

Therapeutic Oligonucleotides: An Outlook on Chemical Strategies to Improve Endosomal Trafficking

Abstract
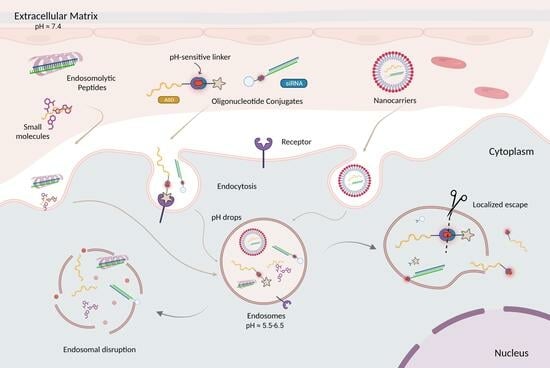
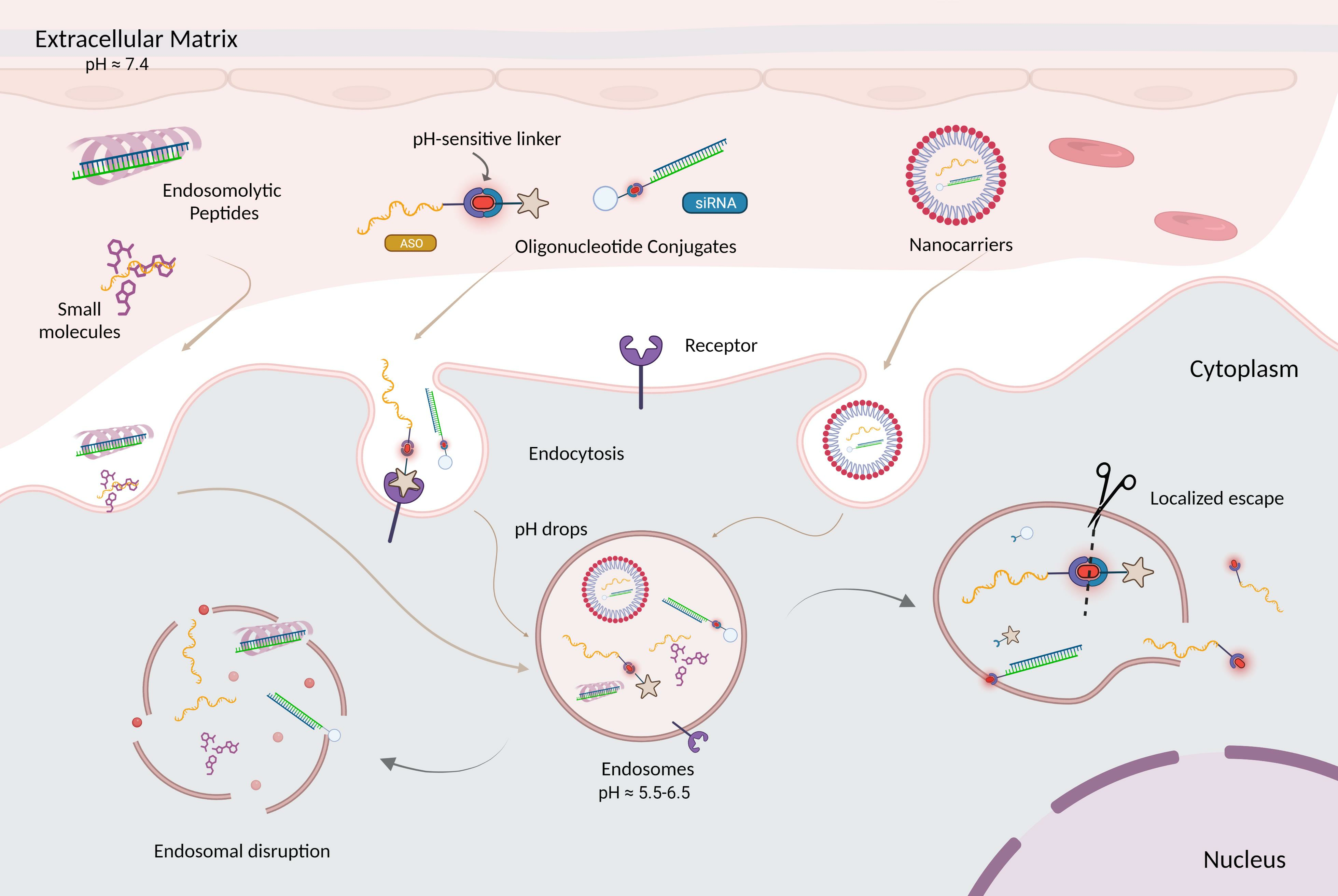
:
1. Introduction
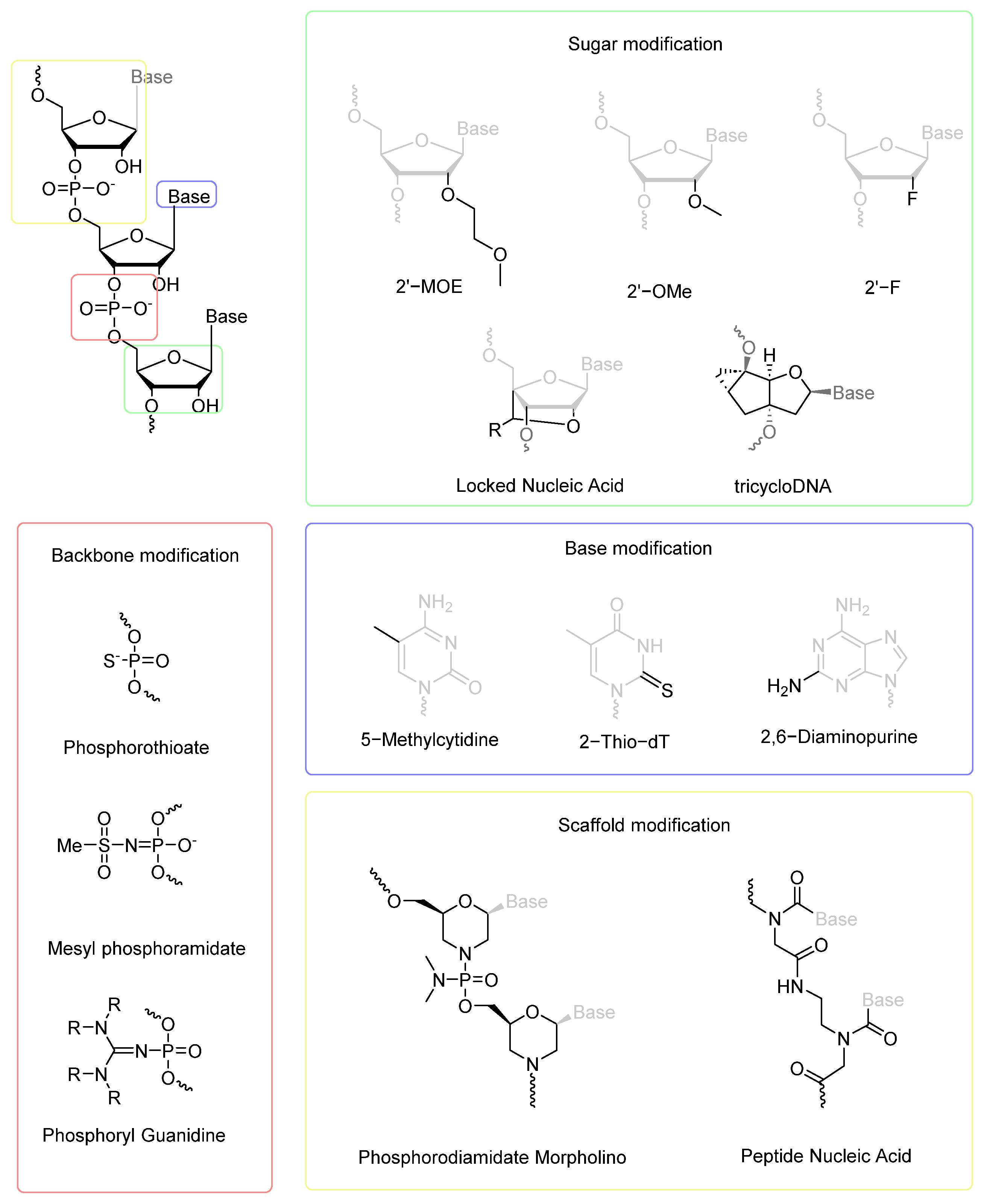
2. Chemical Modifications to Improve Oligonucleotide Drug Properties
2.1. Chemical Modifications in ASOs
2.2. Chemical Modifications in siRNAs
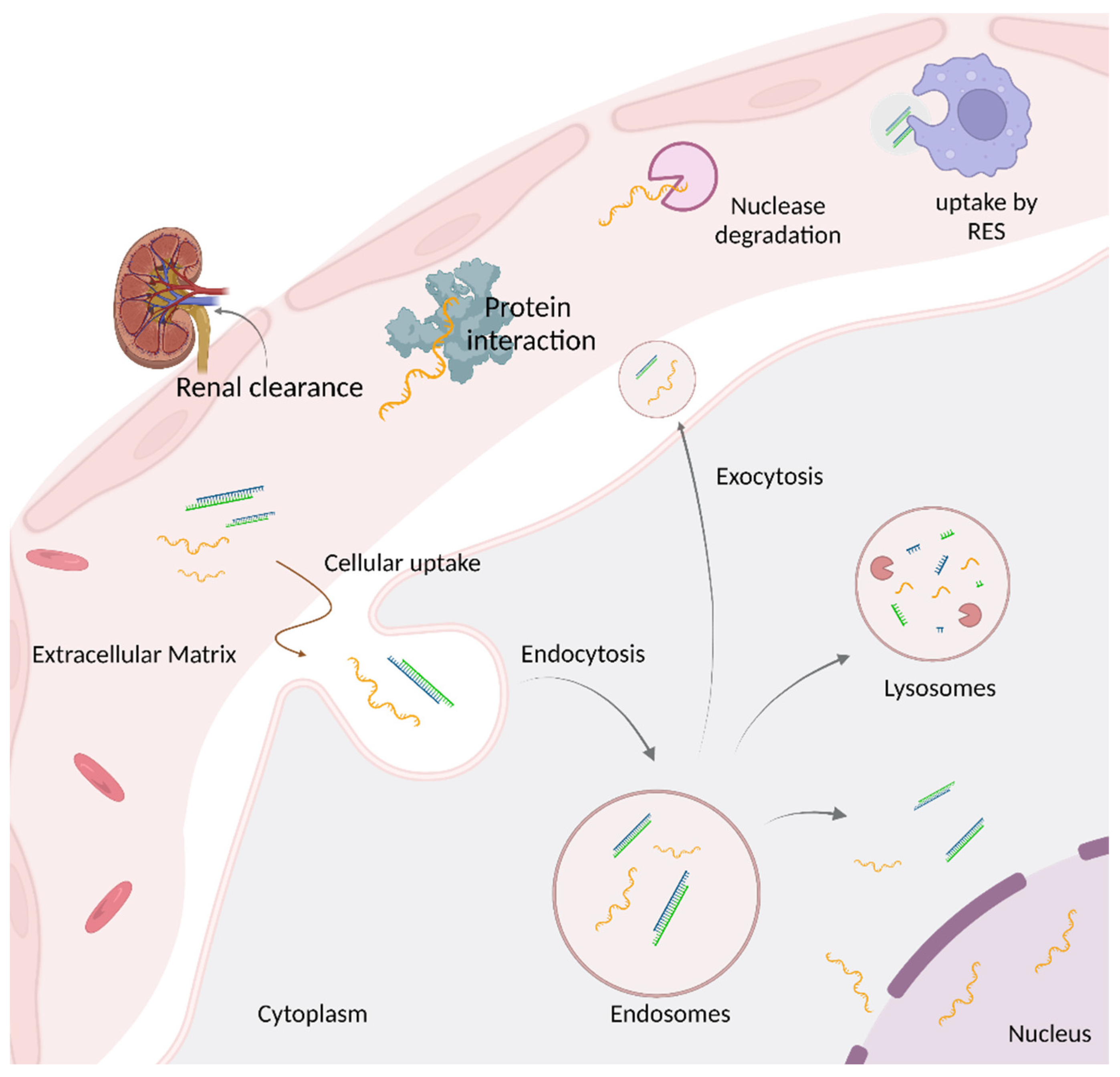
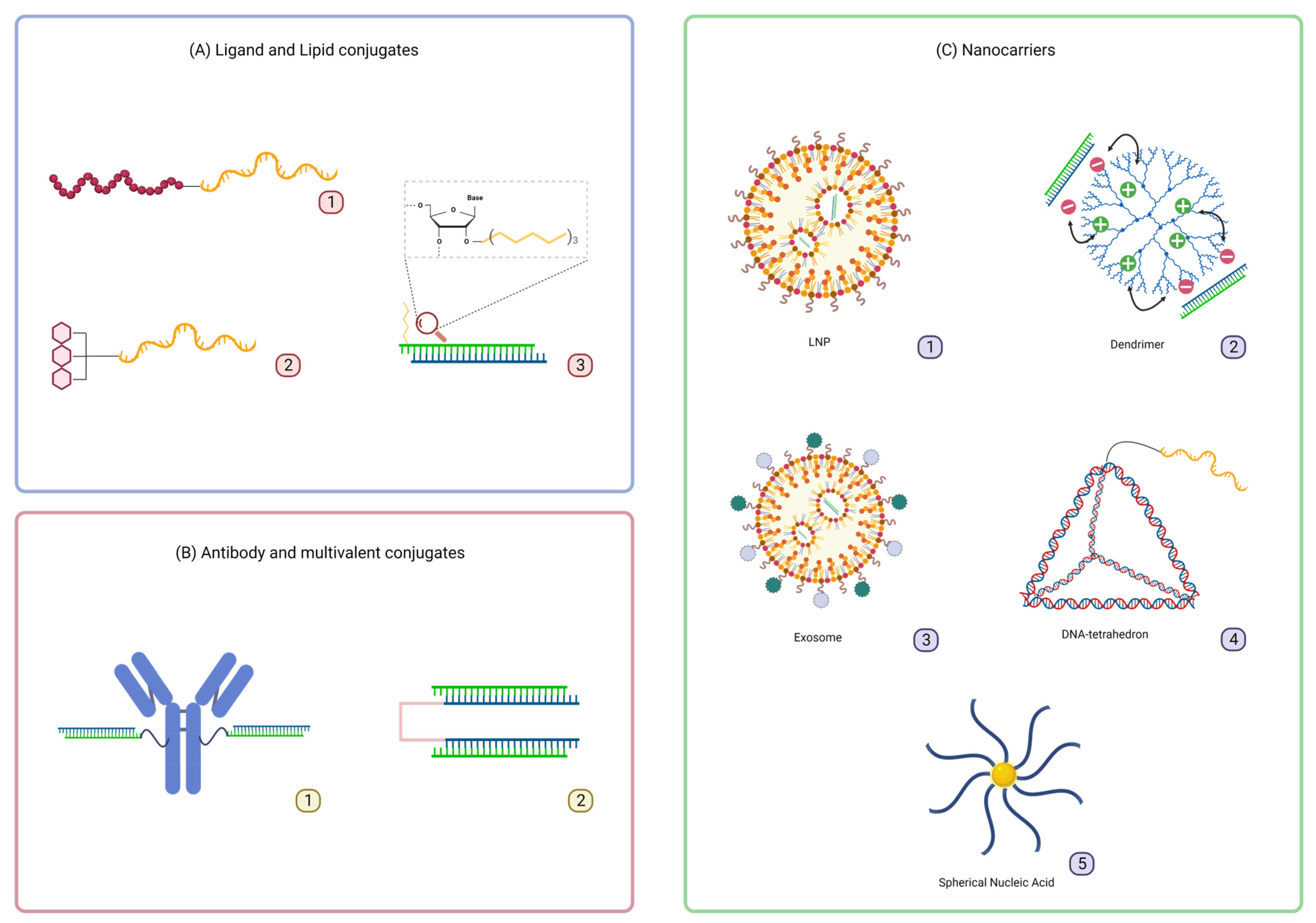
3. Delivery Platforms to Enhance Oligonucleotide Therapeutics Intracellular Uptake
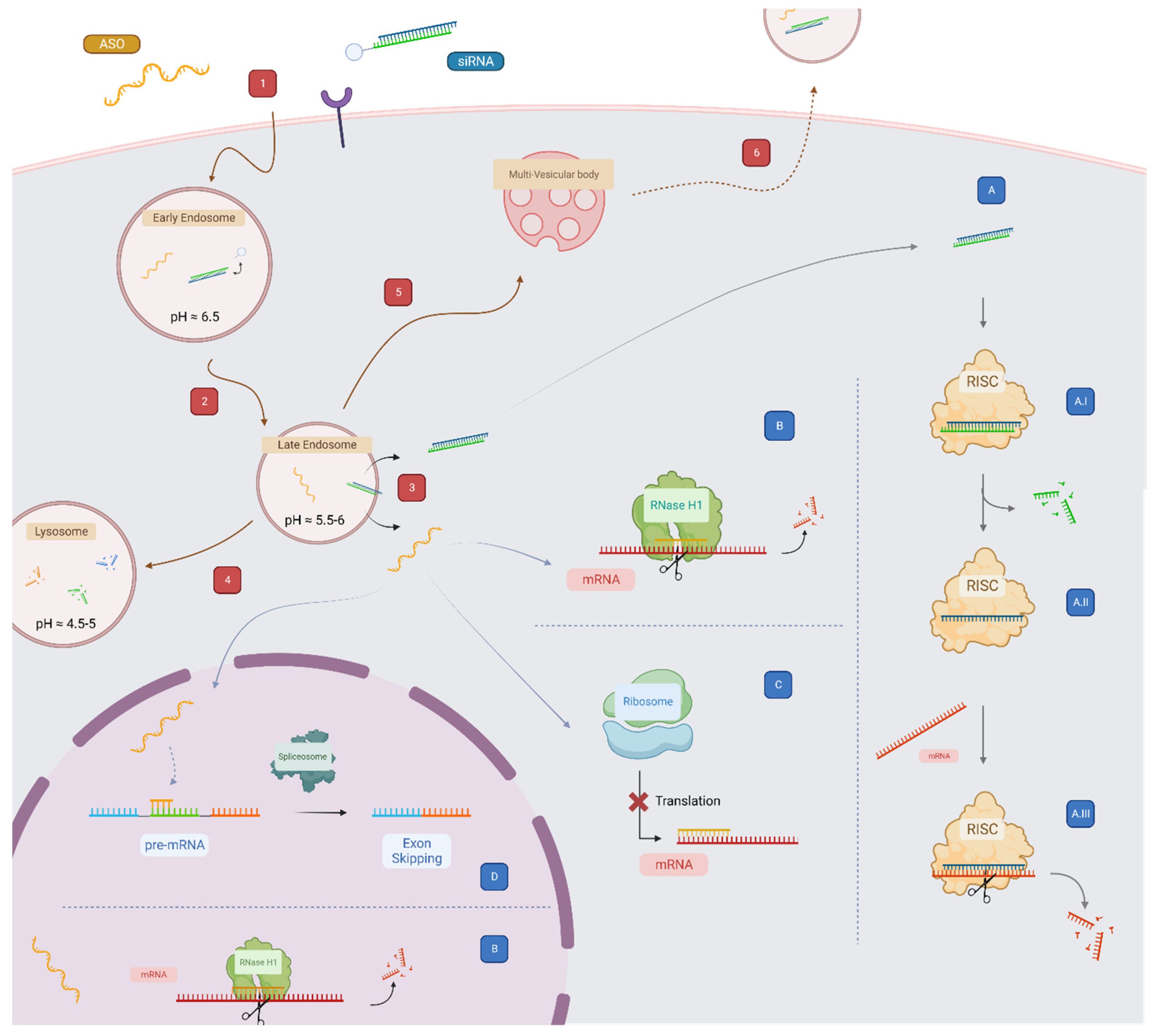
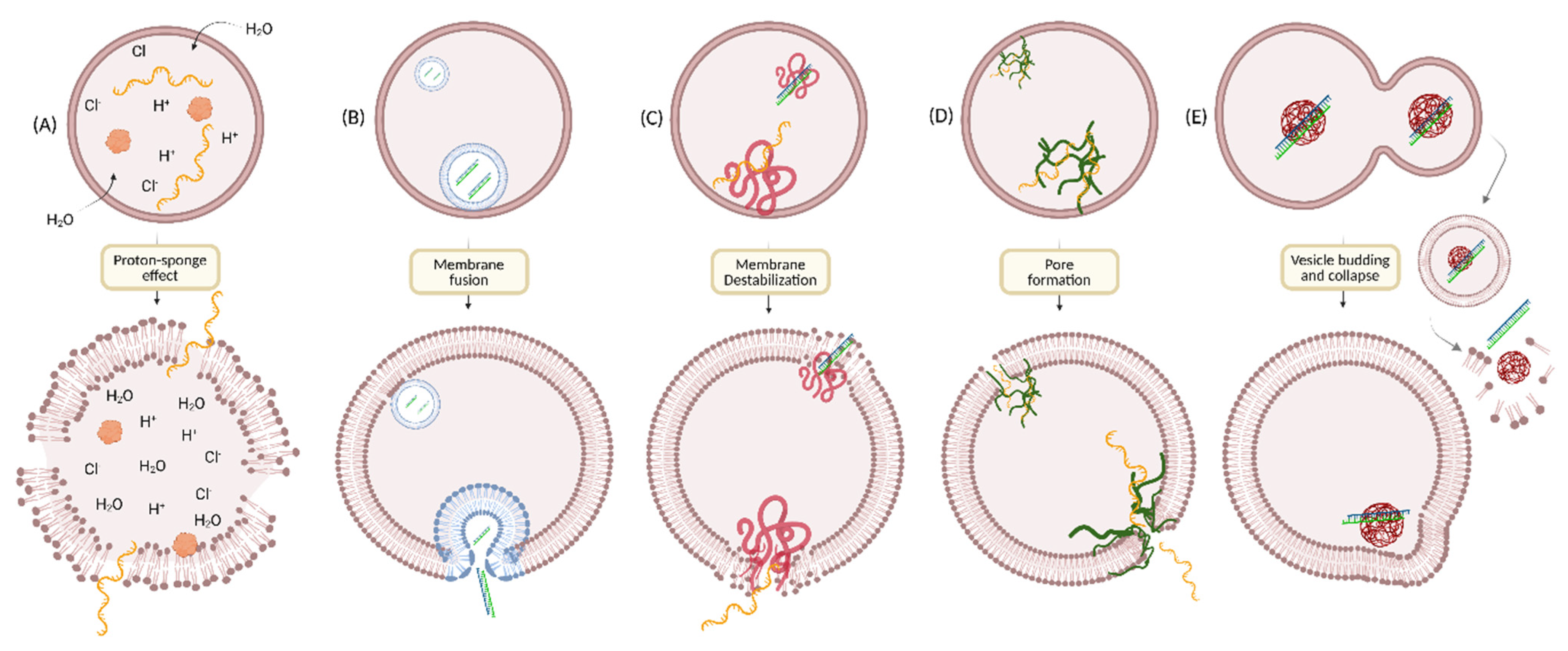
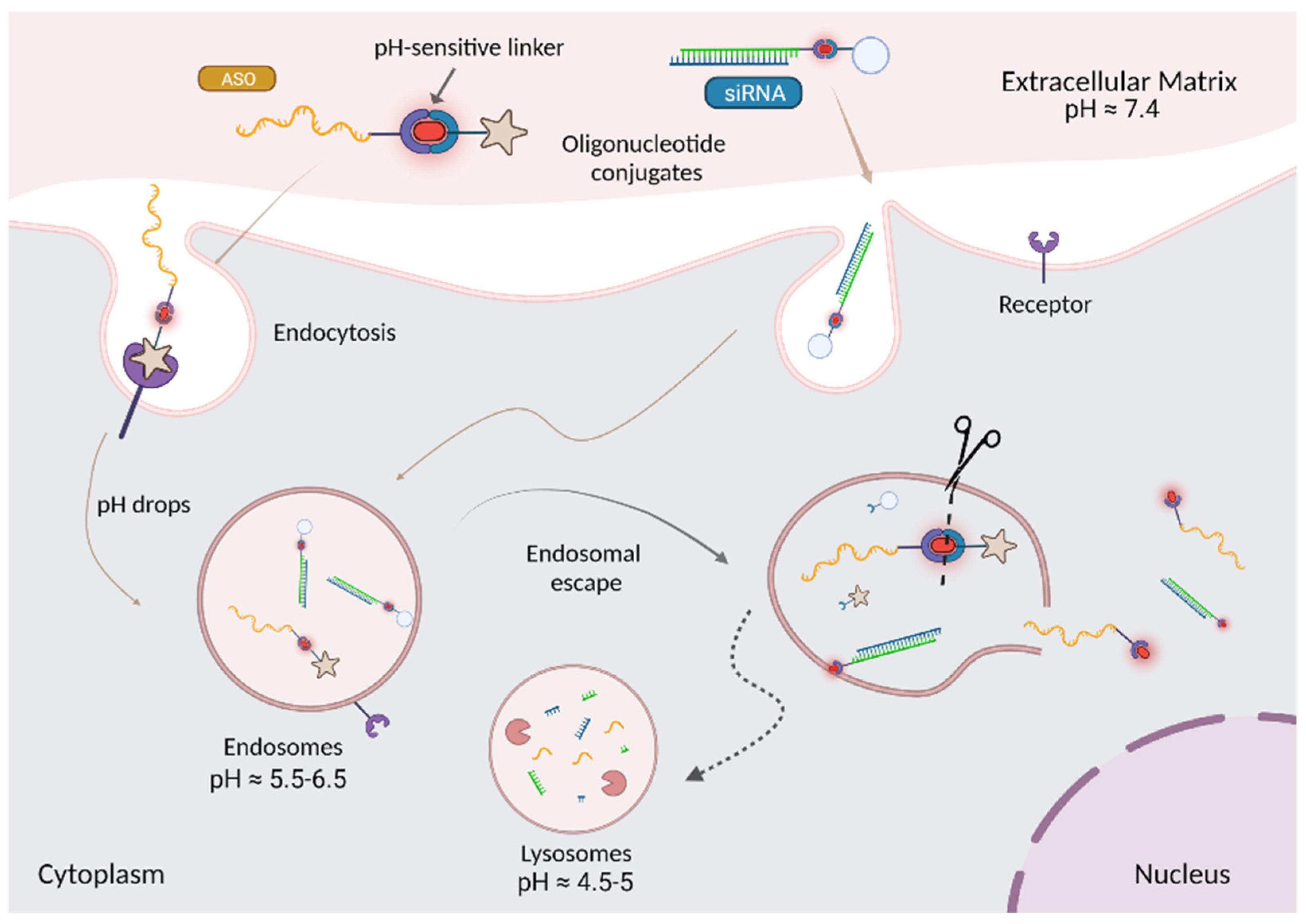
4. Overcoming Endosomal Barrier to Maximize Oligonucleotide Therapeutic Activity
5. Strategies to Enhance Endosomal Escape
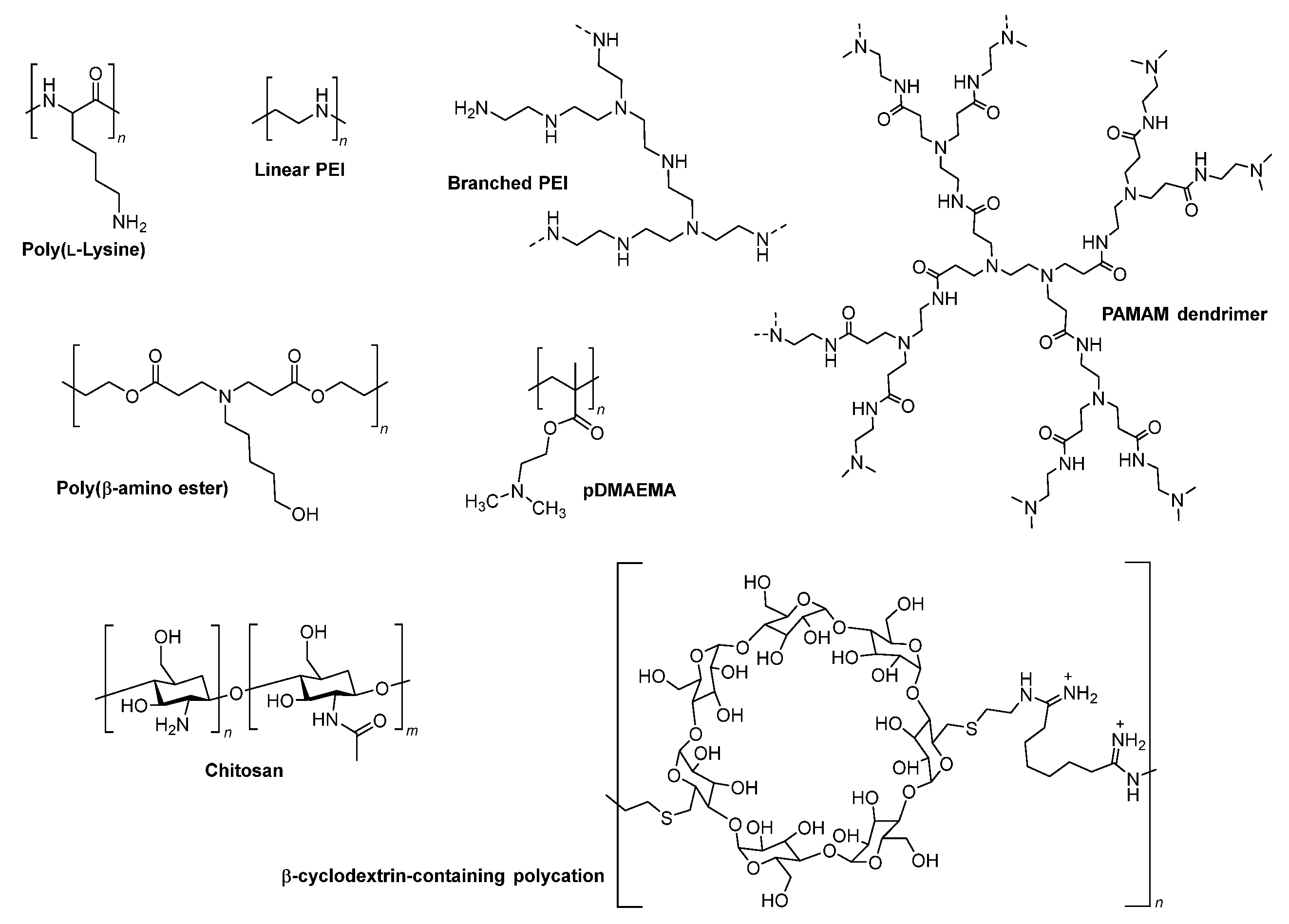
5.1. Endosomal Buffering Polymers
5.2. Cell-Penetrating Peptides (CPPs)/Endosomolytic Peptides (EPs)
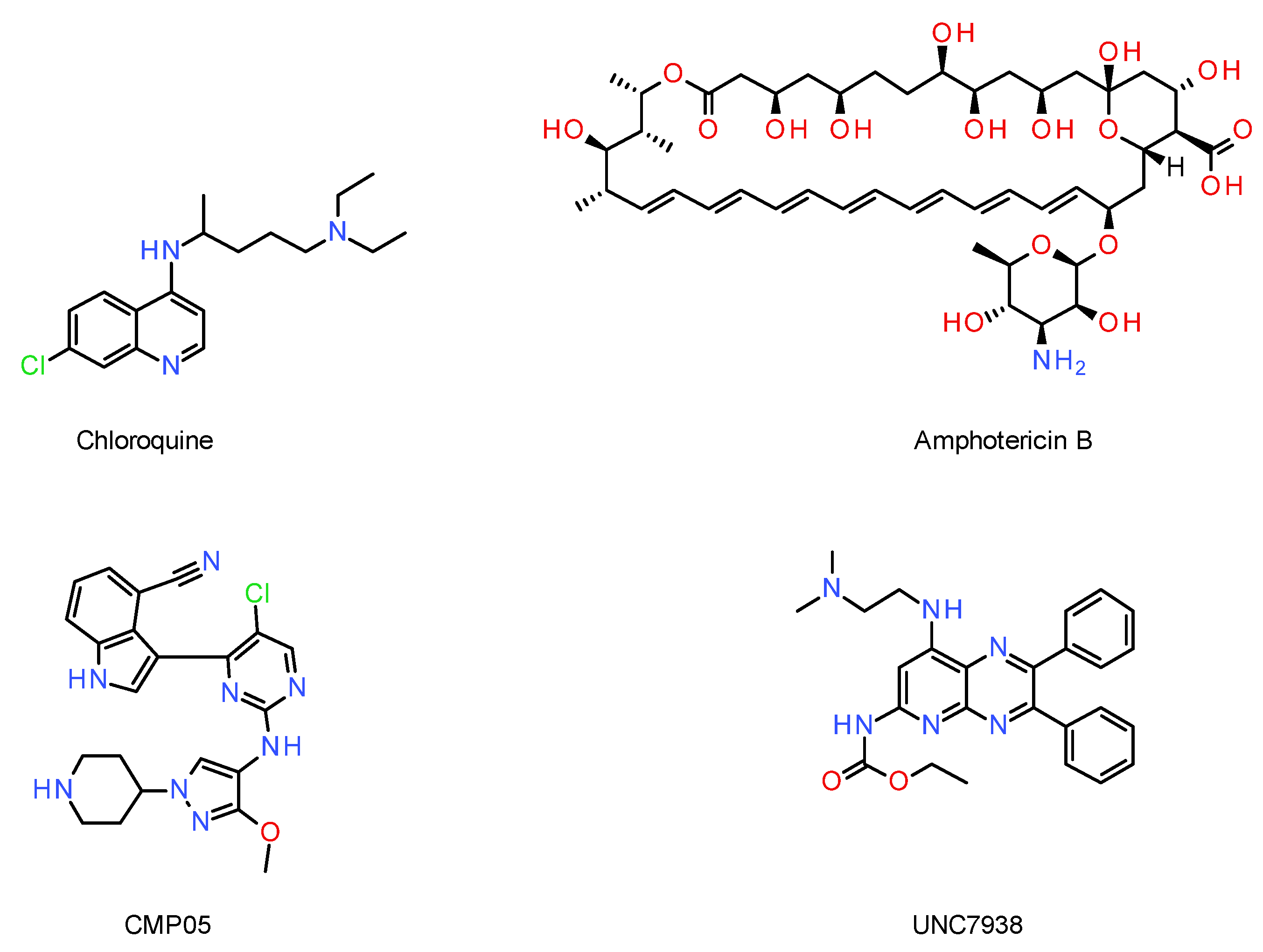
5.3. Small Molecules
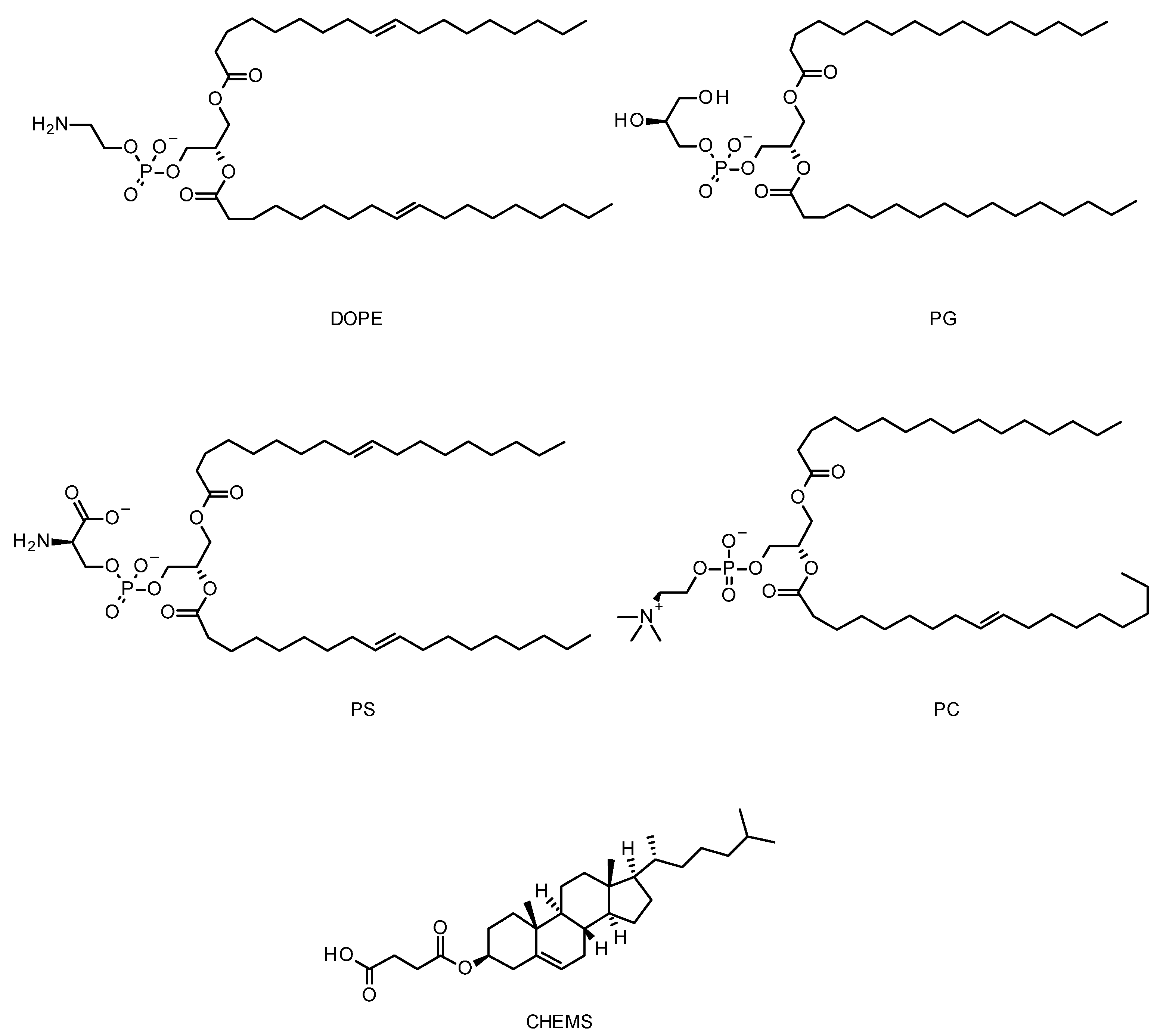
5.4. Cationic Liposomes
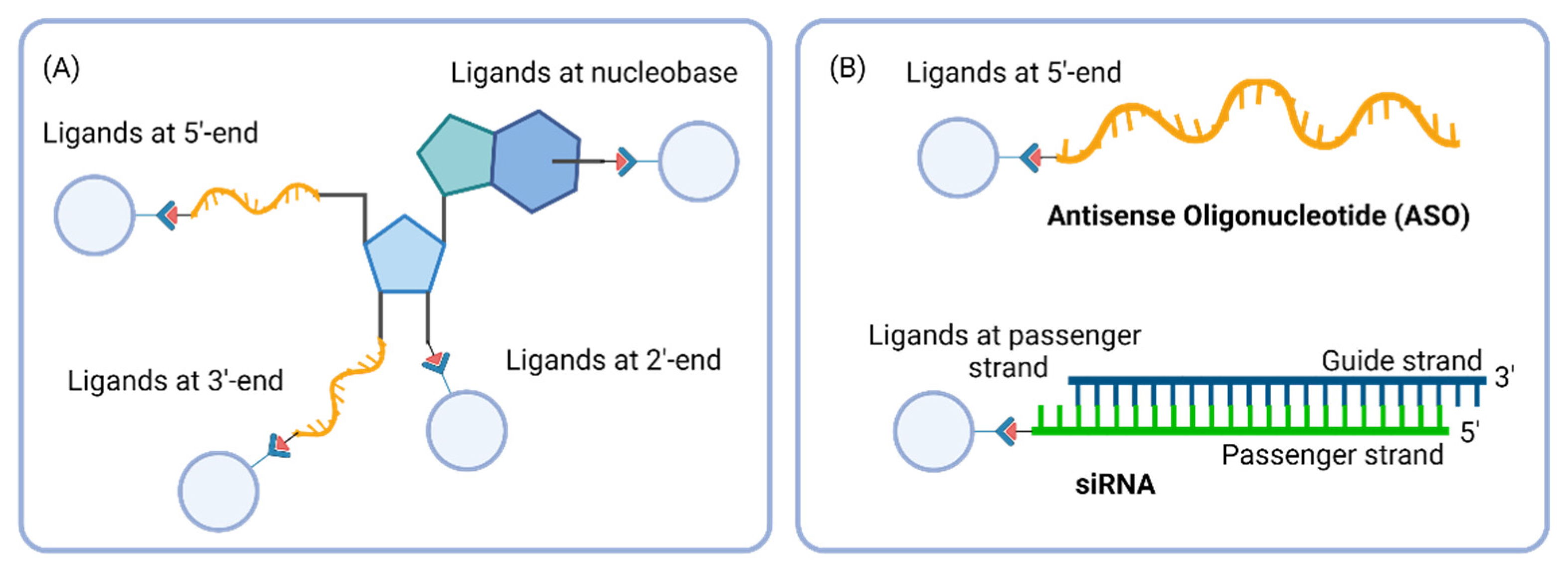
6. Importance of Linker Chemistry in Drug Delivery
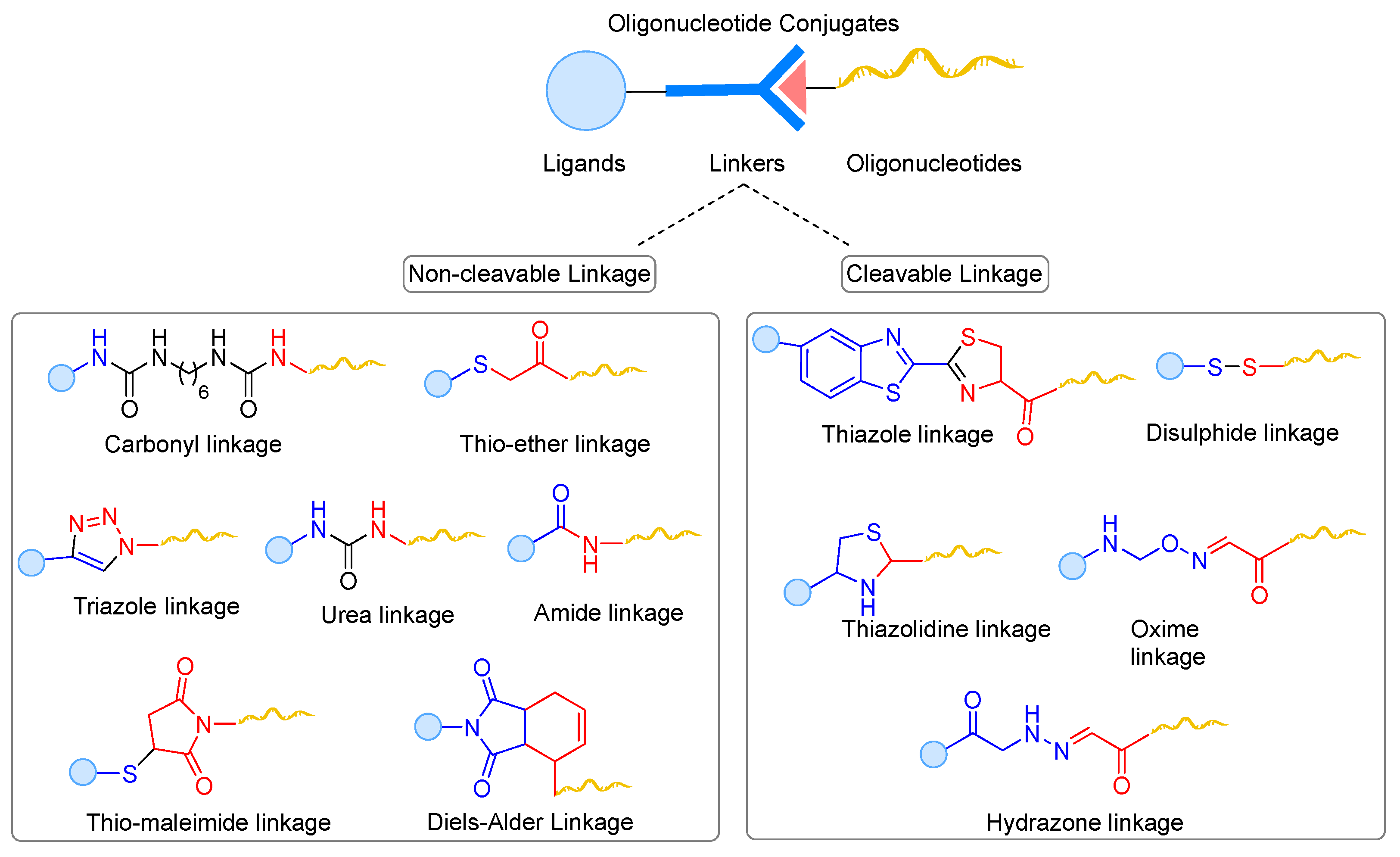
7. Cleavable and Non-Cleavable Linkers in Oligonucleotide Conjugates
7.1. Non-Cleavable Linkages
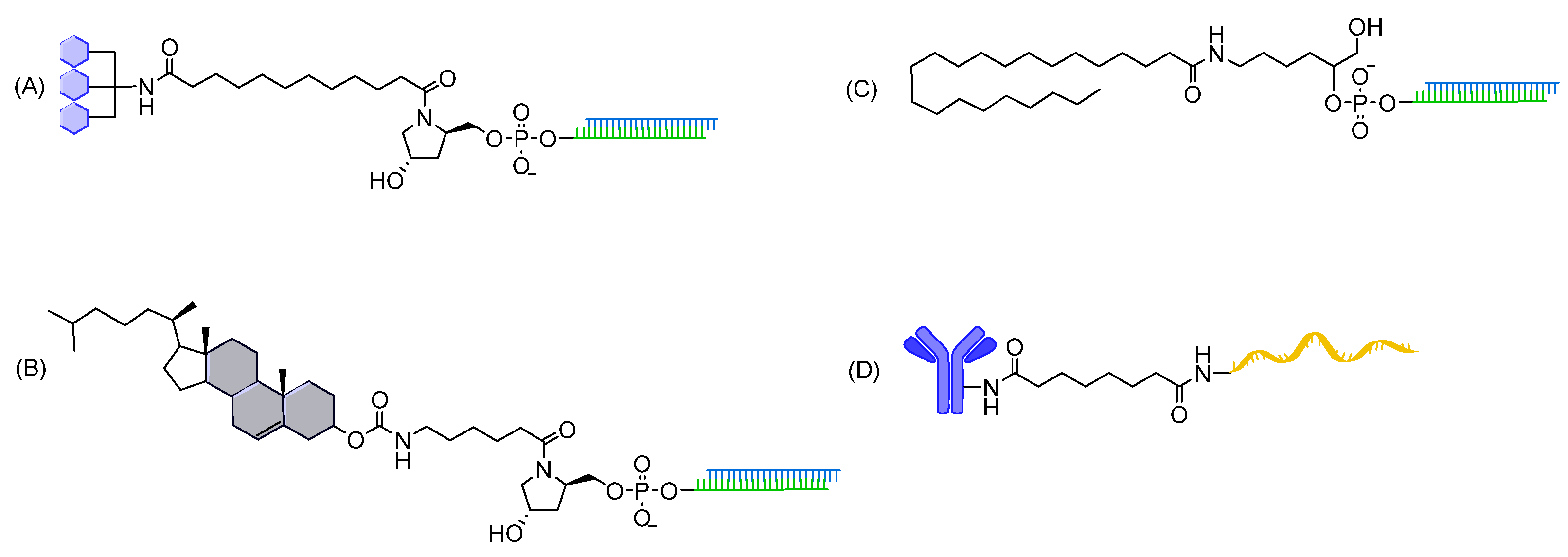
7.1.1. Amide Linkages
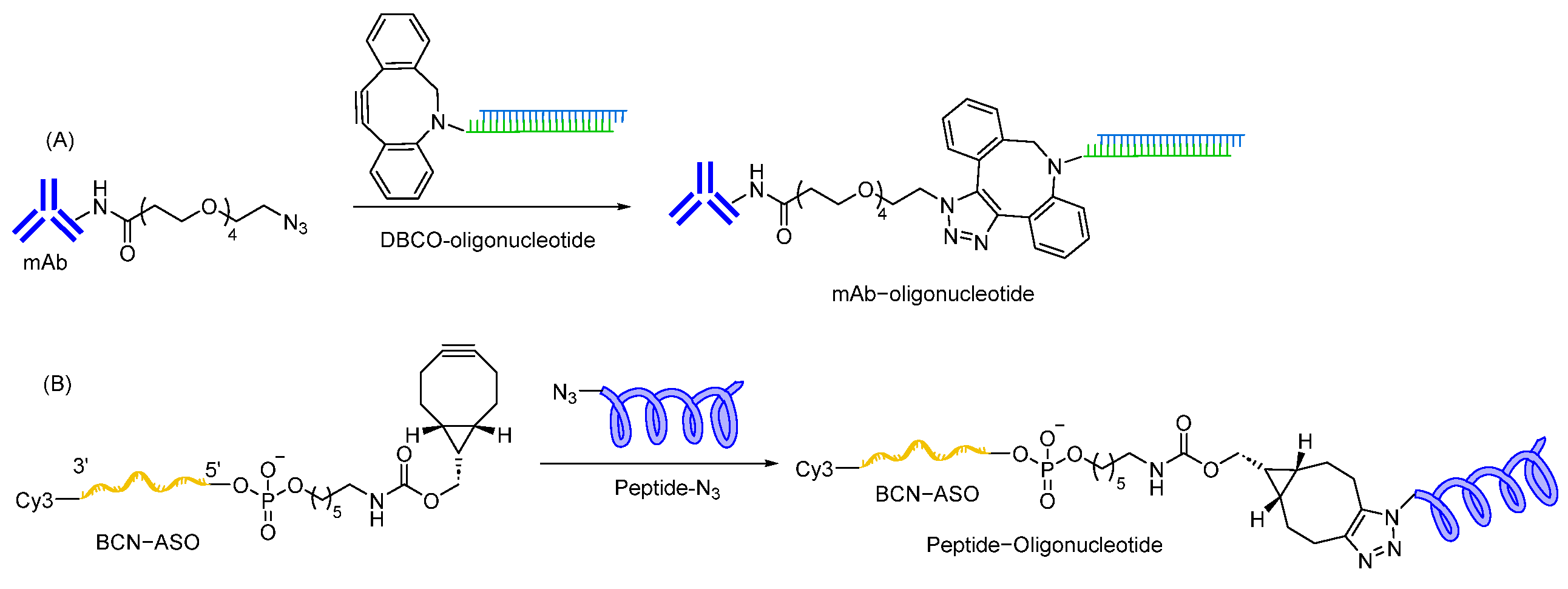
7.1.2. Triazole Linkages
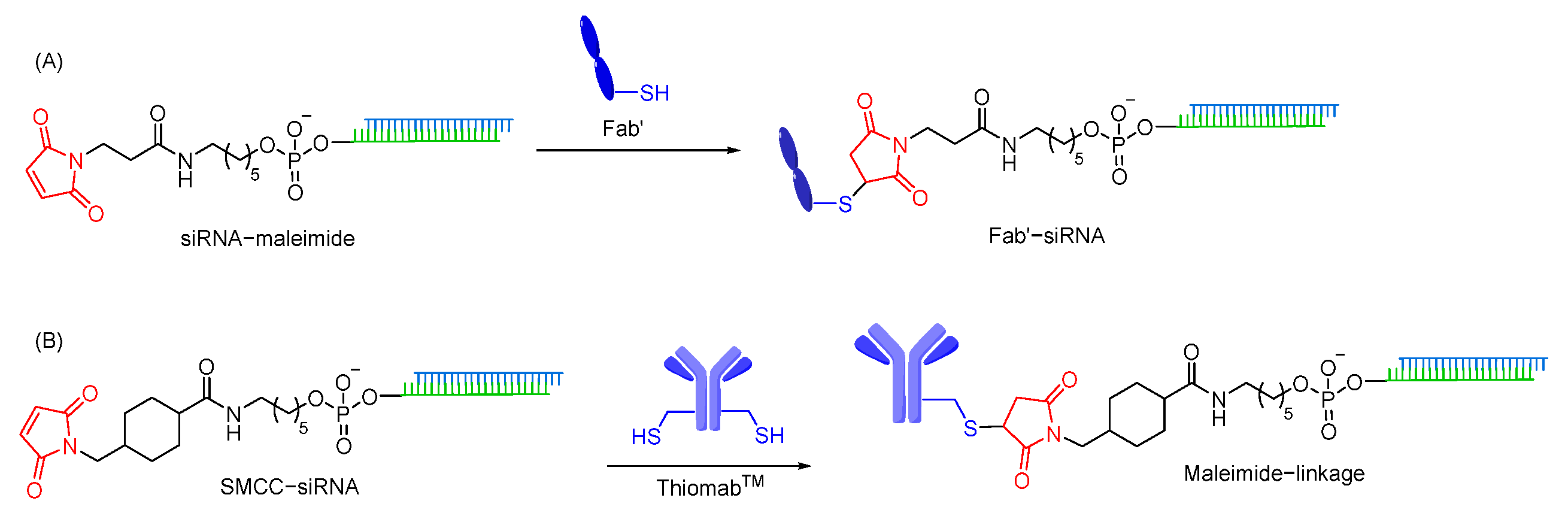
7.1.3. Maleimide Linkages
7.2. Chemically Cleavable Linkages
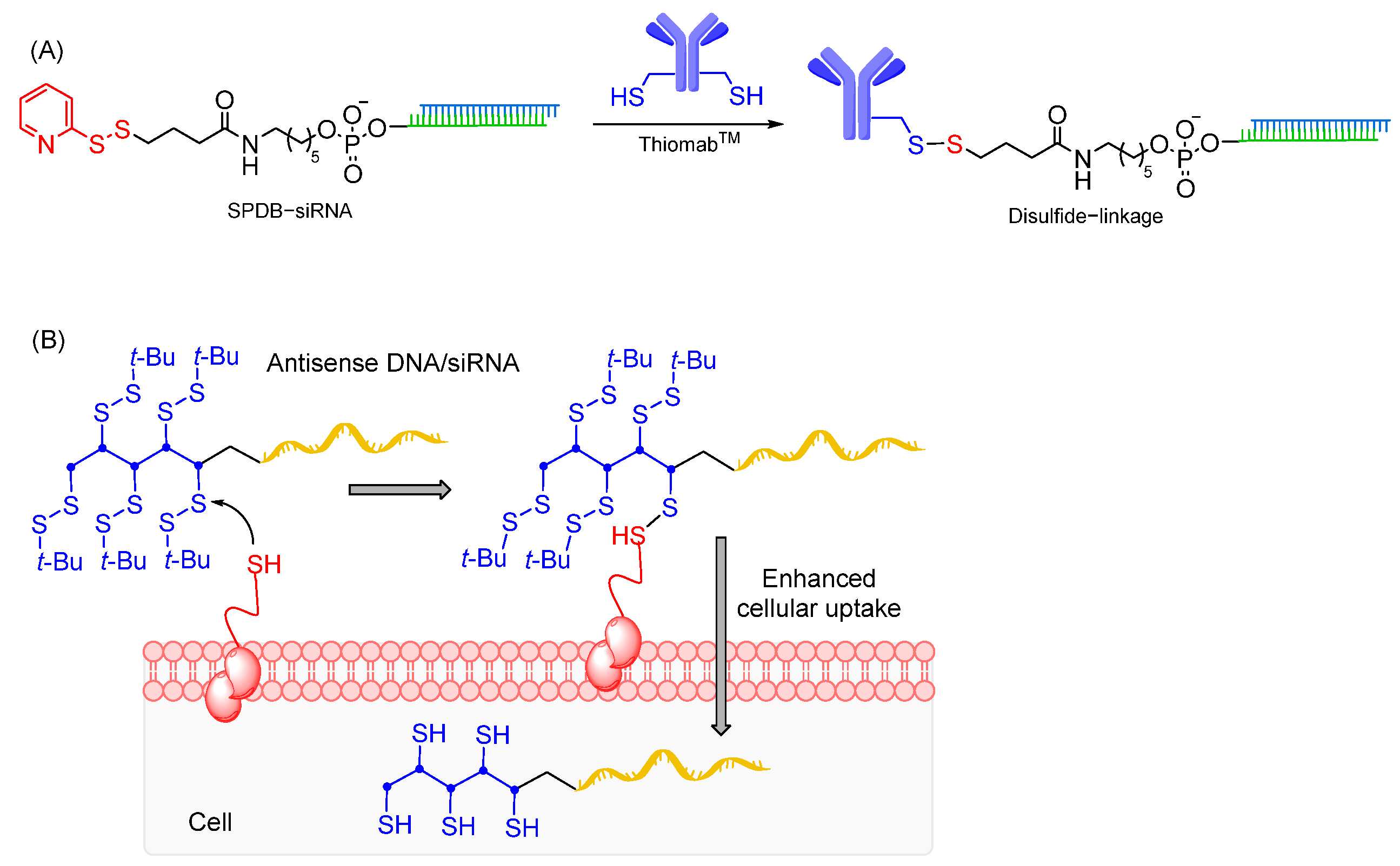
7.2.1. Disulfide Linkages
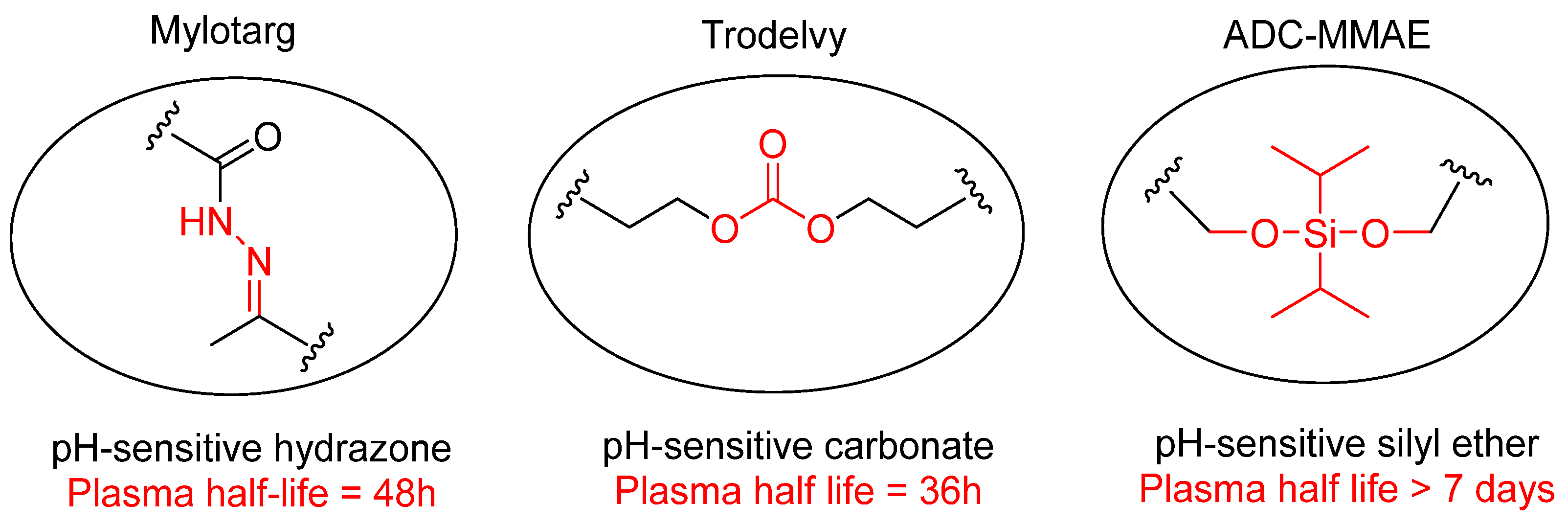
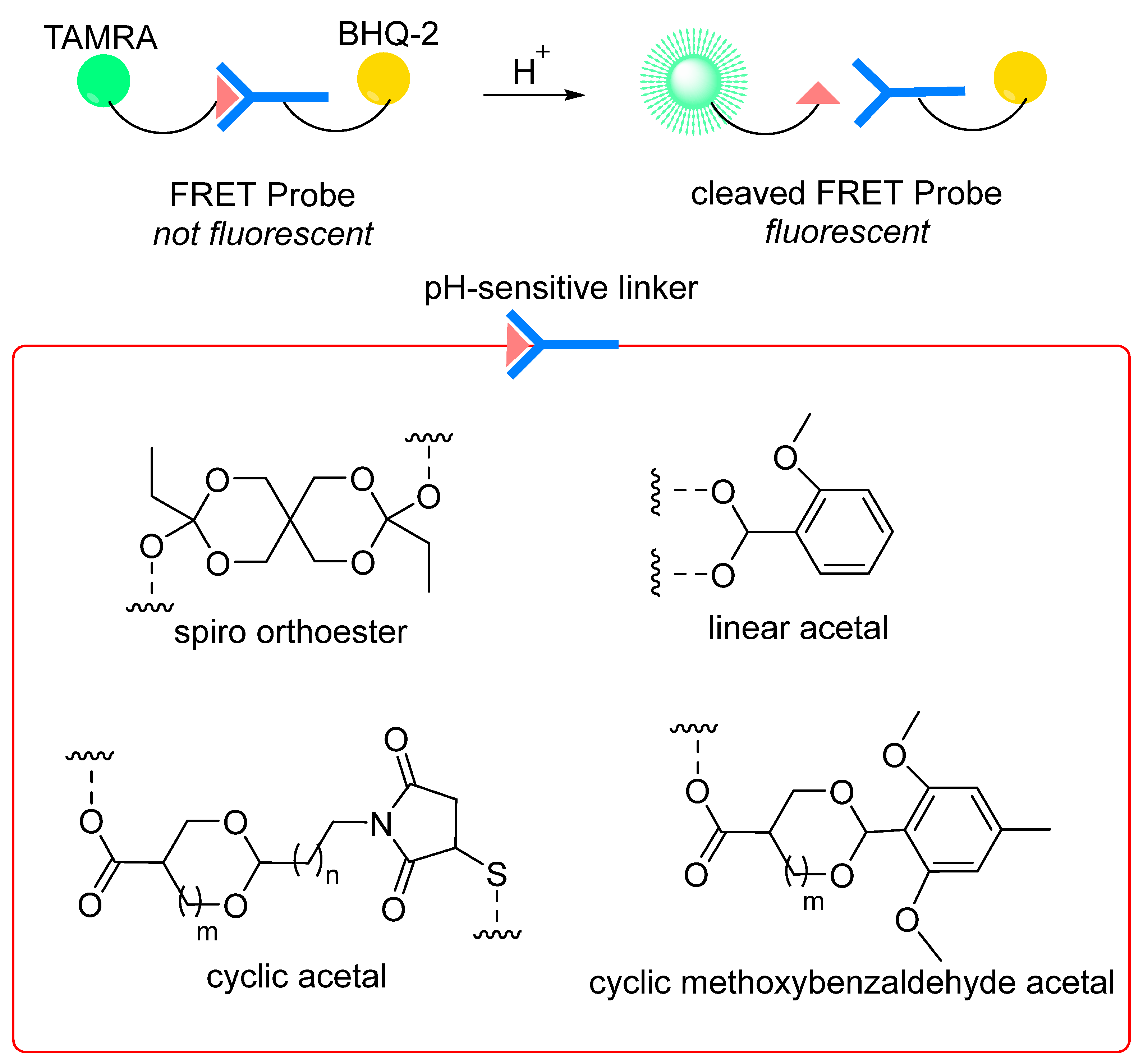
7.2.2. pH-Sensitive Linkages
8. Summary and Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Roberts, T.C.; Langer, R.; Wood, M.J.A. Advances in oligonucleotide drug delivery. Nat. Rev. Drug Discov. 2020, 19, 673–694. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, J.A.; Witzigmann, D.; Thomson, S.B.; Chen, S.; Leavitt, B.R.; Cullis, P.R.; van der Meel, R. The current landscape of nucleic acid therapeutics. Nat. Nanotechnol. 2021, 16, 630–643. [Google Scholar] [CrossRef]
- Juliano, R.; Bauman, J.; Kang, H.; Ming, X. Biological barriers to therapy with antisense and siRNA oligonucleotides. Mol. Pharm. 2009, 6, 686–695. [Google Scholar] [CrossRef]
- Geary, R.S.; Norris, D.; Yu, R.; Bennett, C.F. Pharmacokinetics, biodistribution and cell uptake of antisense oligonucleotides. Adv. Drug Deliv. Rev. 2015, 87, 46–51. [Google Scholar] [CrossRef]
- Kim, Y. Drug Discovery Perspectives of Antisense Oligonucleotides. Biomol. Ther. 2023, 31, 241–252. [Google Scholar] [CrossRef]
- Crooke, S.T.; Baker, B.F.; Crooke, R.M.; Liang, X.H. Antisense technology: An overview and prospectus. Nat. Rev. Drug Discov. 2021, 20, 427–453. [Google Scholar] [CrossRef]
- Dowdy, S.F. Overcoming cellular barriers for RNA therapeutics. Nat. Biotechnol. 2017, 35, 222–229. [Google Scholar] [CrossRef] [PubMed]
- Khvorova, A.; Watts, J.K. The chemical evolution of oligonucleotide therapies of clinical utility. Nat. Biotechnol. 2017, 35, 238–248. [Google Scholar] [CrossRef] [PubMed]
- Egli, M.; Manoharan, M. Chemistry, structure and function of approved oligonucleotide therapeutics. Nucleic Acids Res. 2023, 51, 2529–2573. [Google Scholar] [CrossRef]
- Hammond, S.M.; Aartsma-Rus, A.; Alves, S.; Borgos, S.E.; Buijsen, R.A.M.; Collin, R.W.J.; Covello, G.; Denti, M.A.; Desviat, L.R.; Echevarria, L.; et al. Delivery of oligonucleotide-based therapeutics: Challenges and opportunities. EMBO Mol. Med. 2021, 13, e13243. [Google Scholar] [CrossRef]
- Corey, D.R.; Damha, M.J.; Manoharan, M. Challenges and Opportunities for Nucleic Acid Therapeutics. Nucleic Acid Ther. 2022, 32, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Hache, M.; Swoboda, K.J.; Sethna, N.; Farrow-Gillespie, A.; Khandji, A.; Xia, S.; Bishop, K.M. Intrathecal Injections in Children With Spinal Muscular Atrophy: Nusinersen Clinical Trial Experience. J. Child Neurol. 2016, 31, 899–906. [Google Scholar] [CrossRef]
- Cideciyan, A.V.; Jacobson, S.G.; Drack, A.V.; Ho, A.C.; Charng, J.; Garafalo, A.V.; Roman, A.J.; Sumaroka, A.; Han, I.C.; Hochstedler, M.D.; et al. Effect of an intravitreal antisense oligonucleotide on vision in Leber congenital amaurosis due to a photoreceptor cilium defect. Nat. Med. 2019, 25, 225–228. [Google Scholar] [CrossRef]
- Adams, D.; Gonzalez-Duarte, A.; O’Riordan, W.D.; Yang, C.C.; Ueda, M.; Kristen, A.V.; Tournev, I.; Schmidt, H.H.; Coelho, T.; Berk, J.L.; et al. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N. Engl. J. Med. 2018, 379, 11–21. [Google Scholar] [CrossRef]
- Kristen, A.V.; Ajroud-Driss, S.; Conceicao, I.; Gorevic, P.; Kyriakides, T.; Obici, L. Patisiran, an RNAi therapeutic for the treatment of hereditary transthyretin-mediated amyloidosis. Neurodegener. Dis. Manag. 2019, 9, 5–23. [Google Scholar] [CrossRef]
- Garrelfs, S.F.; Frishberg, Y.; Hulton, S.A.; Koren, M.J.; O’Riordan, W.D.; Cochat, P.; Deschenes, G.; Shasha-Lavsky, H.; Saland, J.M.; Van’t Hoff, W.G.; et al. Lumasiran, an RNAi Therapeutic for Primary Hyperoxaluria Type 1. N. Engl. J. Med. 2021, 384, 1216–1226. [Google Scholar] [CrossRef]
- Balwani, M.; Sardh, E.; Ventura, P.; Peiro, P.A.; Rees, D.C.; Stolzel, U.; Bissell, D.M.; Bonkovsky, H.L.; Windyga, J.; Anderson, K.E.; et al. Phase 3 Trial of RNAi Therapeutic Givosiran for Acute Intermittent Porphyria. N. Engl. J. Med. 2020, 382, 2289–2301. [Google Scholar] [CrossRef]
- Raal, F.J.; Kallend, D.; Ray, K.K.; Turner, T.; Koenig, W.; Wright, R.S.; Wijngaard, P.L.J.; Curcio, D.; Jaros, M.J.; Leiter, L.A.; et al. Inclisiran for the Treatment of Heterozygous Familial Hypercholesterolemia. N. Engl. J. Med. 2020, 382, 1520–1530. [Google Scholar] [CrossRef] [PubMed]
- Ray, K.K.; Wright, R.S.; Kallend, D.; Koenig, W.; Leiter, L.A.; Raal, F.J.; Bisch, J.A.; Richardson, T.; Jaros, M.; Wijngaard, P.L.J.; et al. Two Phase 3 Trials of Inclisiran in Patients with Elevated LDL Cholesterol. N. Engl. J. Med. 2020, 382, 1507–1519. [Google Scholar] [CrossRef] [PubMed]
- Dowdy, S.F. Endosomal escape of RNA therapeutics: How do we solve this rate-limiting problem? RNA 2023, 29, 396–401. [Google Scholar] [CrossRef]
- Biscans, A.; Coles, A.; Haraszti, R.; Echeverria, D.; Hassler, M.; Osborn, M.; Khvorova, A. Diverse lipid conjugates for functional extra-hepatic siRNA delivery in vivo. Nucleic Acids Res. 2019, 47, 1082–1096. [Google Scholar] [CrossRef]
- Biscans, A.; Caiazzi, J.; McHugh, N.; Hariharan, V.; Muhuri, M.; Khvorova, A. Docosanoic acid conjugation to siRNA enables functional and safe delivery to skeletal and cardiac muscles. Mol. Ther. 2021, 29, 1382–1394. [Google Scholar] [CrossRef] [PubMed]
- Prakash, T.P.; Mullick, A.E.; Lee, R.G.; Yu, J.; Yeh, S.T.; Low, A.; Chappell, A.E.; Ostergaard, M.E.; Murray, S.; Gaus, H.J.; et al. Fatty acid conjugation enhances potency of antisense oligonucleotides in muscle. Nucleic Acids Res. 2019, 47, 6029–6044. [Google Scholar] [CrossRef]
- Brown, K.M.; Nair, J.K.; Janas, M.M.; Anglero-Rodriguez, Y.I.; Dang, L.T.H.; Peng, H.; Theile, C.S.; Castellanos-Rizaldos, E.; Brown, C.; Foster, D.; et al. Expanding RNAi therapeutics to extrahepatic tissues with lipophilic conjugates. Nat. Biotechnol. 2022, 40, 1500–1508. [Google Scholar] [CrossRef] [PubMed]
- Wolfrum, C.; Shi, S.; Jayaprakash, K.N.; Jayaraman, M.; Wang, G.; Pandey, R.K.; Rajeev, K.G.; Nakayama, T.; Charrise, K.; Ndungo, E.M.; et al. Mechanisms and optimization of in vivo delivery of lipophilic siRNAs. Nat. Biotechnol. 2007, 25, 1149–1157. [Google Scholar] [CrossRef] [PubMed]
- Osborn, M.F.; Coles, A.H.; Biscans, A.; Haraszti, R.A.; Roux, L.; Davis, S.; Ly, S.; Echeverria, D.; Hassler, M.R.; Godinho, B.; et al. Hydrophobicity drives the systemic distribution of lipid-conjugated siRNAs via lipid transport pathways. Nucleic Acids Res. 2019, 47, 1070–1081. [Google Scholar] [CrossRef]
- Doherty, G.J.; McMahon, H.T. Mechanisms of endocytosis. Annu. Rev. Biochem. 2009, 78, 857–902. [Google Scholar] [CrossRef]
- He, C.; Migawa, M.T.; Chen, K.; Weston, T.A.; Tanowitz, M.; Song, W.; Guagliardo, P.; Iyer, K.S.; Bennett, C.F.; Fong, L.G.; et al. High-resolution visualization and quantification of nucleic acid-based therapeutics in cells and tissues using Nanoscale secondary ion mass spectrometry (NanoSIMS). Nucleic Acids Res. 2021, 49, 1–14. [Google Scholar] [CrossRef]
- Dowdy, S.F.; Setten, R.L.; Cui, X.S.; Jadhav, S.G. Delivery of RNA Therapeutics: The Great Endosomal Escape! Nucleic Acid Ther. 2022, 32, 361–368. [Google Scholar] [CrossRef]
- Juliano, R.L. Chemical Manipulation of the Endosome Trafficking Machinery: Implications for Oligonucleotide Delivery. Biomedicines 2021, 9, 512. [Google Scholar] [CrossRef]
- Juliano, R.L. Intracellular Trafficking and Endosomal Release of Oligonucleotides: What We Know and What We Don’t. Nucleic Acid Ther. 2018, 28, 166–177. [Google Scholar] [CrossRef]
- Hassler, M.R.; Turanov, A.A.; Alterman, J.F.; Haraszti, R.A.; Coles, A.H.; Osborn, M.F.; Echeverria, D.; Nikan, M.; Salomon, W.E.; Roux, L.; et al. Comparison of partially and fully chemically-modified siRNA in conjugate-mediated delivery in vivo. Nucleic Acids Res. 2018, 46, 2185–2196. [Google Scholar] [CrossRef]
- Berdis, A. Nucleobase-modified nucleosides and nucleotides: Applications in biochemistry, synthetic biology, and drug discovery. Front. Chem. 2022, 10, 1051525. [Google Scholar] [CrossRef]
- Zamecnik, P.C.; Stephenson, M.L. Inhibition of Rous sarcoma virus replication and cell transformation by a specific oligodeoxynucleotide. Proc. Natl. Acad. Sci. USA 1978, 75, 280–284. [Google Scholar] [CrossRef] [PubMed]
- Crooke, S.T.; Liang, X.H.; Baker, B.F.; Crooke, R.M. Antisense technology: A review. J. Biol. Chem. 2021, 296, 100416. [Google Scholar] [CrossRef] [PubMed]
- Eckstein, F. Nucleoside Phosphorothioates. J. Am. Chem. Soc. 1966, 88, 4292–4294. [Google Scholar] [CrossRef]
- Stein, C.A.; Subasinghe, C.; Shinozuka, K.; Cohen, J.S. Physicochemical properties of phosphorothioate oligodeoxynucleotides. Nucleic Acids Res. 1988, 16, 3209–3221. [Google Scholar] [CrossRef] [PubMed]
- Gaus, H.J.; Gupta, R.; Chappell, A.E.; Ostergaard, M.E.; Swayze, E.E.; Seth, P.P. Characterization of the interactions of chemically-modified therapeutic nucleic acids with plasma proteins using a fluorescence polarization assay. Nucleic Acids Res. 2019, 47, 1110–1122. [Google Scholar] [CrossRef]
- Crooke, S.T.; Vickers, T.A.; Liang, X.H. Phosphorothioate modified oligonucleotide-protein interactions. Nucleic Acids Res. 2020, 48, 5235–5253. [Google Scholar] [CrossRef]
- Eckstein, F. Phosphorothioates, essential components of therapeutic oligonucleotides. Nucleic Acid Ther. 2014, 24, 374–387. [Google Scholar] [CrossRef]
- Crooke, S.T.; Wang, S.; Vickers, T.A.; Shen, W.; Liang, X.H. Cellular uptake and trafficking of antisense oligonucleotides. Nat. Biotechnol. 2017, 35, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Laurent, Q.; Martinent, R.; Moreau, D.; Winssinger, N.; Sakai, N.; Matile, S. Oligonucleotide Phosphorothioates Enter Cells by Thiol-Mediated Uptake. Angew. Chem. Int. Ed. Engl. 2021, 60, 19102–19106. [Google Scholar] [CrossRef]
- Piascik, P. Fomiversen sodium approved to treat CMV retinitis. J. Am. Pharm. Assoc. 1999, 39, 84–85. [Google Scholar] [CrossRef]
- Kupryushkin, M.S.; Pyshnyi, D.V.; Stetsenko, D.A. Phosphoryl guanidines: A new type of nucleic Acid analogues. Acta Naturae 2014, 6, 116–118. [Google Scholar] [CrossRef]
- Skvortsova, Y.V.; Salina, E.G.; Burakova, E.A.; Bychenko, O.S.; Stetsenko, D.A.; Azhikina, T.L. A New Antisense Phosphoryl Guanidine Oligo-2′-O-Methylribonucleotide Penetrates Into Intracellular Mycobacteria and Suppresses Target Gene Expression. Front. Pharmacol. 2019, 10, 1049. [Google Scholar] [CrossRef]
- Miroshnichenko, S.K.; Patutina, O.A.; Burakova, E.A.; Chelobanov, B.P.; Fokina, A.A.; Vlassov, V.V.; Altman, S.; Zenkova, M.A.; Stetsenko, D.A. Mesyl phosphoramidate antisense oligonucleotides as an alternative to phosphorothioates with improved biochemical and biological properties. Proc. Natl. Acad. Sci. USA 2019, 116, 1229–1234. [Google Scholar] [CrossRef] [PubMed]
- Anderson, B.A.; Freestone, G.C.; Low, A.; De-Hoyos, C.L.; Iii, W.J.D.; Ostergaard, M.E.; Migawa, M.T.; Fazio, M.; Wan, W.B.; Berdeja, A.; et al. Towards next generation antisense oligonucleotides: Mesylphosphoramidate modification improves therapeutic index and duration of effect of gapmer antisense oligonucleotides. Nucleic Acids Res. 2021, 49, 9026–9041. [Google Scholar] [CrossRef]
- Hammond, S.M.; Sergeeva, O.V.; Melnikov, P.A.; Goli, L.; Stoodley, J.; Zatsepin, T.S.; Stetsenko, D.A.; Wood, M.J.A. Mesyl Phosphoramidate Oligonucleotides as Potential Splice-Switching Agents: Impact of Backbone Structure on Activity and Intracellular Localization. Nucleic Acid Ther. 2021, 31, 190–200. [Google Scholar] [CrossRef]
- Goyenvalle, A.; Griffith, G.; Babbs, A.; El Andaloussi, S.; Ezzat, K.; Avril, A.; Dugovic, B.; Chaussenot, R.; Ferry, A.; Voit, T.; et al. Functional correction in mouse models of muscular dystrophy using exon-skipping tricyclo-DNA oligomers. Nat. Med. 2015, 21, 270–275. [Google Scholar] [CrossRef] [PubMed]
- Wan, W.B.; Seth, P.P. The Medicinal Chemistry of Therapeutic Oligonucleotides. J. Med. Chem. 2016, 59, 9645–9667. [Google Scholar] [CrossRef]
- Ostergaard, M.E.; Southwell, A.L.; Kordasiewicz, H.; Watt, A.T.; Skotte, N.H.; Doty, C.N.; Vaid, K.; Villanueva, E.B.; Swayze, E.E.; Bennett, C.F.; et al. Rational design of antisense oligonucleotides targeting single nucleotide polymorphisms for potent and allele selective suppression of mutant Huntingtin in the CNS. Nucleic Acids Res. 2013, 41, 9634–9650. [Google Scholar] [CrossRef]
- Madaoui, M.; Datta, D.; Wassarman, K.; Zlatev, I.; Egli, M.; Ross, B.S.; Manoharan, M. A Chemical Approach to Introduce 2,6-Diaminopurine and 2-Aminoadenine Conjugates into Oligonucleotides without Need for Protecting Groups. Org. Lett. 2022, 24, 6111–6116. [Google Scholar] [CrossRef]
- Hoy, S.M. Nusinersen: First Global Approval. Drugs 2017, 77, 473–479. [Google Scholar] [CrossRef]
- Hair, P.; Cameron, F.; McKeage, K. Mipomersen sodium: First global approval. Drugs 2013, 73, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Aartsma-Rus, A.; Krieg, A.M. FDA Approves Eteplirsen for Duchenne Muscular Dystrophy: The Next Chapter in the Eteplirsen Saga. Nucleic Acid Ther. 2017, 27, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Heo, Y.A. Golodirsen: First Approval. Drugs 2020, 80, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, S. Viltolarsen: First Approval. Drugs 2020, 80, 1027–1031. [Google Scholar] [CrossRef] [PubMed]
- Shirley, M. Casimersen: First Approval. Drugs 2021, 81, 875–879. [Google Scholar] [CrossRef]
- Carthew, R.W.; Sontheimer, E.J. Origins and Mechanisms of miRNAs and siRNAs. Cell 2009, 136, 642–655. [Google Scholar] [CrossRef]
- Liu, J.; Carmell, M.A.; Rivas, F.V.; Marsden, C.G.; Thomson, J.M.; Song, J.J.; Hammond, S.M.; Joshua-Tor, L.; Hannon, G.J. Argonaute2 is the catalytic engine of mammalian RNAi. Science 2004, 305, 1437–1441. [Google Scholar] [CrossRef]
- Bartlett, D.W.; Davis, M.E. Effect of siRNA nuclease stability on the in vitro and in vivo kinetics of siRNA-mediated gene silencing. Biotechnol. Bioeng. 2007, 97, 909–921. [Google Scholar] [CrossRef] [PubMed]
- Layzer, J.M.; McCaffrey, A.P.; Tanner, A.K.; Huang, Z.; Kay, M.A.; Sullenger, B.A. In vivo activity of nuclease-resistant siRNAs. RNA 2004, 10, 766–771. [Google Scholar] [CrossRef]
- Allerson, C.R.; Sioufi, N.; Jarres, R.; Prakash, T.P.; Naik, N.; Berdeja, A.; Wanders, L.; Griffey, R.H.; Swayze, E.E.; Bhat, B. Fully 2′-modified oligonucleotide duplexes with improved in vitro potency and stability compared to unmodified small interfering RNA. J. Med. Chem. 2005, 48, 901–904. [Google Scholar] [CrossRef]
- Braasch, D.A.; Jensen, S.; Liu, Y.; Kaur, K.; Arar, K.; White, M.A.; Corey, D.R. RNA interference in mammalian cells by chemically-modified RNA. Biochemistry 2003, 42, 7967–7975. [Google Scholar] [CrossRef]
- Biscans, A.; Caiazzi, J.; Davis, S.; McHugh, N.; Sousa, J.; Khvorova, A. The chemical structure and phosphorothioate content of hydrophobically modified siRNAs impact extrahepatic distribution and efficacy. Nucleic Acids Res. 2020, 48, 7665–7680. [Google Scholar] [CrossRef] [PubMed]
- Keam, S.J. Vutrisiran: First Approval. Drugs 2022, 82, 1419–1425. [Google Scholar] [CrossRef]
- Stein, C.A.; Hansen, J.B.; Lai, J.; Wu, S.; Voskresenskiy, A.; Hog, A.; Worm, J.; Hedtjarn, M.; Souleimanian, N.; Miller, P.; et al. Efficient gene silencing by delivery of locked nucleic acid antisense oligonucleotides, unassisted by transfection reagents. Nucleic Acids Res. 2010, 38, e3. [Google Scholar] [CrossRef] [PubMed]
- Geary, R.S. Antisense oligonucleotide pharmacokinetics and metabolism. Expert Opin. Drug Metab. Toxicol. 2009, 5, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Anwar, S.; Mir, F.; Yokota, T. Enhancing the Effectiveness of Oligonucleotide Therapeutics Using Cell-Penetrating Peptide Conjugation, Chemical Modification, and Carrier-Based Delivery Strategies. Pharmaceutics 2023, 15, 1130. [Google Scholar] [CrossRef]
- Knerr, L.; Prakash, T.P.; Lee, R.; Drury Iii, W.J.; Nikan, M.; Fu, W.; Pirie, E.; Maria, L.; Valeur, E.; Hayen, A.; et al. Glucagon Like Peptide 1 Receptor Agonists for Targeted Delivery of Antisense Oligonucleotides to Pancreatic Beta Cell. J. Am. Chem. Soc. 2021, 143, 3416–3429. [Google Scholar] [CrossRef]
- Ammala, C.; Drury, W.J., 3rd; Knerr, L.; Ahlstedt, I.; Stillemark-Billton, P.; Wennberg-Huldt, C.; Andersson, E.M.; Valeur, E.; Jansson-Lofmark, R.; Janzen, D.; et al. Targeted delivery of antisense oligonucleotides to pancreatic beta-cells. Sci. Adv. 2018, 4, eaat3386. [Google Scholar] [CrossRef] [PubMed]
- Osborn, M.F.; Khvorova, A. Improving siRNA Delivery In Vivo Through Lipid Conjugation. Nucleic Acid Ther. 2018, 28, 128–136. [Google Scholar] [CrossRef]
- Tran, P.; Weldemichael, T.; Liu, Z.; Li, H.Y. Delivery of Oligonucleotides: Efficiency with Lipid Conjugation and Clinical Outcome. Pharmaceutics 2022, 14, 342. [Google Scholar] [CrossRef] [PubMed]
- Biscans, A.; Coles, A.; Echeverria, D.; Khvorova, A. The valency of fatty acid conjugates impacts siRNA pharmacokinetics, distribution, and efficacy in vivo. J. Control. Release 2019, 302, 116–125. [Google Scholar] [CrossRef]
- Ostergaard, M.E.; Jackson, M.; Low, A.; Chappell, A.E.; Lee, R.G.; Peralta, R.Q.; Yu, J.; Kinberger, G.A.; Dan, A.; Carty, R.; et al. Conjugation of hydrophobic moieties enhances potency of antisense oligonucleotides in the muscle of rodents and non-human primates. Nucleic Acids Res. 2019, 47, 6045–6058. [Google Scholar] [CrossRef]
- Mullard, A. Antibody-oligonucleotide conjugates enter the clinic. Nat. Rev. Drug Discov. 2022, 21, 6–8. [Google Scholar] [CrossRef]
- Hammond, S.M.; Abendroth, F.; Goli, L.; Stoodley, J.; Burrell, M.; Thom, G.; Gurrell, I.; Ahlskog, N.; Gait, M.J.; Wood, M.J.; et al. Antibody-oligonucleotide conjugate achieves CNS delivery in animal models for spinal muscular atrophy. JCI Insight 2022, 7, e154142. [Google Scholar] [CrossRef]
- Desjardins, C.A.; Yao, M.; Hall, J.; O’Donnell, E.; Venkatesan, R.; Spring, S.; Wen, A.; Hsia, N.; Shen, P.; Russo, R.; et al. Enhanced exon skipping and prolonged dystrophin restoration achieved by TfR1-targeted delivery of antisense oligonucleotide using FORCE conjugation in mdx mice. Nucleic Acids Res. 2022, 50, 11401–11414. [Google Scholar] [CrossRef]
- Song, E.; Zhu, P.; Lee, S.K.; Chowdhury, D.; Kussman, S.; Dykxhoorn, D.M.; Feng, Y.; Palliser, D.; Weiner, D.B.; Shankar, P.; et al. Antibody mediated in vivo delivery of small interfering RNAs via cell-surface receptors. Nat. Biotechnol. 2005, 23, 709–717. [Google Scholar] [CrossRef]
- Study of AOC 1001 in Adult Myotonic Dystrophy Type 1 (DM1) Patients (MARINA). Available online: https://classic.clinicaltrials.gov/ct2/show/NCT05027269 (accessed on 14 June 2023).
- Malecova, B.; Burke, R.S.; Cochran, M.; Hood, M.D.; Johns, R.; Kovach, P.R.; Doppalapudi, V.R.; Erdogan, G.; Arias, J.D.; Darimont, B.; et al. Targeted tissue delivery of RNA therapeutics using antibody-oligonucleotide conjugates (AOCs). Nucleic Acids Res. 2023, 51, 5901–5910. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.J.; Wang, L.J.; Han, D.H.; Wu, J.H.; Jiao, D.; Zhang, K.L.; Chen, J.W.; Li, Y.; Yang, F.; Zhang, J.L.; et al. Therapeutic effects of human monoclonal PSMA antibody-mediated TRIM24 siRNA delivery in PSMA-positive castration-resistant prostate cancer. Theranostics 2019, 9, 1247–1263. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.M.; Dahlman, J.E.; Neuman, K.K.; Prata, C.A.H.; Krampert, M.C.; Hadwiger, P.M.; Vornlocher, H.P. Ligand Conjugated Multimeric siRNAs Enable Enhanced Uptake and Multiplexed Gene Silencing. Nucleic Acid Ther. 2019, 29, 231–244. [Google Scholar] [CrossRef]
- Rahman Chowdhury, T.; Taufiq, T.; Ishida, K.; Ariful Islam, M.; Kasahara, Y.; Osawa, T.; Obika, S. Synthesis and biophysical properties of tetravalent PEG-conjugated antisense oligonucleotide. Bioorg. Med. Chem. 2023, 78, 117149. [Google Scholar] [CrossRef] [PubMed]
- Hariharan, V.N.; Shin, M.; Chang, C.W.; O’Reilly, D.; Biscans, A.; Yamada, K.; Guo, Z.; Somasundaran, M.; Tang, Q.; Monopoli, K.; et al. Divalent siRNAs are bioavailable in the lung and efficiently block SARS-CoV-2 infection. Proc. Natl. Acad. Sci. USA 2023, 120, e2219523120. [Google Scholar] [CrossRef] [PubMed]
- Hoy, S.M. Patisiran: First Global Approval. Drugs 2018, 78, 1625–1631. [Google Scholar] [CrossRef]
- Andaloussi, S.E.; Lehto, T.; Mager, I.; Rosenthal-Aizman, K.; Oprea, I.I.; Simonson, O.E.; Sork, H.; Ezzat, K.; Copolovici, D.M.; Kurrikoff, K.; et al. Design of a peptide-based vector, PepFect6, for efficient delivery of siRNA in cell culture and systemically in vivo. Nucleic Acids Res. 2011, 39, 3972–3987. [Google Scholar] [CrossRef]
- Chen, J.; Ellert-Miklaszewska, A.; Garofalo, S.; Dey, A.K.; Tang, J.; Jiang, Y.; Clement, F.; Marche, P.N.; Liu, X.; Kaminska, B.; et al. Synthesis and use of an amphiphilic dendrimer for siRNA delivery into primary immune cells. Nat. Protoc. 2021, 16, 327–351. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, Q.; Zhang, X.; Huang, H.; Tang, S.; Chai, Y.; Xu, Z.; Li, M.; Chen, X.; Liu, J.; et al. Recent advances in exosome-mediated nucleic acid delivery for cancer therapy. J. Nanobiotechnol. 2022, 20, 279. [Google Scholar] [CrossRef]
- Barnaby, S.N.; Sita, T.L.; Petrosko, S.H.; Stegh, A.H.; Mirkin, C.A. Therapeutic applications of spherical nucleic acids. In Nanotechnology-Based Precision Tools for the Detection and Treatment of Cancer; Mirkin, C.A., Meade, T.J., Petrosko, S.H., Stegh, A.H., Eds.; Springer: Cham, Switzerland, 2015; Volume 166, pp. 23–50. [Google Scholar] [CrossRef]
- Bousmail, D.; Amrein, L.; Fakhoury, J.J.; Fakih, H.H.; Hsu, J.C.C.; Panasci, L.; Sleiman, H.F. Precision spherical nucleic acids for delivery of anticancer drugs. Chem. Sci. 2017, 8, 6218–6229. [Google Scholar] [CrossRef]
- Fakih, H.H.; Katolik, A.; Malek-Adamian, E.; Fakhoury, J.J.; Kaviani, S.; Damha, M.J.; Sleiman, H.F. Design and enhanced gene silencing activity of spherical 2’-fluoroarabinose nucleic acids (FANA-SNAs). Chem. Sci. 2021, 12, 2993–3003. [Google Scholar] [CrossRef]
- Yang, J.; Jiang, Q.; He, L.; Zhan, P.; Liu, Q.; Liu, S.; Fu, M.; Liu, J.; Li, C.; Ding, B. Self-Assembled Double-Bundle DNA Tetrahedron for Efficient Antisense Delivery. ACS Appl. Mater. Interfaces 2018, 10, 23693–23699. [Google Scholar] [CrossRef]
- Huang, Y.; Wang, J.; Jiang, K.; Chung, E.J. Improving kidney targeting: The influence of nanoparticle physicochemical properties on kidney interactions. J. Control. Release 2021, 334, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Giljohann, D.A.; Seferos, D.S.; Patel, P.C.; Millstone, J.E.; Rosi, N.L.; Mirkin, C.A. Oligonucleotide loading determines cellular uptake of DNA-modified gold nanoparticles. Nano Lett. 2007, 7, 3818–3821. [Google Scholar] [CrossRef]
- Ye, Y.; Zhang, X.; Xie, F.; Xu, B.; Xie, P.; Yang, T.; Shi, Q.; Zhang, C.Y.; Zhang, Y.; Chen, J.; et al. An engineered exosome for delivering sgRNA:Cas9 ribonucleoprotein complex and genome editing in recipient cells. Biomater. Sci. 2020, 8, 2966–2976. [Google Scholar] [CrossRef]
- Munagala, R.; Aqil, F.; Jeyabalan, J.; Kandimalla, R.; Wallen, M.; Tyagi, N.; Wilcher, S.; Yan, J.; Schultz, D.J.; Spencer, W.; et al. Exosome-mediated delivery of RNA and DNA for gene therapy. Cancer Lett. 2021, 505, 58–72. [Google Scholar] [CrossRef]
- Klumperman, J.; Raposo, G. The complex ultrastructure of the endolysosomal system. Cold Spring Harb. Perspect. Biol. 2014, 6, a016857. [Google Scholar] [CrossRef] [PubMed]
- Luzio, J.P.; Pryor, P.R.; Bright, N.A. Lysosomes: Fusion and function. Nat. Rev. Mol. Cell Biol. 2007, 8, 622–632. [Google Scholar] [CrossRef]
- Scott, C.C.; Vacca, F.; Gruenberg, J. Endosome maturation, transport and functions. Semin. Cell Dev. Biol. 2014, 31, 2–10. [Google Scholar] [CrossRef]
- Sahay, G.; Querbes, W.; Alabi, C.; Eltoukhy, A.; Sarkar, S.; Zurenko, C.; Karagiannis, E.; Love, K.; Chen, D.; Zoncu, R.; et al. Efficiency of siRNA delivery by lipid nanoparticles is limited by endocytic recycling. Nat. Biotechnol. 2013, 31, 653–658. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.R.; Gupta, S.; Qin, J.; Racie, T.; He, G.; Lentini, S.; Malone, R.; Yu, M.; Matsuda, S.; Shulga-Morskaya, S.; et al. Investigating the pharmacodynamic durability of GalNAc-siRNA conjugates. Nucleic Acids Res. 2020, 48, 11827–11844. [Google Scholar] [CrossRef]
- Kendall, G.C.; Mokhonova, E.I.; Moran, M.; Sejbuk, N.E.; Wang, D.W.; Silva, O.; Wang, R.T.; Martinez, L.; Lu, Q.L.; Damoiseaux, R.; et al. Dantrolene enhances antisense-mediated exon skipping in human and mouse models of Duchenne muscular dystrophy. Sci. Transl. Med. 2012, 4, 164ra160. [Google Scholar] [CrossRef]
- Ming, X.; Carver, K.; Fisher, M.; Noel, R.; Cintrat, J.C.; Gillet, D.; Barbier, J.; Cao, C.; Bauman, J.; Juliano, R.L. The small molecule Retro-1 enhances the pharmacological actions of antisense and splice switching oligonucleotides. Nucleic Acids Res. 2013, 41, 3673–3687. [Google Scholar] [CrossRef]
- Osborn, M.F.; Alterman, J.F.; Nikan, M.; Cao, H.; Didiot, M.C.; Hassler, M.R.; Coles, A.H.; Khvorova, A. Guanabenz (Wytensin) selectively enhances uptake and efficacy of hydrophobically modified siRNAs. Nucleic Acids Res. 2015, 43, 8664–8672. [Google Scholar] [CrossRef]
- Yang, B.; Ming, X.; Cao, C.; Laing, B.; Yuan, A.; Porter, M.A.; Hull-Ryde, E.A.; Maddry, J.; Suto, M.; Janzen, W.P.; et al. High-throughput screening identifies small molecules that enhance the pharmacological effects of oligonucleotides. Nucleic Acids Res. 2015, 43, 1987–1996. [Google Scholar] [CrossRef] [PubMed]
- Gilleron, J.; Paramasivam, P.; Zeigerer, A.; Querbes, W.; Marsico, G.; Andree, C.; Seifert, S.; Amaya, P.; Stoter, M.; Koteliansky, V.; et al. Identification of siRNA delivery enhancers by a chemical library screen. Nucleic Acids Res. 2015, 43, 7984–8001. [Google Scholar] [CrossRef]
- Marsh, M.; Helenius, A. Virus entry: Open sesame. Cell 2006, 124, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Gruenberg, J.; van der Goot, F.G. Mechanisms of pathogen entry through the endosomal compartments. Nat. Rev. Mol. Cell Biol. 2006, 7, 495–504. [Google Scholar] [CrossRef]
- Rothman, J.E.; Wieland, F.T. Protein sorting by transport vesicles. Science 1996, 272, 227–234. [Google Scholar] [CrossRef]
- Esser, A.F. The membrane attack complex of complement. Assembly, structure and cytotoxic activity. Toxicology 1994, 87, 229–247. [Google Scholar] [CrossRef] [PubMed]
- Pei, D.; Buyanova, M. Overcoming Endosomal Entrapment in Drug Delivery. Bioconjug. Chem. 2019, 30, 273–283. [Google Scholar] [CrossRef]
- Wittrup, A.; Ai, A.; Liu, X.; Hamar, P.; Trifonova, R.; Charisse, K.; Manoharan, M.; Kirchhausen, T.; Lieberman, J. Visualizing lipid-formulated siRNA release from endosomes and target gene knockdown. Nat. Biotechnol. 2015, 33, 870–876. [Google Scholar] [CrossRef]
- Zelphati, O.; Szoka, F.C., Jr. Mechanism of oligonucleotide release from cationic liposomes. Proc. Natl. Acad. Sci. USA 1996, 93, 11493–11498. [Google Scholar] [CrossRef] [PubMed]
- Tilley, S.J.; Saibil, H.R. The mechanism of pore formation by bacterial toxins. Curr. Opin. Struct. Biol. 2006, 16, 230–236. [Google Scholar] [CrossRef]
- Herce, H.D.; Garcia, A.E.; Litt, J.; Kane, R.S.; Martin, P.; Enrique, N.; Rebolledo, A.; Milesi, V. Arginine-rich peptides destabilize the plasma membrane, consistent with a pore formation translocation mechanism of cell-penetrating peptides. Biophys. J. 2009, 97, 1917–1925. [Google Scholar] [CrossRef] [PubMed]
- Behr, J.-P. The Proton Sponge: A Trick to Enter Cells the Viruses Did Not Exploit. Chimia 1997, 51, 34. [Google Scholar] [CrossRef]
- Sonawane, N.D.; Szoka, F.C., Jr.; Verkman, A.S. Chloride accumulation and swelling in endosomes enhances DNA transfer by polyamine-DNA polyplexes. J. Biol. Chem. 2003, 278, 44826–44831. [Google Scholar] [CrossRef]
- Ur Rehman, Z.; Hoekstra, D.; Zuhorn, I.S. Mechanism of polyplex- and lipoplex-mediated delivery of nucleic acids: Real-time visualization of transient membrane destabilization without endosomal lysis. ACS Nano 2013, 7, 3767–3777. [Google Scholar] [CrossRef] [PubMed]
- Varkouhi, A.K.; Scholte, M.; Storm, G.; Haisma, H.J. Endosomal escape pathways for delivery of biologicals. J. Control. Release 2011, 151, 220–228. [Google Scholar] [CrossRef]
- Bus, T.; Traeger, A.; Schubert, U.S. The great escape: How cationic polyplexes overcome the endosomal barrier. J. Mater. Chem. B 2018, 6, 6904–6918. [Google Scholar] [CrossRef]
- Sahni, A.; Qian, Z.; Pei, D. Cell-Penetrating Peptides Escape the Endosome by Inducing Vesicle Budding and Collapse. ACS Chem. Biol. 2020, 15, 2485–2492. [Google Scholar] [CrossRef]
- Ofridam, F.; Tarhini, M.; Lebaz, N.; Gagnière, É.; Mangin, D.; Elaissari, A. pH-sensitive polymers: Classification and some fine potential applications. Polym. Adv. Technol. 2021, 32, 1455–1484. [Google Scholar] [CrossRef]
- Yin, H.; Kanasty, R.L.; Eltoukhy, A.A.; Vegas, A.J.; Dorkin, J.R.; Anderson, D.G. Non-viral vectors for gene-based therapy. Nat. Rev. Genet. 2014, 15, 541–555. [Google Scholar] [CrossRef] [PubMed]
- Wagner, E.; Ogris, M.; Zauner, W. Polylysine-based transfection systems utilizing receptor-mediated delivery. Adv. Drug Deliv. Rev. 1998, 30, 97–113. [Google Scholar] [CrossRef] [PubMed]
- Creusat, G.; Rinaldi, A.S.; Weiss, E.; Elbaghdadi, R.; Remy, J.S.; Mulherkar, R.; Zuber, G. Proton sponge trick for pH-sensitive disassembly of polyethylenimine-based siRNA delivery systems. Bioconjug. Chem. 2010, 21, 994–1002. [Google Scholar] [CrossRef]
- Patnaik, S.; Gupta, K.C. Novel polyethylenimine-derived nanoparticles for in vivo gene delivery. Expert Opin. Drug Deliv. 2013, 10, 215–228. [Google Scholar] [CrossRef] [PubMed]
- Klemm, A.R.; Young, D.; Lloyd, J.B. Effects of polyethyleneimine on endocytosis and lysosome stability. Biochem. Pharmacol. 1998, 56, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.W. Polylysine copolymers for gene delivery. Cold Spring Harb. Protoc. 2012, 2012, 433–438. [Google Scholar] [CrossRef]
- Alexis, F.; Pridgen, E.; Molnar, L.K.; Farokhzad, O.C. Factors affecting the clearance and biodistribution of polymeric nanoparticles. Mol. Pharm. 2008, 5, 505–515. [Google Scholar] [CrossRef]
- Petersen, H.; Fechner, P.M.; Martin, A.L.; Kunath, K.; Stolnik, S.; Roberts, C.J.; Fischer, D.; Davies, M.C.; Kissel, T. Polyethylenimine-graft-poly(ethylene glycol) copolymers: Influence of copolymer block structure on DNA complexation and biological activities as gene delivery system. Bioconjug. Chem. 2002, 13, 845–854. [Google Scholar] [CrossRef]
- Sun, Y.; Jiao, Y.; Wang, Y.; Lu, D.; Yang, W. The strategy to improve gene transfection efficiency and biocompatibility of hyperbranched PAMAM with the cooperation of PEGylated hyperbranched PAMAM. Int. J. Pharm. 2014, 465, 112–119. [Google Scholar] [CrossRef]
- Yu, G.S.; Bae, Y.M.; Choi, H.; Kong, B.; Choi, I.S.; Choi, J.S. Synthesis of PAMAM dendrimer derivatives with enhanced buffering capacity and remarkable gene transfection efficiency. Bioconjug. Chem. 2011, 22, 1046–1055. [Google Scholar] [CrossRef] [PubMed]
- Lynn, D.M.; Langer, R. Degradable Poly(β-amino esters): Synthesis, Characterization, and Self-Assembly with Plasmid DNA. J. Am. Chem. Soc. 2000, 122, 10761–10768. [Google Scholar] [CrossRef]
- Pack, D.W.; Hoffman, A.S.; Pun, S.; Stayton, P.S. Design and development of polymers for gene delivery. Nat. Rev. Drug Discov. 2005, 4, 581–593. [Google Scholar] [CrossRef]
- Mintzer, M.A.; Simanek, E.E. Nonviral vectors for gene delivery. Chem. Rev. 2009, 109, 259–302. [Google Scholar] [CrossRef] [PubMed]
- Schmid, S.L.; Sorkin, A.; Zerial, M. Endocytosis: Past, present, and future. Cold Spring Harb. Perspect. Biol. 2014, 6, a022509. [Google Scholar] [CrossRef] [PubMed]
- Mudhakir, D.; Harashima, H. Learning from the viral journey: How to enter cells and how to overcome intracellular barriers to reach the nucleus. AAPS J. 2009, 11, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Green, M.; Loewenstein, P.M. Autonomous functional domains of chemically synthesized human immunodeficiency virus tat trans-activator protein. Cell 1988, 55, 1179–1188. [Google Scholar] [CrossRef]
- Frankel, A.D.; Pabo, C.O. Cellular uptake of the tat protein from human immunodeficiency virus. Cell 1988, 55, 1189–1193. [Google Scholar] [CrossRef]
- Wadia, J.S.; Dowdy, S.F. Transmembrane delivery of protein and peptide drugs by TAT-mediated transduction in the treatment of cancer. Adv. Drug Deliv. Rev. 2005, 57, 579–596. [Google Scholar] [CrossRef]
- Berry, C.C. Intracellular delivery of nanoparticles via the HIV-1 tat peptide. Nanomedicine 2008, 3, 357–365. [Google Scholar] [CrossRef]
- Aqeel Ahmad, J.M.K. pH-sensitive endosomolytic peptides in gene and drug delivery: Endosomal escape and current challenges. J. Drug Deliv. Sci. Technol. 2022, 76, 103786. [Google Scholar] [CrossRef]
- Ahmad, A.; Khan, J.M.; Haque, S. Strategies in the design of endosomolytic agents for facilitating endosomal escape in nanoparticles. Biochimie 2019, 160, 61–75. [Google Scholar] [CrossRef] [PubMed]
- Reissmann, S.; Filatova, M.P. New generation of cell-penetrating peptides: Functionality and potential clinical application. J. Pept. Sci. 2021, 27, e3300. [Google Scholar] [CrossRef]
- Xie, J.; Bi, Y.; Zhang, H.; Dong, S.; Teng, L.; Lee, R.J.; Yang, Z. Cell-Penetrating Peptides in Diagnosis and Treatment of Human Diseases: From Preclinical Research to Clinical Application. Front. Pharmacol. 2020, 11, 697. [Google Scholar] [CrossRef]
- Teo, S.L.Y.; Rennick, J.J.; Yuen, D.; Al-Wassiti, H.; Johnston, A.P.R.; Pouton, C.W. Unravelling cytosolic delivery of cell penetrating peptides with a quantitative endosomal escape assay. Nat. Commun. 2021, 12, 3721. [Google Scholar] [CrossRef] [PubMed]
- LeCher, J.C.; Nowak, S.J.; McMurry, J.L. Breaking in and busting out: Cell-penetrating peptides and the endosomal escape problem. Biomol. Concepts 2017, 8, 131–141. [Google Scholar] [CrossRef]
- El-Andaloussi, S.; Holm, T.; Langel, U. Cell-penetrating peptides: Mechanisms and applications. Curr. Pharm. Des. 2005, 11, 3597–3611. [Google Scholar] [CrossRef] [PubMed]
- Kichler, A.; Mason, A.J.; Bechinger, B. Cationic amphipathic histidine-rich peptides for gene delivery. Biochim. Biophys. Acta 2006, 1758, 301–307. [Google Scholar] [CrossRef]
- Moschos, S.A.; Jones, S.W.; Perry, M.M.; Williams, A.E.; Erjefalt, J.S.; Turner, J.J.; Barnes, P.J.; Sproat, B.S.; Gait, M.J.; Lindsay, M.A. Lung delivery studies using siRNA conjugated to TAT(48-60) and penetratin reveal peptide induced reduction in gene expression and induction of innate immunity. Bioconjug. Chem. 2007, 18, 1450–1459. [Google Scholar] [CrossRef]
- McClorey, G.; Banerjee, S. Cell-Penetrating Peptides to Enhance Delivery of Oligonucleotide-Based Therapeutics. Biomedicines 2018, 6, 51. [Google Scholar] [CrossRef] [PubMed]
- Lam, J.K.; Liang, W.; Lan, Y.; Chaudhuri, P.; Chow, M.Y.; Witt, K.; Kudsiova, L.; Mason, A.J. Effective endogenous gene silencing mediated by pH responsive peptides proceeds via multiple pathways. J. Control. Release 2012, 158, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Nicol, F.; Szoka, F.C., Jr. GALA: A designed synthetic pH-responsive amphipathic peptide with applications in drug and gene delivery. Adv. Drug Deliv. Rev. 2004, 56, 967–985. [Google Scholar] [CrossRef]
- Lundberg, P.; El-Andaloussi, S.; Sutlu, T.; Johansson, H.; Langel, U. Delivery of short interfering RNA using endosomolytic cell-penetrating peptides. FASEB J. 2007, 21, 2664–2671. [Google Scholar] [CrossRef] [PubMed]
- Goormaghtigh, E.; De Meutter, J.; Szoka, F.; Cabiaux, V.; Parente, R.A.; Ruysschaert, J.M. Secondary structure and orientation of the amphipathic peptide GALA in lipid structures. An infrared-spectroscopic approach. Eur. J. Biochem. 1991, 195, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Cesbron, Y.; Shaheen, U.; Free, P.; Levy, R. TAT and HA2 facilitate cellular uptake of gold nanoparticles but do not lead to cytosolic localisation. PLoS ONE 2015, 10, e0121683. [Google Scholar] [CrossRef] [PubMed]
- Santiwarangkool, S.; Akita, H.; Khalil, I.A.; Abd Elwakil, M.M.; Sato, Y.; Kusumoto, K.; Harashima, H. A study of the endocytosis mechanism and transendothelial activity of lung-targeted GALA-modified liposomes. J. Control. Release 2019, 307, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Vives, E.; Brodin, P.; Lebleu, B. A truncated HIV-1 Tat protein basic domain rapidly translocates through the plasma membrane and accumulates in the cell nucleus. J. Biol. Chem. 1997, 272, 16010–16017. [Google Scholar] [CrossRef]
- Rogers, F.A.; Manoharan, M.; Rabinovitch, P.; Ward, D.C.; Glazer, P.M. Peptide conjugates for chromosomal gene targeting by triplex-forming oligonucleotides. Nucleic Acids Res. 2004, 32, 6595–6604. [Google Scholar] [CrossRef]
- Pichon, C.; Freulon, I.; Midoux, P.; Mayer, R.; Monsigny, M.; Roche, A.C. Cytosolic and nuclear delivery of oligonucleotides mediated by an amphiphilic anionic peptide. Antisense Nucleic Acid Drug Dev. 1997, 7, 335–343. [Google Scholar] [CrossRef]
- Mastrobattista, E.; Koning, G.A.; van Bloois, L.; Filipe, A.C.; Jiskoot, W.; Storm, G. Functional characterization of an endosome-disruptive peptide and its application in cytosolic delivery of immunoliposome-entrapped proteins. J. Biol. Chem. 2002, 277, 27135–27143. [Google Scholar] [CrossRef]
- Takayama, K.; Nakase, I.; Michiue, H.; Takeuchi, T.; Tomizawa, K.; Matsui, H.; Futaki, S. Enhanced intracellular delivery using arginine-rich peptides by the addition of penetration accelerating sequences (Pas). J. Control. Release 2009, 138, 128–133. [Google Scholar] [CrossRef] [PubMed]
- Lonn, P.; Kacsinta, A.D.; Cui, X.S.; Hamil, A.S.; Kaulich, M.; Gogoi, K.; Dowdy, S.F. Enhancing Endosomal Escape for Intracellular Delivery of Macromolecular Biologic Therapeutics. Sci. Rep. 2016, 6, 32301. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.; Pellois, J.P. Hydrophobicity is a key determinant in the activity of arginine-rich cell penetrating peptides. Sci. Rep. 2022, 12, 15981. [Google Scholar] [CrossRef]
- Allen, J.K.; Sutherland, T.C.; Prater, A.R.; Geoffroy, C.G.; Pellois, J.P. In vivo peptide-based delivery of a gene-modifying enzyme into cells of the central nervous system. Sci. Adv. 2022, 8, eabo2954. [Google Scholar] [CrossRef]
- Juliano, R.L.; Wang, L.; Tavares, F.; Brown, E.G.; James, L.; Ariyarathna, Y.; Ming, X.; Mao, C.; Suto, M. Structure-activity relationships and cellular mechanism of action of small molecules that enhance the delivery of oligonucleotides. Nucleic Acids Res. 2018, 46, 1601–1613. [Google Scholar] [CrossRef] [PubMed]
- Bost, J.P.; Ojansivu, M.; Munson, M.J.; Wesen, E.; Gallud, A.; Gupta, D.; Gustafsson, O.; Saher, O.; Radler, J.; Higgins, S.G.; et al. Novel endosomolytic compounds enable highly potent delivery of antisense oligonucleotides. Commun. Biol. 2022, 5, 185. [Google Scholar] [CrossRef]
- Du Rietz, H.; Hedlund, H.; Wilhelmson, S.; Nordenfelt, P.; Wittrup, A. Imaging small molecule-induced endosomal escape of siRNA. Nat. Commun. 2020, 11, 1809. [Google Scholar] [CrossRef]
- Allen, J.; Najjar, K.; Erazo-Oliveras, A.; Kondow-McConaghy, H.M.; Brock, D.J.; Graham, K.; Hager, E.C.; Marschall, A.L.J.; Dubel, S.; Juliano, R.L.; et al. Cytosolic Delivery of Macromolecules in Live Human Cells Using the Combined Endosomal Escape Activities of a Small Molecule and Cell Penetrating Peptides. ACS Chem. Biol. 2019, 14, 2641–2651. [Google Scholar] [CrossRef]
- Yu, H.; Zou, Y.; Wang, Y.; Huang, X.; Huang, G.; Sumer, B.D.; Boothman, D.A.; Gao, J. Overcoming endosomal barrier by amphotericin B-loaded dual pH-responsive PDMA-b-PDPA micelleplexes for siRNA delivery. ACS Nano 2011, 5, 9246–9255. [Google Scholar] [CrossRef]
- FDA Approves First-of-Its Kind Targeted RNA-Based Therapy to Treat a Rare Disease. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-first-its-kind-targeted-rna-based-therapy-treat-rare-disease (accessed on 15 June 2023).
- FDA Approves First COVID-19 Vaccine. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-first-covid-19-vaccine (accessed on 15 June 2023).
- Moderna COVID-19 Vaccines. Available online: https://www.fda.gov/vaccines-blood-biologics/coronavirus-covid-19-cber-regulated-biologics/moderna-covid-19-vaccines (accessed on 15 June 2023).
- Leung, A.K.; Hafez, I.M.; Baoukina, S.; Belliveau, N.M.; Zhigaltsev, I.V.; Afshinmanesh, E.; Tieleman, D.P.; Hansen, C.L.; Hope, M.J.; Cullis, P.R. Lipid Nanoparticles Containing siRNA Synthesized by Microfluidic Mixing Exhibit an Electron-Dense Nanostructured Core. J. Phys. Chem. C Nanomater. Interfaces 2012, 116, 18440–18450. [Google Scholar] [CrossRef]
- Kimura, N.; Maeki, M.; Sato, Y.; Note, Y.; Ishida, A.; Tani, H.; Harashima, H.; Tokeshi, M. Development of the iLiNP Device: Fine Tuning the Lipid Nanoparticle Size within 10 nm for Drug Delivery. ACS Omega 2018, 3, 5044–5051. [Google Scholar] [CrossRef]
- Hashiba, K.; Sato, Y.; Taguchi, M.; Sakamoto, S.; Otsu, A.; Maeda, Y.; Shishido, T.; Murakawa, M.; Okazaki, A.; Harashima, H. Branching Ionizable Lipids Can Enhance the Stability, Fusogenicity, and Functional Delivery of mRNA. Small Sci. 2023, 3, 2200071–2200083. [Google Scholar] [CrossRef]
- Paliwal, S.R.; Paliwal, R.; Vyas, S.P. A review of mechanistic insight and application of pH-sensitive liposomes in drug delivery. Drug Deliv. 2015, 22, 231–242. [Google Scholar] [CrossRef]
- Budker, V.; Gurevich, V.; Hagstrom, J.E.; Bortzov, F.; Wolff, J.A. pH-sensitive, cationic liposomes: A new synthetic virus-like vector. Nat. Biotechnol. 1996, 14, 760–764. [Google Scholar] [CrossRef]
- Rayamajhi, S.; Marchitto, J.; Nguyen, T.D.T.; Marasini, R.; Celia, C.; Aryal, S. pH-responsive cationic liposome for endosomal escape mediated drug delivery. Colloids Surf. B Biointerfaces 2020, 188, 110804. [Google Scholar] [CrossRef]
- Koynova, R.; Wang, L.; MacDonald, R.C. An intracellular lamellar-nonlamellar phase transition rationalizes the superior performance of some cationic lipid transfection agents. Proc. Natl. Acad. Sci. USA 2006, 103, 14373–14378. [Google Scholar] [CrossRef]
- Wolff, J.A.; Rozema, D.B. Breaking the bonds: Non-viral vectors become chemically dynamic. Mol. Ther. 2008, 16, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Hatakeyama, H.; Sakurai, Y.; Hyodo, M.; Akita, H.; Harashima, H. A pH-sensitive cationic lipid facilitates the delivery of liposomal siRNA and gene silencing activity in vitro and in vivo. J. Control. Release 2012, 163, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Dirin, M.; Winkler, J. Influence of diverse chemical modifications on the ADME characteristics and toxicology of antisense oligonucleotides. Expert Opin. Biol. Ther. 2013, 13, 875–888. [Google Scholar] [CrossRef]
- Koller, E.; Vincent, T.M.; Chappell, A.; De, S.; Manoharan, M.; Bennett, C.F. Mechanisms of single-stranded phosphorothioate modified antisense oligonucleotide accumulation in hepatocytes. Nucleic Acids Res. 2011, 39, 4795–4807. [Google Scholar] [CrossRef] [PubMed]
- Chappell, A.E.; Gaus, H.J.; Berdeja, A.; Gupta, R.; Jo, M.; Prakash, T.P.; Oestergaard, M.; Swayze, E.E.; Seth, P.P. Mechanisms of palmitic acid-conjugated antisense oligonucleotide distribution in mice. Nucleic Acids Res. 2020, 48, 4382–4395. [Google Scholar] [CrossRef] [PubMed]
- Betts, C.; Saleh, A.F.; Arzumanov, A.A.; Hammond, S.M.; Godfrey, C.; Coursindel, T.; Gait, M.J.; Wood, M.J. Pip6-PMO, A New Generation of Peptide-oligonucleotide Conjugates With Improved Cardiac Exon Skipping Activity for DMD Treatment. Mol. Ther. Nucleic Acids 2012, 1, e38. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.R.; Ming, X.; Fisher, M.; Lackey, J.G.; Rajeev, K.G.; Manoharan, M.; Juliano, R.L. Multivalent cyclic RGD conjugates for targeted delivery of small interfering RNA. Bioconjug. Chem. 2011, 22, 1673–1681. [Google Scholar] [CrossRef] [PubMed]
- Gan, L.; Wu, L.C.L.; Wood, J.A.; Yao, M.; Treleaven, C.M.; Estrella, N.L.; Wentworth, B.M.; Hanson, G.J.; Passini, M.A. A cell-penetrating peptide enhances delivery and efficacy of phosphorodiamidate morpholino oligomers in mdx mice. Mol. Ther. Nucleic Acids 2022, 30, 17–27. [Google Scholar] [CrossRef]
- McNamara, J.O., 2nd; Andrechek, E.R.; Wang, Y.; Viles, K.D.; Rempel, R.E.; Gilboa, E.; Sullenger, B.A.; Giangrande, P.H. Cell type-specific delivery of siRNAs with aptamer-siRNA chimeras. Nat. Biotechnol. 2006, 24, 1005–1015. [Google Scholar] [CrossRef]
- Dovgan, I.; Koniev, O.; Kolodych, S.; Wagner, A. Antibody-Oligonucleotide Conjugates as Therapeutic, Imaging, and Detection Agents. Bioconjug. Chem. 2019, 30, 2483–2501. [Google Scholar] [CrossRef]
- Matsuda, S.; Keiser, K.; Nair, J.K.; Charisse, K.; Manoharan, R.M.; Kretschmer, P.; Peng, C.G.; Kel’in, A.V.; Kandasamy, P.; Willoughby, J.L.; et al. siRNA conjugates carrying sequentially assembled trivalent N-acetylgalactosamine linked through nucleosides elicit robust gene silencing in vivo in hepatocytes. ACS Chem. Biol. 2015, 10, 1181–1187. [Google Scholar] [CrossRef]
- Nair, J.K.; Willoughby, J.L.; Chan, A.; Charisse, K.; Alam, M.R.; Wang, Q.; Hoekstra, M.; Kandasamy, P.; Kel’in, A.V.; Milstein, S.; et al. Multivalent N-acetylgalactosamine-conjugated siRNA localizes in hepatocytes and elicits robust RNAi-mediated gene silencing. J. Am. Chem. Soc. 2014, 136, 16958–16961. [Google Scholar] [CrossRef]
- Huang, Y. Preclinical and Clinical Advances of GalNAc-Decorated Nucleic Acid Therapeutics. Mol. Ther. Nucleic Acids 2017, 6, 116–132. [Google Scholar] [CrossRef]
- Hammond, S.M.; Hazell, G.; Shabanpoor, F.; Saleh, A.F.; Bowerman, M.; Sleigh, J.N.; Meijboom, K.E.; Zhou, H.; Muntoni, F.; Talbot, K.; et al. Systemic peptide-mediated oligonucleotide therapy improves long-term survival in spinal muscular atrophy. Proc. Natl. Acad. Sci. USA 2016, 113, 10962–10967. [Google Scholar] [CrossRef]
- Bien-Ly, N.; Yu, Y.J.; Bumbaca, D.; Elstrott, J.; Boswell, C.A.; Zhang, Y.; Luk, W.; Lu, Y.; Dennis, M.S.; Weimer, R.M.; et al. Transferrin receptor (TfR) trafficking determines brain uptake of TfR antibody affinity variants. J. Exp. Med. 2014, 211, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Wiley, D.T.; Webster, P.; Gale, A.; Davis, M.E. Transcytosis and brain uptake of transferrin-containing nanoparticles by tuning avidity to transferrin receptor. Proc. Natl. Acad. Sci. USA 2013, 110, 8662–8667. [Google Scholar] [CrossRef]
- Winter, G.E.; Buckley, D.L.; Paulk, J.; Roberts, J.M.; Souza, A.; Dhe-Paganon, S.; Bradner, J.E. DRUG DEVELOPMENT. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 2015, 348, 1376–1381. [Google Scholar] [CrossRef] [PubMed]
- Guillier, F.; Orain, D.; Bradley, M. Linkers and cleavage strategies in solid-phase organic synthesis and combinatorial chemistry. Chem. Rev. 2000, 100, 3859. [Google Scholar] [CrossRef]
- Kostova, V.; Désos, P.; Starck, J.-B.; Kotschy, A. The Chemistry Behind ADCs. Pharmaceuticals 2021, 14, 442. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Xiao, D.; Xie, F.; Liu, L.; Wang, Y.; Fan, S.; Zhou, X.; Li, S. Antibody-drug conjugates: Recent advances in linker chemistry. Acta Pharm. Sin. B 2021, 11, 3889–3907. [Google Scholar] [CrossRef]
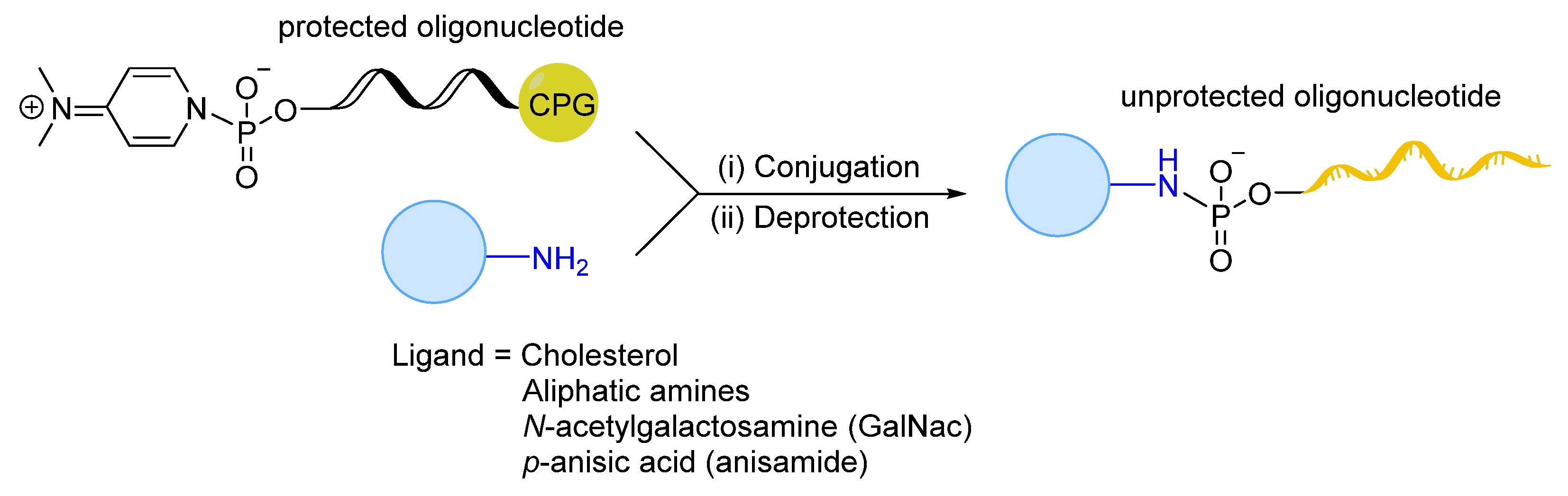
- Singh, Y.; Murat, P.; Defrancq, E. Recent developments in oligonucleotide conjugation. Chem. Soc. Rev. 2010, 39, 2054–2070. [Google Scholar] [CrossRef] [PubMed]
- Patil, N.A. Conjugation Approaches for Peptide-Mediated Delivery of Oligonucleotides Therapeutics. Aust. J. Chem. 2021, 75, 24–33. [Google Scholar] [CrossRef]
- Soutschek, J.; Akinc, A.; Bramlage, B.; Charisse, K.; Constien, R.; Donoghue, M.; Elbashir, S.; Geick, A.; Hadwiger, P.; Harborth, J.; et al. Therapeutic silencing of an endogenous gene by systemic administration of modified siRNAs. Nature 2004, 432, 173–178. [Google Scholar] [CrossRef]
- Lorenz, C.; Hadwiger, P.; John, M.; Vornlocher, H.P.; Unverzagt, C. Steroid and lipid conjugates of siRNAs to enhance cellular uptake and gene silencing in liver cells. Bioorg. Med. Chem. Lett. 2004, 14, 4975–4977. [Google Scholar] [CrossRef]
- Li, G.; Moellering, R.E. A Concise, Modular Antibody-Oligonucleotide Conjugation Strategy Based on Disuccinimidyl Ester Activation Chemistry. ChemBioChem 2019, 20, 1599–1605. [Google Scholar] [CrossRef] [PubMed]
- Winkler, J. Oligonucleotide conjugates for therapeutic applications. Ther. Deliv. 2013, 4, 791–809. [Google Scholar] [CrossRef]
- El-Sagheer, A.H.; Brown, T. Click chemistry with DNA. Chem. Soc. Rev. 2010, 39, 1388–1405. [Google Scholar] [CrossRef] [PubMed]
- Willibald, J.; Harder, J.; Sparrer, K.; Conzelmann, K.K.; Carell, T. Click-modified anandamide siRNA enables delivery and gene silencing in neuronal and immune cells. J. Am. Chem. Soc. 2012, 134, 12330–12333. [Google Scholar] [CrossRef] [PubMed]
- Dieterich, D.C.; Hodas, J.J.; Gouzer, G.; Shadrin, I.Y.; Ngo, J.T.; Triller, A.; Tirrell, D.A.; Schuman, E.M. In situ visualization and dynamics of newly synthesized proteins in rat hippocampal neurons. Nat. Neurosci. 2010, 13, 897–905. [Google Scholar] [CrossRef]
- Satake, N.; Duong, C.; Yoshida, S.; Oestergaard, M.; Chen, C.; Peralta, R.; Guo, S.; Seth, P.P.; Li, Y.; Beckett, L.; et al. Novel Targeted Therapy for Precursor B Cell Acute Lymphoblastic Leukemia: Anti-CD22 Antibody-MXD3 Antisense Oligonucleotide Conjugate. Mol. Med. 2016, 22, 632–642. [Google Scholar] [CrossRef]
- Arnold, A.E.; Malek-Adamian, E.; Le, P.U.; Meng, A.; Martinez-Montero, S.; Petrecca, K.; Damha, M.J.; Shoichet, M.S. Antibody-Antisense Oligonucleotide Conjugate Downregulates a Key Gene in Glioblastoma Stem Cells. Mol. Ther. Nucleic Acids 2018, 11, 518–527. [Google Scholar] [CrossRef]
- Gong, H.; Holcomb, I.; Ooi, A.; Wang, X.; Majonis, D.; Unger, M.A.; Ramakrishnan, R. Simple Method To Prepare Oligonucleotide-Conjugated Antibodies and Its Application in Multiplex Protein Detection in Single Cells. Bioconjug. Chem. 2016, 27, 217–225. [Google Scholar] [CrossRef]
- Shen, L.; Wu, Y.; Xie, W.; Chen, G.; Xing, H. Chemical Biology Approaches toward Precise Structure Control of IgG-Based Antibody-Oligonucleotide Conjugates. ChemBioChem 2023, 24, e202300077. [Google Scholar] [CrossRef]
- Maerle, A.V.; Simonova, M.A.; Pivovarov, V.D.; Voronina, D.V.; Drobyazina, P.E.; Trofimov, D.Y.; Alekseev, L.P.; Zavriev, S.K.; Ryazantsev, D.Y. Development of the covalent antibody-DNA conjugates technology for detection of IgE and IgM antibodies by immuno-PCR. PLoS ONE 2019, 14, e0209860. [Google Scholar] [CrossRef]
- Zieba, A.; Ponten, F.; Uhlen, M.; Landegren, U. In situ protein detection with enhanced specificity using DNA-conjugated antibodies and proximity ligation. Mod. Pathol. 2018, 31, 253–263. [Google Scholar] [CrossRef]
- Kuo, C.; Nikan, M.; Yeh, S.T.; Chappell, A.E.; Tanowitz, M.; Seth, P.P.; Prakash, T.P.; Mullick, A.E. Targeted Delivery of Antisense Oligonucleotides Through Angiotensin Type 1 Receptor. Nucleic Acid Ther. 2022, 32, 300–311. [Google Scholar] [CrossRef]
- Dugal-Tessier, J.; Thirumalairajan, S.; Jain, N. Antibody-Oligonucleotide Conjugates: A Twist to Antibody-Drug Conjugates. J. Clin. Med. 2021, 10, 838. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Li, R.; Pei, X.; Chai, M.; Sun, L.; Huang, Y.; Wang, J.; Barth, S.; Yu, F.; He, H. Antibody–siRNA conjugates (ARC): Emerging siRNA drug formulation. Med. Drug Discov. 2022, 15, 100128. [Google Scholar] [CrossRef]
- Dastpeyman, M.; Sharifi, R.; Amin, A.; Karas, J.A.; Cuic, B.; Pan, Y.; Nicolazzo, J.A.; Turner, B.J.; Shabanpoor, F. Endosomal escape cell-penetrating peptides significantly enhance pharmacological effectiveness and CNS activity of systemically administered antisense oligonucleotides. Int. J. Pharm. 2021, 599, 120398. [Google Scholar] [CrossRef] [PubMed]
- Dastpeyman, M.; Karas, J.A.; Amin, A.; Turner, B.J.; Shabanpoor, F. Modular Synthesis of Trifunctional Peptide-oligonucleotide Conjugates via Native Chemical Ligation. Front. Chem. 2021, 9, 627329. [Google Scholar] [CrossRef]
- Baldwin, A.D.; Kiick, K.L. Tunable degradation of maleimide-thiol adducts in reducing environments. Bioconjug. Chem. 2011, 22, 1946–1953. [Google Scholar] [CrossRef]
- Patil, N.A.; Karas, J.A.; Turner, B.J.; Shabanpoor, F. Thiol-Cyanobenzothiazole Ligation for the Efficient Preparation of Peptide-PNA Conjugates. Bioconjug. Chem. 2019, 30, 793–799. [Google Scholar] [CrossRef]
- Wu, H.; LeValley, P.J.; Luo, T.; Kloxin, A.M.; Kiick, K.L. Manipulation of Glutathione-Mediated Degradation of Thiol-Maleimide Conjugates. Bioconjug. Chem. 2018, 29, 3595–3605. [Google Scholar] [CrossRef]
- Dovgan, I.; Kolodych, S.; Koniev, O.; Wagner, A. 2-(Maleimidomethyl)-1,3-Dioxanes (MD): A Serum-Stable Self-hydrolysable Hydrophilic Alternative to Classical Maleimide Conjugation. Sci. Rep. 2016, 6, 30835. [Google Scholar] [CrossRef]
- Sugo, T.; Terada, M.; Oikawa, T.; Miyata, K.; Nishimura, S.; Kenjo, E.; Ogasawara-Shimizu, M.; Makita, Y.; Imaichi, S.; Murata, S.; et al. Development of antibody-siRNA conjugate targeted to cardiac and skeletal muscles. J. Control. Release 2016, 237, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Cuellar, T.L.; Barnes, D.; Nelson, C.; Tanguay, J.; Yu, S.F.; Wen, X.; Scales, S.J.; Gesch, J.; Davis, D.; van Brabant Smith, A.; et al. Systematic evaluation of antibody-mediated siRNA delivery using an industrial platform of THIOMAB-siRNA conjugates. Nucleic Acids Res. 2015, 43, 1189–1203. [Google Scholar] [CrossRef]
- Saito, G.; Swanson, J.A.; Lee, K.D. Drug delivery strategy utilizing conjugation via reversible disulfide linkages: Role and site of cellular reducing activities. Adv. Drug Deliv. Rev. 2003, 55, 199–215. [Google Scholar] [CrossRef] [PubMed]
- Uckun, F.M.; Qazi, S.; Dibirdik, I.; Myers, D.E. Rational design of an immunoconjugate for selective knock-down of leukemia-specific E2A-PBX1 fusion gene expression in human Pre-B leukemia. Integr. Biol. 2013, 5, 122–132. [Google Scholar] [CrossRef] [PubMed]
- Pontarelli, A.; Liu, J.T.; Movasat, H.; Menard, S.; Oh, J.K.; Wilds, C.J. Synthesis of a Convertible Linker Containing a Disulfide Group for Oligonucleotide Functionalization. Org. Lett. 2022, 24, 5579–5583. [Google Scholar] [CrossRef]
- Zatsepin, T.S.; Turner, J.J.; Oretskaya, T.S.; Gait, M.J. Conjugates of oligonucleotides and analogues with cell penetrating peptides as gene silencing agents. Curr. Pharm. Des. 2005, 11, 3639–3654. [Google Scholar] [CrossRef]
- Klabenkova, K.; Fokina, A.; Stetsenko, D. Chemistry of Peptide-Oligonucleotide Conjugates: A Review. Molecules 2021, 26, 5420. [Google Scholar] [CrossRef]
- Shu, Z.; Tanaka, I.; Ota, A.; Fushihara, D.; Abe, N.; Kawaguchi, S.; Nakamoto, K.; Tomoike, F.; Tada, S.; Ito, Y.; et al. Disulfide-Unit Conjugation Enables Ultrafast Cytosolic Internalization of Antisense DNA and siRNA. Angew. Chem. Int. Ed. Engl. 2019, 58, 6611–6615. [Google Scholar] [CrossRef]
- Hiraoka, H.; Shu, Z.; Tri Le, B.; Masuda, K.; Nakamoto, K.; Fangjie, L.; Abe, N.; Hashiya, F.; Kimura, Y.; Shimizu, Y.; et al. Antisense Oligonucleotide Modified with Disulfide Units Induces Efficient Exon Skipping in mdx Myotubes through Enhanced Membrane Permeability and Nucleus Internalization. ChemBioChem 2021, 22, 3437–3442. [Google Scholar] [CrossRef]
- Danial, M.; Postma, A. Disulfide conjugation chemistry: A mixed blessing for therapeutic drug delivery? Ther. Deliv. 2017, 8, 359–362. [Google Scholar] [CrossRef]
- Stasinska, A.R.; Putaj, P.; Chmielewski, M.K. Disulfide bridge as a linker in nucleic acids’ bioconjugation. Part II: A summary of practical applications. Bioorg. Chem. 2020, 95, 103518. [Google Scholar] [CrossRef] [PubMed]
- Stasinska, A.R.; Putaj, P.; Chmielewski, M.K. Disulfide bridge as a linker in nucleic acids’ bioconjugation. Part I: An overview of synthetic strategies. Bioorg. Chem. 2019, 92, 103223. [Google Scholar] [CrossRef]
- Bargh, J.D.; Isidro-Llobet, A.; Parker, J.S.; Spring, D.R. Cleavable linkers in antibody-drug conjugates. Chem. Soc. Rev. 2019, 48, 4361–4374. [Google Scholar] [CrossRef] [PubMed]
- Casey, J.R.; Grinstein, S.; Orlowski, J. Sensors and regulators of intracellular pH. Nat. Rev. Mol. Cell Biol. 2010, 11, 50–61. [Google Scholar] [CrossRef]
- Othus, M.; Appelbaum, F.R.; Petersdorf, S.H.; Kopecky, K.J.; Slovak, M.; Nevill, T.; Brandwein, J.; Larson, R.A.; Stiff, P.J.; Walter, R.B.; et al. Fate of patients with newly diagnosed acute myeloid leukemia who fail primary induction therapy. Biol. Blood Marrow Transplant. 2015, 21, 559–564. [Google Scholar] [CrossRef]
- Lamb, Y.N. Inotuzumab Ozogamicin: First Global Approval. Drugs 2017, 77, 1603–1610. [Google Scholar] [CrossRef]
- Hamann, P.R.; Hinman, L.M.; Hollander, I.; Beyer, C.F.; Lindh, D.; Holcomb, R.; Hallett, W.; Tsou, H.R.; Upeslacis, J.; Shochat, D.; et al. Gemtuzumab ozogamicin, a potent and selective anti-CD33 antibody-calicheamicin conjugate for treatment of acute myeloid leukemia. Bioconjug. Chem. 2002, 13, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Holz, E.; Darwish, M.; Tesar, D.B.; Shatz-Binder, W. A Review of Protein- and Peptide-Based Chemical Conjugates: Past, Present, and Future. Pharmaceutics 2023, 15, 600. [Google Scholar] [CrossRef]
- Santi, D.V.; Cabel, L.; Bidard, F.C. Does sacituzumab-govitecan act as a conventional antibody drug conjugate (ADC), a prodrug of SN-38 or both? Ann. Transl. Med. 2021, 9, 1113. [Google Scholar] [CrossRef]
- Govindan, S.V.; Cardillo, T.M.; Sharkey, R.M.; Tat, F.; Gold, D.V.; Goldenberg, D.M. Milatuzumab-SN-38 conjugates for the treatment of CD74+ cancers. Mol. Cancer Ther. 2013, 12, 968–978. [Google Scholar] [CrossRef]
- Wang, Y.; Fan, S.; Xiao, D.; Xie, F.; Li, W.; Zhong, W.; Zhou, X. Novel Silyl Ether-Based Acid-Cleavable Antibody-MMAE Conjugates with Appropriate Stability and Efficacy. Cancers 2019, 11, 957. [Google Scholar] [CrossRef]
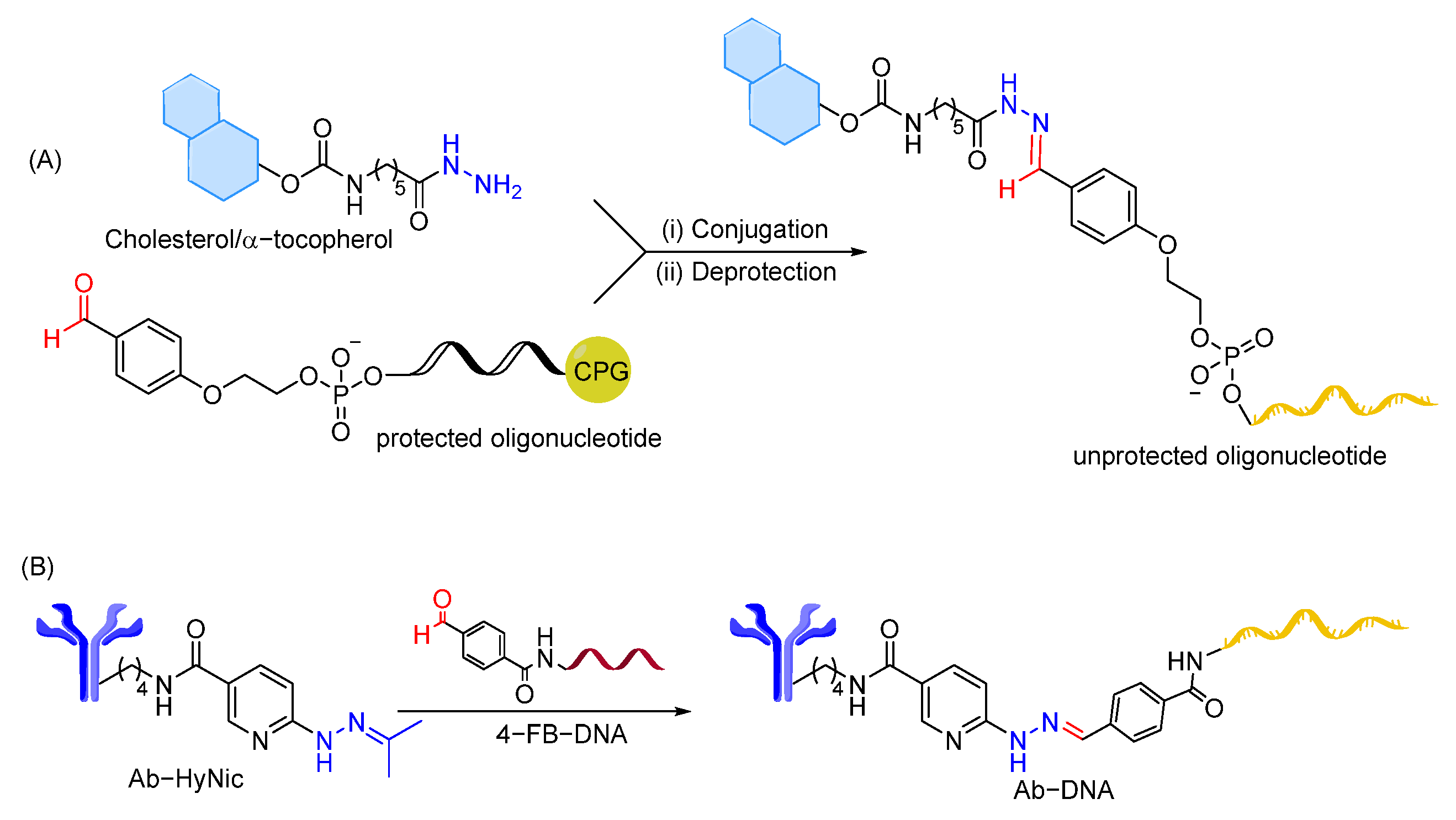
- Meschaninova, M.I.; Entelis, N.S.; Chernolovskaya, E.L.; Venyaminova, A.G. A Versatile Solid-Phase Approach to the Synthesis of Oligonucleotide Conjugates with Biodegradable Hydrazone Linker. Molecules 2021, 26, 2119. [Google Scholar] [CrossRef]
- Kozlov, I.A.; Melnyk, P.C.; Stromsborg, K.E.; Chee, M.S.; Barker, D.L.; Zhao, C. Efficient strategies for the conjugation of oligonucleotides to antibodies enabling highly sensitive protein detection. Biopolymers 2004, 73, 621–630. [Google Scholar] [CrossRef]
- Ollivier, N.; Olivier, C.; Gouyette, C.; Huynh-Dinh, T.; Gras-Masse, H.; Melnyk, O. Synthesis of oligonucleotide–peptide conjugates using hydrazone chemical ligation. Tetrahedron Lett. 2002, 43, 997–999. [Google Scholar] [CrossRef]
- Leriche, G.; Nothisen, M.; Baumlin, N.; Muller, C.D.; Bagnard, D.; Remy, J.S.; Jacques, S.A.; Wagner, A. Spiro Diorthoester (SpiDo), a Human Plasma Stable Acid-Sensitive Cleavable Linker for Lysosomal Release. Bioconjug. Chem. 2015, 26, 1461–1465. [Google Scholar] [CrossRef]
- Jacques, S.A.; Leriche, G.; Mosser, M.; Nothisen, M.; Muller, C.D.; Remy, J.S.; Wagner, A. From solution to in-cell study of the chemical reactivity of acid sensitive functional groups: A rational approach towards improved cleavable linkers for biospecific endosomal release. Org. Biomol. Chem. 2016, 14, 4794–4803. [Google Scholar] [CrossRef]
- Tobaldi, E.; Dovgan, I.; Mosser, M.; Becht, J.M.; Wagner, A. Structural investigation of cyclo-dioxo maleimide cross-linkers for acid and serum stability. Org. Biomol. Chem. 2017, 15, 9305–9310. [Google Scholar] [CrossRef]
- Rady, T.; Turelli, L.; Nothisen, M.; Tobaldi, E.; Erb, S.; Thoreau, F.; Hernandez-Alba, O.; Cianferani, S.; Daubeuf, F.; Wagner, A.; et al. A Novel Family of Acid-Cleavable Linker Based on Cyclic Acetal Motifs for the Production of Antibody-Drug Conjugates with High Potency and Selectivity. Bioconjug. Chem. 2022, 33, 1860–1866. [Google Scholar] [CrossRef] [PubMed]
- Dal Corso, A.; Pignataro, L.; Belvisi, L.; Gennari, C. Innovative Linker Strategies for Tumor-Targeted Drug Conjugates. Chemistry 2019, 25, 14740–14757. [Google Scholar] [CrossRef] [PubMed]
- Cal, P.M.; Vicente, J.B.; Pires, E.; Coelho, A.V.; Veiros, L.F.; Cordeiro, C.; Gois, P.M. Iminoboronates: A new strategy for reversible protein modification. J. Am. Chem. Soc. 2012, 134, 10299–10305. [Google Scholar] [CrossRef] [PubMed]
- Dilek, O.; Lei, Z.; Mukherjee, K.; Bane, S. Rapid formation of a stable boron-nitrogen heterocycle in dilute, neutral aqueous solution for bioorthogonal coupling reactions. Chem. Commun. 2015, 51, 16992–16995. [Google Scholar] [CrossRef]
- Antonio, J.P.M.; Russo, R.; Carvalho, C.P.; Cal, P.; Gois, P.M.P. Boronic acids as building blocks for the construction of therapeutically useful bioconjugates. Chem. Soc. Rev. 2019, 48, 3513–3536. [Google Scholar] [CrossRef] [PubMed]
- Gu, H.; Chio, T.I.; Lei, Z.; Staples, R.J.; Hirschi, J.S.; Bane, S. Formation of hydrazones and stabilized boron-nitrogen heterocycles in aqueous solution from carbohydrazides and ortho-formylphenylboronic acids. Org. Biomol. Chem. 2017, 15, 7543–7548. [Google Scholar] [CrossRef] [PubMed]
- Chio, T.I.; Gu, H.; Mukherjee, K.; Tumey, L.N.; Bane, S.L. Site-Specific Bioconjugation and Multi-Bioorthogonal Labeling via Rapid Formation of a Boron-Nitrogen Heterocycle. Bioconjug. Chem. 2019, 30, 1554–1564. [Google Scholar] [CrossRef]
- Gu, H.; Ghosh, S.; Staples, R.J.; Bane, S.L. beta-Hydroxy-Stabilized Boron-Nitrogen Heterocycles Enable Rapid and Efficient C-Terminal Protein Modification. Bioconjug. Chem. 2019, 30, 2604–2613. [Google Scholar] [CrossRef]
- Roy, J.; Kaake, M.; Srinivasarao, M.; Low, P.S. Targeted Tubulysin B Hydrazide Conjugate for the Treatment of Luteinizing Hormone-Releasing Hormone Receptor-Positive Cancers. Bioconjug. Chem. 2018, 29, 2208–2214. [Google Scholar] [CrossRef]
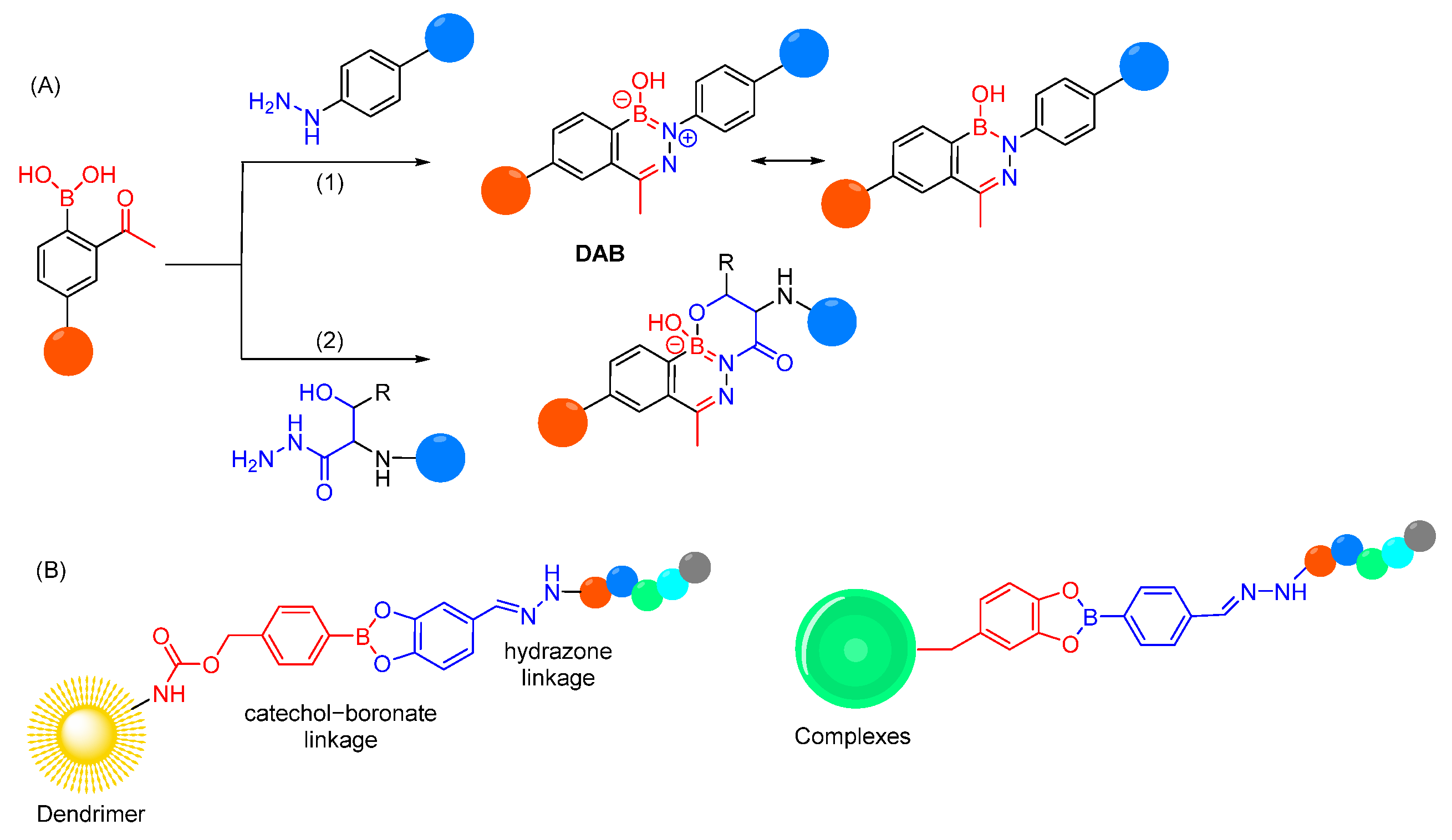
- Rong, G.; Chen, L.; Zhu, F.; Tan, E.; Cheng, Y. Polycatechols with Robust Efficiency in Cytosolic Peptide Delivery via Catechol-Boronate Chemistry. Nano Lett. 2022, 22, 6245–6253. [Google Scholar] [CrossRef]
- Gao, X.; Yuan, C.; Tan, E.; Li, Z.; Cheng, Y.; Xiao, J.; Rong, G. Dual-responsive bioconjugates bearing a bifunctional adaptor for robust cytosolic peptide delivery. J. Control. Release 2023, 355, 675–684. [Google Scholar] [CrossRef]
- Lee, Y.; Ishii, T.; Cabral, H.; Kim, H.J.; Seo, J.H.; Nishiyama, N.; Oshima, H.; Osada, K.; Kataoka, K. Charge-conversional polyionic complex micelles-efficient nanocarriers for protein delivery into cytoplasm. Angew. Chem. Int. Ed. Engl. 2009, 48, 5309–5312. [Google Scholar] [CrossRef]
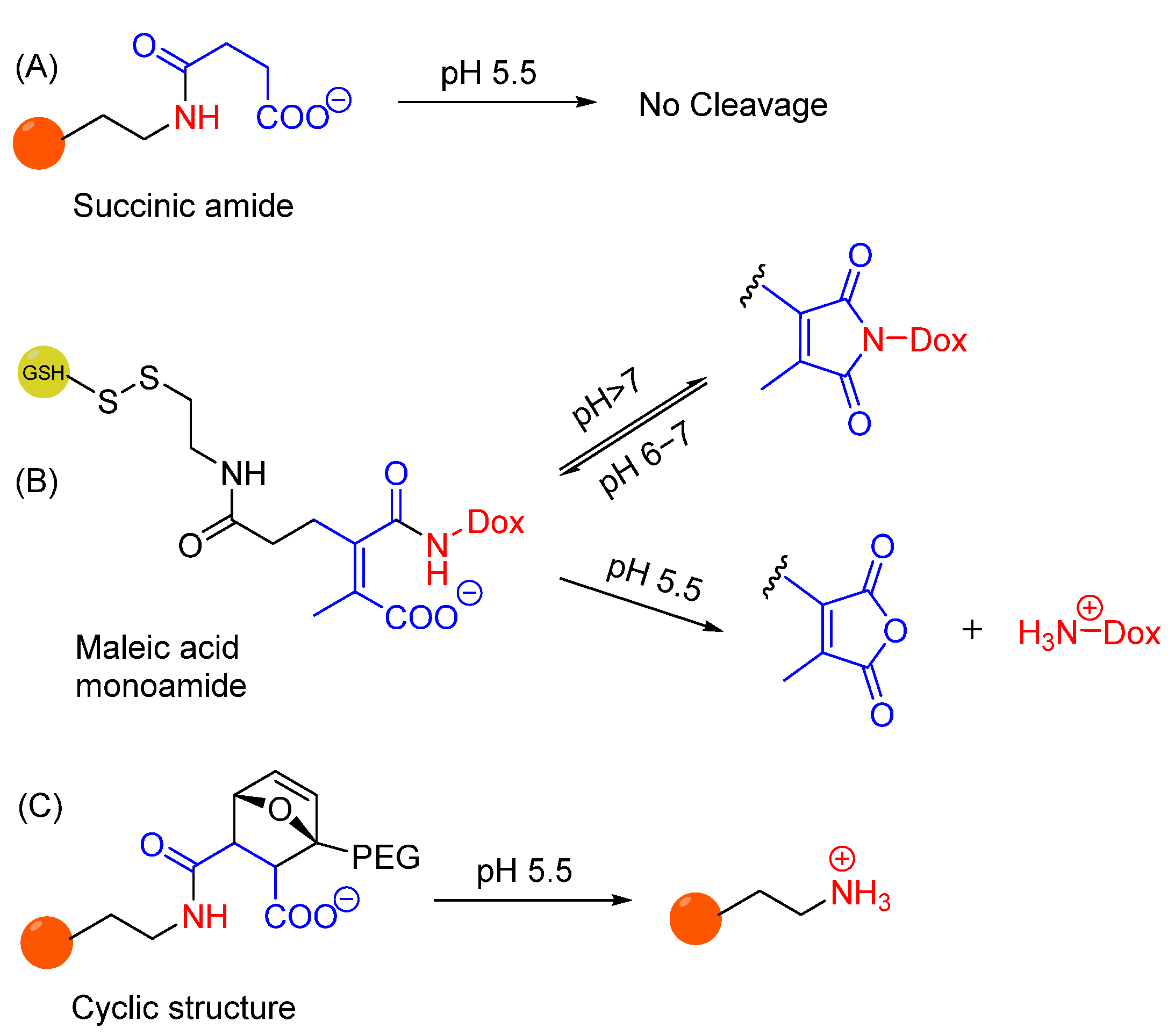
- Zhang, A.; Yao, L.; An, M. Reversing the undesirable pH-profile of doxorubicin via activation of a di-substituted maleamic acid prodrug at tumor acidity. Chem. Commun. 2017, 53, 12826–12829. [Google Scholar] [CrossRef]
- Melodia, D.; Di Pietro, Z.; Cao, C.; Stenzel, M.H.; Chapman, R. Traceless pH-Sensitive Antibody Conjugation Inspired by Citraconic Anhydride. Biomacromolecules 2022, 23, 5322–5329. [Google Scholar] [CrossRef] [PubMed]
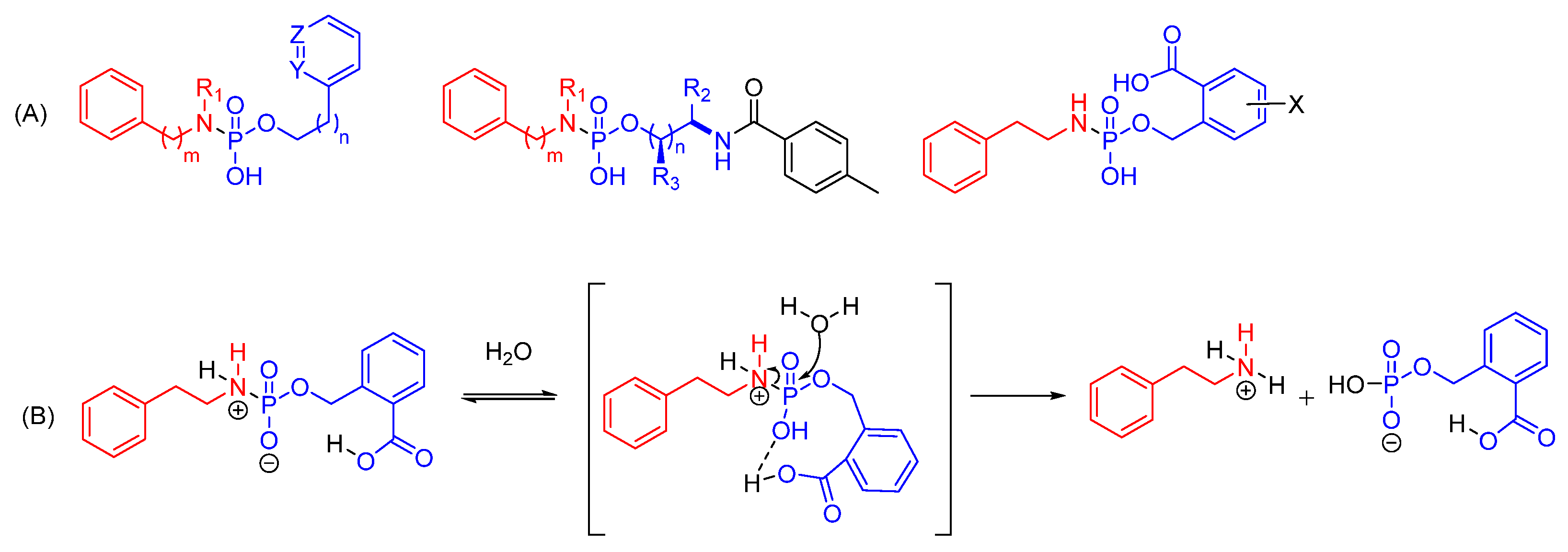
- Choy, C.J.; Geruntho, J.J.; Davis, A.L.; Berkman, C.E. Tunable pH-Sensitive Linker for Controlled Release. Bioconjug. Chem. 2016, 27, 824–830. [Google Scholar] [CrossRef]
- Choy, C.J.; Ley, C.R.; Davis, A.L.; Backer, B.S.; Geruntho, J.J.; Clowers, B.H.; Berkman, C.E. Second-Generation Tunable pH-Sensitive Phosphoramidate-Based Linkers for Controlled Release. Bioconjug. Chem. 2016, 27, 2206–2213. [Google Scholar] [CrossRef]
- Backer, B.S.; Choy, C.J.; Davis, A.L.; Browne, Z.S.; Berkman, C.E. Tunable pH-Sensitive 2-Carboxybenzyl Phosphoramidate Cleavable Linkers. Tetrahedron Lett. 2020, 61, 151650. [Google Scholar] [CrossRef] [PubMed]
- Kropacheva, N.O.; Golyshkin, A.A.; Vorobyeva, M.A.; Meschaninova, M.I. Convenient Solid-Phase Attachment of Small-Molecule Ligands to Oligonucleotides via a Biodegradable Acid-Labile P-N-Bond. Molecules 2023, 28, 1904. [Google Scholar] [CrossRef] [PubMed]
- Migawa, M.T.; Shen, W.; Wan, W.B.; Vasquez, G.; Oestergaard, M.E.; Low, A.; De Hoyos, C.L.; Gupta, R.; Murray, S.; Tanowitz, M.; et al. Site-specific replacement of phosphorothioate with alkyl phosphonate linkages enhances the therapeutic profile of gapmer ASOs by modulating interactions with cellular proteins. Nucleic Acids Res. 2019, 47, 5465–5479. [Google Scholar] [CrossRef]
- Patutina, O.A.; Gaponova Miroshnichenko, S.K.; Sen’kova, A.V.; Savin, I.A.; Gladkikh, D.V.; Burakova, E.A.; Fokina, A.A.; Maslov, M.A.; Shmendel, E.V.; Wood, M.J.A.; et al. Mesyl phosphoramidate backbone modified antisense oligonucleotides targeting miR-21 with enhanced in vivo therapeutic potency. Proc. Natl. Acad. Sci. USA 2020, 117, 32370–32379. [Google Scholar] [CrossRef]
- Kandasamy, P.; Liu, Y.; Aduda, V.; Akare, S.; Alam, R.; Andreucci, A.; Boulay, D.; Bowman, K.; Byrne, M.; Cannon, M.; et al. Impact of guanidine-containing backbone linkages on stereopure antisense oligonucleotides in the CNS. Nucleic Acids Res. 2022, 50, 5401–5423. [Google Scholar] [CrossRef]
- Baker, Y.R.; Thorpe, C.; Chen, J.; Poller, L.M.; Cox, L.; Kumar, P.; Lim, W.F.; Lie, L.; McClorey, G.; Epple, S.; et al. An LNA-amide modification that enhances the cell uptake and activity of phosphorothioate exon-skipping oligonucleotides. Nat. Commun. 2022, 13, 4036. [Google Scholar] [CrossRef] [PubMed]
- Dysko, A.; Baker, Y.R.; McClorey, G.; Wood, M.J.A.; Fenner, S.; Williams, G.; El-Sagheer, A.; Brown, T. Covalently attached intercalators restore duplex stability and splice-switching activity to triazole-modified oligonucleotides. RSC Chem. Biol. 2022, 3, 765–772. [Google Scholar] [CrossRef]
- Morita, K.; Hasegawa, C.; Kaneko, M.; Tsutsumi, S.; Sone, J.; Ishikawa, T.; Imanishi, T.; Koizumi, M. 2’-O,4’-C-ethylene-bridged nucleic acids (ENA): Highly nuclease-resistant and thermodynamically stable oligonucleotides for antisense drug. Bioorg. Med. Chem. Lett. 2002, 12, 73–76. [Google Scholar] [CrossRef] [PubMed]
- Koizumi, M. 2’-O,4’-C-ethylene-bridged nucleic acids (ENA) as next-generation antisense and antigene agents. Biol. Pharm. Bull. 2004, 27, 453–456. [Google Scholar] [CrossRef]
- Koizumi, M. ENA oligonucleotides as therapeutics. Curr. Opin. Mol. Ther. 2006, 8, 144–149. [Google Scholar]
- Schlegel, M.K.; Janas, M.M.; Jiang, Y.; Barry, J.D.; Davis, W.; Agarwal, S.; Berman, D.; Brown, C.R.; Castoreno, A.; LeBlanc, S.; et al. From bench to bedside: Improving the clinical safety of GalNAc-siRNA conjugates using seed-pairing destabilization. Nucleic Acids Res. 2022, 50, 6656–6670. [Google Scholar] [CrossRef]
- Haraszti, R.A.; Roux, L.; Coles, A.H.; Turanov, A.A.; Alterman, J.F.; Echeverria, D.; Godinho, B.; Aronin, N.; Khvorova, A. 5′-Vinylphosphonate improves tissue accumulation and efficacy of conjugated siRNAs in vivo. Nucleic Acids Res. 2017, 45, 7581–7592. [Google Scholar] [CrossRef]
- Wang, S.; Allen, N.; Prakash, T.P.; Liang, X.H.; Crooke, S.T. Lipid Conjugates Enhance Endosomal Release of Antisense Oligonucleotides Into Cells. Nucleic Acid Ther. 2019, 29, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Guidry, E.N.; Farand, J.; Soheili, A.; Parish, C.A.; Kevin, N.J.; Pipik, B.; Calati, K.B.; Ikemoto, N.; Waldman, J.H.; Latham, A.H.; et al. Improving the in vivo therapeutic index of siRNA polymer conjugates through increasing pH responsiveness. Bioconjug. Chem. 2014, 25, 296–307. [Google Scholar] [CrossRef]
- Rozema, D.B.; Lewis, D.L.; Wakefield, D.H.; Wong, S.C.; Klein, J.J.; Roesch, P.L.; Bertin, S.L.; Reppen, T.W.; Chu, Q.; Blokhin, A.V.; et al. Dynamic PolyConjugates for targeted in vivo delivery of siRNA to hepatocytes. Proc. Natl. Acad. Sci. USA 2007, 104, 12982–12987. [Google Scholar] [CrossRef] [PubMed]























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chemical Modification | Drug Name | Oligonucleotide Class | Indication | Advantages and Disadvantages of Modification |
|---|---|---|---|---|
| Approved Oligonucleotide Therapeutics | ||||
| PS (DNA) | VITRAVENE® (fomivirsen) | ASO | CMV retinitis (1998 withdrawn) | PS backbone modification:
|
| 2′-OMe/2′-F, PS | GIVLAARI® (givosiran) | siRNA | Acute hepatic porphyria (2019) | 2′-sugar modification:
|
| OXLUMO® (lumasiran) | siRNA | Primary hyperoxluria (2020) | ||
| LEQVIO® (inclisiran) | siRNA | Heterozygous familial hypercholesterolemia (HeFH) or clinical atherosclerotic cardiovascular disease (ASCVD) (2020, EMA), (2021, FDA) | ||
| AMVUTTRA® (vuttrisiran) | siRNA | Hereditary ATTR (hATTR) (2022) | ||
| 2′-F/2′-OMe | MACUGEN® (pegaptanib) | Aptamer | Neovascular AMD (2004, withdrawn) | |
| 2′-OMe | ONPATTRO® (patisiran) | siRNA | Hereditary ATTR (hATTR) (2018) | |
| 5′-Me-C, PS, 2′-O-MOE, | KYNAMRO® (mipomersen) | ASO | Homozygous familial hypercholesterolemia (HoFH) (2013 withdrawn) | 5′-Me-C nucleobase modification:
|
| SPINRAZA® (nusinersen) | SSO | Spinal muscular atrophy (SMA) (2016) | ||
| WAYLIVRA® (volanesorsen) | ASO | Familial chlylomicronemia syndrome (FCS) (2019) | ||
| TEGSEDI® (inotersen) | ASO | Hereditary ATTR (hATTR) (2018) | ||
| QALSODY® (Tofersen) | ASO | Amyotrophic lateral sclerosis (ALS) (2023) | ||
| PMO | EXONDYS 51® (eteplirsen) | SSO | Duchene muscular dystrophy (DMD) (2016) | PMO scaffold modification:
|
| VYONDYS 53® (golodirsen) | SSO | Duchene muscular dystrophy (DMD) (2019) | ||
| VILTEPSO® (viltolarsen) | SSO | Duchene muscular dystrophy (DMD) (2020) | ||
| AMONDYS 45® (casimersen) | SSO | Duchene muscular dystrophy (DMD) (2021) | ||
| Other chemical modifications under clinical investigation | ||||
| PN | WVE-N531 * | SSO | Duchene muscular dystrophy (DMD) (Phase 1/2) | PN backbone modification:
|
| WVE-003 * | ASO | Huntington’s Disease (Phase 1/2) | ||
| tcDNA | SQY51 | SSO | Duchene muscular dystrophy (DMD) (Phase 1/2) | tcDNA sugar modification:
|
| Family | Peptide | Sequence | Reference |
|---|---|---|---|
| Cationic | Tat-(43–60) | LGISYGRKKRRQRRRPPQ | [159] |
| Tat-(48–60) | GRKKRRQRRRPPQ | [159] | |
| LAH-L1 (cationic amphipatic) | KKALLAHALHLLALLALHLAHALKKA | [150] | |
| Amphipathic anionic | Penetratin | RQIKIWFQNRRMKWKK | [160] |
| E5 (derived from HA2) | GLFEAIAEFIEGGWEGLIEGCA | [161] | |
| INF7 (derived from HA2) | GLFEAIEGFIENGWEGMIDGWYGC (dimer) | [162] | |
| GALA | WEAALAEALAEALAEHLAEALAEALEALAA | [154] | |
| Penetration Accelerating Sequence-Hydrophobic | Cathepsin D domain | GKPILFF | [163] |
| EED6 | GFWFG | [164] | |
| EED7 | GWWG | [164] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mangla, P.; Vicentini, Q.; Biscans, A. Therapeutic Oligonucleotides: An Outlook on Chemical Strategies to Improve Endosomal Trafficking. Cells 2023, 12, 2253. https://doi.org/10.3390/cells12182253
Mangla P, Vicentini Q, Biscans A. Therapeutic Oligonucleotides: An Outlook on Chemical Strategies to Improve Endosomal Trafficking. Cells. 2023; 12(18):2253. https://doi.org/10.3390/cells12182253
Chicago/Turabian StyleMangla, Priyanka, Quentin Vicentini, and Annabelle Biscans. 2023. "Therapeutic Oligonucleotides: An Outlook on Chemical Strategies to Improve Endosomal Trafficking" Cells 12, no. 18: 2253. https://doi.org/10.3390/cells12182253