
The Dual Role of the Airway Epithelium in Asthma: Active Barrier and Regulator of Inflammation

Abstract
:1. Introduction
2. Cellular Architecture of the Airway Epithelial Lining

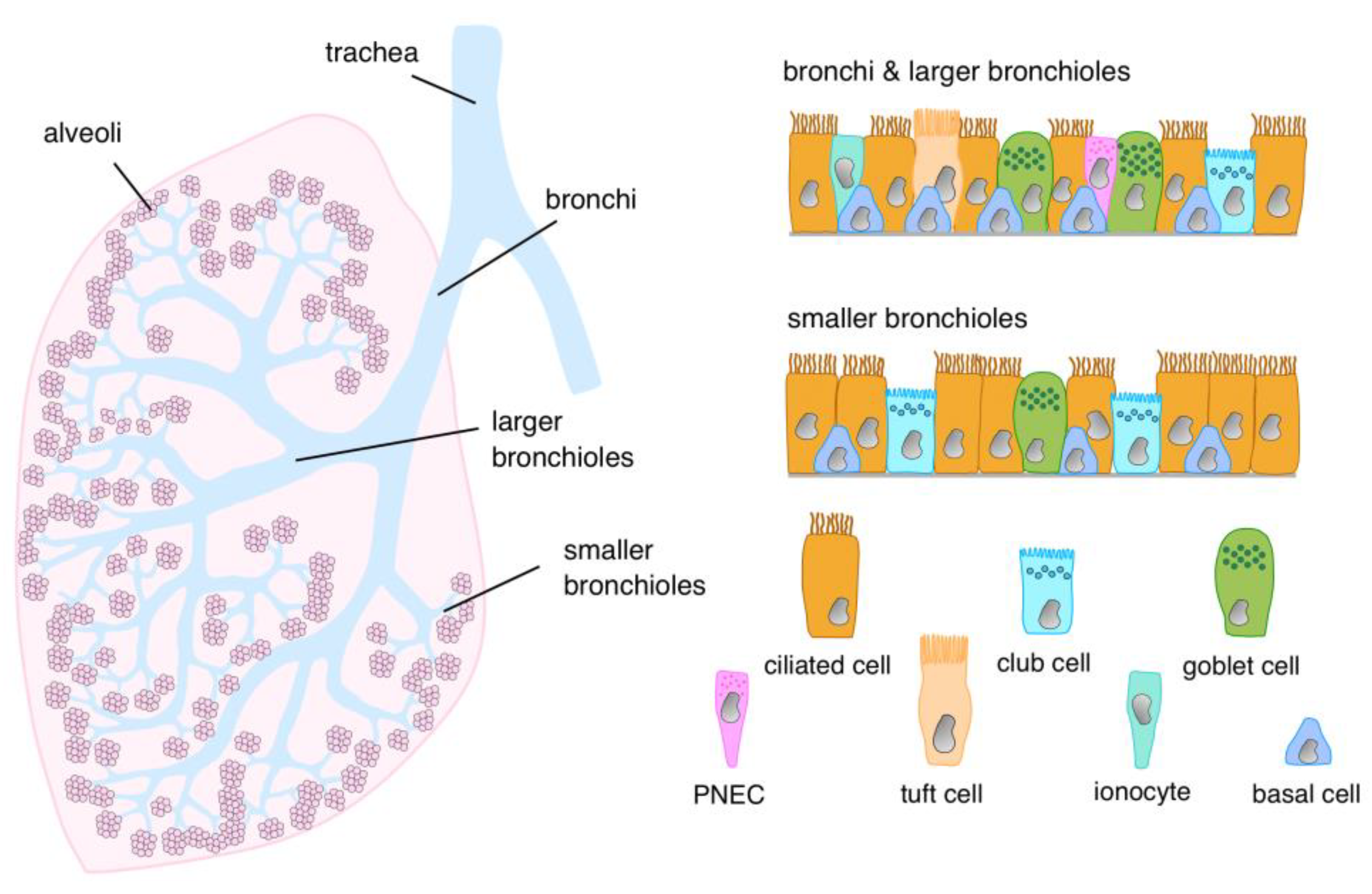{kind=link}
{kind=link}
{kind=link}
| Cell Type | Bronchi | Larger Bronchioles | Smaller Bronchioles | Reference |
|---|---|---|---|---|
| Basal | 27–30 | 27–30 | 4–23 | Crystal 2014 [16] Boers 1998 [13] Walters 2014 [44] |
| 8 → 50 | 2–22 | Okuda 2021 [15] Zuo 2020 [45] Deprez 2020 [20] Braga 2019 [18] Travaglini 2020 [17] | ||
| Ciliated | 53 | 62–71 | Staudt 2014 [46] Walters 2014 [44] | |
| 5 → 50 | 30–60 | Okuda 2021 [47] Zuo 2020 [45] Deprez 2020 [20] Braga 2019 [18] Travaglini 2020 [17] Paranjapye 2022 [48] | ||
| Goblet | 11 | 1–10 | 0–2 | Lumsden 1984 [14] Boers 1999 [15] |
| Club Cell (“Secretory Cell”) | 0 | 1–30 | 10–40 | Lumsden 1984 [14] Boers 1999 [15] Okuda 2021 [47] Zuo 2020 [45] Deprez 2020 [20] Braga 2019 [18] Travaglini 2020 [17] Paranjapye 2022 [48] |
| Neuroendocrine | “rare” (0.2–0.5) | Boers 1996 [34] Knight 2003 [33] | ||
| “abundant” (2.5–10) | Weichselbaum 2005 [35] | |||
| Ionocytes | “rare” (<1–2) | Plasschaert 2018 [43] Braga 2019 [18] Scudieri 2020 [24] Travaglini 2020 [17] Paranjapye 2022 [48] | ||
| Tuft Cell (Brush Cell) | “rare” | Goldfarbmuren 2020 [49] Ualiyeva 2020 [31] Deprez 2020 [20] | ||
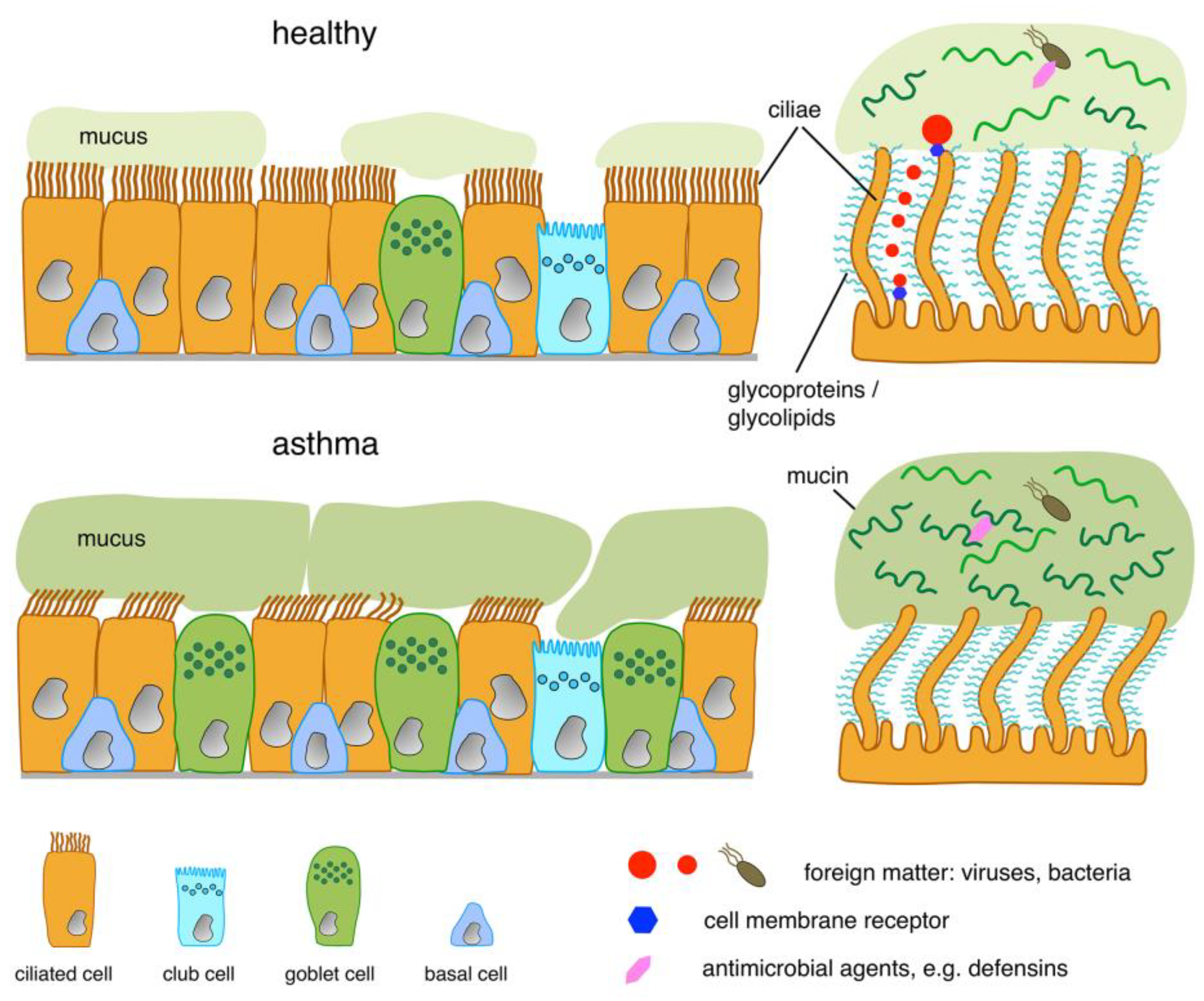
3. Structure of the Luminal Boundary
4. Molecular Function of the Boundary and the Epithelial Cells Constituting It
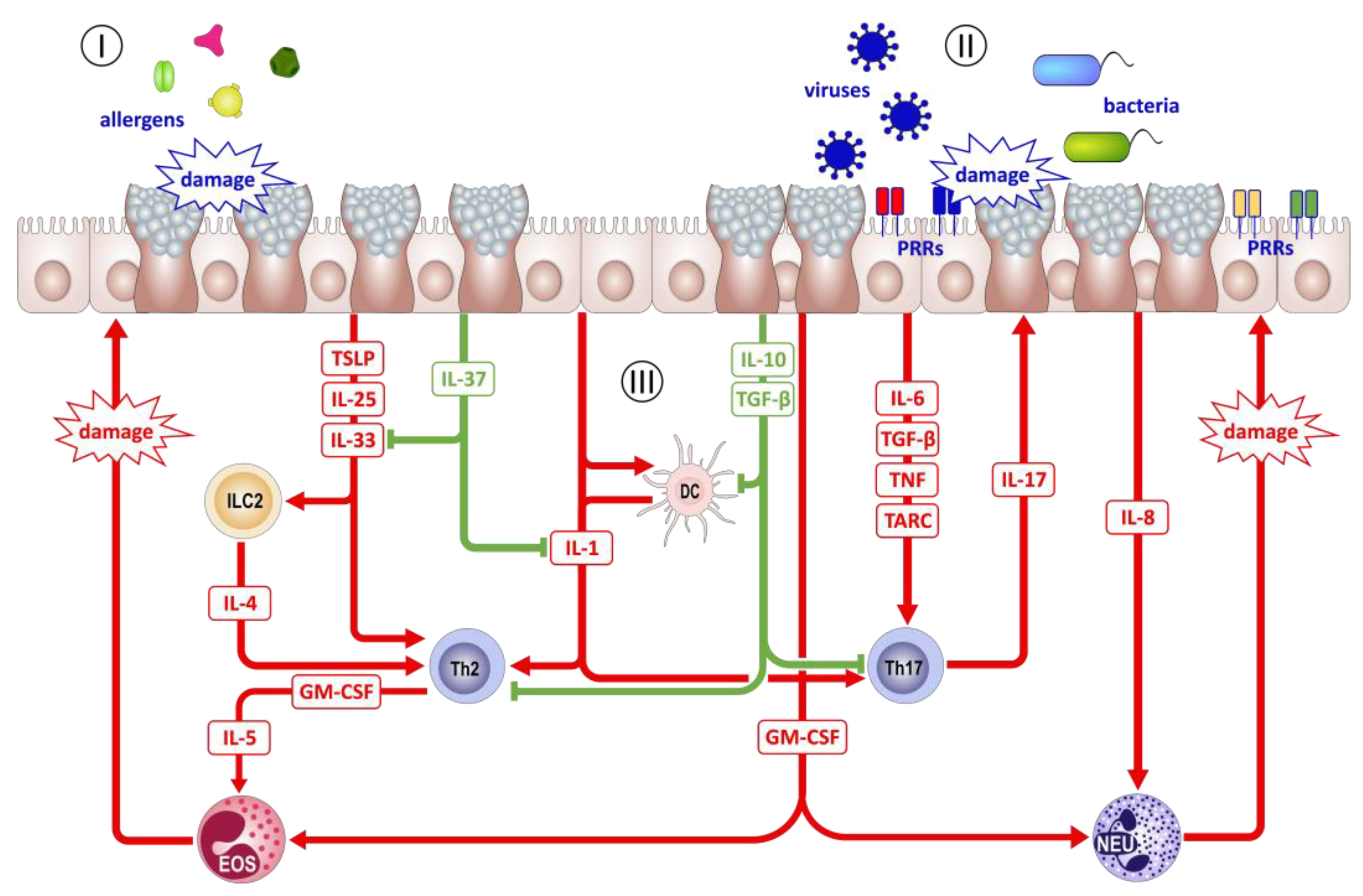
5. AECs as Sentinels of the Environment
6. AECs as Regulators of Inflammation
7. Trained Immunity of the Airway Epithelium
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- GBD 2019 Diseases and Injuries Collaborators Global Burden of 369 Diseases and Injuries in 204 Countries and Territories. 1990–2019: A Systematic Analysis for the Global Burden of Disease Study 2019. Lancet 2020, 396, 1204–1222. [CrossRef] [PubMed]
- Chung, K.F.; Wenzel, S.E.; Brozek, J.L.; Bush, A.; Castro, M.; Sterk, P.J.; Adcock, I.M.; Bateman, E.D.; Bel, E.H.; Bleecker, E.R.; et al. International ERS/ATS Guidelines on Definition, Evaluation and Treatment of Severe Asthma. Eur. Respir. J. 2014, 43, 343–373. [Google Scholar] [CrossRef] [PubMed]
- Papi, A.; Brightling, C.; Pedersen, S.E.; Reddel, H.K. Asthma. Lancet 2018, 391, 783–800. [Google Scholar] [CrossRef] [PubMed]
- Dunnill, M.S. The Pathology of Asthma, with Special Reference to Changes in the Bronchial Mucosa. J. Clin. Pathol. 1960, 13, 27–33. [Google Scholar] [CrossRef]
- Yaneva, M.; Darlenski, R. The Link between Atopic Dermatitis and Asthma- Immunological Imbalance and Beyond. Asthma Res. Pract. 2021, 7, 16. [Google Scholar] [CrossRef]
- Tsuge, M.; Ikeda, M.; Matsumoto, N.; Yorifuji, T.; Tsukahara, H. Current Insights into Atopic March. Children 2021, 8, 1067. [Google Scholar] [CrossRef]
- Chetta, A.; Foresi, A.; Del Donno, M.; Consigli, G.F.; Bertorelli, G.; Pesci, A.; Barbee, R.A.; Olivieri, D. Bronchial Responsiveness to Distilled Water and Methacholine and Its Relationship to Inflammation and Remodeling of the Airways in Asthma. Am. J. Respir. Crit. Care Med. 1996, 153, 910–917. [Google Scholar] [CrossRef]
- Wasserman, S.I. Basic Mechanisms in Asthma. Ann. Allergy 1988, 60, 477–482. [Google Scholar]
- Nadel, J.A. Inflammation and Asthma. J. Allergy Clin. Immunol. 1984, 73, 651–653. [Google Scholar] [CrossRef]
- Ricciardolo, F.L.M.; Carriero, V.; Bertolini, F. Which Therapy for Non-Type(T)2/T2-Low Asthma. J. Pers. Med. 2021, 12, 10. [Google Scholar] [CrossRef]
- Green, R.H.; Brightling, C.E.; Woltmann, G.; Parker, D.; Wardlaw, A.J.; Pavord, I.D. Analysis of Induced Sputum in Adults with Asthma: Identification of Subgroup with Isolated Sputum Neutrophilia and Poor Response to Inhaled Corticosteroids. Thorax 2002, 57, 875–879. [Google Scholar] [CrossRef] [PubMed]
- Staudt, M.R.; Buro-Auriemma, L.J.; Walters, M.S.; Salit, J.; Vincent, T.; Shaykhiev, R.; Mezey, J.G.; Tilley, A.E.; Kaner, R.J.; Ho, M.W.Y.; et al. Airway Basal Stem/Progenitor Cells Have Diminished Capacity to Regenerate Airway Epithelium in Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2014, 190, 955–958. [Google Scholar] [CrossRef]
- Boers, J.E.; Ambergen, A.W.; Thunnissen, F.B. Number and Proliferation of Basal and Parabasal Cells in Normal Human Airway Epithelium. Am. J. Respir. Crit. Care Med. 1998, 157, 2000–2006. [Google Scholar] [CrossRef] [PubMed]
- Lumsden, A.B.; McLean, A.; Lamb, D. Goblet and Clara Cells of Human Distal Airways: Evidence for Smoking Induced Changes in Their Numbers. Thorax 1984, 39, 844–849. [Google Scholar] [CrossRef] [PubMed]
- Boers, J.E.; Ambergen, A.W.; Thunnissen, F.B. Number and Proliferation of Clara Cells in Normal Human Airway Epithelium. Am. J. Respir. Crit. Care Med. 1999, 159, 1585–1591. [Google Scholar] [CrossRef]
- Crystal, R.G. Airway Basal Cells. The “Smoking Gun” of Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2014, 190, 1355–1362. [Google Scholar] [CrossRef] [PubMed]
- Travaglini, K.J.; Nabhan, A.N.; Penland, L.; Sinha, R.; Gillich, A.; Sit, R.V.; Chang, S.; Conley, S.D.; Mori, Y.; Seita, J.; et al. A Molecular Cell Atlas of the Human Lung from Single-Cell RNA Sequencing. Nature 2020, 587, 619–625. [Google Scholar] [CrossRef]
- Vieira Braga, F.A.; Kar, G.; Berg, M.; Carpaij, O.A.; Polanski, K.; Simon, L.M.; Brouwer, S.; Gomes, T.; Hesse, L.; Jiang, J.; et al. A Cellular Census of Human Lungs Identifies Novel Cell States in Health and in Asthma. Nat. Med. 2019, 25, 1153–1163. [Google Scholar] [CrossRef]
- Hewitt, R.J.; Lloyd, C.M. Regulation of Immune Responses by the Airway Epithelial Cell Landscape. Nat. Rev. Immunol. 2021, 21, 347–362. [Google Scholar] [CrossRef]
- Deprez, M.; Zaragosi, L.-E.; Truchi, M.; Becavin, C.; Ruiz García, S.; Arguel, M.-J.; Plaisant, M.; Magnone, V.; Lebrigand, K.; Abelanet, S.; et al. A Single-Cell Atlas of the Human Healthy Airways. Am. J. Respir. Crit. Care Med. 2020, 202, 1636–1645. [Google Scholar] [CrossRef]
- Kadur Lakshminarasimha Murthy, P.; Sontake, V.; Tata, A.; Kobayashi, Y.; Macadlo, L.; Okuda, K.; Conchola, A.S.; Nakano, S.; Gregory, S.; Miller, L.A.; et al. Human Distal Lung Maps and Lineage Hierarchies Reveal a Bipotent Progenitor. Nature 2022, 604, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Mizuno, T.; Sridharan, A.; Du, Y.; Guo, M.; Tang, J.; Wikenheiser-Brokamp, K.A.; Perl, A.-K.T.; Funari, V.A.; Gokey, J.J.; et al. Single-Cell RNA Sequencing Identifies Diverse Roles of Epithelial Cells in Idiopathic Pulmonary Fibrosis. JCI Insight 2016, 1, e90558. [Google Scholar] [CrossRef] [PubMed]
- Ravindra, N.G.; Alfajaro, M.M.; Gasque, V.; Huston, N.C.; Wan, H.; Szigeti-Buck, K.; Yasumoto, Y.; Greaney, A.M.; Habet, V.; Chow, R.D.; et al. Single-Cell Longitudinal Analysis of SARS-CoV-2 Infection in Human Airway Epithelium Identifies Target Cells, Alterations in Gene Expression, and Cell State Changes. PLoS Biol. 2021, 19, e3001143. [Google Scholar] [CrossRef] [PubMed]
- Scudieri, P.; Musante, I.; Venturini, A.; Guidone, D.; Genovese, M.; Cresta, F.; Caci, E.; Palleschi, A.; Poeta, M.; Santamaria, F.; et al. Ionocytes and CFTR Chloride Channel Expression in Normal and Cystic Fibrosis Nasal and Bronchial Epithelial Cells. Cells 2020, 9, 2090. [Google Scholar] [CrossRef] [PubMed]
- Basil, M.C.; Cardenas-Diaz, F.L.; Kathiriya, J.J.; Morley, M.P.; Carl, J.; Brumwell, A.N.; Katzen, J.; Slovik, K.J.; Babu, A.; Zhou, S.; et al. Human Distal Airways Contain a Multipotent Secretory Cell That Can Regenerate Alveoli. Nature 2022, 604, 120–126. [Google Scholar] [CrossRef]
- Sikkema, L.; Ramírez-Suástegui, C.; Strobl, D.C.; Gillett, T.E.; Zappia, L.; Madissoon, E.; Markov, N.S.; Zaragosi, L.-E.; Ji, Y.; Ansari, M.; et al. An Integrated Cell Atlas of the Lung in Health and Disease. Nat. Med. 2023, 29, 1563–1577. [Google Scholar] [CrossRef]
- Davis, J.D.; Wypych, T.P. Cellular and Functional Heterogeneity of the Airway Epithelium. Mucosal Immunol. 2021, 14, 978–990. [Google Scholar] [CrossRef]
- Rock, J.R.; Randell, S.H.; Hogan, B.L.M. Airway Basal Stem Cells: A Perspective on Their Roles in Epithelial Homeostasis and Remodeling. Dis. Model. Mech. 2010, 3, 545–556. [Google Scholar] [CrossRef]
- Tata, P.R.; Rajagopal, J. Plasticity in the Lung: Making and Breaking Cell Identity. Development 2017, 144, 755–766. [Google Scholar] [CrossRef]
- Jeffery, P.K. Morphologic Features of Airway Surface Epithelial Cells and Glands. Am. Rev. Respir. Dis. 1983, 128, S14–S20. [Google Scholar] [CrossRef]
- Ualiyeva, S.; Hallen, N.; Kanaoka, Y.; Ledderose, C.; Matsumoto, I.; Junger, W.G.; Barrett, N.A.; Bankova, L.G. Airway Brush Cells Generate Cysteinyl Leukotrienes through the ATP Sensor P2Y2. Sci. Immunol. 2020, 5, eaax7224. [Google Scholar] [CrossRef]
- Sell, E.A.; Ortiz-Carpena, J.F.; Herbert, D.R.; Cohen, N.A. Tuft Cells in the Pathogenesis of Chronic Rhinosinusitis with Nasal Polyps and Asthma. Ann. Allergy Asthma Immunol. 2021, 126, 143–151. [Google Scholar] [CrossRef]
- Knight, D.A.; Holgate, S.T. The Airway Epithelium: Structural and Functional Properties in Health and Disease. Respirology 2003, 8, 432–446. [Google Scholar] [CrossRef] [PubMed]
- Boers, J.E.; den Brok, J.L.; Koudstaal, J.; Arends, J.W.; Thunnissen, F.B. Number and Proliferation of Neuroendocrine Cells in Normal Human Airway Epithelium. Am. J. Respir. Crit. Care Med. 1996, 154, 758–763. [Google Scholar] [CrossRef] [PubMed]
- Weichselbaum, M.; Sparrow, M.P.; Hamilton, E.J.; Thompson, P.J.; Knight, D.A. A Confocal Microscopic Study of Solitary Pulmonary Neuroendocrine Cells in Human Airway Epithelium. Respir. Res. 2005, 6, 115. [Google Scholar] [CrossRef] [PubMed]
- Brouns, I.; Oztay, F.; Pintelon, I.; De Proost, I.; Lembrechts, R.; Timmermans, J.-P.; Adriaensen, D. Neurochemical Pattern of the Complex Innervation of Neuroepithelial Bodies in Mouse Lungs. Histochem. Cell. Biol. 2009, 131, 55–74. [Google Scholar] [CrossRef]
- Gu, X.; Karp, P.H.; Brody, S.L.; Pierce, R.A.; Welsh, M.J.; Holtzman, M.J.; Ben-Shahar, Y. Chemosensory Functions for Pulmonary Neuroendocrine Cells. Am. J. Respir. Cell. Mol. Biol. 2014, 50, 637–646. [Google Scholar] [CrossRef]
- Branchfield, K.; Nantie, L.; Verheyden, J.M.; Sui, P.; Wienhold, M.D.; Sun, X. Pulmonary Neuroendocrine Cells Function as Airway Sensors to Control Lung Immune Response. Science 2016, 351, 707–710. [Google Scholar] [CrossRef]
- Zhang, N.; Xu, J.; Jiang, C.; Lu, S. Neuro-Immune Regulation in Inflammation and Airway Remodeling of Allergic Asthma. Front. Immunol. 2022, 13, 894047. [Google Scholar] [CrossRef]
- Xu, J.; Yu, H.; Sun, X. Less Is More: Rare Pulmonary Neuroendocrine Cells Function as Critical Sensors in Lung. Dev. Cell 2020, 55, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Sui, P.; Wiesner, D.L.; Xu, J.; Zhang, Y.; Lee, J.; Van Dyken, S.; Lashua, A.; Yu, C.; Klein, B.S.; Locksley, R.M.; et al. Pulmonary Neuroendocrine Cells Amplify Allergic Asthma Responses. Science 2018, 360, eaan8546. [Google Scholar] [CrossRef] [PubMed]
- Barrios, J.; Kho, A.T.; Aven, L.; Mitchel, J.A.; Park, J.-A.; Randell, S.H.; Miller, L.A.; Tantisira, K.G.; Ai, X. Pulmonary Neuroendocrine Cells Secrete γ-Aminobutyric Acid to Induce Goblet Cell Hyperplasia in Primate Models. Am. J. Respir. Cell Mol. Biol. 2019, 60, 687–694. [Google Scholar] [CrossRef] [PubMed]
- Plasschaert, L.W.; Žilionis, R.; Choo-Wing, R.; Savova, V.; Knehr, J.; Roma, G.; Klein, A.M.; Jaffe, A.B. A Single-Cell Atlas of the Airway Epithelium Reveals the CFTR-Rich Pulmonary Ionocyte. Nature 2018, 560, 377–381. [Google Scholar] [CrossRef] [PubMed]
- Walters, M.S.; De, B.P.; Salit, J.; Buro-Auriemma, L.J.; Wilson, T.; Rogalski, A.M.; Lief, L.; Hackett, N.R.; Staudt, M.R.; Tilley, A.E.; et al. Smoking Accelerates Aging of the Small Airway Epithelium. Respir. Res. 2014, 15, 94. [Google Scholar] [CrossRef]
- Zuo, W.-L.; Rostami, M.R.; Shenoy, S.A.; LeBlanc, M.G.; Salit, J.; Strulovici-Barel, Y.; O’Beirne, S.L.; Kaner, R.J.; Leopold, P.L.; Mezey, J.G.; et al. Cell-Specific Expression of Lung Disease Risk-Related Genes in the Human Small Airway Epithelium. Respir. Res. 2020, 21, 200. [Google Scholar] [CrossRef] [PubMed]
- Staudt, M.R.; Rogalski, A.; Tilley, A.E.; Kaner, R.J.; Harvey, B.-G.; Crystal, R.G. Smoking Is Associated with A Loss of Ciliated Cells Throughout the Airways. In C26. Tobacco: How It Harms Us and More Reasons to Quit; American Thoracic Society International Conference Abstracts; American Thoracic Society: New York, NY, USA, 2014; p. A4097. [Google Scholar]
- Okuda, K.; Dang, H.; Kobayashi, Y.; Carraro, G.; Nakano, S.; Chen, G.; Kato, T.; Asakura, T.; Gilmore, R.C.; Morton, L.C.; et al. Secretory Cells Dominate Airway CFTR Expression and Function in Human Airway Superficial Epithelia. Am. J. Respir. Crit. Care Med. 2021, 203, 1275–1289. [Google Scholar] [CrossRef]
- Paranjapye, A.; Leir, S.-H.; Huang, F.; Kerschner, J.L.; Harris, A. Cell Function and Identity Revealed by Comparative ScRNA-Seq Analysis in Human Nasal, Bronchial and Epididymis Epithelia. Eur. J. Cell. Biol. 2022, 101, 151231. [Google Scholar] [CrossRef]
- Goldfarbmuren, K.C.; Jackson, N.D.; Sajuthi, S.P.; Dyjack, N.; Li, K.S.; Rios, C.L.; Plender, E.G.; Montgomery, M.T.; Everman, J.L.; Bratcher, P.E.; et al. Dissecting the Cellular Specificity of Smoking Effects and Reconstructing Lineages in the Human Airway Epithelium. Nat. Commun. 2020, 11, 2485. [Google Scholar] [CrossRef]
- Hough, K.P.; Curtiss, M.L.; Blain, T.J.; Liu, R.-M.; Trevor, J.; Deshane, J.S.; Thannickal, V.J. Airway Remodeling in Asthma. Front. Med. 2020, 7, 191. [Google Scholar] [CrossRef]
- Siddiqui, S.; Martin, J.G. Structural Aspects of Airway Remodeling in Asthma. Curr. Allergy Asthma Rep. 2008, 8, 540–547. [Google Scholar] [CrossRef]
- Ordoñez, C.L.; Khashayar, R.; Wong, H.H.; Ferrando, R.; Wu, R.; Hyde, D.M.; Hotchkiss, J.A.; Zhang, Y.; Novikov, A.; Dolganov, G.; et al. Mild and Moderate Asthma Is Associated with Airway Goblet Cell Hyperplasia and Abnormalities in Mucin Gene Expression. Am. J. Respir. Crit. Care Med. 2001, 163, 517–523. [Google Scholar] [CrossRef]
- Fahy, J.V. Goblet Cell and Mucin Gene Abnormalities in Asthma. Chest 2002, 122, 320S–326S. [Google Scholar] [CrossRef]
- Blyth, D.I.; Pedrick, M.S.; Savage, T.J.; Hessel, E.M.; Fattah, D. Lung Inflammation and Epithelial Changes in a Murine Model of Atopic Asthma. Am. J. Respir. Cell. Mol. Biol. 1996, 14, 425–438. [Google Scholar] [CrossRef]
- Laitinen, L.A.; Heino, M.; Laitinen, A.; Kava, T.; Haahtela, T. Damage of the Airway Epithelium and Bronchial Reactivity in Patients with Asthma. Am. Rev. Respir. Dis. 1985, 131, 599–606. [Google Scholar] [CrossRef]
- Lambrecht, B.N.; Hammad, H. Death at the Airway Epithelium in Asthma. Cell Res. 2013, 23, 588–589. [Google Scholar] [CrossRef]
- Thomas, B.; Rutman, A.; Hirst, R.A.; Haldar, P.; Wardlaw, A.J.; Bankart, J.; Brightling, C.E.; O’Callaghan, C. Ciliary Dysfunction and Ultrastructural Abnormalities Are Features of Severe Asthma. J. Allergy Clin. Immunol. 2010, 126, 722–729.e2. [Google Scholar] [CrossRef]
- Li, M.; Shang, Y.-X. Ultrastructural Changes in Rat Airway Epithelium in Asthmatic Airway Remodeling. Pathol. Res. Pract. 2014, 210, 1038–1042. [Google Scholar] [CrossRef]
- Leishangthem, G.D.; Mabalirajan, U.; Singh, V.P.; Agrawal, A.; Ghosh, B.; Dinda, A.K. Ultrastructural Changes of Airway in Murine Models of Allergy and Diet-Induced Metabolic Syndrome. ISRN Allergy 2013, 2013, 261297. [Google Scholar] [CrossRef]
- Okuda, K.; Chen, G.; Subramani, D.B.; Wolf, M.; Gilmore, R.C.; Kato, T.; Radicioni, G.; Kesimer, M.; Chua, M.; Dang, H.; et al. Localization of Secretory Mucins MUC5AC and MUC5B in Normal/Healthy Human Airways. Am. J. Respir. Crit. Care Med. 2019, 199, 715–727. [Google Scholar] [CrossRef]
- Kesimer, M.; Ehre, C.; Burns, K.A.; Davis, C.W.; Sheehan, J.K.; Pickles, R.J. Molecular Organization of the Mucins and Glycocalyx Underlying Mucus Transport over Mucosal Surfaces of the Airways. Mucosal Immunol. 2013, 6, 379–392. [Google Scholar] [CrossRef]
- Thornton, D.J.; Rousseau, K.; McGuckin, M.A. Structure and Function of the Polymeric Mucins in Airways Mucus. Annu. Rev. Physiol. 2008, 70, 459–486. [Google Scholar] [CrossRef] [PubMed]
- Houtmeyers, E.; Gosselink, R.; Gayan-Ramirez, G.; Decramer, M. Regulation of Mucociliary Clearance in Health and Disease. Eur. Respir. J. 1999, 13, 1177–1188. [Google Scholar] [CrossRef] [PubMed]
- Button, B.; Cai, L.-H.; Ehre, C.; Kesimer, M.; Hill, D.B.; Sheehan, J.K.; Boucher, R.C.; Rubinstein, M. A Periciliary Brush Promotes the Lung Health by Separating the Mucus Layer from Airway Epithelia. Science 2012, 337, 937–941. [Google Scholar] [CrossRef]
- Knowles, M.R.; Boucher, R.C. Mucus Clearance as a Primary Innate Defense Mechanism for Mammalian Airways. J. Clin. Investig. 2002, 109, 571–577. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Shang, M.; Chen, T.; Ren, P.; Sun, H.; Qu, H.; Lin, Z.; Zhou, L.; Yu, J.; Jiang, H.; et al. The Immunological Characteristics and Probiotic Function of Recombinant Bacillus Subtilis Spore Expressing Clonorchis Sinensis Cysteine Protease. Parasit. Vectors 2016, 9, 648. [Google Scholar] [CrossRef]
- Gustafsson, J.K.; Ermund, A.; Ambort, D.; Johansson, M.E.V.; Nilsson, H.E.; Thorell, K.; Hebert, H.; Sjövall, H.; Hansson, G.C. Bicarbonate and Functional CFTR Channel Are Required for Proper Mucin Secretion and Link Cystic Fibrosis with Its Mucus Phenotype. J. Exp. Med. 2012, 209, 1263–1272. [Google Scholar] [CrossRef]
- Hill, D.B.; Long, R.F.; Kissner, W.J.; Atieh, E.; Garbarine, I.C.; Markovetz, M.R.; Fontana, N.C.; Christy, M.; Habibpour, M.; Tarran, R.; et al. Pathological Mucus and Impaired Mucus Clearance in Cystic Fibrosis Patients Result from Increased Concentration, Not Altered PH. Eur. Respir. J. 2018, 52, 1801297. [Google Scholar] [CrossRef]
- Morrison, C.B.; Shaffer, K.M.; Araba, K.C.; Markovetz, M.R.; Wykoff, J.A.; Quinney, N.L.; Hao, S.; Delion, M.F.; Flen, A.L.; Morton, L.C.; et al. Treatment of Cystic Fibrosis Airway Cells with CFTR Modulators Reverses Aberrant Mucus Properties via Hydration. Eur. Respir. J. 2022, 59, 2100185. [Google Scholar] [CrossRef]
- Wilson, R.; Pitt, T.; Taylor, G.; Watson, D.; MacDermot, J.; Sykes, D.; Roberts, D.; Cole, P. Pyocyanin and 1-Hydroxyphenazine Produced by Pseudomonas Aeruginosa Inhibit the Beating of Human Respiratory Cilia in Vitro. J. Clin. Investig. 1987, 79, 221–229. [Google Scholar] [CrossRef]
- Jorissen, M.; Willems, T. The Secondary Nature of Ciliary (Dis)Orientation in Secondary and Primary Ciliary Dyskinesia. Acta Otolaryngol. 2004, 124, 527–531. [Google Scholar] [CrossRef]
- Münter, M.; Pieper, M.; Kohlfaerber, T.; Bodenstorfer, E.; Ahrens, M.; Winter, C.; Huber, R.; König, P.; Hüttmann, G.; Schulz-Hildebrandt, H. Microscopic Optical Coherence Tomography (MOCT) at 600 KHz for 4D Volumetric Imaging and Dynamic Contrast. Biomed. Opt. Express 2021, 12, 6024–6039. [Google Scholar] [CrossRef]
- Widdicombe, J.H. Regulation of the Depth and Composition of Airway Surface Liquid. J. Anat. 2002, 201, 313–318. [Google Scholar] [CrossRef] [PubMed]
- Tarran, R.; Trout, L.; Donaldson, S.H.; Boucher, R.C. Soluble Mediators, Not Cilia, Determine Airway Surface Liquid Volume in Normal and Cystic Fibrosis Superficial Airway Epithelia. J. Gen. Physiol. 2006, 127, 591–604. [Google Scholar] [CrossRef] [PubMed]
- Kirkham, S.; Sheehan, J.K.; Knight, D.; Richardson, P.S.; Thornton, D.J. Heterogeneity of Airways Mucus: Variations in the Amounts and Glycoforms of the Major Oligomeric Mucins MUC5AC and MUC5B. Biochem. J. 2002, 361, 537–546. [Google Scholar] [CrossRef]
- Welsh, K.G.; Rousseau, K.; Fisher, G.; Bonser, L.R.; Bradding, P.; Brightling, C.E.; Thornton, D.J.; Gaillard, E.A. MUC5AC and a Glycosylated Variant of MUC5B Alter Mucin Composition in Children with Acute Asthma. Chest 2017, 152, 771–779. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Li, H.; Yu, L.; Wang, N.; Li, X.; Chen, W. IL-1β Upregulates Muc5ac Expression via NF-ΚB-Induced HIF-1α in Asthma. Immunol. Lett. 2017, 192, 20–26. [Google Scholar] [CrossRef]
- Joris, L.; Dab, I.; Quinton, P.M. Elemental Composition of Human Airway Surface Fluid in Healthy and Diseased Airways. Am. Rev. Respir. Dis. 1993, 148, 1633–1637. [Google Scholar] [CrossRef]
- Yuan, S.; Hollinger, M.; Lachowicz-Scroggins, M.E.; Kerr, S.C.; Dunican, E.M.; Daniel, B.M.; Ghosh, S.; Erzurum, S.C.; Willard, B.; Hazen, S.L.; et al. Oxidation Increases Mucin Polymer Cross-Links to Stiffen Airway Mucus Gels. Sci. Transl. Med. 2015, 7, 276ra27. [Google Scholar] [CrossRef]
- Innes, A.L.; Carrington, S.D.; Thornton, D.J.; Kirkham, S.; Rousseau, K.; Dougherty, R.H.; Raymond, W.W.; Caughey, G.H.; Muller, S.J.; Fahy, J.V. Ex Vivo Sputum Analysis Reveals Impairment of Protease-Dependent Mucus Degradation by Plasma Proteins in Acute Asthma. Am. J. Respir. Crit. Care Med. 2009, 180, 203–210. [Google Scholar] [CrossRef]
- Olbrich, H.; Horváth, J.; Fekete, A.; Loges, N.T.; Storm van’s Gravesande, K.; Blum, A.; Hörmann, K.; Omran, H. Axonemal Localization of the Dynein Component DNAH5 Is Not Altered in Secondary Ciliary Dyskinesia. Pediatr. Res. 2006, 59, 418–422. [Google Scholar] [CrossRef]
- Laoukili, J.; Perret, E.; Willems, T.; Minty, A.; Parthoens, E.; Houcine, O.; Coste, A.; Jorissen, M.; Marano, F.; Caput, D.; et al. IL-13 Alters Mucociliary Differentiation and Ciliary Beating of Human Respiratory Epithelial Cells. J. Clin. Investig. 2001, 108, 1817–1824. [Google Scholar] [CrossRef]
- König, P.; Krain, B.; Krasteva, G.; Kummer, W. Serotonin Increases Cilia-Driven Particle Transport via an Acetylcholine-Independent Pathway in the Mouse Trachea. PLoS ONE 2009, 4, e4938. [Google Scholar] [CrossRef]
- Klein, M.K.; Haberberger, R.V.; Hartmann, P.; Faulhammer, P.; Lips, K.S.; Krain, B.; Wess, J.; Kummer, W.; König, P. Muscarinic Receptor Subtypes in Cilia-Driven Transport and Airway Epithelial Development. Eur. Respir. J. 2009, 33, 1113–1121. [Google Scholar] [CrossRef]
- Sheehan, J.K.; Richardson, P.S.; Fung, D.C.; Howard, M.; Thornton, D.J. Analysis of Respiratory Mucus Glycoproteins in Asthma: A Detailed Study from a Patient Who Died in Status Asthmaticus. Am. J. Respir. Cell. Mol. Biol. 1995, 13, 748–756. [Google Scholar] [CrossRef]
- Kuyper, L.M.; Paré, P.D.; Hogg, J.C.; Lambert, R.K.; Ionescu, D.; Woods, R.; Bai, T.R. Characterization of Airway Plugging in Fatal Asthma. Am. J. Med. 2003, 115, 6–11. [Google Scholar] [CrossRef]
- Bonser, L.R.; Erle, D.J. Airway Mucus and Asthma: The Role of MUC5AC and MUC5B. J. Clin. Med. 2017, 6, E112. [Google Scholar] [CrossRef] [PubMed]
- Bennett, H.S. Morphological Aspects of Extracellular Polysaccharides. J. Histochem. Cytochem. 1963, 11, 14–23. [Google Scholar] [CrossRef]
- Afzelius, B.A. Glycocalyx and Glycocalyceal Bodies in the Respiratory Epithelium of Nose and Bronchi. Ultrastruct. Pathol. 1984, 7, 1–8. [Google Scholar] [CrossRef]
- Frey, A.; Lunding, L.P.; Ehlers, J.C.; Weckmann, M.; Zissler, U.M.; Wegmann, M. More Than Just a Barrier: The Immune Functions of the Airway Epithelium in Asthma Pathogenesis. Front. Immunol. 2020, 11, 761. [Google Scholar] [CrossRef] [PubMed]
- Frey, A.; Giannasca, K.T.; Weltzin, R.; Giannasca, P.J.; Reggio, H.; Lencer, W.I.; Neutra, M.R. Role of the Glycocalyx in Regulating Access of Microparticles to Apical Plasma Membranes of Intestinal Epithelial Cells: Implications for Microbial Attachment and Oral Vaccine Targeting. J. Exp. Med. 1996, 184, 1045–1059. [Google Scholar] [CrossRef] [PubMed]
- Neutra, M.R.; Frey, A.; Kraehenbuhl, J.P. Epithelial M Cells: Gateways for Mucosal Infection and Immunization. Cell 1996, 86, 345–348. [Google Scholar] [CrossRef]
- Mantis, N.J.; Frey, A.; Neutra, M.R. Accessibility of Glycolipid and Oligosaccharide Epitopes on Rabbit Villus and Follicle-Associated Epithelium. Am. J. Physiol. Gastrointest. Liver Physiol. 2000, 278, G915–G923. [Google Scholar] [CrossRef]
- Excoffon, K.J.D.A.; Koerber, J.T.; Dickey, D.D.; Murtha, M.; Keshavjee, S.; Kaspar, B.K.; Zabner, J.; Schaffer, D.V. Directed Evolution of Adeno-Associated Virus to an Infectious Respiratory Virus. Proc. Natl. Acad. Sci. USA 2009, 106, 3865–3870. [Google Scholar] [CrossRef] [PubMed]
- Summerford, C.; Samulski, R.J. Membrane-Associated Heparan Sulfate Proteoglycan Is a Receptor for Adeno-Associated Virus Type 2 Virions. J. Virol. 1998, 72, 1438–1445. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.T.; Westphal, M.; Burlingham, B.T.; Winterhoff, U.; Doerfler, W. Structure and Composition of the Adenovirus Type 2 Core. J. Virol. 1975, 16, 366–387. [Google Scholar] [CrossRef] [PubMed]
- Walters, R.W.; Grunst, T.; Bergelson, J.M.; Finberg, R.W.; Welsh, M.J.; Zabner, J. Basolateral Localization of Fiber Receptors Limits Adenovirus Infection from the Apical Surface of Airway Epithelia. J. Biol. Chem. 1999, 274, 10219–10226. [Google Scholar] [CrossRef]
- McKay, T.; Patel, M.; Pickles, R.J.; Johnson, L.G.; Olsen, J.C. Influenza M2 Envelope Protein Augments Avian Influenza Hemagglutinin Pseudotyping of Lentiviral Vectors. Gene Ther. 2006, 13, 715–724. [Google Scholar] [CrossRef]
- Griesenbach, U.; Inoue, M.; Meng, C.; Farley, R.; Chan, M.; Newman, N.K.; Brum, A.; You, J.; Kerton, A.; Shoemark, A.; et al. Assessment of F/HN-Pseudotyped Lentivirus as a Clinically Relevant Vector for Lung Gene Therapy. Am. J. Respir. Crit. Care Med. 2012, 186, 846–856. [Google Scholar] [CrossRef]
- McCarron, A.; Cmielewski, P.; Drysdale, V.; Parsons, D.; Donnelley, M. Effective Viral-Mediated Lung Gene Therapy: Is Airway Surface Preparation Necessary? Gene Ther. 2022, 30, 469–477. [Google Scholar] [CrossRef]
- Eggo, R.M.; Scott, J.G.; Galvani, A.P.; Meyers, L.A. Respiratory Virus Transmission Dynamics Determine Timing of Asthma Exacerbation Peaks: Evidence from a Population-Level Model. Proc. Natl. Acad. Sci. USA 2016, 113, 2194–2199. [Google Scholar] [CrossRef]
- Mathia, N.R.; Timoszyk, J.; Stetsko, P.I.; Megill, J.R.; Smith, R.L.; Wall, D.A. Permeability Characteristics of Calu-3 Human Bronchial Epithelial Cells: In Vitro-in Vivo Correlation to Predict Lung Absorption in Rats. J. Drug. Target. 2002, 10, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.Q.; Finkbeiner, W.E.; Wine, J.J.; Mrsny, R.J.; Widdicombe, J.H. Calu-3: A Human Airway Epithelial Cell Line That Shows CAMP-Dependent Cl- Secretion. Am. J. Physiol. 1994, 266, L493–L501. [Google Scholar] [CrossRef] [PubMed]
- Watson, A.J.M.; Chu, S.; Sieck, L.; Gerasimenko, O.; Bullen, T.; Campbell, F.; McKenna, M.; Rose, T.; Montrose, M.H. Epithelial Barrier Function in Vivo Is Sustained despite Gaps in Epithelial Layers. Gastroenterology 2005, 129, 902–912. [Google Scholar] [CrossRef] [PubMed]
- Iwanaga, T.; Han, H.; Adachi, K.; Fujita, T. A Novel Mechanism for Disposing of Effete Epithelial Cells in the Small Intestine of Guinea Pigs. Gastroenterology 1993, 105, 1089–1097. [Google Scholar] [CrossRef]
- Xiao, C.; Puddicombe, S.M.; Field, S.; Haywood, J.; Broughton-Head, V.; Puxeddu, I.; Haitchi, H.M.; Vernon-Wilson, E.; Sammut, D.; Bedke, N.; et al. Defective Epithelial Barrier Function in Asthma. J. Allergy Clin. Immunol. 2011, 128, 549–556.e1-12. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zhou, X.-D.; Xu, R.; Du, X.-Z.; Li, Q.; Li, B.; Zhang, G.-Y.; Chen, L.-X.; Perelman, J.M.; Kolosov, V.P. The Degradation of Airway Epithelial Tight Junctions in Asthma Under High Airway Pressure Is Probably Mediated by Piezo-1. Front. Physiol. 2021, 12, 637790. [Google Scholar] [CrossRef]
- Sajjan, U.; Wang, Q.; Zhao, Y.; Gruenert, D.C.; Hershenson, M.B. Rhinovirus Disrupts the Barrier Function of Polarized Airway Epithelial Cells. Am. J. Respir. Crit. Care Med. 2008, 178, 1271–1281. [Google Scholar] [CrossRef] [PubMed]
- Vinhas, R.; Cortes, L.; Cardoso, I.; Mendes, V.M.; Manadas, B.; Todo-Bom, A.; Pires, E.; Veríssimo, P. Pollen Proteases Compromise the Airway Epithelial Barrier through Degradation of Transmembrane Adhesion Proteins and Lung Bioactive Peptides. Allergy 2011, 66, 1088–1098. [Google Scholar] [CrossRef]
- Gaspar, R.; de Matos, M.R.; Cortes, L.; Nunes-Correia, I.; Todo-Bom, A.; Pires, E.; Veríssimo, P. Pollen Proteases Play Multiple Roles in Allergic Disorders. Int. J. Mol. Sci. 2020, 21, 3578. [Google Scholar] [CrossRef]
- Schmidt, H.; Braubach, P.; Schilpp, C.; Lochbaum, R.; Neuland, K.; Thompson, K.; Jonigk, D.; Frick, M.; Dietl, P.; Wittekindt, O.H. IL-13 Impairs Tight Junctions in Airway Epithelia. Int. J. Mol. Sci. 2019, 20, 3222. [Google Scholar] [CrossRef]
- Felgentreff, K.; Beisswenger, C.; Griese, M.; Gulder, T.; Bringmann, G.; Bals, R. The Antimicrobial Peptide Cathelicidin Interacts with Airway Mucus. Peptides 2006, 27, 3100–3106. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.J.; Kofonow, J.M.; Rosen, P.L.; Siebert, A.P.; Chen, B.; Doghramji, L.; Xiong, G.; Adappa, N.D.; Palmer, J.N.; Kennedy, D.W.; et al. Bitter and Sweet Taste Receptors Regulate Human Upper Respiratory Innate Immunity. J. Clin. Investig. 2014, 124, 1393–1405. [Google Scholar] [CrossRef] [PubMed]
- Schutte, B.C.; McCray, P.B. [Beta]-Defensins in Lung Host Defense. Annu. Rev. Physiol. 2002, 64, 709–748. [Google Scholar] [CrossRef] [PubMed]
- Cantin, A.M.; Ouellet, C.; Cloutier, A.; McDonald, P.P. Airway Mucins Inhibit Oxidative and Non-Oxidative Bacterial Killing by Human Neutrophils. Front. Pharmacol. 2020, 11, 554353. [Google Scholar] [CrossRef] [PubMed]
- Yoshii, Y.; Shimizu, K.; Morozumi, M.; Chiba, N.; Ubukata, K.; Uruga, H.; Hanada, S.; Wakui, H.; Minagawa, S.; Hara, H.; et al. Detection of Pathogens by Real-Time PCR in Adult Patients with Acute Exacerbation of Bronchial Asthma. BMC Pulm. Med. 2017, 17, 150. [Google Scholar] [CrossRef] [PubMed]
- Iikura, M.; Hojo, M.; Koketsu, R.; Watanabe, S.; Sato, A.; Chino, H.; Ro, S.; Masaki, H.; Hirashima, J.; Ishii, S.; et al. The Importance of Bacterial and Viral Infections Associated with Adult Asthma Exacerbations in Clinical Practice. PLoS ONE 2015, 10, e0123584. [Google Scholar] [CrossRef]
- Nakamura, Y.; Kimura, S.; Hase, K. M Cell-Dependent Antigen Uptake on Follicle-Associated Epithelium for Mucosal Immune Surveillance. Inflamm. Regen. 2018, 38, 15. [Google Scholar] [CrossRef]
- Rescigno, M.; Urbano, M.; Valzasina, B.; Francolini, M.; Rotta, G.; Bonasio, R.; Granucci, F.; Kraehenbuhl, J.P.; Ricciardi-Castagnoli, P. Dendritic Cells Express Tight Junction Proteins and Penetrate Gut Epithelial Monolayers to Sample Bacteria. Nat. Immunol. 2001, 2, 361–367. [Google Scholar] [CrossRef]
- Jahnsen, F.L.; Strickland, D.H.; Thomas, J.A.; Tobagus, I.T.; Napoli, S.; Zosky, G.R.; Turner, D.J.; Sly, P.D.; Stumbles, P.A.; Holt, P.G. Accelerated Antigen Sampling and Transport by Airway Mucosal Dendritic Cells Following Inhalation of a Bacterial Stimulus. J. Immunol. 2006, 177, 5861–5867. [Google Scholar] [CrossRef]
- Sung, S.-S.J.; Fu, S.M.; Rose, C.E.; Gaskin, F.; Ju, S.-T.; Beaty, S.R. A Major Lung CD103 (AlphaE)-Beta7 Integrin-Positive Epithelial Dendritic Cell Population Expressing Langerin and Tight Junction Proteins. J. Immunol. 2006, 176, 2161–2172. [Google Scholar] [CrossRef]
- Johansen, F.E.; Braathen, R.; Brandtzaeg, P. The J Chain Is Essential for Polymeric Ig Receptor-Mediated Epithelial Transport of IgA. J. Immunol. 2001, 167, 5185–5192. [Google Scholar] [CrossRef]
- Castro, C.D.; Flajnik, M.F. Putting J Chain Back on the Map: How Might Its Expression Define Plasma Cell Development? J. Immunol. 2014, 193, 3248–3255. [Google Scholar] [CrossRef] [PubMed]
- Brandtzaeg, P. Transport Models for Secretory IgA and Secretory IgM. Clin. Exp. Immunol. 1981, 44, 221–232. [Google Scholar] [PubMed]
- Mostov, K.E. Transepithelial Transport of Immunoglobulins. Annu. Rev. Immunol. 1994, 12, 63–84. [Google Scholar] [CrossRef]
- Bomsel, M.; Heyman, M.; Hocini, H.; Lagaye, S.; Belec, L.; Dupont, C.; Desgranges, C. Intracellular Neutralization of HIV Transcytosis across Tight Epithelial Barriers by Anti-HIV Envelope Protein DIgA or IgM. Immunity 1998, 9, 277–287. [Google Scholar] [CrossRef]
- Bartemes, K.R.; Cooper, K.M.; Drain, K.L.; Kita, H. Secretory IgA Induces Antigen-Independent Eosinophil Survival and Cytokine Production without Inducing Effector Functions. J. Allergy Clin. Immunol. 2005, 116, 827–835. [Google Scholar] [CrossRef]
- Iikura, M.; Yamaguchi, M.; Fujisawa, T.; Miyamasu, M.; Takaishi, T.; Morita, Y.; Iwase, T.; Moro, I.; Yamamoto, K.; Hirai, K. Secretory IgA Induces Degranulation of IL-3-Primed Basophils. J. Immunol. 1998, 161, 1510–1515. [Google Scholar] [CrossRef]
- Thomas, L.L.; Xu, W.; Ardon, T.T. Immobilized Lactoferrin Is a Stimulus for Eosinophil Activation. J. Immunol. 2002, 169, 993–999. [Google Scholar] [CrossRef]
- Phalipon, A.; Cardona, A.; Kraehenbuhl, J.P.; Edelman, L.; Sansonetti, P.J.; Corthésy, B. Secretory Component: A New Role in Secretory IgA-Mediated Immune Exclusion in Vivo. Immunity 2002, 17, 107–115. [Google Scholar] [CrossRef]
- Reyneveld, G.I.; Savelkoul, H.F.J.; Parmentier, H.K. Current Understanding of Natural Antibodies and Exploring the Possibilities of Modulation Using Veterinary Models. A Review. Front. Immunol. 2020, 11, 2139. [Google Scholar] [CrossRef]
- Meek, B.; Back, J.W.; Klaren, V.N.A.; Speijer, D.; Peek, R. Conserved Regions of Protein Disulfide Isomerase Are Targeted by Natural IgA Antibodies in Humans. Int. Immunol. 2002, 14, 1291–1301. [Google Scholar] [CrossRef]
- Rosado, M.M.; Aranburu, A.; Capolunghi, F.; Giorda, E.; Cascioli, S.; Cenci, F.; Petrini, S.; Miller, E.; Leanderson, T.; Bottazzo, G.F.; et al. From the Fetal Liver to Spleen and Gut: The Highway to Natural Antibody. Mucosal Immunol. 2009, 2, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Holodick, N.E.; Rodríguez-Zhurbenko, N.; Hernández, A.M. Defining Natural Antibodies. Front. Immunol. 2017, 8, 872. [Google Scholar] [CrossRef] [PubMed]
- Shao, W.; Hu, F.; Ma, J.; Zhang, C.; Liao, Q.; Zhu, Z.; Liu, E.; Qiu, X. Epithelial Cells Are a Source of Natural IgM That Contribute to Innate Immune Responses. Int. J. Biochem. Cell. Biol. 2016, 73, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Winner, L.; Mack, J.; Weltzin, R.; Mekalanos, J.J.; Kraehenbuhl, J.P.; Neutra, M.R. New Model for Analysis of Mucosal Immunity: Intestinal Secretion of Specific Monoclonal Immunoglobulin A from Hybridoma Tumors Protects against Vibrio Cholerae Infection. Infect. Immun. 1991, 59, 977–982. [Google Scholar] [CrossRef]
- Ruggeri, F.M.; Johansen, K.; Basile, G.; Kraehenbuhl, J.P.; Svensson, L. Antirotavirus Immunoglobulin A Neutralizes Virus in Vitro after Transcytosis through Epithelial Cells and Protects Infant Mice from Diarrhea. J. Virol. 1998, 72, 2708–2714. [Google Scholar] [CrossRef]
- Corthésy, B. Role of Secretory Immunoglobulin A and Secretory Component in the Protection of Mucosal Surfaces. Future Microbiol. 2010, 5, 817–829. [Google Scholar] [CrossRef]
- Michetti, P.; Mahan, M.J.; Slauch, J.M.; Mekalanos, J.J.; Neutra, M.R. Monoclonal Secretory Immunoglobulin A Protects Mice against Oral Challenge with the Invasive Pathogen Salmonella Typhimurium. Infect. Immun. 1992, 60, 1786–1792. [Google Scholar] [CrossRef]
- Pilette, C.; Durham, S.R.; Vaerman, J.-P.; Sibille, Y. Mucosal Immunity in Asthma and Chronic Obstructive Pulmonary Disease: A Role for Immunoglobulin A? Proc. Am. Thorac. Soc. 2004, 1, 125–135. [Google Scholar] [CrossRef]
- Arnaboldi, P.M.; Behr, M.J.; Metzger, D.W. Mucosal B Cell Deficiency in IgA-/- Mice Abrogates the Development of Allergic Lung Inflammation. J. Immunol. 2005, 175, 1276–1285. [Google Scholar] [CrossRef]
- Iankov, I.D.; Petrov, D.P.; Mladenov, I.V.; Haralambieva, I.H.; Kalev, O.K.; Balabanova, M.S.; Mitov, I.G. Protective Efficacy of IgA Monoclonal Antibodies to O and H Antigens in a Mouse Model of Intranasal Challenge with Salmonella Enterica Serotype Enteritidis. Microbes Infect. 2004, 6, 901–910. [Google Scholar] [CrossRef]
- Richmond, B.W.; Brucker, R.M.; Han, W.; Du, R.-H.; Zhang, Y.; Cheng, D.-S.; Gleaves, L.; Abdolrasulnia, R.; Polosukhina, D.; Clark, P.E.; et al. Airway Bacteria Drive a Progressive COPD-like Phenotype in Mice with Polymeric Immunoglobulin Receptor Deficiency. Nat. Commun. 2016, 7, 11240. [Google Scholar] [CrossRef] [PubMed]
- Johansen, F.E.; Pekna, M.; Norderhaug, I.N.; Haneberg, B.; Hietala, M.A.; Krajci, P.; Betsholtz, C.; Brandtzaeg, P. Absence of Epithelial Immunoglobulin A Transport, with Increased Mucosal Leakiness, in Polymeric Immunoglobulin Receptor/Secretory Component-Deficient Mice. J. Exp. Med. 1999, 190, 915–922. [Google Scholar] [CrossRef] [PubMed]
- Arakawa, S.; Suzukawa, M.; Watanabe, K.; Kobayashi, K.; Matsui, H.; Nagai, H.; Nagase, T.; Ohta, K. Secretory Immunoglobulin A Induces Human Lung Fibroblasts to Produce Inflammatory Cytokines and Undergo Activation. Clin. Exp. Immunol. 2019, 195, 287–301. [Google Scholar] [CrossRef]
- Kobayashi, K.; Suzukawa, M.; Watanabe, K.; Arakawa, S.; Igarashi, S.; Asari, I.; Hebisawa, A.; Matsui, H.; Nagai, H.; Nagase, T.; et al. Secretory IgA Accumulated in the Airspaces of Idiopathic Pulmonary Fibrosis and Promoted VEGF, TGF-β and IL-8 Production by A549 Cells. Clin. Exp. Immunol. 2020, 199, 326–336. [Google Scholar] [CrossRef] [PubMed]
- Hupin, C.; Rombaux, P.; Bowen, H.; Gould, H.; Lecocq, M.; Pilette, C. Downregulation of Polymeric Immunoglobulin Receptor and Secretory IgA Antibodies in Eosinophilic Upper Airway Diseases. Allergy 2013, 68, 1589–1597. [Google Scholar] [CrossRef] [PubMed]
- Ladjemi, M.Z.; Gras, D.; Dupasquier, S.; Detry, B.; Lecocq, M.; Garulli, C.; Fregimilicka, C.; Bouzin, C.; Gohy, S.; Chanez, P.; et al. Bronchial Epithelial IgA Secretion Is Impaired in Asthma. Role of IL-4/IL-13. Am. J. Respir. Crit. Care Med. 2018, 197, 1396–1409. [Google Scholar] [CrossRef]
- Gohy, S.T.; Detry, B.R.; Lecocq, M.; Bouzin, C.; Weynand, B.A.; Amatngalim, G.D.; Sibille, Y.M.; Pilette, C. Polymeric Immunoglobulin Receptor Down-Regulation in Chronic Obstructive Pulmonary Disease. Persistence in the Cultured Epithelium and Role of Transforming Growth Factor-β. Am. J. Respir. Crit. Care Med. 2014, 190, 509–521. [Google Scholar] [CrossRef]
- Ratajczak, C.; Guisset, A.; Detry, B.; Sibille, Y.; Pilette, C. Dual Effect of Neutrophils on PIgR/Secretory Component in Human Bronchial Epithelial Cells: Role of TGF-Beta. J. Biomed. Biotechnol. 2010, 2010, 428618. [Google Scholar] [CrossRef]
- Jaffar, Z.; Ferrini, M.E.; Herritt, L.A.; Roberts, K. Cutting Edge: Lung Mucosal Th17-Mediated Responses Induce Polymeric Ig Receptor Expression by the Airway Epithelium and Elevate Secretory IgA Levels. J. Immunol. 2009, 182, 4507–4511. [Google Scholar] [CrossRef]
- Pouwels, S.D.; Heijink, I.H.; ten Hacken, N.H.T.; Vandenabeele, P.; Krysko, D.V.; Nawijn, M.C.; van Oosterhout, A.J.M. DAMPs Activating Innate and Adaptive Immune Responses in COPD. Mucosal Immunol. 2014, 7, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. Toll-like Receptors and Their Crosstalk with Other Innate Receptors in Infection and Immunity. Immunity 2011, 34, 637–650. [Google Scholar] [CrossRef] [PubMed]
- Poynter, M.E.; Irvin, C.G.; Janssen-Heininger, Y.M.W. Rapid Activation of Nuclear Factor-KappaB in Airway Epithelium in a Murine Model of Allergic Airway Inflammation. Am. J. Pathol. 2002, 160, 1325–1334. [Google Scholar] [CrossRef] [PubMed]
- Hertz, C.J.; Wu, Q.; Porter, E.M.; Zhang, Y.J.; Weismüller, K.-H.; Godowski, P.J.; Ganz, T.; Randell, S.H.; Modlin, R.L. Activation of Toll-like Receptor 2 on Human Tracheobronchial Epithelial Cells Induces the Antimicrobial Peptide Human Beta Defensin-2. J. Immunol. 2003, 171, 6820–6826. [Google Scholar] [CrossRef]
- Kool, M.; Pétrilli, V.; Smedt, T.D.; Rolaz, A.; Hammad, H.; van Nimwegen, M.; Bergen, I.M.; Castillo, R.; Lambrecht, B.N.; Tschopp, J. Cutting Edge: Alum Adjuvant Stimulates Inflammatory Dendritic Cells through Activation of the NALP3 Inflammasome. J. Immunol. 2008, 181, 3755–3759. [Google Scholar] [CrossRef]
- Eisenbarth, S.C.; Colegio, O.R.; O’Connor, W.; Sutterwala, F.S.; Flavell, R.A. Crucial Role for the Nalp3 Inflammasome in the Immunostimulatory Properties of Aluminium Adjuvants. Nature 2008, 453, 1122–1126. [Google Scholar] [CrossRef]
- Parker, D.; Prince, A. Type I Interferon Response to Extracellular Bacteria in the Airway Epithelium. Trends Immunol. 2011, 32, 582–588. [Google Scholar] [CrossRef]
- Kang, J.H.; Hwang, S.M.; Chung, I.Y. S100A8, S100A9 and S100A12 Activate Airway Epithelial Cells to Produce MUC5AC via Extracellular Signal-Regulated Kinase and Nuclear Factor-ΚB Pathways. Immunology 2015, 144, 79–90. [Google Scholar] [CrossRef]
- Ellson, C.D.; Dunmore, R.; Hogaboam, C.M.; Sleeman, M.A.; Murray, L.A. Danger-Associated Molecular Patterns and Danger Signals in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Cell. Mol. Biol. 2014, 51, 163–168. [Google Scholar] [CrossRef]
- Takeuchi, O.; Akira, S. Pattern Recognition Receptors and Inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef]
- Guryanova, S.V.; Gigani, O.B.; Gudima, G.O.; Kataeva, A.M.; Kolesnikova, N.V. Dual Effect of Low-Molecular-Weight Bioregulators of Bacterial Origin in Experimental Model of Asthma. Life 2022, 12, 192. [Google Scholar] [CrossRef] [PubMed]
- Parker, D.; Prince, A. Innate Immunity in the Respiratory Epithelium. Am. J. Respir. Cell. Mol. Biol. 2011, 45, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Gay, N.J.; Keith, F.J. Drosophila Toll and IL-1 Receptor. Nature 1991, 351, 355–356. [Google Scholar] [CrossRef] [PubMed]
- Slack, J.L.; Schooley, K.; Bonnert, T.P.; Mitcham, J.L.; Qwarnstrom, E.E.; Sims, J.E.; Dower, S.K. Identification of Two Major Sites in the Type I Interleukin-1 Receptor Cytoplasmic Region Responsible for Coupling to pro-Inflammatory Signaling Pathways. J. Biol. Chem. 2000, 275, 4670–4678. [Google Scholar] [CrossRef]
- Takeuchi, O.; Kawai, T.; Mühlradt, P.F.; Morr, M.; Radolf, J.D.; Zychlinsky, A.; Takeda, K.; Akira, S. Discrimination of Bacterial Lipoproteins by Toll-like Receptor 6. Int. Immunol. 2001, 13, 933–940. [Google Scholar] [CrossRef]
- Takeuchi, O.; Sato, S.; Horiuchi, T.; Hoshino, K.; Takeda, K.; Dong, Z.; Modlin, R.L.; Akira, S. Cutting Edge: Role of Toll-like Receptor 1 in Mediating Immune Response to Microbial Lipoproteins. J. Immunol. 2002, 169, 10–14. [Google Scholar] [CrossRef]
- Aliprantis, A.O.; Yang, R.B.; Mark, M.R.; Suggett, S.; Devaux, B.; Radolf, J.D.; Klimpel, G.R.; Godowski, P.; Zychlinsky, A. Cell Activation and Apoptosis by Bacterial Lipoproteins through Toll-like Receptor-2. Science 1999, 285, 736–739. [Google Scholar] [CrossRef]
- Schwandner, R.; Dziarski, R.; Wesche, H.; Rothe, M.; Kirschning, C.J. Peptidoglycan- and Lipoteichoic Acid-Induced Cell Activation Is Mediated by Toll-like Receptor 2. J. Biol. Chem. 1999, 274, 17406–17409. [Google Scholar] [CrossRef]
- Poltorak, A.; He, X.; Smirnova, I.; Liu, M.Y.; Van Huffel, C.; Du, X.; Birdwell, D.; Alejos, E.; Silva, M.; Galanos, C.; et al. Defective LPS Signaling in C3H/HeJ and C57BL/10ScCr Mice: Mutations in Tlr4 Gene. Science 1998, 282, 2085–2088. [Google Scholar] [CrossRef]
- Hayashi, F.; Smith, K.D.; Ozinsky, A.; Hawn, T.R.; Yi, E.C.; Goodlett, D.R.; Eng, J.K.; Akira, S.; Underhill, D.M.; Aderem, A. The Innate Immune Response to Bacterial Flagellin Is Mediated by Toll-like Receptor 5. Nature 2001, 410, 1099–1103. [Google Scholar] [CrossRef]
- Hemmi, H.; Takeuchi, O.; Kawai, T.; Kaisho, T.; Sato, S.; Sanjo, H.; Matsumoto, M.; Hoshino, K.; Wagner, H.; Takeda, K.; et al. A Toll-like Receptor Recognizes Bacterial DNA. Nature 2000, 408, 740–745. [Google Scholar] [CrossRef] [PubMed]
- Alexopoulou, L.; Holt, A.C.; Medzhitov, R.; Flavell, R.A. Recognition of Double-Stranded RNA and Activation of NF-KappaB by Toll-like Receptor 3. Nature 2001, 413, 732–738. [Google Scholar] [CrossRef] [PubMed]
- Diebold, S.S.; Kaisho, T.; Hemmi, H.; Akira, S.; Reis e Sousa, C. Innate Antiviral Responses by Means of TLR7-Mediated Recognition of Single-Stranded RNA. Science 2004, 303, 1529–1531. [Google Scholar] [CrossRef]
- Heil, F.; Hemmi, H.; Hochrein, H.; Ampenberger, F.; Kirschning, C.; Akira, S.; Lipford, G.; Wagner, H.; Bauer, S. Species-Specific Recognition of Single-Stranded RNA via Toll-like Receptor 7 and 8. Science 2004, 303, 1526–1529. [Google Scholar] [CrossRef]
- Oosting, M.; Cheng, S.-C.; Bolscher, J.M.; Vestering-Stenger, R.; Plantinga, T.S.; Verschueren, I.C.; Arts, P.; Garritsen, A.; van Eenennaam, H.; Sturm, P.; et al. Human TLR10 Is an Anti-Inflammatory Pattern-Recognition Receptor. Proc. Natl. Acad. Sci. USA 2014, 111, E4478–E4484. [Google Scholar] [CrossRef]
- Henrick, B.M.; Yao, X.-D.; Zahoor, M.A.; Abimiku, A.; Osawe, S.; Rosenthal, K.L. TLR10 Senses HIV-1 Proteins and Significantly Enhances HIV-1 Infection. Front. Immunol. 2019, 10, 482. [Google Scholar] [CrossRef]
- Zhang, D.; Zhang, G.; Hayden, M.S.; Greenblatt, M.B.; Bussey, C.; Flavell, R.A.; Ghosh, S. A Toll-like Receptor That Prevents Infection by Uropathogenic Bacteria. Science 2004, 303, 1522–1526. [Google Scholar] [CrossRef]
- Yarovinsky, F.; Zhang, D.; Andersen, J.F.; Bannenberg, G.L.; Serhan, C.N.; Hayden, M.S.; Hieny, S.; Sutterwala, F.S.; Flavell, R.A.; Ghosh, S.; et al. TLR11 Activation of Dendritic Cells by a Protozoan Profilin-like Protein. Science 2005, 308, 1626–1629. [Google Scholar] [CrossRef]
- Kim, Y.-M.; Brinkmann, M.M.; Paquet, M.-E.; Ploegh, H.L. UNC93B1 Delivers Nucleotide-Sensing Toll-like Receptors to Endolysosomes. Nature 2008, 452, 234–238. [Google Scholar] [CrossRef]
- Robinson, M.J.; Sancho, D.; Slack, E.C.; LeibundGut-Landmann, S.; Reis e Sousa, C. Myeloid C-Type Lectins in Innate Immunity. Nat. Immunol. 2006, 7, 1258–1265. [Google Scholar] [CrossRef]
- Lee, H.-M.; Yuk, J.-M.; Shin, D.-M.; Jo, E.-K. Dectin-1 Is Inducible and Plays an Essential Role for Mycobacteria-Induced Innate Immune Responses in Airway Epithelial Cells. J. Clin. Immunol. 2009, 29, 795–805. [Google Scholar] [CrossRef]
- Sun, W.-K.; Lu, X.; Li, X.; Sun, Q.-Y.; Su, X.; Song, Y.; Sun, H.-M.; Shi, Y. Dectin-1 Is Inducible and Plays a Crucial Role in Aspergillus-Induced Innate Immune Responses in Human Bronchial Epithelial Cells. Eur. J. Clin. Microbiol. Infect. Dis. 2012, 31, 2755–2764. [Google Scholar] [CrossRef] [PubMed]
- Nathan, A.T.; Peterson, E.A.; Chakir, J.; Wills-Karp, M. Innate Immune Responses of Airway Epithelium to House Dust Mite Are Mediated through Beta-Glucan-Dependent Pathways. J. Allergy Clin. Immunol. 2009, 123, 612–618. [Google Scholar] [CrossRef] [PubMed]
- Al-Ghouleh, A.; Johal, R.; Sharquie, I.K.; Emara, M.; Harrington, H.; Shakib, F.; Ghaemmaghami, A.M. The Glycosylation Pattern of Common Allergens: The Recognition and Uptake of Der p 1 by Epithelial and Dendritic Cells Is Carbohydrate Dependent. PLoS ONE 2012, 7, e33929. [Google Scholar] [CrossRef]
- Sow, F.B.; Nandakumar, S.; Velu, V.; Kellar, K.L.; Schlesinger, L.S.; Amara, R.R.; Lafuse, W.P.; Shinnick, T.M.; Sable, S.B. Mycobacterium Tuberculosis Components Stimulate Production of the Antimicrobial Peptide Hepcidin. Tuberculosis 2011, 91, 314–321. [Google Scholar] [CrossRef]
- Michael, C.F.; Waters, C.M.; LeMessurier, K.S.; Samarasinghe, A.E.; Song, C.Y.; Malik, K.U.; Lew, D.B. Airway Epithelial Repair by a Prebiotic Mannan Derived from Saccharomyces Cerevisiae. J. Immunol. Res. 2017, 2017, 8903982. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, S.; Rosenstiel, P. Debug Your Bugs—How NLRs Shape Intestinal Host-Microbe Interactions. Front. Immunol. 2013, 4, 479. [Google Scholar] [CrossRef]
- Chamaillard, M.; Hashimoto, M.; Horie, Y.; Masumoto, J.; Qiu, S.; Saab, L.; Ogura, Y.; Kawasaki, A.; Fukase, K.; Kusumoto, S.; et al. An Essential Role for NOD1 in Host Recognition of Bacterial Peptidoglycan Containing Diaminopimelic Acid. Nat. Immunol. 2003, 4, 702–707. [Google Scholar] [CrossRef]
- Girardin, S.E.; Boneca, I.G.; Carneiro, L.A.M.; Antignac, A.; Jéhanno, M.; Viala, J.; Tedin, K.; Taha, M.-K.; Labigne, A.; Zähringer, U.; et al. Nod1 Detects a Unique Muropeptide from Gram-Negative Bacterial Peptidoglycan. Science 2003, 300, 1584–1587. [Google Scholar] [CrossRef]
- Girardin, S.E.; Boneca, I.G.; Viala, J.; Chamaillard, M.; Labigne, A.; Thomas, G.; Philpott, D.J.; Sansonetti, P.J. Nod2 Is a General Sensor of Peptidoglycan through Muramyl Dipeptide (MDP) Detection. J. Biol. Chem. 2003, 278, 8869–8872. [Google Scholar] [CrossRef]
- Matikainen, S.; Sirén, J.; Tissari, J.; Veckman, V.; Pirhonen, J.; Severa, M.; Sun, Q.; Lin, R.; Meri, S.; Uzé, G.; et al. Tumor Necrosis Factor Alpha Enhances Influenza A Virus-Induced Expression of Antiviral Cytokines by Activating RIG-I Gene Expression. J. Virol. 2006, 80, 3515–3522. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Nagarkar, D.R.; Bowman, E.R.; Schneider, D.; Gosangi, B.; Lei, J.; Zhao, Y.; McHenry, C.L.; Burgens, R.V.; Miller, D.J.; et al. Role of Double-Stranded RNA Pattern Recognition Receptors in Rhinovirus-Induced Airway Epithelial Cell Responses. J. Immunol. 2009, 183, 6989–6997. [Google Scholar] [CrossRef] [PubMed]
- Lambrecht, B.N.; Hammad, H. The Airway Epithelium in Asthma. Nat. Med. 2012, 18, 684–692. [Google Scholar] [CrossRef] [PubMed]
- Denney, L.; Ho, L.-P. The Role of Respiratory Epithelium in Host Defence against Influenza Virus Infection. Biomed. J. 2018, 41, 218–233. [Google Scholar] [CrossRef] [PubMed]
- Tizaoui, K.; Kaabachi, W.; Hamzaoui, K.; Hamzaoui, A. Association of Single Nucleotide Polymorphisms in Toll-like Receptor Genes With Asthma Risk: A Systematic Review and Meta-Analysis. Allergy Asthma Immunol. Res. 2015, 7, 130–140. [Google Scholar] [CrossRef]
- Valette, K.; Li, Z.; Bon-Baret, V.; Chignon, A.; Bérubé, J.-C.; Eslami, A.; Lamothe, J.; Gaudreault, N.; Joubert, P.; Obeidat, M.; et al. Prioritization of Candidate Causal Genes for Asthma in Susceptibility Loci Derived from UK Biobank. Commun. Biol. 2021, 4, 700. [Google Scholar] [CrossRef]
- Schurman, S.H.; Bravo, M.A.; Innes, C.L.; Jackson, W.B.; McGrath, J.A.; Miranda, M.L.; Garantziotis, S. Toll-like Receptor 4 Pathway Polymorphisms Interact with Pollution to Influence Asthma Diagnosis and Severity. Sci. Rep. 2018, 8, 12713. [Google Scholar] [CrossRef]
- Cho, H.J.; Kim, S.-H.; Kim, J.-H.; Choi, H.; Son, J.-K.; Hur, G.-Y.; Park, H.-S. Effect of Toll-like Receptor 4 Gene Polymorphisms on Work-Related Respiratory Symptoms and Sensitization to Wheat Flour in Bakery Workers. Ann. Allergy Asthma Immunol. 2011, 107, 57–64. [Google Scholar] [CrossRef]
- Zhang, Q.; Qian, F.H.; Zhou, L.F.; Wei, G.Z.; Jin, G.F.; Bai, J.L.; Yin, K.S. Polymorphisms in Toll-like Receptor 4 Gene Are Associated with Asthma Severity but Not Susceptibility in a Chinese Han Population. J. Investig. Allergol. Clin. Immunol. 2011, 21, 370–377. [Google Scholar]
- Gao, Z.; Dosman, J.A.; Rennie, D.C.; Schwartz, D.A.; Yang, I.V.; Beach, J.; Senthilselvan, A. Association of Toll-like Receptor 2 Gene Polymorphisms with Lung Function in Workers in Swine Operations. Ann. Allergy Asthma Immunol. 2013, 110, 44–50.e1. [Google Scholar] [CrossRef]
- Qian, F.H.; Zhang, Q.; Zhou, L.F.; Jin, G.F.; Bai, J.L.; Yin, K.S. Polymorphisms in the Toll-like Receptor 2 Subfamily and Risk of Asthma: A Case-Control Analysis in a Chinese Population. J. Investig. Allergol. Clin. Immunol. 2010, 20, 340–346. [Google Scholar]
- Ellis, A.K.; Tsitoura, D.C.; Quint, D.; Powley, W.; Lee, L.A. Safety and Pharmacodynamics of Intranasal GSK2245035, a TLR7 Agonist for Allergic Rhinitis: A Randomized Trial. Clin. Exp. Allergy 2017, 47, 1193–1203. [Google Scholar] [CrossRef]
- Tsitoura, D.; Ambery, C.; Price, M.; Powley, W.; Garthside, S.; Biggadike, K.; Quint, D. Early Clinical Evaluation of the Intranasal TLR7 Agonist GSK2245035: Use of Translational Biomarkers to Guide Dosing and Confirm Target Engagement. Clin. Pharmacol. Ther. 2015, 98, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Greiff, L.; Ahlström-Emanuelsson, C.; Alenäs, M.; Almqvist, G.; Andersson, M.; Cervin, A.; Dolata, J.; Lindgren, S.; Mårtensson, A.; Young, B.; et al. Biological Effects and Clinical Efficacy of a Topical Toll-like Receptor 7 Agonist in Seasonal Allergic Rhinitis: A Parallel Group Controlled Phase IIa Study. Inflamm. Res. 2015, 64, 903–915. [Google Scholar] [CrossRef] [PubMed]
- Horak, F. VTX-1463, a Novel TLR8 Agonist for the Treatment of Allergic Rhinitis. Expert Opin. Investig. Drugs 2011, 20, 981–986. [Google Scholar] [CrossRef] [PubMed]
- Worm, M.; Higenbottam, T.; Pfaar, O.; Mösges, R.; Aberer, W.; Gunawardena, K.; Wessiepe, D.; Lee, D.; Kramer, M.F.; Skinner, M.; et al. Randomized Controlled Trials Define Shape of Dose Response for Pollinex Quattro Birch Allergoid Immunotherapy. Allergy 2018, 73, 1812–1822. [Google Scholar] [CrossRef] [PubMed]
- Zielen, S.; Gabrielpillai, J.; Herrmann, E.; Schulze, J.; Schubert, R.; Rosewich, M. Long-Term Effect of Monophosphoryl Lipid A Adjuvanted Specific Immunotherapy in Patients with Grass Pollen Allergy. Immunotherapy 2018, 10, 529–536. [Google Scholar] [CrossRef]
- Beeh, K.-M.; Kanniess, F.; Wagner, F.; Schilder, C.; Naudts, I.; Hammann-Haenni, A.; Willers, J.; Stocker, H.; Mueller, P.; Bachmann, M.F.; et al. The Novel TLR-9 Agonist QbG10 Shows Clinical Efficacy in Persistent Allergic Asthma. J. Allergy Clin. Immunol. 2013, 131, 866–874. [Google Scholar] [CrossRef]
- Casale, T.B.; Cole, J.; Beck, E.; Vogelmeier, C.F.; Willers, J.; Lassen, C.; Hammann-Haenni, A.; Trokan, L.; Saudan, P.; Wechsler, M.E. CYT003, a TLR9 Agonist, in Persistent Allergic Asthma—A Randomized Placebo-Controlled Phase 2b Study. Allergy 2015, 70, 1160–1168. [Google Scholar] [CrossRef]
- Silkoff, P.E.; Flavin, S.; Gordon, R.; Loza, M.J.; Sterk, P.J.; Lutter, R.; Diamant, Z.; Turner, R.B.; Lipworth, B.J.; Proud, D.; et al. Toll-like Receptor 3 Blockade in Rhinovirus-Induced Experimental Asthma Exacerbations: A Randomized Controlled Study. J. Allergy Clin. Immunol. 2018, 141, 1220–1230. [Google Scholar] [CrossRef]
- Lambrecht, B.N.; Hammad, H.; Fahy, J.V. The Cytokines of Asthma. Immunity 2019, 50, 975–991. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, J.I.; Bergman, R.; Birnbaum, H.G.; Colice, G.L.; Silverman, R.A.; McLaurin, K. Effect of Asthma Exacerbations on Health Care Costs among Asthmatic Patients with Moderate and Severe Persistent Asthma. J. Allergy Clin. Immunol. 2012, 129, 1229–1235. [Google Scholar] [CrossRef] [PubMed]
- Costantini, C.; Micheletti, A.; Calzetti, F.; Perbellini, O.; Pizzolo, G.; Cassatella, M.A. Neutrophil Activation and Survival Are Modulated by Interaction with NK Cells. Int. Immunol. 2010, 22, 827–838. [Google Scholar] [CrossRef] [PubMed]
- Rossios, C.; Pavlidis, S.; Hoda, U.; Kuo, C.-H.; Wiegman, C.; Russell, K.; Sun, K.; Loza, M.J.; Baribaud, F.; Durham, A.L.; et al. Sputum Transcriptomics Reveal Upregulation of IL-1 Receptor Family Members in Patients with Severe Asthma. J. Allergy Clin. Immunol. 2018, 141, 560–570. [Google Scholar] [CrossRef]
- Kim, R.Y.; Pinkerton, J.W.; Essilfie, A.T.; Robertson, A.A.B.; Baines, K.J.; Brown, A.C.; Mayall, J.R.; Ali, M.K.; Starkey, M.R.; Hansbro, N.G.; et al. Role for NLRP3 Inflammasome-Mediated, IL-1β-Dependent Responses in Severe, Steroid-Resistant Asthma. Am. J. Respir. Crit. Care Med. 2017, 196, 283–297. [Google Scholar] [CrossRef]
- Gibson, P.G.; Saltos, N.; Fakes, K. Acute Anti-Inflammatory Effects of Inhaled Budesonide in Asthma: A Randomized Controlled Trial. Am. J. Respir. Crit. Care Med. 2001, 163, 32–36. [Google Scholar] [CrossRef]
- Hastie, A.T.; Moore, W.C.; Meyers, D.A.; Vestal, P.L.; Li, H.; Peters, S.P.; Bleecker, E.R. National Heart, Lung, and Blood Institute Severe Asthma Research Program Analyses of Asthma Severity Phenotypes and Inflammatory Proteins in Subjects Stratified by Sputum Granulocytes. J. Allergy Clin. Immunol. 2010, 125, 1028–1036.e13. [Google Scholar] [CrossRef]
- Lee, Y.; Quoc, Q.L.; Park, H.S. Biomarkers for Severe Asthma: Lessons From Longitudinal Cohort Studies. Allergy Asthma Immunol. Res. 2021, 13, 375–389. [Google Scholar] [CrossRef]
- Hellings, P.W.; Kasran, A.; Liu, Z.; Vandekerckhove, P.; Wuyts, A.; Overbergh, L.; Mathieu, C.; Ceuppens, J.L. Interleukin-17 Orchestrates the Granulocyte Influx into Airways after Allergen Inhalation in a Mouse Model of Allergic Asthma. Am. J. Respir. Cell. Mol. Biol. 2003, 28, 42–50. [Google Scholar] [CrossRef]
- Bullens, D.M.A.; Truyen, E.; Coteur, L.; Dilissen, E.; Hellings, P.W.; Dupont, L.J.; Ceuppens, J.L. IL-17 MRNA in Sputum of Asthmatic Patients: Linking T Cell Driven Inflammation and Granulocytic Influx? Respir. Res. 2006, 7, 135. [Google Scholar] [CrossRef]
- Fei, X.; Zhang, P.-Y.; Zhang, X.; Zhang, G.-Q.; Bao, W.-P.; Zhang, Y.-Y.; Zhang, M.; Zhou, X. IL-17A Monoclonal Antibody Partly Reverses the Glucocorticoids Insensitivity in Mice Exposed to Ozonec. Inflammation 2017, 40, 788–797. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.E.; Chan, K. Interleukin-17 Stimulates the Expression of Interleukin-8, Growth-Related Oncogene-Alpha, and Granulocyte-Colony-Stimulating Factor by Human Airway Epithelial Cells. Am. J. Respir. Cell. Mol. Biol. 2002, 26, 748–753. [Google Scholar] [CrossRef]
- Laan, M.; Lötvall, J.; Chung, K.F.; Lindén, A. IL-17-Induced Cytokine Release in Human Bronchial Epithelial Cells in Vitro: Role of Mitogen-Activated Protein (MAP) Kinases. Br. J. Pharmacol. 2001, 133, 200–206. [Google Scholar] [CrossRef]
- Berin, M.C.; Eckmann, L.; Broide, D.H.; Kagnoff, M.F. Regulated Production of the T Helper 2-Type T-Cell Chemoattractant TARC by Human Bronchial Epithelial Cells in Vitro and in Human Lung Xenografts. Am. J. Respir. Cell. Mol. Biol. 2001, 24, 382–389. [Google Scholar] [CrossRef]
- Monick, M.M.; Powers, L.S.; Hassan, I.; Groskreutz, D.; Yarovinsky, T.O.; Barrett, C.W.; Castilow, E.M.; Tifrea, D.; Varga, S.M.; Hunninghake, G.W. Respiratory Syncytial Virus Synergizes with Th2 Cytokines to Induce Optimal Levels of TARC/CCL17. J. Immunol. 2007, 179, 1648–1658. [Google Scholar] [CrossRef] [PubMed]
- Heijink, I.H.; Marcel Kies, P.; van Oosterhout, A.J.M.; Postma, D.S.; Kauffman, H.F.; Vellenga, E. Der p, IL-4, and TGF-Beta Cooperatively Induce EGFR-Dependent TARC Expression in Airway Epithelium. Am. J. Respir. Cell. Mol. Biol. 2007, 36, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Cianferoni, A.; Spergel, J. The Importance of TSLP in Allergic Disease and Its Role as a Potential Therapeutic Target. Expert. Rev. Clin. Immunol. 2014, 10, 1463–1474. [Google Scholar] [CrossRef]
- Sokol, C.L.; Barton, G.M.; Farr, A.G.; Medzhitov, R. A Mechanism for the Initiation of Allergen-Induced T Helper Type 2 Responses. Nat. Immunol. 2008, 9, 310–318. [Google Scholar] [CrossRef]
- Pandey, A.; Ozaki, K.; Baumann, H.; Levin, S.D.; Puel, A.; Farr, A.G.; Ziegler, S.F.; Leonard, W.J.; Lodish, H.F. Cloning of a Receptor Subunit Required for Signaling by Thymic Stromal Lymphopoietin. Nat. Immunol. 2000, 1, 59–64. [Google Scholar] [CrossRef]
- Ito, T.; Wang, Y.-H.; Duramad, O.; Hori, T.; Delespesse, G.J.; Watanabe, N.; Qin, F.X.-F.; Yao, Z.; Cao, W.; Liu, Y.-J. TSLP-Activated Dendritic Cells Induce an Inflammatory T Helper Type 2 Cell Response through OX40 Ligand. J. Exp. Med. 2005, 202, 1213–1223. [Google Scholar] [CrossRef]
- Ochiai, S.; Roediger, B.; Abtin, A.; Shklovskaya, E.; Fazekas de St Groth, B.; Yamane, H.; Weninger, W.; Le Gros, G.; Ronchese, F. CD326loCD103loCD11blo Dermal Dendritic Cells Are Activated by Thymic Stromal Lymphopoietin during Contact Sensitization in Mice. J. Immunol. 2014, 193, 2504–2511. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, N.; Hanabuchi, S.; Soumelis, V.; Yuan, W.; Ho, S.; de Waal Malefyt, R.; Liu, Y.-J. Human Thymic Stromal Lymphopoietin Promotes Dendritic Cell-Mediated CD4+ T Cell Homeostatic Expansion. Nat. Immunol. 2004, 5, 426–434. [Google Scholar] [CrossRef]
- Zhou, B.; Comeau, M.R.; De Smedt, T.; Liggitt, H.D.; Dahl, M.E.; Lewis, D.B.; Gyarmati, D.; Aye, T.; Campbell, D.J.; Ziegler, S.F. Thymic Stromal Lymphopoietin as a Key Initiator of Allergic Airway Inflammation in Mice. Nat. Immunol. 2005, 6, 1047–1053. [Google Scholar] [CrossRef] [PubMed]
- Kitajima, M.; Lee, H.-C.; Nakayama, T.; Ziegler, S.F. TSLP Enhances the Function of Helper Type 2 Cells. Eur. J. Immunol. 2011, 41, 1862–1871. [Google Scholar] [CrossRef]
- Yao, W.; Zhang, Y.; Jabeen, R.; Nguyen, E.T.; Wilkes, D.S.; Tepper, R.S.; Kaplan, M.H.; Zhou, B. Interleukin-9 Is Required for Allergic Airway Inflammation Mediated by the Cytokine Thymic Stromal Lymphopoietin. Immunity 2013, 38, 360–372. [Google Scholar] [CrossRef]
- Wang, Q.; Du, J.; Zhu, J.; Yang, X.; Zhou, B. Thymic Stromal Lymphopoietin Signaling in CD4+ T Cells Is Required for TH2 Memory. J. Allergy Clin. Immunol. 2015, 135, 781–791.e3. [Google Scholar] [CrossRef]
- Omori, M.; Ziegler, S. Induction of IL-4 Expression in CD4+ T Cells by Thymic Stromal Lymphopoietin. J. Immunol. 2007, 178, 1396–1404. [Google Scholar] [CrossRef]
- Rochman, I.; Watanabe, N.; Arima, K.; Liu, Y.-J.; Leonard, W.J. Cutting Edge: Direct Action of Thymic Stromal Lymphopoietin on Activated Human CD4+ T Cells. J. Immunol. 2007, 178, 6720–6724. [Google Scholar] [CrossRef]
- Rochman, Y.; Dienger-Stambaugh, K.; Richgels, P.K.; Lewkowich, I.P.; Kartashov, A.V.; Barski, A.; Khurana Hershey, G.K.; Leonard, W.J.; Singh, H. TSLP Signaling in CD4+ T Cells Programs a Pathogenic T Helper 2 Cell State. Sci. Signal. 2018, 11, eaam8858. [Google Scholar] [CrossRef]
- Kang, C.-M.; Jang, A.-S.; Ahn, M.-H.; Shin, J.-A.; Kim, J.-H.; Choi, Y.-S.; Rhim, T.-Y.; Park, C.-S. Interleukin-25 and Interleukin-13 Production by Alveolar Macrophages in Response to Particles. Am. J. Respir. Cell. Mol. Biol. 2005, 33, 290–296. [Google Scholar] [CrossRef]
- Ikeda, K.; Nakajima, H.; Suzuki, K.; Kagami, S.; Hirose, K.; Suto, A.; Saito, Y.; Iwamoto, I. Mast Cells Produce Interleukin-25 upon Fc Epsilon RI-Mediated Activation. Blood 2003, 101, 3594–3596. [Google Scholar] [CrossRef]
- Corrigan, C.J.; Wang, W.; Meng, Q.; Fang, C.; Eid, G.; Caballero, M.R.; Lv, Z.; An, Y.; Wang, Y.-H.; Liu, Y.-J.; et al. Allergen-Induced Expression of IL-25 and IL-25 Receptor in Atopic Asthmatic Airways and Late-Phase Cutaneous Responses. J. Allergy Clin. Immunol. 2011, 128, 116–124. [Google Scholar] [CrossRef]
- Xu, M.; Dong, C. IL-25 in Allergic Inflammation. Immunol. Rev. 2017, 278, 185–191. [Google Scholar] [CrossRef]
- Tamachi, T.; Maezawa, Y.; Ikeda, K.; Kagami, S.-I.; Hatano, M.; Seto, Y.; Suto, A.; Suzuki, K.; Watanabe, N.; Saito, Y.; et al. IL-25 Enhances Allergic Airway Inflammation by Amplifying a TH2 Cell-Dependent Pathway in Mice. J. Allergy Clin. Immunol. 2006, 118, 606–614. [Google Scholar] [CrossRef]
- Gregory, L.G.; Jones, C.P.; Walker, S.A.; Sawant, D.; Gowers, K.H.C.; Campbell, G.A.; McKenzie, A.N.J.; Lloyd, C.M. IL-25 Drives Remodelling in Allergic Airways Disease Induced by House Dust Mite. Thorax 2013, 68, 82–90. [Google Scholar] [CrossRef]
- Stock, P.; Lombardi, V.; Kohlrautz, V.; Akbari, O. Induction of Airway Hyperreactivity by IL-25 Is Dependent on a Subset of Invariant NKT Cells Expressing IL-17RB. J. Immunol. 2009, 182, 5116–5122. [Google Scholar] [CrossRef]
- Angkasekwinai, P.; Park, H.; Wang, Y.-H.; Wang, Y.-H.; Chang, S.H.; Corry, D.B.; Liu, Y.-J.; Zhu, Z.; Dong, C. Interleukin 25 Promotes the Initiation of Proallergic Type 2 Responses. J. Exp. Med. 2007, 204, 1509–1517. [Google Scholar] [CrossRef]
- Saenz, S.A.; Siracusa, M.C.; Monticelli, L.A.; Ziegler, C.G.K.; Kim, B.S.; Brestoff, J.R.; Peterson, L.W.; Wherry, E.J.; Goldrath, A.W.; Bhandoola, A.; et al. IL-25 Simultaneously Elicits Distinct Populations of Innate Lymphoid Cells and Multipotent Progenitor Type 2 (MPPtype2) Cells. J. Exp. Med. 2013, 210, 1823–1837. [Google Scholar] [CrossRef]
- Klein Wolterink, R.G.J.; Kleinjan, A.; van Nimwegen, M.; Bergen, I.; de Bruijn, M.; Levani, Y.; Hendriks, R.W. Pulmonary Innate Lymphoid Cells Are Major Producers of IL-5 and IL-13 in Murine Models of Allergic Asthma. Eur. J. Immunol. 2012, 42, 1106–1116. [Google Scholar] [CrossRef]
- Moussion, C.; Ortega, N.; Girard, J.-P. The IL-1-Like Cytokine IL-33 Is Constitutively Expressed in the Nucleus of Endothelial Cells and Epithelial Cells In Vivo: A Novel ‘Alarmin’? PLoS ONE 2008, 3, e3331. [Google Scholar] [CrossRef]
- Gudbjartsson, D.F.; Bjornsdottir, U.S.; Halapi, E.; Helgadottir, A.; Sulem, P.; Jonsdottir, G.M.; Thorleifsson, G.; Helgadottir, H.; Steinthorsdottir, V.; Stefansson, H.; et al. Sequence Variants Affecting Eosinophil Numbers Associate with Asthma and Myocardial Infarction. Nat. Genet. 2009, 41, 342–347. [Google Scholar] [CrossRef]
- Moffatt, M.F.; Gut, I.G.; Demenais, F.; Strachan, D.P.; Bouzigon, E.; Heath, S.; von Mutius, E.; Farrall, M.; Lathrop, M.; Cookson, W.O.C.M. A Large-Scale, Consortium-Based Genomewide Association Study of Asthma. N. Engl. J. Med. 2010, 363, 1211–1221. [Google Scholar] [CrossRef]
- Torgerson, D.G.; Ampleford, E.J.; Chiu, G.Y.; Gauderman, W.J.; Gignoux, C.R.; Graves, P.E.; Himes, B.E.; Levin, A.M.; Mathias, R.A.; Hancock, D.B.; et al. Meta-Analysis of Genome-Wide Association Studies of Asthma in Ethnically Diverse North American Populations. Nat. Genet. 2011, 43, 887–892. [Google Scholar] [CrossRef]
- Grotenboer, N.S.; Ketelaar, M.E.; Koppelman, G.H.; Nawijn, M.C. Decoding Asthma: Translating Genetic Variation in IL33 and IL1RL1 into Disease Pathophysiology. J. Allergy Clin. Immunol. 2013, 131, 856–865. [Google Scholar] [CrossRef]
- Bønnelykke, K.; Sleiman, P.; Nielsen, K.; Kreiner-Møller, E.; Mercader, J.M.; Belgrave, D.; den Dekker, H.T.; Husby, A.; Sevelsted, A.; Faura-Tellez, G.; et al. A Genome-Wide Association Study Identifies CDHR3 as a Susceptibility Locus for Early Childhood Asthma with Severe Exacerbations. Nat. Genet. 2014, 46, 51–55. [Google Scholar] [CrossRef]
- Lefrançais, E.; Roga, S.; Gautier, V.; Gonzalez-de-Peredo, A.; Monsarrat, B.; Girard, J.-P.; Cayrol, C. IL-33 Is Processed into Mature Bioactive Forms by Neutrophil Elastase and Cathepsin G. Proc. Natl. Acad. Sci. USA 2012, 109, 1673–1678. [Google Scholar] [CrossRef]
- Lefrançais, E.; Duval, A.; Mirey, E.; Roga, S.; Espinosa, E.; Cayrol, C.; Girard, J.-P. Central Domain of IL-33 Is Cleaved by Mast Cell Proteases for Potent Activation of Group-2 Innate Lymphoid Cells. Proc. Natl. Acad. Sci. USA 2014, 111, 15502–15507. [Google Scholar] [CrossRef]
- Clancy, D.M.; Sullivan, G.P.; Moran, H.B.T.; Henry, C.M.; Reeves, E.P.; McElvaney, N.G.; Lavelle, E.C.; Martin, S.J. Extracellular Neutrophil Proteases Are Efficient Regulators of IL-1, IL-33, and IL-36 Cytokine Activity but Poor Effectors of Microbial Killing. Cell Rep. 2018, 22, 2937–2950. [Google Scholar] [CrossRef]
- Lüthi, A.U.; Cullen, S.P.; McNeela, E.A.; Duriez, P.J.; Afonina, I.S.; Sheridan, C.; Brumatti, G.; Taylor, R.C.; Kersse, K.; Vandenabeele, P.; et al. Suppression of Interleukin-33 Bioactivity through Proteolysis by Apoptotic Caspases. Immunity 2009, 31, 84–98. [Google Scholar] [CrossRef]
- Cayrol, C.; Girard, J.-P. The IL-1-like Cytokine IL-33 Is Inactivated after Maturation by Caspase-1. Proc. Natl. Acad. Sci. USA 2009, 106, 9021–9026. [Google Scholar] [CrossRef]
- Cayrol, C.; Duval, A.; Schmitt, P.; Roga, S.; Camus, M.; Stella, A.; Burlet-Schiltz, O.; Gonzalez-de-Peredo, A.; Girard, J.-P. Environmental Allergens Induce Allergic Inflammation through Proteolytic Maturation of IL-33. Nat. Immunol. 2018, 19, 375–385. [Google Scholar] [CrossRef] [PubMed]
- Pecaric-Petkovic, T.; Didichenko, S.A.; Kaempfer, S.; Spiegl, N.; Dahinden, C.A. Human Basophils and Eosinophils Are the Direct Target Leukocytes of the Novel IL-1 Family Member IL-33. Blood 2009, 113, 1526–1534. [Google Scholar] [CrossRef] [PubMed]
- Mjösberg, J.M.; Trifari, S.; Crellin, N.K.; Peters, C.P.; van Drunen, C.M.; Piet, B.; Fokkens, W.J.; Cupedo, T.; Spits, H. Human IL-25- and IL-33-Responsive Type 2 Innate Lymphoid Cells Are Defined by Expression of CRTH2 and CD161. Nat. Immunol. 2011, 12, 1055–1062. [Google Scholar] [CrossRef] [PubMed]
- Bonilla, W.V.; Fröhlich, A.; Senn, K.; Kallert, S.; Fernandez, M.; Johnson, S.; Kreutzfeldt, M.; Hegazy, A.N.; Schrick, C.; Fallon, P.G.; et al. The Alarmin Interleukin-33 Drives Protective Antiviral CD8+ T Cell Responses. Science 2012, 335, 984–989. [Google Scholar] [CrossRef]
- Schiering, C.; Krausgruber, T.; Chomka, A.; Fröhlich, A.; Adelmann, K.; Wohlfert, E.A.; Pott, J.; Griseri, T.; Bollrath, J.; Hegazy, A.N.; et al. The Alarmin IL-33 Promotes Regulatory T-Cell Function in the Intestine. Nature 2014, 513, 564–568. [Google Scholar] [CrossRef]
- Baumann, C.; Bonilla, W.V.; Fröhlich, A.; Helmstetter, C.; Peine, M.; Hegazy, A.N.; Pinschewer, D.D.; Löhning, M. T-Bet- and STAT4-Dependent IL-33 Receptor Expression Directly Promotes Antiviral Th1 Cell Responses. Proc. Natl. Acad. Sci. USA 2015, 112, 4056–4061. [Google Scholar] [CrossRef]
- Molofsky, A.B.; Savage, A.K.; Locksley, R.M. Interleukin-33 in Tissue Homeostasis, Injury, and Inflammation. Immunity 2015, 42, 1005–1019. [Google Scholar] [CrossRef]
- Endo, Y.; Hirahara, K.; Iinuma, T.; Shinoda, K.; Tumes, D.J.; Asou, H.K.; Matsugae, N.; Obata-Ninomiya, K.; Yamamoto, H.; Motohashi, S.; et al. The Interleukin-33-P38 Kinase Axis Confers Memory T Helper 2 Cell Pathogenicity in the Airway. Immunity 2015, 42, 294–308. [Google Scholar] [CrossRef]
- Morimoto, Y.; Hirahara, K.; Kiuchi, M.; Wada, T.; Ichikawa, T.; Kanno, T.; Okano, M.; Kokubo, K.; Onodera, A.; Sakurai, D.; et al. Amphiregulin-Producing Pathogenic Memory T Helper 2 Cells Instruct Eosinophils to Secrete Osteopontin and Facilitate Airway Fibrosis. Immunity 2018, 49, 134–150.e6. [Google Scholar] [CrossRef]
- Moro, K.; Yamada, T.; Tanabe, M.; Takeuchi, T.; Ikawa, T.; Kawamoto, H.; Furusawa, J.-I.; Ohtani, M.; Fujii, H.; Koyasu, S. Innate Production of TH2 Cytokines by Adipose Tissue-Associated c-Kit+Sca-1+ Lymphoid Cells. Nature 2010, 463, 540–544. [Google Scholar] [CrossRef]
- Salimi, M.; Barlow, J.L.; Saunders, S.P.; Xue, L.; Gutowska-Owsiak, D.; Wang, X.; Huang, L.-C.; Johnson, D.; Scanlon, S.T.; McKenzie, A.N.J.; et al. A Role for IL-25 and IL-33-Driven Type-2 Innate Lymphoid Cells in Atopic Dermatitis. J. Exp. Med. 2013, 210, 2939–2950. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, C.; Khan, A.R.; Floudas, A.; Saunders, S.P.; Hams, E.; Rodewald, H.-R.; McKenzie, A.N.J.; Fallon, P.G. ILC2s Regulate Adaptive Th2 Cell Functions via PD-L1 Checkpoint Control. J. Exp. Med. 2017, 214, 2507–2521. [Google Scholar] [CrossRef] [PubMed]
- Halim, T.Y.F.; Rana, B.M.J.; Walker, J.A.; Kerscher, B.; Knolle, M.D.; Jolin, H.E.; Serrao, E.M.; Haim-Vilmovsky, L.; Teichmann, S.A.; Rodewald, H.-R.; et al. Tissue-Restricted Adaptive Type 2 Immunity Is Orchestrated by Expression of the Costimulatory Molecule OX40L on Group 2 Innate Lymphoid Cells. Immunity 2018, 48, 1195–1207.e6. [Google Scholar] [CrossRef]
- Halim, T.Y.F.; Steer, C.A.; Mathä, L.; Gold, M.J.; Martinez-Gonzalez, I.; McNagny, K.M.; McKenzie, A.N.J.; Takei, F. Group 2 Innate Lymphoid Cells Are Critical for the Initiation of Adaptive T Helper 2 Cell-Mediated Allergic Lung Inflammation. Immunity 2014, 40, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Halim, T.Y.F.; Hwang, Y.Y.; Scanlon, S.T.; Zaghouani, H.; Garbi, N.; Fallon, P.G.; McKenzie, A.N.J. Group 2 Innate Lymphoid Cells License Dendritic Cells to Potentiate Memory TH2 Cell Responses. Nat. Immunol. 2016, 17, 57–64. [Google Scholar] [CrossRef]
- Obata-Ninomiya, K.; Ishiwata, K.; Nakano, H.; Endo, Y.; Ichikawa, T.; Onodera, A.; Hirahara, K.; Okamoto, Y.; Kanuka, H.; Nakayama, T. CXCR6+ST2+ Memory T Helper 2 Cells Induced the Expression of Major Basic Protein in Eosinophils to Reduce the Fecundity of Helminth. Proc. Natl. Acad. Sci. USA 2018, 115, E9849–E9858. [Google Scholar] [CrossRef]
- Hammad, H.; Lambrecht, B.N. The Basic Immunology of Asthma. Cell 2021, 184, 2521–2522. [Google Scholar] [CrossRef]
- Fahy, J.V.; Dickey, B.F. Airway Mucus Function and Dysfunction. N. Engl. J. Med. 2010, 363, 2233–2247. [Google Scholar] [CrossRef]
- Zissler, U.M.; Chaker, A.M.; Effner, R.; Ulrich, M.; Guerth, F.; Piontek, G.; Dietz, K.; Regn, M.; Knapp, B.; Theis, F.J.; et al. Interleukin-4 and Interferon-γ Orchestrate an Epithelial Polarization in the Airways. Mucosal Immunol. 2016, 9, 917–926. [Google Scholar] [CrossRef]
- Massagué, J. TGFβ Signalling in Context. Nat. Rev. Mol. Cell. Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef]
- Yu, X.; Buttgereit, A.; Lelios, I.; Utz, S.G.; Cansever, D.; Becher, B.; Greter, M. The Cytokine TGF-β Promotes the Development and Homeostasis of Alveolar Macrophages. Immunity 2017, 47, 903–912.e4. [Google Scholar] [CrossRef]
- Denney, L.; Byrne, A.J.; Shea, T.J.; Buckley, J.S.; Pease, J.E.; Herledan, G.M.F.; Walker, S.A.; Gregory, L.G.; Lloyd, C.M. Pulmonary Epithelial Cell-Derived Cytokine TGF-Β1 Is a Critical Cofactor for Enhanced Innate Lymphoid Cell Function. Immunity 2015, 43, 945–958. [Google Scholar] [CrossRef] [PubMed]
- Travis, M.A.; Sheppard, D. TGF-β Activation and Function in Immunity. Annu. Rev. Immunol. 2014, 32, 51–82. [Google Scholar] [CrossRef] [PubMed]
- Kelly, A.; Houston, S.A.; Sherwood, E.; Casulli, J.; Travis, M.A. Regulation of Innate and Adaptive Immunity by TGFβ. Adv. Immunol. 2017, 134, 137–233. [Google Scholar] [CrossRef]
- Kariyawasam, H.H.; Pegorier, S.; Barkans, J.; Xanthou, G.; Aizen, M.; Ying, S.; Kay, A.B.; Lloyd, C.M.; Robinson, D.S. Activin and Transforming Growth Factor-Beta Signaling Pathways Are Activated after Allergen Challenge in Mild Asthma. J. Allergy Clin. Immunol. 2009, 124, 454–462. [Google Scholar] [CrossRef] [PubMed]
- Hansen, G.; McIntire, J.J.; Yeung, V.P.; Berry, G.; Thorbecke, G.J.; Chen, L.; DeKruyff, R.H.; Umetsu, D.T. CD4+ T Helper Cells Engineered to Produce Latent TGF-Beta1 Reverse Allergen-Induced Airway Hyperreactivity and Inflammation. J. Clin. Investig. 2000, 105, 61–70. [Google Scholar] [CrossRef]
- Kulkarni, A.B.; Huh, C.G.; Becker, D.; Geiser, A.; Lyght, M.; Flanders, K.C.; Roberts, A.B.; Sporn, M.B.; Ward, J.M.; Karlsson, S. Transforming Growth Factor Beta 1 Null Mutation in Mice Causes Excessive Inflammatory Response and Early Death. Proc. Natl. Acad. Sci. USA 1993, 90, 770–774. [Google Scholar] [CrossRef]
- Scherf, W.; Burdach, S.; Hansen, G. Reduced Expression of Transforming Growth Factor Beta 1 Exacerbates Pathology in an Experimental Asthma Model. Eur. J. Immunol. 2005, 35, 198–206. [Google Scholar] [CrossRef]
- Gregory, L.G.; Mathie, S.A.; Walker, S.A.; Pegorier, S.; Jones, C.P.; Lloyd, C.M. Overexpression of Smad2 Drives House Dust Mite–Mediated Airway Remodeling and Airway Hyperresponsiveness via Activin and IL-25. Am. J. Respir. Crit. Care Med. 2010, 182, 143–154. [Google Scholar] [CrossRef]
- Gregory, L.G.; Jones, C.P.; Mathie, S.A.; Pegorier, S.; Lloyd, C.M. Endothelin-1 Directs Airway Remodeling and Hyper-Reactivity in a Murine Asthma Model. Allergy 2013, 68, 1579–1588. [Google Scholar] [CrossRef]
- Nold, M.F.; Nold-Petry, C.A.; Zepp, J.A.; Palmer, B.E.; Bufler, P.; Dinarello, C.A. IL-37 Is a Fundamental Inhibitor of Innate Immunity. Nat. Immunol. 2010, 11, 1014–1022. [Google Scholar] [CrossRef]
- Kim, M.S.; Baek, A.R.; Lee, J.H.; Jang, A.S.; Kim, D.J.; Chin, S.S.; Park, S.W. IL-37 Attenuates Lung Fibrosis by Inducing Autophagy and Regulating TGF-Β1 Production in Mice. J. Immunol. 2019, 203, 2265–2275. [Google Scholar] [CrossRef]
- Nold-Petry, C.A.; Lo, C.Y.; Rudloff, I.; Elgass, K.D.; Li, S.; Gantier, M.P.; Lotz-Havla, A.S.; Gersting, S.W.; Cho, S.X.; Lao, J.C.; et al. IL-37 Requires the Receptors IL-18Rα and IL-1R8 (SIGIRR) to Carry out Its Multifaceted Anti-Inflammatory Program upon Innate Signal Transduction. Nat. Immunol. 2015, 16, 354–365. [Google Scholar] [CrossRef]
- Lunding, L.; Webering, S.; Vock, C.; Schröder, A.; Raedler, D.; Schaub, B.; Fehrenbach, H.; Wegmann, M. IL-37 Requires IL-18Rα and SIGIRR/IL-1R8 to Diminish Allergic Airway Inflammation in Mice. Allergy 2015, 70, 366–373. [Google Scholar] [CrossRef]
- Schröder, A.; Lunding, L.P.; Zissler, U.M.; Vock, C.; Webering, S.; Ehlers, J.C.; Orinska, Z.; Chaker, A.; Schmidt-Weber, C.B.; Lang, N.J.; et al. IL-37 Regulates Allergic Inflammation by Counterbalancing pro-Inflammatory IL-1 and IL-33. Allergy 2022, 77, 856–869. [Google Scholar] [CrossRef]
- Charrad, R.; Berraïes, A.; Hamdi, B.; Ammar, J.; Hamzaoui, K.; Hamzaoui, A. Anti-Inflammatory Activity of IL-37 in Asthmatic Children: Correlation with Inflammatory Cytokines TNF-α, IL-β, IL-6 and IL-17A. Immunobiology 2016, 221, 182–187. [Google Scholar] [CrossRef]
- Abushouk, A.; Alkhalaf, H.; Aldamegh, M.; Bin Shigair, S.; Mahabbat, N.; Hakami, M.; Abu-Jaffal, A.S.; Nasr, A. IL-35 and IL-37 Are Negatively Correlated with High IgE Production among Children with Asthma in Saudi Arabia. J. Asthma 2022, 59, 655–662. [Google Scholar] [CrossRef]
- Huang, N.; Liu, K.; Liu, J.; Gao, X.; Zeng, Z.; Zhang, Y.; Chen, J. Interleukin-37 Alleviates Airway Inflammation and Remodeling in Asthma via Inhibiting the Activation of NF-ΚB and STAT3 Signalings. Int. Immunopharmacol. 2018, 55, 198–204. [Google Scholar] [CrossRef]
- Meng, P.; Chen, Z.-G.; Zhang, T.-T.; Liang, Z.-Z.; Zou, X.-L.; Yang, H.-L.; Li, H.-T. IL-37 Alleviates House Dust Mite-Induced Chronic Allergic Asthma by Targeting TSLP through the NF-ΚB and ERK1/2 Signaling Pathways. Immunol. Cell. Biol. 2019, 97, 403–415. [Google Scholar] [CrossRef]
- Lv, J.; Xiong, Y.; Li, W.; Cui, X.; Cheng, X.; Leng, Q.; He, R. IL-37 Inhibits IL-4/IL-13-Induced CCL11 Production and Lung Eosinophilia in Murine Allergic Asthma. Allergy 2018, 73, 1642–1652. [Google Scholar] [CrossRef]
- Netea, M.G.; Joosten, L.A.B.; Latz, E.; Mills, K.H.G.; Natoli, G.; Stunnenberg, H.G.; O’Neill, L.A.J.; Xavier, R.J. Trained Immunity: A Program of Innate Immune Memory in Health and Disease. Science 2016, 352, aaf1098. [Google Scholar] [CrossRef]
- Loske, J.; Röhmel, J.; Lukassen, S.; Stricker, S.; Magalhães, V.G.; Liebig, J.; Chua, R.L.; Thürmann, L.; Messingschlager, M.; Seegebarth, A.; et al. Pre-Activated Antiviral Innate Immunity in the Upper Airways Controls Early SARS-CoV-2 Infection in Children. Nat. Biotechnol. 2021, 40, 319–324. [Google Scholar] [CrossRef]
- Rawlins, E.L.; Hogan, B.L.M. Ciliated Epithelial Cell Lifespan in the Mouse Trachea and Lung. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 295, L231–L234. [Google Scholar] [CrossRef]
- Schneider, C.; Nobs, S.P.; Kurrer, M.; Rehrauer, H.; Thiele, C.; Kopf, M. Induction of the Nuclear Receptor PPAR-γ by the Cytokine GM-CSF Is Critical for the Differentiation of Fetal Monocytes into Alveolar Macrophages. Nat. Immunol. 2014, 15, 1026–1037. [Google Scholar] [CrossRef]
- Gibbings, S.L.; Thomas, S.M.; Atif, S.M.; McCubbrey, A.L.; Desch, A.N.; Danhorn, T.; Leach, S.M.; Bratton, D.L.; Henson, P.M.; Janssen, W.J.; et al. Three Unique Interstitial Macrophages in the Murine Lung at Steady State. Am. J. Respir. Cell. Mol. Biol. 2017, 57, 66–76. [Google Scholar] [CrossRef]
- Engler, A.E.; Ysasi, A.B.; Pihl, R.M.F.; Villacorta-Martin, C.; Heston, H.M.; Richardson, H.M.K.; Thapa, B.R.; Moniz, N.R.; Belkina, A.C.; Mazzilli, S.A.; et al. Airway-Associated Macrophages in Homeostasis and Repair. Cell. Rep. 2020, 33, 108553. [Google Scholar] [CrossRef]
- Naik, S.; Larsen, S.B.; Gomez, N.C.; Alaverdyan, K.; Sendoel, A.; Yuan, S.; Polak, L.; Kulukian, A.; Chai, S.; Fuchs, E. Inflammatory Memory Sensitizes Skin Epithelial Stem Cells to Tissue Damage. Nature 2017, 550, 475–480. [Google Scholar] [CrossRef]
- Ordovas-Montanes, J.; Dwyer, D.F.; Nyquist, S.K.; Buchheit, K.M.; Vukovic, M.; Deb, C.; Wadsworth, M.H.; Hughes, T.K.; Kazer, S.W.; Yoshimoto, E.; et al. Allergic Inflammatory Memory in Human Respiratory Epithelial Progenitor Cells. Nature 2018, 560, 649–654. [Google Scholar] [CrossRef]
- Beyaz, S.; Mana, M.D.; Roper, J.; Kedrin, D.; Saadatpour, A.; Hong, S.-J.; Bauer-Rowe, K.E.; Xifaras, M.E.; Akkad, A.; Arias, E.; et al. High-Fat Diet Enhances Stemness and Tumorigenicity of Intestinal Progenitors. Nature 2016, 531, 53–58. [Google Scholar] [CrossRef]
- Bigot, J.; Guillot, L.; Guitard, J.; Ruffin, M.; Corvol, H.; Chignard, M.; Hennequin, C.; Balloy, V. Respiratory Epithelial Cells Can Remember Infection: A Proof-of-Concept Study. J. Infect. Dis. 2020, 221, 1000–1005. [Google Scholar] [CrossRef]
- Carlier, F.M.; de Fays, C.; Pilette, C. Epithelial Barrier Dysfunction in Chronic Respiratory Diseases. Front. Physiol. 2021, 12, 691227. [Google Scholar] [CrossRef]
- Kim, Y.; Kugler, M.C.; Wei, Y.; Kim, K.K.; Li, X.; Brumwell, A.N.; Chapman, H.A. Integrin Alpha3beta1-Dependent Beta-Catenin Phosphorylation Links Epithelial Smad Signaling to Cell Contacts. J. Cell. Biol. 2009, 184, 309–322. [Google Scholar] [CrossRef]
- Boland, S.; Boisvieux-Ulrich, E.; Houcine, O.; Baeza-Squiban, A.; Pouchelet, M.; Schoëvaërt, D.; Marano, F. TGF Beta 1 Promotes Actin Cytoskeleton Reorganization and Migratory Phenotype in Epithelial Tracheal Cells in Primary Culture. J. Cell. Sci. 1996, 109 Pt 9, 2207–2219. [Google Scholar] [CrossRef]
- Crosby, L.M.; Waters, C.M. Epithelial Repair Mechanisms in the Lung. Am. J. Physiol. Lung Cell. Mol. Physiol. 2010, 298, L715–L731. [Google Scholar] [CrossRef]
- Seibold, M.A. Interleukin-13 Stimulation Reveals the Cellular and Functional Plasticity of the Airway Epithelium. Ann. Am. Thorac. Soc. 2018, 15, S98–S102. [Google Scholar] [CrossRef]
- Gomi, K.; Tang, Y.; Arbelaez, V.; Crystal, R.G.; Walters, M.S. Endothelial Cell Mediated Promotion of Ciliated Cell Differentiation of Human Airway Basal Cells via Insulin and Insulin-Like Growth Factor 1 Receptor Mediated Signaling. Stem Cell Rev. Rep. 2017, 13, 309–317. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frey, A.; Lunding, L.P.; Wegmann, M. The Dual Role of the Airway Epithelium in Asthma: Active Barrier and Regulator of Inflammation. Cells 2023, 12, 2208. https://doi.org/10.3390/cells12182208
Frey A, Lunding LP, Wegmann M. The Dual Role of the Airway Epithelium in Asthma: Active Barrier and Regulator of Inflammation. Cells. 2023; 12(18):2208. https://doi.org/10.3390/cells12182208
Chicago/Turabian StyleFrey, Andreas, Lars P. Lunding, and Michael Wegmann. 2023. "The Dual Role of the Airway Epithelium in Asthma: Active Barrier and Regulator of Inflammation" Cells 12, no. 18: 2208. https://doi.org/10.3390/cells12182208