The Relevancy of Data Regarding the Metabolism of Iron to Our Understanding of Deregulated Mechanisms in ALS; Hypotheses and Pitfalls. Front. Neurosci. 2018, 12, 1031. [Google Scholar] [CrossRef] [PubMed]
- Mezzaroba, L.; Alfieri, D.F.; Colado Simão, A.N.; Vissoci Reiche, E.M. The role of zinc, copper, manganese and iron in neurodegenerative diseases. Neurotoxicology 2019, 74, 230–241. [Google Scholar] [CrossRef]
- Gangania, M.K.; Batra, J.; Kushwaha, S.; Agarwal, R. Role of Iron and Copper in the Pathogenesis of Parkinson’s Disease. Indian. J. Clin. Biochem. 2017, 32, 353–356. [Google Scholar] [CrossRef]
- Konno, T.; Melo, E.P.; Chambers, J.E.; Avezov, E. Intracellular Sources of ROS/H(2)O(2) in Health and Neurodegeneration: Spotlight on Endoplasmic Reticulum. Cells 2021, 10, 233. [Google Scholar] [CrossRef]
- Behl, C.; Davis, J.B.; Lesley, R.; Schubert, D. Hydrogen peroxide mediates amyloid beta protein toxicity. Cell 1994, 77, 817–827. [Google Scholar] [CrossRef]
- Sebastià, J.; Pertusa, M.; Vílchez, D.; Planas, A.M.; Verbeek, R.; Rodríguez-Farré, E.; Cristòfol, R.; Sanfeliu, C. Carboxyl-terminal fragment of amyloid precursor protein and hydrogen peroxide induce neuronal cell death through different pathways. J. Neural. Transm. 2006, 113, 1837–1845. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Li, K.; Qin, W.; Zhu, B.; Zhou, Z.; Shi, J.; Wang, K.; Hu, J.; Fan, C.; Li, D. Unraveling the role of hydrogen peroxide in α-synuclein aggregation using an ultrasensitive nanoplasmonic probe. Anal. Chem. 2015, 87, 1968–1973. [Google Scholar] [CrossRef] [PubMed]
- Turnbull, S.; Tabner, B.J.; El-Agnaf, O.M.; Moore, S.; Davies, Y.; Allsop, D. alpha-Synuclein implicated in Parkinson’s disease catalyses the formation of hydrogen peroxide in vitro. Free. Radic. Biol. Med. 2001, 30, 1163–1170. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.C.; Liang, J.Z.; Li, C.; He, Z.X.; Yuan, H.Y.; Huang, B.Y.; Liu, X.L.; Tang, B.; Pang, D.W.; Du, H.N.; et al. Pathological hydrogen peroxide triggers the fibrillization of wild-type SOD1 via sulfenic acid modification of Cys-111. Cell Death Dis. 2018, 9, 67. [Google Scholar] [CrossRef]
- Koo, B.K.; Munroe, W.; Gralla, E.B.; Valentine, J.S.; Whitelegge, J.P. A Novel SOD1 Intermediate Oligomer, Role of Free Thiols and Disulfide Exchange. Front. Neurosci. 2020, 14, 619279. [Google Scholar] [CrossRef]
- Chen, X.; Shang, H.; Qiu, X.; Fujiwara, N.; Cui, L.; Li, X.M.; Gao, T.M.; Kong, J. Oxidative modification of cysteine 111 promotes disulfide bond-independent aggregation of SOD1. Neurochem. Res. 2012, 37, 835–845. [Google Scholar] [CrossRef]
- Sanghai, N.; Tranmer, G.K. Hydrogen Peroxide and Amyotrophic Lateral Sclerosis: From Biochemistry to Pathophysiology. Antioxidants 2021, 11, 52. [Google Scholar] [CrossRef]
- Cohen, T.J.; Hwang, A.W.; Unger, T.; Trojanowski, J.Q.; Lee, V.M. Redox signalling directly regulates TDP-43 via cysteine oxidation and disulphide cross-linking. EMBO J. 2012, 31, 1241–1252. [Google Scholar] [CrossRef]
- Sidorova, Y.; Domanskyi, A. Detecting Oxidative Stress Biomarkers in Neurodegenerative Disease Models and Patients. Methods Protoc. 2020, 3, 66. [Google Scholar] [CrossRef]
- Frijhoff, J.; Winyard, P.G.; Zarkovic, N.; Davies, S.S.; Stocker, R.; Cheng, D.; Knight, A.R.; Taylor, E.L.; Oettrich, J.; Ruskovska, T.; et al. Clinical Relevance of Biomarkers of Oxidative Stress. Antioxid. Redox Signal. 2015, 23, 1144–1170. [Google Scholar] [CrossRef]
- Marrocco, I.; Altieri, F.; Peluso, I. Measurement and Clinical Significance of Biomarkers of Oxidative Stress in Humans. Oxid. Med. Cell Longev. 2017, 2017, 6501046. [Google Scholar] [CrossRef] [PubMed]
- Obata, T. Role of hydroxyl radical formation in neurotoxicity as revealed by in vivo free radical trapping. Toxicol. Lett. 2002, 132, 83–93. [Google Scholar] [CrossRef]
- Gutteridge, J.M. Hydroxyl radicals, iron, oxidative stress, and neurodegeneration. Ann. N. Y. Acad. Sci. 1994, 738, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Jomova, K.; Valko, M. Advances in metal-induced oxidative stress and human disease. Toxicology 2011, 283, 65–87. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B.; Adhikary, A.; Dingfelder, M.; Dizdaroglu, M. Hydroxyl radical is a significant player in oxidative DNA damage in vivo. Chem. Soc. Rev. 2021, 50, 8355–8360. [Google Scholar] [CrossRef] [PubMed]
- Selley, M.L.; Close, D.R.; Stern, S.E. The effect of increased concentrations of homocysteine on the concentration of (E)-4-hydroxy-2-nonenal in the plasma and cerebrospinal fluid of patients with Alzheimer’s disease. Neurobiol. Aging 2002, 23, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Dexter, D.T.; Carter, C.J.; Wells, F.R.; Javoy-Agid, F.; Agid, Y.; Lees, A.; Jenner, P.; Marsden, C.D. Basal lipid peroxidation in substantia nigra is increased in Parkinson’s disease. J. Neurochem. 1989, 52, 381–389. [Google Scholar] [CrossRef]
- Pedersen, W.A.; Fu, W.; Keller, J.N.; Markesbery, W.R.; Appel, S.; Smith, R.G.; Kasarskis, E.; Mattson, M.P. Protein modification by the lipid peroxidation product 4-hydroxynonenal in the spinal cords of amyotrophic lateral sclerosis patients. Ann. Neurol. 1998, 44, 819–824. [Google Scholar] [CrossRef]
- Lovell, M.A.; Ehmann, W.D.; Butler, S.M.; Markesbery, W.R. Elevated thiobarbituric acid-reactive substances and antioxidant enzyme activity in the brain in Alzheimer’s disease. Neurology 1995, 45, 1594–1601. [Google Scholar] [CrossRef]
- Younes-Mhenni, S.; Frih-Ayed, M.; Kerkeni, A.; Bost, M.; Chazot, G. Peripheral blood markers of oxidative stress in Parkinson’s disease. Eur. Neurol. 2007, 58, 78–83. [Google Scholar] [CrossRef]
- Bonnefont-Rousselot, D.; Lacomblez, L.; Jaudon, M.; Lepage, S.; Salachas, F.; Bensimon, G.; Bizard, C.; Doppler, V.; Delattre, J.; Meininger, V. Blood oxidative stress in amyotrophic lateral sclerosis. J. Neurol. Sci. 2000, 178, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wang, Z.; Zeng, B.; Zheng, M.; Xiao, N.; Zhao, Z. Fenton-like reaction of the iron(II)-histidine complex generates hydroxyl radicals: Implications for oxidative stress and Alzheimer’s disease. Chem. Commun. 2021, 57, 12293–12296. [Google Scholar] [CrossRef] [PubMed]
- Rakhit, R.; Cunningham, P.; Furtos-Matei, A.; Dahan, S.; Qi, X.F.; Crow, J.P.; Cashman, N.R.; Kondejewski, L.H.; Chakrabartty, A. Oxidation-induced misfolding and aggregation of superoxide dismutase and its implications for amyotrophic lateral sclerosis. J. Biol. Chem. 2002, 277, 47551–47556. [Google Scholar] [CrossRef]
- Herrera, A.; Muñoz, P.; Steinbusch, H.W.M.; Segura-Aguilar, J. Are Dopamine Oxidation Metabolites Involved in the Loss of Dopaminergic Neurons in the Nigrostriatal System in Parkinson’s Disease? ACS Chem. Neurosci. 2017, 8, 702–711. [Google Scholar] [CrossRef] [PubMed]
- Nunomura, A.; Moreira, P.I.; Castellani, R.J.; Lee, H.G.; Zhu, X.; Smith, M.A.; Perry, G. Oxidative damage to RNA in aging and neurodegenerative disorders. Neurotox. Res. 2012, 22, 231–248. [Google Scholar] [CrossRef]
- Nakabeppu, Y.; Tsuchimoto, D.; Yamaguchi, H.; Sakumi, K. Oxidative damage in nucleic acids and Parkinson’s disease. J. Neurosci. Res. 2007, 85, 919–934. [Google Scholar] [CrossRef] [PubMed]
- Konopka, A.; Atkin, J.D. DNA Damage, Defective DNA Repair, and Neurodegeneration in Amyotrophic Lateral Sclerosis. Front. Aging Neurosci. 2022, 14, 786420. [Google Scholar] [CrossRef] [PubMed]
- Dalle-Donne, I.; Rossi, R.; Giustarini, D.; Milzani, A.; Colombo, R. Protein carbonyl groups as biomarkers of oxidative stress. Clin. Chim. Acta 2003, 329, 23–38. [Google Scholar] [CrossRef]
- Alam, Z.I.; Daniel, S.E.; Lees, A.J.; Marsden, D.C.; Jenner, P.; Halliwell, B. A generalised increase in protein carbonyls in the brain in Parkinson’s but not incidental Lewy body disease. J. Neurochem. 1997, 69, 1326–1329. [Google Scholar] [CrossRef]
- Mitsumoto, H.; Santella, R.M.; Liu, X.; Bogdanov, M.; Zipprich, J.; Wu, H.C.; Mahata, J.; Kilty, M.; Bednarz, K.; Bell, D.; et al. Oxidative stress biomarkers in sporadic ALS. Amyotroph. Lateral Scler. 2008, 9, 177–183. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Reed, T.T.; Perluigi, M.; De Marco, C.; Coccia, R.; Keller, J.N.; Markesbery, W.R.; Sultana, R. Elevated levels of 3-nitrotyrosine in brain from subjects with amnestic mild cognitive impairment: Implications for the role of nitration in the progression of Alzheimer’s disease. Brain Res. 2007, 1148, 243–248. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Espejo, E.; Rodríguez de Fonseca, F.; Suárez, J.; Tolosa, E.; Vilas, D.; Aldecoa, I.; Berenguer, J.; Damas-Hermoso, F. Native α-Synuclein, 3-Nitrotyrosine Proteins, and Patterns of Nitro-α-Synuclein-Immunoreactive Inclusions in Saliva and Submandibulary Gland in Parkinson’s Disease. Antioxidants 2021, 10, 715. [Google Scholar] [CrossRef] [PubMed]
- Beal, M.F.; Ferrante, R.J.; Browne, S.E.; Matthews, R.T.; Kowall, N.W.; Brown, R.H., Jr. Increased 3-nitrotyrosine in both sporadic and familial amyotrophic lateral sclerosis. Ann. Neurol. 1997, 42, 644–654. [Google Scholar] [CrossRef] [PubMed]
- Bandookwala, M.; Sengupta, P. 3-Nitrotyrosine: A versatile oxidative stress biomarker for major neurodegenerative diseases. Int. J. Neurosci. 2020, 130, 1047–1062. [Google Scholar] [CrossRef]
- Ahsan, H. 3-Nitrotyrosine: A biomarker of nitrogen free radical species modified proteins in systemic autoimmunogenic conditions. Hum. Immunol. 2013, 74, 1392–1399. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, C.C.; Braverman, E.R. Zinc, the brain and behavior. Biol. Psychiatry 1982, 17, 513–532. [Google Scholar]
- Marreiro, D.D.; Cruz, K.J.; Morais, J.B.; Beserra, J.B.; Severo, J.S.; de Oliveira, A.R. Zinc and Oxidative Stress: Current Mechanisms. Antioxidants 2017, 6, 24. [Google Scholar] [CrossRef]
- Choi, S.; Hong, D.K.; Choi, B.Y.; Suh, S.W. Zinc in the Brain: Friend or Foe? Int. J. Mol. Sci. 2020, 21, 8941. [Google Scholar] [CrossRef]
- Lee, J.Y.; Cole, T.B.; Palmiter, R.D.; Suh, S.W.; Koh, J.Y. Contribution by synaptic zinc to the gender-disparate plaque formation in human Swedish mutant APP transgenic mice. Proc. Natl. Acad. Sci. USA 2002, 99, 7705–7710. [Google Scholar] [CrossRef]
- Forsleff, L.; Schauss, A.G.; Bier, I.D.; Stuart, S. Evidence of functional zinc deficiency in Parkinson’s disease. J. Altern. Complement. Med. 1999, 5, 57–64. [Google Scholar] [CrossRef]
- Du, K.; Liu, M.Y.; Zhong, X.; Wei, M.J. Decreased circulating Zinc levels in Parkinson’s disease: A meta-analysis study. Sci. Rep. 2017, 7, 3902. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.P.; Lee, N.M. Role of zinc in ALS. Amyotroph. Lateral Scler. 2007, 8, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Hilton, J.B.; White, A.R.; Crouch, P.J. Metal-deficient SOD1 in amyotrophic lateral sclerosis. J. Mol. Med. 2015, 93, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Trist, B.G.; Genoud, S.; Roudeau, S.; Rookyard, A.; Abdeen, A.; Cottam, V.; Hare, D.J.; White, M.; Altvater, J.; Fifita, J.A.; et al. Altered SOD1 maturation and post-translational modification in amyotrophic lateral sclerosis spinal cord. Brain 2022, 145, 3108–3130. [Google Scholar] [CrossRef]
- Vermot, A.; Petit-Härtlein, I.; Smith, S.M.E.; Fieschi, F. NADPH Oxidases (NOX): An Overview from Discovery, Molecular Mechanisms to Physiology and Pathology. Antioxidants 2021, 10, 890. [Google Scholar] [CrossRef]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: Role and Response of Short Guanine Tracts at Genomic Locations. Int. J. Mol. Sci. 2019, 20, 4258. [Google Scholar] [CrossRef]
- Fairman, W.A.; Amara, S.G. Functional diversity of excitatory amino acid transporters: Ion channel and transport modes. Am. J. Physiol. 1999, 277, F481–F486. [Google Scholar] [CrossRef]
- Bano, D.; Young, K.W.; Guerin, C.J.; Lefeuvre, R.; Rothwell, N.J.; Naldini, L.; Rizzuto, R.; Carafoli, E.; Nicotera, P. Cleavage of the plasma membrane Na+/Ca2+ exchanger in excitotoxicity. Cell 2005, 120, 275–285. [Google Scholar] [CrossRef]
- Wang, R.; Reddy, P.H. Role of Glutamate and NMDA Receptors in Alzheimer’s Disease. J. Alzheimers Dis. 2017, 57, 1041–1048. [Google Scholar] [CrossRef]
- Iovino, L.; Tremblay, M.E.; Civiero, L. Glutamate-induced excitotoxicity in Parkinson’s disease: The role of glial cells. J. Pharmacol. Sci. 2020, 144, 151–164. [Google Scholar] [CrossRef]
- Foran, E.; Trotti, D. Glutamate transporters and the excitotoxic path to motor neuron degeneration in amyotrophic lateral sclerosis. Antioxid. Redox Signal. 2009, 11, 1587–1602. [Google Scholar] [CrossRef] [PubMed]
- Connolly, N.M.; Prehn, J.H. The metabolic response to excitotoxicity—Lessons from single-cell imaging. J. Bioenerg. Biomembr. 2015, 47, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Granzotto, A.; Canzoniero, L.M.T.; Sensi, S.L. A Neurotoxic Ménage-à-trois: Glutamate, Calcium, and Zinc in the Excitotoxic Cascade. Front. Mol. Neurosci. 2020, 13, 600089. [Google Scholar] [CrossRef] [PubMed]
- Gleitze, S.; Paula-Lima, A.; Núñez, M.T.; Hidalgo, C. The calcium-iron connection in ferroptosis-mediated neuronal death. Free. Radic. Biol. Med. 2021, 175, 28–41. [Google Scholar] [CrossRef] [PubMed]
- Doble, A. The role of excitotoxicity in neurodegenerative disease: Implications for therapy. Pharmacol. Ther. 1999, 81, 163–221. [Google Scholar] [CrossRef]
- Danbolt, N.C. Glutamate uptake. Prog. Neurobiol. 2001, 65, 1–105. [Google Scholar] [CrossRef]
- Mahmoud, S.; Gharagozloo, M.; Simard, C.; Gris, D. Astrocytes Maintain Glutamate Homeostasis in the CNS by Controlling the Balance between Glutamate Uptake and Release. Cells 2019, 8, 184. [Google Scholar] [CrossRef]
- Armbruster, M.; Hanson, E.; Dulla, C.G. Glutamate Clearance Is Locally Modulated by Presynaptic Neuronal Activity in the Cerebral Cortex. J. Neurosci. 2016, 36, 10404–10415. [Google Scholar] [CrossRef]
- Pehar, M.; Harlan, B.A.; Killoy, K.M.; Vargas, M.R. Role and Therapeutic Potential of Astrocytes in Amyotrophic Lateral Sclerosis. Curr. Pharm. Des. 2017, 23, 5010–5021. [Google Scholar] [CrossRef]
- van den Berg, C.J.; Garfinkel, D. A simulation study of brain compartments. Metabolism of glutamate and related substances in mouse brain. Biochem. J. 1971, 123, 211–218. [Google Scholar] [CrossRef]
- Ottersen, O.P.; Zhang, N.; Walberg, F. Metabolic compartmentation of glutamate and glutamine: Morphological evidence obtained by quantitative immunocytochemistry in rat cerebellum. Neuroscience 1992, 46, 519–534. [Google Scholar] [CrossRef] [PubMed]
- Magistretti, P.J.; Allaman, I. A cellular perspective on brain energy metabolism and functional imaging. Neuron 2015, 86, 883–901. [Google Scholar] [CrossRef] [PubMed]
- Schousboe, A.; Westergaard, N.; Waagepetersen, H.S.; Larsson, O.M.; Bakken, I.J.; Sonnewald, U. Trafficking between glia and neurons of TCA cycle intermediates and related metabolites. Glia 1997, 21, 99–105. [Google Scholar] [CrossRef]
- Jiang, L.L.; Zhu, B.; Zhao, Y.; Li, X.; Liu, T.; Pina-Crespo, J.; Zhou, L.; Xu, W.; Rodriguez, M.J.; Yu, H.; et al. Membralin deficiency dysregulates astrocytic glutamate homeostasis leading to ALS-like impairment. J. Clin. Investig. 2019, 129, 3103–3120. [Google Scholar] [CrossRef]
- Sultan, S.; Li, L.; Moss, J.; Petrelli, F.; Cassé, F.; Gebara, E.; Lopatar, J.; Pfrieger, F.W.; Bezzi, P.; Bischofberger, J.; et al. Synaptic Integration of Adult-Born Hippocampal Neurons Is Locally Controlled by Astrocytes. Neuron 2015, 88, 957–972. [Google Scholar] [CrossRef]
- González-Reyes, R.E.; Nava-Mesa, M.O.; Vargas-Sánchez, K.; Ariza-Salamanca, D.; Mora-Muñoz, L. Involvement of Astrocytes in Alzheimer’s Disease from a Neuroinflammatory and Oxidative Stress Perspective. Front. Mol. Neurosci. 2017, 10, 427. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Giraldo, M.; González-Reyes, R.E.; Ramírez-Guerrero, S.; Bonilla-Trilleras, C.E.; Guardo-Maya, S.; Nava-Mesa, M.O. Astrocytes as a Therapeutic Target in Alzheimer’s Disease-Comprehensive Review and Recent Developments. Int. J. Mol. Sci. 2022, 23, 13630. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, I.; Asanuma, M. Neuron-Astrocyte Interactions in Parkinson’s Disease. Cells 2020, 9, 2623. [Google Scholar] [CrossRef]
- Bancroft, E.A.; Srinivasan, R. Emerging Roles for Aberrant Astrocytic Calcium Signals in Parkinson’s Disease. Front. Physiol. 2021, 12, 812212. [Google Scholar] [CrossRef]
- Bruijn, L.I.; Becher, M.W.; Lee, M.K.; Anderson, K.L.; Jenkins, N.A.; Copeland, N.G.; Sisodia, S.S.; Rothstein, J.D.; Borchelt, D.R.; Price, D.L.; et al. ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron 1997, 18, 327–338. [Google Scholar] [CrossRef]
- Yamanaka, K.; Komine, O. The multi-dimensional roles of astrocytes in ALS. Neurosci. Res. 2018, 126, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Verma, M.; Lizama, B.N.; Chu, C.T. Excitotoxicity, calcium and mitochondria: A triad in synaptic neurodegeneration. Transl. Neurodegener. 2022, 11, 3. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.; Alavi, M.V.; Kim, K.Y.; Kang, T.; Scott, R.T.; Noh, Y.H.; Lindsey, J.D.; Wissinger, B.; Ellisman, M.H.; Weinreb, R.N.; et al. A new vicious cycle involving glutamate excitotoxicity, oxidative stress and mitochondrial dynamics. Cell Death Dis. 2011, 2, e240. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.Z.; Weiss, J.H. Marked synergism between mutant SOD1 and glutamate transport inhibition in the induction of motor neuronal degeneration in spinal cord slice cultures. Brain Res. 2012, 1448, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Musarò, A. State of the art and the dark side of amyotrophic lateral sclerosis. World J. Biol. Chem. 2010, 1, 62–68. [Google Scholar] [CrossRef]
- Van Den Bosch, L.; Van Damme, P.; Bogaert, E.; Robberecht, W. The role of excitotoxicity in the pathogenesis of amyotrophic lateral sclerosis. Biochim. Biophys. Acta 2006, 1762, 1068–1082. [Google Scholar] [CrossRef]
- Jiang, T.; Handley, E.; Brizuela, M.; Dawkins, E.; Lewis, K.E.A.; Clark, R.M.; Dickson, T.C.; Blizzard, C.A. Amyotrophic lateral sclerosis mutant TDP-43 may cause synaptic dysfunction through altered dendritic spine function. Dis. Model. Mech. 2019, 12, dmm038109. [Google Scholar] [CrossRef]
- Pirie, E.; Oh, C.K.; Zhang, X.; Han, X.; Cieplak, P.; Scott, H.R.; Deal, A.K.; Ghatak, S.; Martinez, F.J.; Yeo, G.W.; et al. S-nitrosylated TDP-43 triggers aggregation, cell-to-cell spread, and neurotoxicity in hiPSCs and in vivo models of ALS/FTD. Proc. Natl. Acad. Sci. USA 2021, 118, e2021368118. [Google Scholar] [CrossRef]
- Olney, J.W.; Wozniak, D.F.; Farber, N.B. Excitotoxic neurodegeneration in Alzheimer disease. New hypothesis and new therapeutic strategies. Arch. Neurol. 1997, 54, 1234–1240. [Google Scholar] [CrossRef]
- Reddy, P.H.; Beal, M.F. Amyloid beta, mitochondrial dysfunction and synaptic damage: Implications for cognitive decline in aging and Alzheimer’s disease. Trends Mol. Med. 2008, 14, 45–53. [Google Scholar] [CrossRef]
- Liu, B.; Zhu, Y.; Zhou, J.; Wei, Y.; Long, C.; Chen, M.; Ling, Y.; Ge, J.; Zhuo, Y. Endoplasmic reticulum stress promotes amyloid-beta peptides production in RGC-5 cells. Cell Stress. Chaperones 2014, 19, 827–835. [Google Scholar] [CrossRef] [PubMed]
- Ghemrawi, R.; Khair, M. Endoplasmic Reticulum Stress and Unfolded Protein Response in Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 6127. [Google Scholar] [CrossRef] [PubMed]
- Puspita, L.; Chung, S.Y.; Shim, J.W. Oxidative stress and cellular pathologies in Parkinson’s disease. Mol. Brain 2017, 10, 53. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Suaga, P.; Bravo-San Pedro, J.M.; González-Polo, R.A.; Fuentes, J.M.; Niso-Santano, M. ER-mitochondria signaling in Parkinson’s disease. Cell Death Dis. 2018, 9, 337. [Google Scholar] [CrossRef] [PubMed]
- Höhn, A.; Tramutola, A.; Cascella, R. Proteostasis Failure in Neurodegenerative Diseases: Focus on Oxidative Stress. Oxid. Med. Cell Longev. 2020, 2020, 5497046. [Google Scholar] [CrossRef]
- Yerbury, J.J.; Ooi, L.; Dillin, A.; Saunders, D.N.; Hatters, D.M.; Beart, P.M.; Cashman, N.R.; Wilson, M.R.; Ecroyd, H. Walking the tightrope: Proteostasis and neurodegenerative disease. J. Neurochem. 2016, 137, 489–505. [Google Scholar] [CrossRef]
- Hetz, C. Adapting the proteostasis capacity to sustain brain healthspan. Cell 2021, 184, 1545–1560. [Google Scholar] [CrossRef]
- Klaips, C.L.; Jayaraj, G.G.; Hartl, F.U. Pathways of cellular proteostasis in aging and disease. J. Cell Biol. 2018, 217, 51–63. [Google Scholar] [CrossRef]
- Goedert, M.; Eisenberg, D.S.; Crowther, R.A. Propagation of Tau Aggregates and Neurodegeneration. Annu. Rev. Neurosci. 2017, 40, 189–210. [Google Scholar] [CrossRef]
- Xilouri, M.; Stefanis, L. Autophagy in the central nervous system: Implications for neurodegenerative disorders. CNS Neurol. Disord. Drug Targets 2010, 9, 701–719. [Google Scholar] [CrossRef]
- Kim, Y.C.; Guan, K.L. mTOR: A pharmacologic target for autophagy regulation. J. Clin. Investig. 2015, 125, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Nazio, F.; Strappazzon, F.; Antonioli, M.; Bielli, P.; Cianfanelli, V.; Bordi, M.; Gretzmeier, C.; Dengjel, J.; Piacentini, M.; Fimia, G.M.; et al. mTOR inhibits autophagy by controlling ULK1 ubiquitylation, self-association and function through AMBRA1 and TRAF6. Nat. Cell Biol. 2013, 15, 406–416. [Google Scholar] [CrossRef] [PubMed]
- Webb, J.L.; Ravikumar, B.; Atkins, J.; Skepper, J.N.; Rubinsztein, D.C. Alpha-Synuclein is degraded by both autophagy and the proteasome. J. Biol. Chem. 2003, 278, 25009–25013. [Google Scholar] [CrossRef]
- Barmada, S.J.; Serio, A.; Arjun, A.; Bilican, B.; Daub, A.; Ando, D.M.; Tsvetkov, A.; Pleiss, M.; Li, X.; Peisach, D.; et al. Autophagy induction enhances TDP43 turnover and survival in neuronal ALS models. Nat. Chem. Biol. 2014, 10, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, L. Targeting Autophagy for the Treatment of Alzheimer’s Disease: Challenges and Opportunities. Front. Mol. Neurosci. 2019, 12, 203. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A.; Yang, D.S. Autophagy failure in Alzheimer’s disease--locating the primary defect. Neurobiol. Dis. 2011, 43, 38–45. [Google Scholar] [CrossRef]
- Hou, X.; Watzlawik, J.O.; Fiesel, F.C.; Springer, W. Autophagy in Parkinson’s Disease. J. Mol. Biol. 2020, 432, 2651–2672. [Google Scholar] [CrossRef]
- Arotcarena, M.L.; Teil, M.; Dehay, B. Autophagy in Synucleinopathy: The Overwhelmed and Defective Machinery. Cells 2019, 8, 565. [Google Scholar] [CrossRef]
- Lee, H.J.; Cho, E.D.; Lee, K.W.; Kim, J.H.; Cho, S.G.; Lee, S.J. Autophagic failure promotes the exocytosis and intercellular transfer of α-synuclein. Exp. Mol. Med. 2013, 45, e22. [Google Scholar] [CrossRef]
- Chen, S.; Zhang, X.; Song, L.; Le, W. Autophagy dysregulation in amyotrophic lateral sclerosis. Brain Pathol. 2012, 22, 110–116. [Google Scholar] [CrossRef]
- Ramesh, N.; Pandey, U.B. Autophagy Dysregulation in ALS: When Protein Aggregates Get Out of Hand. Front. Mol. Neurosci. 2017, 10, 263. [Google Scholar] [CrossRef] [PubMed]
- Vicencio, E.; Beltrán, S.; Labrador, L.; Manque, P.; Nassif, M.; Woehlbier, U. Implications of Selective Autophagy Dysfunction for ALS Pathology. Cells 2020, 9, 381. [Google Scholar] [CrossRef] [PubMed]
- Ramesh Babu, J.; Lamar Seibenhener, M.; Peng, J.; Strom, A.L.; Kemppainen, R.; Cox, N.; Zhu, H.; Wooten, M.C.; Diaz-Meco, M.T.; Moscat, J.; et al. Genetic inactivation of p62 leads to accumulation of hyperphosphorylated tau and neurodegeneration. J. Neurochem. 2008, 106, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Tanji, K.; Odagiri, S.; Miki, Y.; Maruyama, A.; Nikaido, Y.; Mimura, J.; Mori, F.; Warabi, E.; Yanagawa, T.; Ueno, S.; et al. p62 Deficiency Enhances α-Synuclein Pathology in Mice. Brain Pathol. 2015, 25, 552–564. [Google Scholar] [CrossRef] [PubMed]
- Hiji, M.; Takahashi, T.; Fukuba, H.; Yamashita, H.; Kohriyama, T.; Matsumoto, M. White matter lesions in the brain with frontotemporal lobar degeneration with motor neuron disease: TDP-43-immunopositive inclusions co-localize with p62, but not ubiquitin. Acta Neuropathol. 2008, 116, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Gal, J.; Ström, A.L.; Kwinter, D.M.; Kilty, R.; Zhang, J.; Shi, P.; Fu, W.; Wooten, M.W.; Zhu, H. Sequestosome 1/p62 links familial ALS mutant SOD1 to LC3 via an ubiquitin-independent mechanism. J. Neurochem. 2009, 111, 1062–1073. [Google Scholar] [CrossRef] [PubMed]
- Pickford, F.; Masliah, E.; Britschgi, M.; Lucin, K.; Narasimhan, R.; Jaeger, P.A.; Small, S.; Spencer, B.; Rockenstein, E.; Levine, B.; et al. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J. Clin. Investig. 2008, 118, 2190–2199. [Google Scholar] [CrossRef]
- Shi, P.; Ström, A.L.; Gal, J.; Zhu, H. Effects of ALS-related SOD1 mutants on dynein- and KIF5-mediated retrograde and anterograde axonal transport. Biochim. Biophys. Acta 2010, 1802, 707–716. [Google Scholar] [CrossRef]
- Puls, I.; Jonnakuty, C.; LaMonte, B.H.; Holzbaur, E.L.; Tokito, M.; Mann, E.; Floeter, M.K.; Bidus, K.; Drayna, D.; Oh, S.J.; et al. Mutant dynactin in motor neuron disease. Nat. Genet. 2003, 33, 455–456. [Google Scholar] [CrossRef]
- Bercier, V.; Hubbard, J.M.; Fidelin, K.; Duroure, K.; Auer, T.O.; Revenu, C.; Wyart, C.; Del Bene, F. Dynactin1 depletion leads to neuromuscular synapse instability and functional abnormalities. Mol. Neurodegener. 2019, 14, 27. [Google Scholar] [CrossRef]
- Cozzi, M.; Ferrari, V. Autophagy Dysfunction in ALS: From Transport to Protein Degradation. J. Mol. Neurosci. 2022, 72, 1456–1481. [Google Scholar] [CrossRef] [PubMed]
- LaMonte, B.H.; Wallace, K.E.; Holloway, B.A.; Shelly, S.S.; Ascaño, J.; Tokito, M.; Van Winkle, T.; Howland, D.S.; Holzbaur, E.L. Disruption of dynein/dynactin inhibits axonal transport in motor neurons causing late-onset progressive degeneration. Neuron 2002, 34, 715–727. [Google Scholar] [CrossRef] [PubMed]
- Kuźma-Kozakiewicz, M.; Chudy, A.; Kaźmierczak, B.; Dziewulska, D.; Usarek, E.; Barańczyk-Kuźma, A. Dynactin Deficiency in the CNS of Humans with Sporadic ALS and Mice with Genetically Determined Motor Neuron Degeneration. Neurochem. Res. 2013, 38, 2463–2473. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Xue, H.; Yue, Y.; Hao, S.; Huang, S.H.; Zhang, Z. Role of mitophagy in the neurodegenerative diseases and its pharmacological advances: A review. Front. Mol. Neurosci. 2022, 15, 1014251. [Google Scholar] [CrossRef]
- Fivenson, E.M.; Lautrup, S.; Sun, N.; Scheibye-Knudsen, M.; Stevnsner, T.; Nilsen, H.; Bohr, V.A.; Fang, E.F. Mitophagy in neurodegeneration and aging. Neurochem. Int. 2017, 109, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Chan, D.C. Mitochondrial dynamics—Fusion, fission, movement, and mitophagy—In neurodegenerative diseases. Hum. Mol. Genet. 2009, 18, R169–R176. [Google Scholar] [CrossRef]
- Magalhães, J.D.; Fão, L.; Vilaça, R.; Cardoso, S.M.; Rego, A.C. Macroautophagy and Mitophagy in Neurodegenerative Disorders: Focus on Therapeutic Interventions. Biomedicines 2021, 9, 1625. [Google Scholar] [CrossRef] [PubMed]
- Lionaki, E.; Markaki, M.; Palikaras, K.; Tavernarakis, N. Mitochondria, autophagy and age-associated neurodegenerative diseases: New insights into a complex interplay. Biochim. Biophys. Acta 2015, 1847, 1412–1423. [Google Scholar] [CrossRef]
- Fang, E.F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Hasan-Olive, M.M.; Caponio, D.; Dan, X.; et al. Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 2019, 22, 401–412. [Google Scholar] [CrossRef]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.; Harvey, K.; Gispert, S.; Ali, Z.; Del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef]
- Goiran, T.; Eldeeb, M.A.; Zorca, C.E.; Fon, E.A. Hallmarks and Molecular Tools for the Study of Mitophagy in Parkinson’s Disease. Cells 2022, 11, 2097. [Google Scholar] [CrossRef] [PubMed]
- Rogers, R.S.; Tungtur, S.; Tanaka, T.; Nadeau, L.L.; Badawi, Y.; Wang, H.; Ni, H.M.; Ding, W.X.; Nishimune, H. Impaired Mitophagy Plays a Role in Denervation of Neuromuscular Junctions in ALS Mice. Front. Neurosci. 2017, 11, 473. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Duan, Y.; Qin, C.; Li, J.C.; Duan, G.; Deng, X.; Ni, J.; Cao, X.; Xiang, K.; Tian, K.; et al. Distinct multilevel misregulations of Parkin and PINK1 revealed in cell and animal models of TDP-43 proteinopathy. Cell Death Dis. 2018, 9, 953. [Google Scholar] [CrossRef] [PubMed]
- Salvadores, N.; Gerónimo-Olvera, C.; Court, F.A. Axonal Degeneration in AD: The Contribution of Aβ and Tau. Front. Aging Neurosci. 2020, 12, 581767. [Google Scholar] [CrossRef] [PubMed]
- Stokin, G.B.; Goldstein, L.S. Axonal transport and Alzheimer’s disease. Annu. Rev. Biochem. 2006, 75, 607–627. [Google Scholar] [CrossRef] [PubMed]
- Kanaan, N.M.; Pigino, G.F.; Brady, S.T.; Lazarov, O.; Binder, L.I.; Morfini, G.A. Axonal degeneration in Alzheimer’s disease: When signaling abnormalities meet the axonal transport system. Exp. Neurol. 2013, 246, 44–53. [Google Scholar] [CrossRef]
- Chu, Y.; Morfini, G.A.; Langhamer, L.B.; He, Y.; Brady, S.T.; Kordower, J.H. Alterations in axonal transport motor proteins in sporadic and experimental Parkinson’s disease. Brain 2012, 135, 2058–2073. [Google Scholar] [CrossRef]
- Prots, I.; Grosch, J.; Brazdis, R.M.; Simmnacher, K.; Veber, V.; Havlicek, S.; Hannappel, C.; Krach, F.; Krumbiegel, M.; Schütz, O.; et al. α-Synuclein oligomers induce early axonal dysfunction in human iPSC-based models of synucleinopathies. Proc. Natl. Acad. Sci. USA 2018, 115, 7813–7818. [Google Scholar] [CrossRef]
- Roy, S.; Zhang, B.; Lee, V.M.; Trojanowski, J.Q. Axonal transport defects: A common theme in neurodegenerative diseases. Acta Neuropathol. 2005, 109, 5–13. [Google Scholar] [CrossRef]
- Morfini, G.A.; Burns, M.; Binder, L.I.; Kanaan, N.M.; LaPointe, N.; Bosco, D.A.; Brown, R.H., Jr.; Brown, H.; Tiwari, A.; Hayward, L.; et al. Axonal transport defects in neurodegenerative diseases. J. Neurosci. 2009, 29, 12776–12786. [Google Scholar] [CrossRef]
- De Vos, K.J.; Hafezparast, M. Neurobiology of axonal transport defects in motor neuron diseases: Opportunities for translational research? Neurobiol. Dis. 2017, 105, 283–299. [Google Scholar] [CrossRef] [PubMed]
- Ikenaka, K.; Katsuno, M.; Kawai, K.; Ishigaki, S.; Tanaka, F.; Sobue, G. Disruption of axonal transport in motor neuron diseases. Int. J. Mol. Sci. 2012, 13, 1225–1238. [Google Scholar] [CrossRef] [PubMed]
- Sleigh, J.N.; Tosolini, A.P.; Gordon, D.; Devoy, A.; Fratta, P.; Fisher, E.M.C.; Talbot, K.; Schiavo, G. Mice Carrying ALS Mutant TDP-43, but Not Mutant FUS, Display In Vivo Defects in Axonal Transport of Signaling Endosomes. Cell Rep. 2020, 30, 3655–3662.e2. [Google Scholar] [CrossRef] [PubMed]
- Mandelkow, E.M.; Stamer, K.; Vogel, R.; Thies, E.; Mandelkow, E. Clogging of axons by tau, inhibition of axonal traffic and starvation of synapses. Neurobiol. Aging 2003, 24, 1079–1085. [Google Scholar] [CrossRef] [PubMed]
- Clark, C.; Lewczuk, P.; Kornhuber, J.; Richiardi, J.; Maréchal, B.; Karikari, T.K.; Blennow, K.; Zetterberg, H.; Popp, J. Plasma neurofilament light and phosphorylated tau 181 as biomarkers of Alzheimer’s disease pathology and clinical disease progression. Alzheimers Res. Ther. 2021, 13, 65. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; Cao, L.; Dai, Y.P. Role of Neurofilament Light Chain as a Potential Biomarker for Alzheimer’s Disease: A Correlative Meta-Analysis. Front. Aging Neurosci. 2019, 11, 254. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhang, W.; Liu, F.; Mao, C.; Liu, C.F.; Cheng, W.; Feng, J. Association of Cerebrospinal Fluid Neurofilament Heavy Protein Levels With Clinical Progression in Patients With Parkinson Disease. JAMA Netw. Open 2022, 5, e2223821. [Google Scholar] [CrossRef]
- Frigerio, I.; Laansma, M.A.; Lin, C.P.; Hermans, E.J.M.; Bouwman, M.M.A.; Bol, J.; Galis-de Graaf, Y.; Hepp, D.H.; Rozemuller, A.J.M.; Barkhof, F.; et al. Neurofilament light chain is increased in the parahippocampal cortex and associates with pathological hallmarks in Parkinson’s disease dementia. Transl. Neurodegener. 2023, 12, 3. [Google Scholar] [CrossRef]
- Heckler, I.; Venkataraman, I. Phosphorylated neurofilament heavy chain: A potential diagnostic biomarker in amyotrophic lateral sclerosis. J. Neurophysiol. 2022, 127, 737–745. [Google Scholar] [CrossRef]
- Behzadi, A.; Pujol-Calderón, F.; Tjust, A.E.; Wuolikainen, A.; Höglund, K.; Forsberg, K.; Portelius, E.; Blennow, K.; Zetterberg, H.; Andersen, P.M. Neurofilaments can differentiate ALS subgroups and ALS from common diagnostic mimics. Sci. Rep. 2021, 11, 22128. [Google Scholar] [CrossRef]
- Zhou, W.; Zhang, J.; Ye, F.; Xu, G.; Su, H.; Su, Y.; Zhang, X. Plasma neurofilament light chain levels in Alzheimer’s disease. Neurosci. Lett. 2017, 650, 60–64. [Google Scholar] [CrossRef] [PubMed]
- Heshmatollah, A.; Fani, L.; Koudstaal, P.J.; Ghanbari, M.; Ikram, M.A.; Ikram, M.K. Plasma β-Amyloid, Total-Tau, and Neurofilament Light Chain Levels and the Risk of Stroke: A Prospective Population-Based Study. Neurology 2022, 98, e1729–e1737. [Google Scholar] [CrossRef] [PubMed]
- Mollenhauer, B.; Dakna, M.; Kruse, N.; Galasko, D.; Foroud, T.; Zetterberg, H.; Schade, S.; Gera, R.G.; Wang, W.; Gao, F.; et al. Validation of Serum Neurofilament Light Chain as a Biomarker of Parkinson’s Disease Progression. Mov. Disord. 2020, 35, 1999–2008. [Google Scholar] [CrossRef] [PubMed]
- Batzu, L.; Rota, S.; Hye, A.; Heslegrave, A.; Trivedi, D.; Gibson, L.L.; Farrell, C.; Zinzalias, P.; Rizos, A.; Zetterberg, H.; et al. Plasma p-tau181, neurofilament light chain and association with cognition in Parkinson’s disease. NPJ Park. Dis. 2022, 8, 154. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Ji, Y.; Wang, W.; Zhang, L.; Chen, Z.; Yu, M.; Shen, Y.; Ding, F.; Gu, X.; Sun, H. Amyotrophic Lateral Sclerosis: Molecular Mechanisms, Biomarkers, and Therapeutic Strategies. Antioxidants 2021, 10, 1012. [Google Scholar] [CrossRef]
- Poesen, K.; Van Damme, P. Diagnostic and Prognostic Performance of Neurofilaments in ALS. Front. Neurol. 2018, 9, 1167. [Google Scholar] [CrossRef]
- Wyss-Coray, T.; Mucke, L. Inflammation in neurodegenerative disease—A double-edged sword. Neuron 2002, 35, 419–432. [Google Scholar] [CrossRef]
- Stephenson, J.; Nutma, E.; van der Valk, P.; Amor, S. Inflammation in CNS neurodegenerative diseases. Immunology 2018, 154, 204–219. [Google Scholar] [CrossRef]
- Russo, M.V.; McGavern, D.B. Inflammatory neuroprotection following traumatic brain injury. Science 2016, 353, 783–785. [Google Scholar] [CrossRef]
- Kwon, H.S.; Koh, S.-H. Neuroinflammation in neurodegenerative disorders: The roles of microglia and astrocytes. Transl. Neurodegener. 2020, 9, 42. [Google Scholar] [CrossRef]
- Baufeld, C.; O’Loughlin, E.; Calcagno, N.; Madore, C.; Butovsky, O. Differential contribution of microglia and monocytes in neurodegenerative diseases. J. Neural Transm. 2018, 125, 809–826. [Google Scholar] [CrossRef] [PubMed]
- Glass, C.K.; Saijo, K.; Winner, B.; Marchetto, M.C.; Gage, F.H. Mechanisms underlying inflammation in neurodegeneration. Cell 2010, 140, 918–934. [Google Scholar] [CrossRef] [PubMed]
- Colombo, E.; Farina, C. Astrocytes: Key regulators of neuroinflammation. Trends Immunol. 2016, 37, 608–620. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Barres, B.A. Reactive astrocytes: Production, function, and therapeutic potential. Immunity 2017, 46, 957–967. [Google Scholar] [CrossRef]
- Di Benedetto, G.; Burgaletto, C.; Bellanca, C.M.; Munafò, A.; Bernardini, R.; Cantarella, G. Role of Microglia and astrocytes in Alzheimer’s disease: From neuroinflammation to Ca2+ homeostasis dysregulation. Cells 2022, 11, 2728. [Google Scholar] [CrossRef] [PubMed]
- Nagele, R.G.; Wegiel, J.; Venkataraman, V.; Imaki, H.; Wang, K.-C.; Wegiel, J. Contribution of glial cells to the development of amyloid plaques in Alzheimer’s disease. Neurobiol. Aging 2004, 25, 663–674. [Google Scholar] [CrossRef]
- Ferreira, S.A.; Romero-Ramos, M. Microglia response during Parkinson’s disease: Alpha-synuclein intervention. Front. Cell. Neurosci. 2018, 12, 247. [Google Scholar] [CrossRef]
- Vila, M.; Jackson-Lewis, V.; Guégan, C.; Teismann, P.; Choi, D.-K.; Tieu, K.; Przedborski, S. The role of glial cells in Parkinson’s disease. Curr. Opin. Neurol. 2001, 14, 483–489. [Google Scholar] [CrossRef]
- Grosskreutz, J.; Van Den Bosch, L.; Keller, B.U. Calcium dysregulation in amyotrophic lateral sclerosis. Cell Calcium 2010, 47, 165–174. [Google Scholar] [CrossRef]
- Philips, T.; Rothstein, J.D. Glial cells in amyotrophic lateral sclerosis. Exp. Neurol. 2014, 262, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Jha, N.K.; Jha, S.K.; Kar, R.; Nand, P.; Swati, K.; Goswami, V.K. Nuclear factor-kappa β as a therapeutic target for Alzheimer’s disease. J. Neurochem. 2019, 150, 113–137. [Google Scholar] [CrossRef] [PubMed]
- Bellucci, A.; Bubacco, L.; Longhena, F.; Parrella, E.; Faustini, G.; Porrini, V.; Bono, F.; Missale, C.; Pizzi, M. Nuclear factor-κB dysregulation and α-synuclein pathology: Critical interplay in the pathogenesis of Parkinson’s disease. Front. Aging Neurosci. 2020, 12, 68. [Google Scholar] [CrossRef] [PubMed]
- Källstig, E.; McCabe, B.D.; Schneider, B.L. The Links between ALS and NF-κB. Int. J. Mol. Sci. 2021, 22, 3875. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P.; Camandola, S. NF-κB in neuronal plasticity and neurodegenerative disorders. J. Clin. Investig. 2001, 107, 247–254. [Google Scholar] [CrossRef]
- Singh, A.; Ansari, V.A.; Mahmood, T.; Ahsan, F.; Wasim, R. Neurodegeneration: Microglia: Nf-Kappab Signaling Pathways. Drug Res. 2022, 72, 496–499. [Google Scholar] [CrossRef]
- Kempuraj, D.; Thangavel, R.; Natteru, P.; Selvakumar, G.; Saeed, D.; Zahoor, H.; Zaheer, S.; Iyer, S.; Zaheer, A. Neuroinflammation induces neurodegeneration. J. Neurol. Neurosurg. Spine 2016, 1, 1003. [Google Scholar]
- Choi, S.S.; Lee, H.J.; Lim, I.; Satoh, J.-i.; Kim, S.U. Human astrocytes: Secretome profiles of cytokines and chemokines. PLoS ONE 2014, 9, e92325. [Google Scholar] [CrossRef]
- Ma³ek, R.; Borowicz, K.K.; Jargie³³o, M.g.; Czuczwar, S.A.J. Role of nuclear factor kB in the central nervous system. Pharmacol. Rep. 2007, 59, 25–33. [Google Scholar]
- Dutta, K.; Thammisetty, S.S.; Boutej, H.; Bareil, C.; Julien, J.-P. Mitigation of ALS pathology by neuron-specific inhibition of nuclear factor kappa B signaling. J. Neurosci. 2020, 40, 5137–5154. [Google Scholar] [CrossRef]
- Jover-Mengual, T.; Hwang, J.-Y.; Byun, H.-R.; Court-Vazquez, B.L.; Centeno, J.M.; Burguete, M.C.; Zukin, R.S. The role of NF-κB triggered inflammation in cerebral ischemia. Front. Cell. Neurosci. 2021, 15, 633610. [Google Scholar] [CrossRef] [PubMed]
- Shih, R.-H.; Wang, C.-Y.; Yang, C.-M. NF-kappaB signaling pathways in neurological inflammation: A mini review. Front. Mol. Neurosci. 2015, 8, 77. [Google Scholar] [CrossRef] [PubMed]
- Kaltschmidt, B.; Uherek, M.; Volk, B.; Baeuerle, P.A.; Kaltschmidt, C. Transcription factor NF-κB is activated in primary neurons by amyloid β peptides and in neurons surrounding early plaques from patients with Alzheimer disease. Proc. Natl. Acad. Sci. USA 1997, 94, 2642–2647. [Google Scholar] [CrossRef] [PubMed]
- Martorana, F.; Foti, M.; Virtuoso, A.; Gaglio, D.; Aprea, F.; Latronico, T.; Rossano, R.; Riccio, P.; Papa, M.; Alberghina, L. Differential modulation of NF-κB in neurons and astrocytes underlies neuroprotection and antigliosis activity of natural antioxidant molecules. Oxidative Med. Cell Longev. 2019, 2019, 8056904. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.; Meffert, M. Roles for NF-κB in nerve cell survival, plasticity, and disease. Cell Death Differ. 2006, 13, 852–860. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sanghai, N.; Tranmer, G.K. Biochemical and Molecular Pathways in Neurodegenerative Diseases: An Integrated View. Cells 2023, 12, 2318. https://doi.org/10.3390/cells12182318
Sanghai N, Tranmer GK. Biochemical and Molecular Pathways in Neurodegenerative Diseases: An Integrated View. Cells. 2023; 12(18):2318. https://doi.org/10.3390/cells12182318
Chicago/Turabian StyleSanghai, Nitesh, and Geoffrey K. Tranmer. 2023. "Biochemical and Molecular Pathways in Neurodegenerative Diseases: An Integrated View" Cells 12, no. 18: 2318. https://doi.org/10.3390/cells12182318