
A Review of Single-Cell RNA-Seq Annotation, Integration, and Cell–Cell Communication

Abstract
:1. Introduction
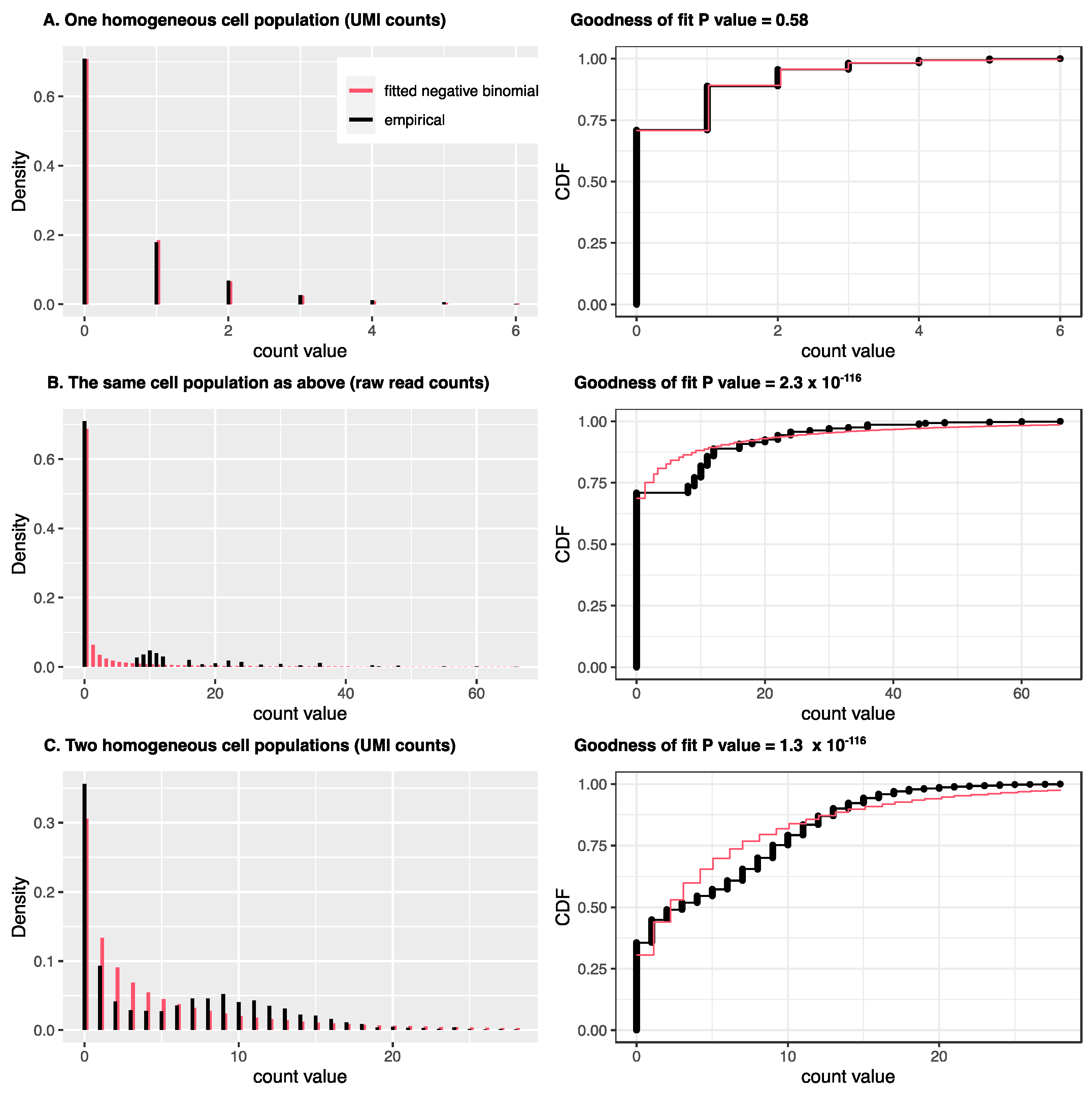
2. Statistical Count Modeling for scRNA-Seq and Spatial Transcriptomics
3. Cell-Type Annotation
3.1. Cell Annotation by Signature Scoring
3.1.1. Signature Database
3.1.2. Scoring Method
- Cell marker databases are compiled from diverse data sources generated using different technologies, each with its own technical biases such as sensitivity, dropouts, and cell population purity. The derived signatures for the same cell type can therefore vary across technologies. Additionally, signatures obtained from bulk RNA-seq or microarray data may not accurately annotate cell types in single-cell data.
- There is a lack of consistent criteria or methods for curating signatures. Gene sets can be derived experimentally, computationally, or manually curated from the literature. Even computational selection methods, such as differential expression analysis, can result in different gene sets due to arbitrary cutoffs (e.g., log2 fold change, false discovery rate, top number of genes).
- The size of gene sets (i.e., the number of genes they contain) varies greatly, making it difficult to compare the scores of different signatures. Smaller gene sets (e.g., size < 20) are more likely to yield cells with unstable scores, while larger gene sets (e.g., size > 100) can provide greater stability for detection and evaluation. It is often observed that the signature scores of large random gene sets follow an approximately normal distribution, abiding by the central limit theorem.
- Redundancy across gene sets is common in large databases. Since gene sets may share a significant proportion of their constituent genes, scoring results can be dominated by long lists of candidate cell types associated with overlapping signatures, potentially obscuring meaningful cell types that possess only a few marker genes.
- Most databases adopt a flat structure, treating each cell type equally and independently. While this approach can effectively distinguish major cell types, it may struggle to identify cell subtypes due to the lack of relationships between cell types. Hierarchical cell type databases could enhance discrimination of specific cell types or subtypes [77].
- Unstandardized cell nomenclature in certain publications can lead to overlapping or ambiguous anatomy terms or identifiers for cell types. To address this, collaborative efforts such as the Cell Ontology (CL) and The Human Cell Atlas (HCA) have begun to build a high-dimensional compendium of cell information.
- Assess the reliability of cell annotation by plotting the score histogram of a specific gene set and examining the distribution of scores within cell types in the dataset.
- Visualize the signature scores or average expression of a gene set in a two-dimensional plot. Calculating the mean expression with library-size normalization provides an intuitive approach.
- Some methods are sensitive to the number of detected genes or dropout rates. Checking marker gene expression through dot plots or stacked violin plots can help to identify potential issues.
- Employ a confusion matrix or mosaic plot to evaluate the final assignment of cell type labels.
3.2. Cell Annotation by Supervised Learning
3.2.1. Feature Selection
3.2.2. Prediction Model (Classifier)
- Accuracy: This metric captures the ratio of correctly classified cell types to the total number of cells, providing a broad view of model performance.
- Adjusted Rand Index (ARI): ARI allows for the comparison of clustering patterns between the predicted and actual (ground truth) classifications. It offers an insight into how closely the model’s clustering aligns with the actual data.
- F1 score: The F1 score offers a robust measure of a model’s classification accuracy. It amalgamates precision and recall into a single measure by averaging the individual F1 scores for each class. It provides a more nuanced view of model performance, especially in scenarios where class imbalances exist.
- Normalized Mutual Information (NMI): NMI is a metric that quantifies the shared information between the predicted and ground truth distributions. By normalizing against the maximum possible mutual information value, it gives a relative perspective on how much the predicted labels reveal about the actual labels, which is particularly useful in clustering contexts.
- Variation of Information (VI): VI evaluates the degree of difference between predicted and actual labels. It effectively gauges how much the model’s classification deviates from the true label distribution.
3.3. Other Cell Annotation Methods
3.3.1. Cell-Integration-Based Label Transfer
3.3.2. Semi-Supervised Annotation
3.4. Perspective
4. Single Cell Data Integration
4.1. Mapping
4.2. Deconvolution
4.3. Multimodality Fusion
4.4. Linear and Non-Linear Modeling
4.5. Batch Correction for Cell-Level Analysis and Gene-Level Analysis
4.6. Available Benchmark Results
4.7. Assumptions, Potential Limitations, and Future Direction
5. Cell–Cell Communication
5.1. Cell–Cell Communication: Genes’ View
5.2. Cell–Cell Communication: Cells’ View
5.3. Cell–Cell Communication: Spatial View
5.4. Cell–Cell Communications: Perspective
6. Summary and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tang, F.; Barbacioru, C.; Wang, Y.; Nordman, E.; Lee, C.; Xu, N.; Wang, X.; Bodeau, J.; Tuch, B.B.; Siddiqui, A.; et al. MRNA-Seq Whole-Transcriptome Analysis of a Single Cell. Nat. Methods 2009, 6, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Sandberg, R. Entering the Era of Single-Cell Transcriptomics in Biology and Medicine. Nat. Methods 2014, 11, 22–24. [Google Scholar] [CrossRef] [Green Version]
- Ståhl, P.L.; Salmén, F.; Vickovic, S.; Lundmark, A.; Navarro, J.F.; Magnusson, J.; Giacomello, S.; Asp, M.; Westholm, J.O.; Huss, M. Visualization and Analysis of Gene Expression in Tissue Sections by Spatial Transcriptomics. Science 2016, 353, 78–82. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.H.; Boettiger, A.N.; Moffitt, J.R.; Wang, S.; Zhuang, X. Spatially Resolved, Highly Multiplexed RNA Profiling in Single Cells. Science 2015, 348, aaa6090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merritt, C.R.; Ong, G.T.; Church, S.E.; Barker, K.; Danaher, P.; Geiss, G.; Hoang, M.; Jung, J.; Liang, Y.; McKay-Fleisch, J. Multiplex Digital Spatial Profiling of Proteins and RNA in Fixed Tissue. Nat. Biotechnol. 2020, 38, 586–599. [Google Scholar] [CrossRef] [PubMed]
- Marx, V. Method of the Year: Spatially Resolved Transcriptomics. Nat. Methods 2021, 18, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Aran, D. Single-Cell RNA Sequencing for Studying Human Cancers. Annu. Rev. Biomed. Data Sci. 2023, 6. [Google Scholar] [CrossRef]
- Chen, H.; Ye, F.; Guo, G. Revolutionizing Immunology with Single-Cell RNA Sequencing. Cell. Mol. Immunol. 2019, 16, 242–249. [Google Scholar] [CrossRef] [Green Version]
- Papalexi, E.; Satija, R. Single-Cell RNA Sequencing to Explore Immune Cell Heterogeneity. Nat. Rev. Immunol. 2018, 18, 35–45. [Google Scholar] [CrossRef]
- Davis-Marcisak, E.F.; Deshpande, A.; Stein-O’Brien, G.L.; Ho, W.J.; Laheru, D.; Jaffee, E.M.; Fertig, E.J.; Kagohara, L.T. From Bench to Bedside: Single-Cell Analysis for Cancer Immunotherapy. Cancer Cell 2021, 39, 1062–1080. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Chen, Y.; Jing, Y.; Green, M.R.; Han, L. Advancing CAR T Cell Therapy through the Use of Multidimensional Omics Data. Nat. Rev. Clin. Oncol. 2023, 20, 211–228. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.X.; Terry, J.M.; Belgrader, P.; Ryvkin, P.; Bent, Z.W.; Wilson, R.; Ziraldo, S.B.; Wheeler, T.D.; McDermott, G.P.; Zhu, J. Massively Parallel Digital Transcriptional Profiling of Single Cells. Nat. Commun. 2017, 8, 14049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramsköld, D.; Luo, S.; Wang, Y.-C.; Li, R.; Deng, Q.; Faridani, O.R.; Daniels, G.A.; Khrebtukova, I.; Loring, J.F.; Laurent, L.C. Full-Length MRNA-Seq from Single-Cell Levels of RNA and Individual Circulating Tumor Cells. Nat. Biotechnol. 2012, 30, 777–782. [Google Scholar] [CrossRef] [Green Version]
- Picelli, S.; Faridani, O.R.; Björklund, Å.K.; Winberg, G.; Sagasser, S.; Sandberg, R. Full-Length RNA-Seq from Single Cells Using Smart-Seq2. Nat. Protoc. 2014, 9, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Ke, R.; Mignardi, M.; Pacureanu, A.; Svedlund, J.; Botling, J.; Wählby, C.; Nilsson, M. In Situ Sequencing for RNA Analysis in Preserved Tissue and Cells. Nat. Methods 2013, 10, 857–860. [Google Scholar] [CrossRef]
- Raj, A.; Van Den Bogaard, P.; Rifkin, S.A.; Van Oudenaarden, A.; Tyagi, S. Imaging Individual MRNA Molecules Using Multiple Singly Labeled Probes. Nat. Methods 2008, 5, 877–879. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Allen, W.E.; Wright, M.A.; Sylwestrak, E.L.; Samusik, N.; Vesuna, S.; Evans, K.; Liu, C.; Ramakrishnan, C.; Liu, J. Three-Dimensional Intact-Tissue Sequencing of Single-Cell Transcriptional States. Science 2018, 361, eaat5691. [Google Scholar] [CrossRef] [Green Version]
- Moffitt, J.R.; Bambah-Mukku, D.; Eichhorn, S.W.; Vaughn, E.; Shekhar, K.; Perez, J.D.; Rubinstein, N.D.; Hao, J.; Regev, A.; Dulac, C.; et al. Molecular, Spatial and Functional Single-Cell Profiling of the Hypothalamic Preoptic Region. Science 2018, 362, eaau5324. [Google Scholar] [CrossRef] [Green Version]
- Lubeck, E.; Coskun, A.F.; Zhiyentayev, T.; Ahmad, M.; Cai, L. Single-Cell in Situ RNA Profiling by Sequential Hybridization. Nat. Methods 2014, 11, 360–361. [Google Scholar] [CrossRef] [Green Version]
- Eng, C.-H.L.; Lawson, M.; Zhu, Q.; Dries, R.; Koulena, N.; Takei, Y.; Yun, J.; Cronin, C.; Karp, C.; Yuan, G.-C.; et al. Transcriptome-Scale Super-Resolved Imaging in Tissues by RNA SeqFISH+. Nature 2019, 568, 235–239. [Google Scholar] [CrossRef]
- Vickovic, S.; Eraslan, G.; Salmén, F.; Klughammer, J.; Stenbeck, L.; Schapiro, D.; Äijö, T.; Bonneau, R.; Bergenstråhle, L.; Navarro, J.F.; et al. High-Definition Spatial Transcriptomics for in Situ Tissue Profiling. Nat. Methods 2019, 16, 987–990. [Google Scholar] [CrossRef]
- Rodriques, S.G.; Stickels, R.R.; Goeva, A.; Martin, C.A.; Murray, E.; Vanderburg, C.R.; Welch, J.; Chen, L.M.; Chen, F.; Macosko, E.Z. Slide-Seq: A Scalable Technology for Measuring Genome-Wide Expression at High Spatial Resolution. Science 2019, 363, 1463–1467. [Google Scholar] [CrossRef]
- Stickels, R.R.; Murray, E.; Kumar, P.; Li, J.; Marshall, J.L.; Di Bella, D.J.; Arlotta, P.; Macosko, E.Z.; Chen, F. Highly Sensitive Spatial Transcriptomics at Near-Cellular Resolution with Slide-SeqV2. Nat. Biotechnol. 2021, 39, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Buenrostro, J.D.; Wu, B.; Litzenburger, U.M.; Ruff, D.; Gonzales, M.L.; Snyder, M.P.; Chang, H.Y.; Greenleaf, W.J. Single-Cell Chromatin Accessibility Reveals Principles of Regulatory Variation. Nature 2015, 523, 486–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satpathy, A.T.; Granja, J.M.; Yost, K.E.; Qi, Y.; Meschi, F.; McDermott, G.P.; Olsen, B.N.; Mumbach, M.R.; Pierce, S.E.; Corces, M.R.; et al. Massively Parallel Single-Cell Chromatin Landscapes of Human Immune Cell Development and Intratumoral T Cell Exhaustion. Nat. Biotechnol. 2019, 37, 925–936. [Google Scholar] [CrossRef]
- Cusanovich, D.A.; Daza, R.; Adey, A.; Pliner, H.A.; Christiansen, L.; Gunderson, K.L.; Steemers, F.J.; Trapnell, C.; Shendure, J. Multiplex Single Cell Profiling of Chromatin Accessibility by Combinatorial Cellular Indexing. Science 2015, 348, 910–914. [Google Scholar] [CrossRef] [Green Version]
- Guo, H.; Zhu, P.; Guo, F.; Li, X.; Wu, X.; Fan, X.; Wen, L.; Tang, F. Profiling DNA Methylome Landscapes of Mammalian Cells with Single-Cell Reduced-Representation Bisulfite Sequencing. Nat. Protoc. 2015, 10, 645–659. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Rivkin, A.; Zhou, J.; Sandoval, J.P.; Kurihara, L.; Lucero, J.; Castanon, R.; Nery, J.R.; Pinto-Duarte, A.; Bui, B.; et al. Robust Single-Cell DNA Methylome Profiling with SnmC-Seq2. Nat. Commun. 2018, 9, 3824. [Google Scholar] [CrossRef] [Green Version]
- Han, L.; Wu, H.-J.; Zhu, H.; Kim, K.-Y.; Marjani, S.L.; Riester, M.; Euskirchen, G.; Zi, X.; Yang, J.; Han, J.; et al. Bisulfite-Independent Analysis of CpG Island Methylation Enables Genome-Scale Stratification of Single Cells. Nucleic Acids Res. 2017, 45, e77. [Google Scholar] [CrossRef] [Green Version]
- Ma, S.; Zhang, B.; LaFave, L.; Earl, A.S.; Chiang, Z.; Hu, Y.; Ding, J.; Brack, A.; Kartha, V.K.; Tay, T.; et al. Chromatin Potential Identified by Shared Single-Cell Profiling of RNA and Chromatin. Cell 2020, 183, 1103–1116.e20. [Google Scholar] [CrossRef]
- Stoeckius, M.; Hafemeister, C.; Stephenson, W.; Houck-Loomis, B.; Chattopadhyay, P.K.; Swerdlow, H.; Satija, R.; Smibert, P. Simultaneous Epitope and Transcriptome Measurement in Single Cells. Nat. Methods 2017, 14, 865–868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mimitou, E.P.; Lareau, C.A.; Chen, K.Y.; Zorzetto-Fernandes, A.L.; Hao, Y.; Takeshima, Y.; Luo, W.; Huang, T.-S.; Yeung, B.Z.; Papalexi, E.; et al. Scalable, Multimodal Profiling of Chromatin Accessibility, Gene Expression and Protein Levels in Single Cells. Nat. Biotechnol. 2021, 39, 1246–1258. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Cao, Y.; Yang, J.Y.H.; Yang, P. Benchmarking Clustering Algorithms on Estimating the Number of Cell Types from Single-Cell RNA-Sequencing Data. Genome Biol. 2022, 23, 49. [Google Scholar] [CrossRef]
- Cheng, C.; Easton, J.; Rosencrance, C.; Li, Y.; Ju, B.; Williams, J.; Mulder, H.L.; Pang, Y.; Chen, W.; Chen, X. Latent Cellular Analysis Robustly Reveals Subtle Diversity in Large-Scale Single-Cell RNA-Seq Data. Nucleic Acids Res. 2019, 47, e143. [Google Scholar] [CrossRef] [Green Version]
- Trapnell, C.; Cacchiarelli, D.; Grimsby, J.; Pokharel, P.; Li, S.; Morse, M.; Lennon, N.J.; Livak, K.J.; Mikkelsen, T.S.; Rinn, J.L. The Dynamics and Regulators of Cell Fate Decisions Are Revealed by Pseudotemporal Ordering of Single Cells. Nat. Biotechnol. 2014, 32, 381–386. [Google Scholar] [CrossRef] [Green Version]
- Cao, J.; Spielmann, M.; Qiu, X.; Huang, X.; Ibrahim, D.M.; Hill, A.J.; Zhang, F.; Mundlos, S.; Christiansen, L.; Steemers, F.J.; et al. The Single-Cell Transcriptional Landscape of Mammalian Organogenesis. Nature 2019, 566, 496–502. [Google Scholar] [CrossRef]
- Qiu, X.; Mao, Q.; Tang, Y.; Wang, L.; Chawla, R.; Pliner, H.A.; Trapnell, C. Reversed Graph Embedding Resolves Complex Single-Cell Trajectories. Nat. Methods 2017, 14, 979–982. [Google Scholar] [CrossRef] [Green Version]
- Comitani, F.; Nash, J.O.; Cohen-Gogo, S.; Chang, A.I.; Wen, T.T.; Maheshwari, A.; Goyal, B.; Tio, E.S.; Tabatabaei, K.; Mayoh, C.; et al. Diagnostic Classification of Childhood Cancer Using Multiscale Transcriptomics. Nat. Med. 2023, 29, 656–666. [Google Scholar] [CrossRef] [PubMed]
- Kleinberg, J. An Impossibility Theorem for Clustering. Adv. Neural Inf. Process. Syst. 2002, 15, 1–8. [Google Scholar]
- Carlsson, G. Topology and Data. Bull. Am. Math. Soc. 2009, 46, 255–308. [Google Scholar] [CrossRef] [Green Version]
- Zeng, H. What Is a Cell Type and How to Define It? Cell 2022, 185, 2739–2755. [Google Scholar] [CrossRef]
- Qiu, X.; Hill, A.; Packer, J.; Lin, D.; Ma, Y.-A.; Trapnell, C. Single-Cell MRNA Quantification and Differential Analysis with Census. Nat. Methods 2017, 14, 309–315. [Google Scholar] [CrossRef]
- Kharchenko, P.V.; Silberstein, L.; Scadden, D.T. Bayesian Approach to Single-Cell Differential Expression Analysis. Nat. Methods 2014, 11, 740–742. [Google Scholar] [CrossRef]
- Chen, W.; Li, Y.; Easton, J.; Finkelstein, D.; Wu, G.; Chen, X. UMI-Count Modeling and Differential Expression Analysis for Single-Cell RNA Sequencing. Genome Biol. 2018, 19, 70. [Google Scholar] [CrossRef] [Green Version]
- Svensson, V. Droplet ScRNA-Seq Is Not Zero-Inflated. Nat. Biotechnol. 2020, 38, 147–150. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.H.; Zhou, X.; Chen, M. Demystifying “Drop-Outs” in Single-Cell UMI Data. Genome Biol. 2020, 21, 196. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, A.; Stephens, M. Separating Measurement and Expression Models Clarifies Confusion in Single-Cell RNA Sequencing Analysis. Nat. Genet. 2021, 53, 770–777. [Google Scholar] [CrossRef] [PubMed]
- Jiang, R.; Sun, T.; Song, D.; Li, J.J. Statistics or Biology: The Zero-Inflation Controversy about ScRNA-Seq Data. Genome Biol. 2022, 23, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Kleshchevnikov, V.; Shmatko, A.; Dann, E.; Aivazidis, A.; King, H.W.; Li, T.; Elmentaite, R.; Lomakin, A.; Kedlian, V.; Gayoso, A. Cell2location Maps Fine-Grained Cell Types in Spatial Transcriptomics. Nat. Biotechnol. 2022, 40, 661–671. [Google Scholar] [CrossRef]
- Andersson, A.; Bergenstråhle, J.; Asp, M.; Bergenstråhle, L.; Jurek, A.; Fernández Navarro, J.; Lundeberg, J. Single-Cell and Spatial Transcriptomics Enables Probabilistic Inference of Cell Type Topography. Commun. Biol. 2020, 3, 565. [Google Scholar] [CrossRef]
- Lopez, R.; Li, B.; Keren-Shaul, H.; Boyeau, P.; Kedmi, M.; Pilzer, D.; Jelinski, A.; Yofe, I.; David, E.; Wagner, A. DestVI Identifies Continuums of Cell Types in Spatial Transcriptomics Data. Nat. Biotechnol. 2022, 40, 1360–1369. [Google Scholar] [CrossRef]
- Lopez, R.; Nazaret, A.; Langevin, M.; Samaran, J.; Regier, J.; Jordan, M.I.; Yosef, N. A Joint Model of Unpaired Data from ScRNA-Seq and Spatial Transcriptomics for Imputing Missing Gene Expression Measurements. arXiv 2019, arXiv:190502269. [Google Scholar]
- Xu, C.; Lopez, R.; Mehlman, E.; Regier, J.; Jordan, M.I.; Yosef, N. Probabilistic Harmonization and Annotation of Single-cell Transcriptomics Data with Deep Generative Models. Mol. Syst. Biol. 2021, 17, e9620. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Zhu, J.; Ma, Y.; Zhou, X. Modeling Zero Inflation Is Not Necessary for Spatial Transcriptomics. Genome Biol. 2022, 23, 118. [Google Scholar] [CrossRef] [PubMed]
- Van der Maaten, L.; Hinton, G. Visualizing Data Using T-SNE. J. Mach. Learn. Res. 2008, 9. [Google Scholar]
- Becht, E.; McInnes, L.; Healy, J.; Dutertre, C.-A.; Kwok, I.W.; Ng, L.G.; Ginhoux, F.; Newell, E.W. Dimensionality Reduction for Visualizing Single-Cell Data Using UMAP. Nat. Biotechnol. 2019, 37, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Satija, R.; Farrell, J.A.; Gennert, D.; Schier, A.F.; Regev, A. Spatial Reconstruction of Single-Cell Gene Expression Data. Nat. Biotechnol. 2015, 33, 495–502. [Google Scholar] [CrossRef] [Green Version]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database Hallmark Gene Set Collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [Green Version]
- Lachmann, A.; Torre, D.; Keenan, A.B.; Jagodnik, K.M.; Lee, H.J.; Wang, L.; Silverstein, M.C.; Ma’ayan, A. Massive Mining of Publicly Available RNA-Seq Data from Human and Mouse. Nat. Commun. 2018, 9, 1366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palasca, O.; Santos, A.; Stolte, C.; Gorodkin, J.; Jensen, L.J. TISSUES 2.0: An Integrative Web Resource on Mammalian Tissue Expression. Database 2018, 2018, bay003. [Google Scholar] [CrossRef]
- Lopez, D.; Montoya, D.; Ambrose, M.; Lam, L.; Briscoe, L.; Adams, C.; Modlin, R.L.; Pellegrini, M. SaVanT: A Web-Based Tool for the Sample-Level Visualization of Molecular Signatures in Gene Expression Profiles. BMC Genom. 2017, 18, 824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aran, D.; Hu, Z.; Butte, A.J. XCell: Digitally Portraying the Tissue Cellular Heterogeneity Landscape. Genome Biol. 2017, 18, 220. [Google Scholar] [CrossRef] [Green Version]
- Aran, D.; Looney, A.P.; Liu, L.; Wu, E.; Fong, V.; Hsu, A.; Chak, S.; Naikawadi, R.P.; Wolters, P.J.; Abate, A.R.; et al. Reference-Based Analysis of Lung Single-Cell Sequencing Reveals a Transitional Profibrotic Macrophage. Nat. Immunol. 2019, 20, 163–172. [Google Scholar] [CrossRef]
- Franzén, O.; Gan, L.-M.; Björkegren, J.L.M. PanglaoDB: A Web Server for Exploration of Mouse and Human Single-Cell RNA Sequencing Data. Database 2019, 2019, baz046. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Lan, Y.; Xu, J.; Quan, F.; Zhao, E.; Deng, C.; Luo, T.; Xu, L.; Liao, G.; Yan, M.; et al. CellMarker: A Manually Curated Resource of Cell Markers in Human and Mouse. Nucleic Acids Res. 2019, 47, D721–D728. [Google Scholar] [CrossRef] [Green Version]
- Hu, C.; Li, T.; Xu, Y.; Zhang, X.; Li, F.; Bai, J.; Chen, J.; Jiang, W.; Yang, K.; Ou, Q.; et al. CellMarker 2.0: An Updated Database of Manually Curated Cell Markers in Human/Mouse and Web Tools Based on ScRNA-Seq Data. Nucleic Acids Res. 2023, 51, D870–D876. [Google Scholar] [CrossRef]
- Shao, X.; Liao, J.; Lu, X.; Xue, R.; Ai, N.; Fan, X. ScCATCH: Automatic Annotation on Cell Types of Clusters from Single-Cell RNA Sequencing Data. iScience 2020, 23, 100882. [Google Scholar] [CrossRef] [Green Version]
- Bard, J.; Rhee, S.Y.; Ashburner, M. An Ontology for Cell Types. Genome Biol. 2005, 6, R21. [Google Scholar] [CrossRef] [Green Version]
- Barbie, D.A.; Tamayo, P.; Boehm, J.S.; Kim, S.Y.; Moody, S.E.; Dunn, I.F.; Schinzel, A.C.; Sandy, P.; Meylan, E.; Scholl, C.; et al. Systematic RNA Interference Reveals That Oncogenic KRAS-Driven Cancers Require TBK1. Nature 2009, 462, 108–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hänzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene Set Variation Analysis for Microarray and RNA-Seq Data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foroutan, M.; Bhuva, D.D.; Lyu, R.; Horan, K.; Cursons, J.; Davis, M.J. Single Sample Scoring of Molecular Phenotypes. BMC Bioinform. 2018, 19, 404. [Google Scholar] [CrossRef] [Green Version]
- Pont, F.; Tosolini, M.; Fournié, J.J. Single-Cell Signature Explorer for Comprehensive Visualization of Single Cell Signatures across ScRNA-Seq Datasets. Nucleic Acids Res. 2019, 47, e133. [Google Scholar] [CrossRef] [Green Version]
- Aibar, S.; González-Blas, C.B.; Moerman, T.; Huynh-Thu, V.A.; Imrichova, H.; Hulselmans, G.; Rambow, F.; Marine, J.-C.; Geurts, P.; Aerts, J.; et al. SCENIC: Single-Cell Regulatory Network Inference and Clustering. Nat. Methods 2017, 14, 1083–1086. [Google Scholar] [CrossRef] [Green Version]
- Andreatta, M.; Carmona, S.J. UCell: Robust and Scalable Single-Cell Gene Signature Scoring. Comput. Struct. Biotechnol. J. 2021, 19, 3796–3798. [Google Scholar] [CrossRef] [PubMed]
- Noureen, N.; Ye, Z.; Chen, Y.; Wang, X.; Zheng, S. Signature-Scoring Methods Developed for Bulk Samples Are Not Adequate for Cancer Single-Cell RNA Sequencing Data. eLife 2022, 11, e71994. [Google Scholar] [CrossRef] [PubMed]
- Ianevski, A.; Giri, A.K.; Aittokallio, T. Fully-Automated and Ultra-Fast Cell-Type Identification Using Specific Marker Combinations from Single-Cell Transcriptomic Data. Nat. Commun. 2022, 13, 1246. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Sheng, Q.; Shyr, Y.; Liu, Q. ScMRMA: Single Cell Multiresolution Marker-Based Annotation. Nucleic Acids Res. 2022, 50, e7. [Google Scholar] [CrossRef]
- Regev, A.; Teichmann, S.A.; Lander, E.S.; Amit, I.; Benoist, C.; Birney, E.; Bodenmiller, B.; Campbell, P.; Carninci, P.; Clatworthy, M.; et al. The Human Cell Atlas. eLife 2017, 6, e27041. [Google Scholar] [CrossRef]
- Schaum, N.; Karkanias, J.; Neff, N.F.; May, A.P.; Quake, S.R.; Wyss-Coray, T.; Darmanis, S.; Batson, J.; Botvinnik, O.; Chen, M.B.; et al. Single-Cell Transcriptomics of 20 Mouse Organs Creates a Tabula Muris. Nature 2018, 562, 367–372. [Google Scholar] [CrossRef]
- Han, X.; Wang, R.; Zhou, Y.; Fei, L.; Sun, H.; Lai, S.; Saadatpour, A.; Zhou, Z.; Chen, H.; Ye, F.; et al. Mapping the Mouse Cell Atlas by Microwell-Seq. Cell 2018, 172, 1091–1107.e17. [Google Scholar] [CrossRef] [Green Version]
- Cortes, C.; Vapnik, V. Support-Vector Networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef]
- Breiman, L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef] [Green Version]
- Cover, T.; Hart, P. Nearest Neighbor Pattern Classification. IEEE Trans. Inf. Theory 1967, 13, 21–27. [Google Scholar] [CrossRef] [Green Version]
- McCulloch, W.S.; Pitts, W. A Logical Calculus of the Ideas Immanent in Nervous Activity. Bull. Math. Biophys. 1943, 5, 115–133. [Google Scholar] [CrossRef]
- LeCun, Y.; Bengio, Y.; Hinton, G. Deep Learning. Nature 2015, 521, 436–444. [Google Scholar] [CrossRef]
- Student. The Probable Error of a Mean. Biometrika 1908, 6, 1–25. [Google Scholar] [CrossRef]
- Wilcoxon, F. Individual Comparisons by Ranking Methods. Biom. Bull. 1945, 1, 80–83. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.I.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma Powers Differential Expression Analyses for RNA-Sequencing and Microarray Studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Bartlett, M.S. Properties of Sufficiency and Statistical Tests. Proc. R. Soc. Lond. Ser.-Math. Phys. Sci. 1937, 160, 268–282. [Google Scholar]
- Li, C.; Liu, B.; Kang, B.; Liu, Z.; Liu, Y.; Chen, C.; Ren, X.; Zhang, Z. SciBet as a Portable and Fast Single Cell Type Identifier. Nat. Commun. 2020, 11, 1818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearson, K.X. On the Criterion That a given System of Deviations from the Probable in the Case of a Correlated System of Variables Is Such That It Can Be Reasonably Supposed to Have Arisen from Random Sampling. Lond. Edinb. Dublin Philos. Mag. J. Sci. 1900, 50, 157–175. [Google Scholar] [CrossRef] [Green Version]
- Massey Jr, F.J. The Kolmogorov-Smirnov Test for Goodness of Fit. J. Am. Stat. Assoc. 1951, 46, 68–78. [Google Scholar] [CrossRef]
- Wang, J.; Wen, S.; Symmans, W.F.; Pusztai, L.; Coombes, K.R. The Bimodality Index: A Criterion for Discovering and Ranking Bimodal Signatures from Cancer Gene Expression Profiling Data. Cancer Inform. 2009, 7, CIN.S2846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, K.; Yu, T.; Wu, H. Accurate Feature Selection Improves Single-Cell RNA-Seq Cell Clustering. Brief. Bioinform. 2021, 22, bbab034. [Google Scholar] [CrossRef]
- Andrews, T.S.; Hemberg, M. M3Drop: Dropout-Based Feature Selection for ScRNASeq. Bioinformatics 2019, 35, 2865–2867. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Cao, Y.; Kim, H.J.; Salim, A.; Speed, T.P.; Lin, D.M.; Yang, P.; Yang, J.Y.H. ScClassify: Sample Size Estimation and Multiscale Classification of Cells Using Single and Multiple Reference. Mol. Syst. Biol. 2020, 16, e9389. [Google Scholar] [CrossRef]
- Alquicira-Hernandez, J.; Sathe, A.; Ji, H.P.; Nguyen, Q.; Powell, J.E. ScPred: Accurate Supervised Method for Cell-Type Classification from Single-Cell RNA-Seq Data. Genome Biol. 2019, 20, 264. [Google Scholar] [CrossRef] [Green Version]
- Tan, Y.; Cahan, P. SingleCellNet: A Computational Tool to Classify Single Cell RNA-Seq Data Across Platforms and Across Species. Cell Syst. 2019, 9, 207–213.e2. [Google Scholar] [CrossRef]
- Ji, X.; Tsao, D.; Bai, K.; Tsao, M.; Xing, L.; Zhang, X. ScAnnotate: An Automated Cell-Type Annotation Tool for Single-Cell RNA-Sequencing Data. Bioinforma. Adv. 2023, 3, vbad030. [Google Scholar] [CrossRef]
- Pliner, H.A.; Shendure, J.; Trapnell, C. Supervised Classification Enables Rapid Annotation of Cell Atlases. Nat. Methods 2019, 16, 983–986. [Google Scholar] [CrossRef] [PubMed]
- Zou, H.; Hastie, T. Regularization and Variable Selection via the Elastic Net. J. R. Stat. Soc. Ser. B Stat. Methodol. 2005, 67, 301–320. [Google Scholar] [CrossRef] [Green Version]
- Domínguez Conde, C.; Xu, C.; Jarvis, L.B.; Rainbow, D.B.; Wells, S.B.; Gomes, T.; Howlett, S.K.; Suchanek, O.; Polanski, K.; King, H.W.; et al. Cross-Tissue Immune Cell Analysis Reveals Tissue-Specific Features in Humans. Science 2022, 376, eabl5197. [Google Scholar] [CrossRef]
- Shao, X.; Yang, H.; Zhuang, X.; Liao, J.; Yang, P.; Cheng, J.; Lu, X.; Chen, H.; Fan, X. ScDeepSort: A Pre-Trained Cell-Type Annotation Method for Single-Cell Transcriptomics Using Deep Learning with a Weighted Graph Neural Network. Nucleic Acids Res. 2021, 49, e122. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.-J.; Wei, L.; Lu, S.; Yang, D.-C.; Gao, G. Searching Large-Scale ScRNA-Seq Databases via Unbiased Cell Embedding with Cell BLAST. Nat. Commun. 2020, 11, 3458. [Google Scholar] [CrossRef]
- Yang, F.; Wang, W.; Wang, F.; Fang, Y.; Tang, D.; Huang, J.; Lu, H.; Yao, J. ScBERT as a Large-Scale Pretrained Deep Language Model for Cell Type Annotation of Single-Cell RNA-Seq Data. Nat. Mach. Intell. 2022, 4, 852–866. [Google Scholar] [CrossRef]
- Devlin, J.; Chang, M.-W.; Lee, K.; Toutanova, K. BERT: Pre-Training of Deep Bidirectional Transformers for Language Understanding. In Proceedings of the 2019 Conference of the North American Chapter of the Association for Computational Linguistics: Human Language Technologies, Volume 1 (Long and Short Papers), Minneapolis, MN, USA, 2–7 June 2019; pp. 4171–4186. [Google Scholar]
- Zhang, A.W.; O’Flanagan, C.; Chavez, E.A.; Lim, J.L.P.; Ceglia, N.; McPherson, A.; Wiens, M.; Walters, P.; Chan, T.; Hewitson, B.; et al. Probabilistic Cell-Type Assignment of Single-Cell RNA-Seq for Tumor Microenvironment Profiling. Nat. Methods 2019, 16, 1007–1015. [Google Scholar] [CrossRef]
- De Kanter, J.K.; Lijnzaad, P.; Candelli, T.; Margaritis, T.; Holstege, F.C.P. CHETAH: A Selective, Hierarchical Cell Type Identification Method for Single-Cell RNA Sequencing. Nucleic Acids Res. 2019, 47, e95. [Google Scholar] [CrossRef] [Green Version]
- Kiselev, V.Y.; Yiu, A.; Hemberg, M. Scmap: Projection of Single-Cell RNA-Seq Data across Data Sets. Nat. Methods 2018. [Google Scholar] [CrossRef]
- Chen, J.; Xu, H.; Tao, W.; Chen, Z.; Zhao, Y.; Han, J.-D.J. Transformer for One Stop Interpretable Cell Type Annotation. Nat. Commun. 2023, 14, 223. [Google Scholar] [CrossRef]
- Hossin, M.; Sulaiman, M.N. A Review on Evaluation Metrics for Data Classification Evaluations. Int. J. Data Min. Knowl. Manag. Process 2015, 5, 1. [Google Scholar]
- Berthelot, D.; Carlini, N.; Goodfellow, I.; Papernot, N.; Oliver, A.; Raffel, C.A. Mixmatch: A Holistic Approach to Semi-Supervised Learning. Adv. Neural Inf. Process. Syst. 2019, 32, 5049–5059. [Google Scholar]
- van Engelen, J.E.; Hoos, H.H. A Survey on Semi-Supervised Learning. Mach. Learn. 2020, 109, 373–440. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.J. Semi-Supervised Learning Literature Survey. Available online: https://pages.cs.wisc.edu/~jerryzhu/pub/ssl_survey.pdf (accessed on 25 May 2023).
- Killamsetty, K.; Zhao, X.; Chen, F.; Iyer, R. Retrieve: Coreset Selection for Efficient and Robust Semi-Supervised Learning. Adv. Neural Inf. Process. Syst. 2021, 34, 14488–14501. [Google Scholar]
- Zhang, Z.; Luo, D.; Zhong, X.; Choi, J.H.; Ma, Y.; Wang, S.; Mahrt, E.; Guo, W.; Stawiski, E.W.; Modrusan, Z.; et al. SCINA: A Semi-Supervised Subtyping Algorithm of Single Cells and Bulk Samples. Genes 2019, 10, 531. [Google Scholar] [CrossRef] [Green Version]
- Kimmel, J.C.; Kelley, D.R. Semisupervised Adversarial Neural Networks for Single-Cell Classification. Genome Res. 2021, 31, 1781–1793. [Google Scholar] [CrossRef]
- Dempster, A.P.; Laird, N.M.; Rubin, D.B. Maximum Likelihood from Incomplete Data via the EM Algorithm. J. R. Stat. Soc. Ser. B Methodol. 1977, 39, 1–22. [Google Scholar]
- Ganin, Y.; Ustinova, E.; Ajakan, H.; Germain, P.; Larochelle, H.; Laviolette, F.; Marchand, M.; Lempitsky, V. Domain-Adversarial Training of Neural Networks. J. Mach. Learn. Res. 2016, 17, 1–35. [Google Scholar]
- Breschi, A.; Muñoz-Aguirre, M.; Wucher, V.; Davis, C.A.; Garrido-Martín, D.; Djebali, S.; Gillis, J.; Pervouchine, D.D.; Vlasova, A.; Dobin, A.; et al. A Limited Set of Transcriptional Programs Define Major Cell Types. Genome Res. 2020, 30, 1047–1059. [Google Scholar] [CrossRef]
- Bakken, T.; Cowell, L.; Aevermann, B.D.; Novotny, M.; Hodge, R.; Miller, J.A.; Lee, A.; Chang, I.; McCorrison, J.; Pulendran, B.; et al. Cell Type Discovery and Representation in the Era of High-Content Single Cell Phenotyping. BMC Bioinform. 2017, 18, 559. [Google Scholar] [CrossRef]
- Qiu, P. Embracing the Dropouts in Single-Cell RNA-Seq Analysis. Nat. Commun. 2020, 11, 1169. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, G.W.; Zhou, Y.; Petrovic, J.; Fasolino, M.; Xu, L.; Shaffer, S.M.; Pear, W.S.; Vahedi, G.; Faryabi, R.B. TooManyCells Identifies and Visualizes Relationships of Single-Cell Clades. Nat. Methods 2020, 17, 405–413. [Google Scholar] [CrossRef]
- Tracy, C.A.; Widom, H. Level-Spacing Distributions and the Airy Kernel. Commun. Math. Phys. 1994, 159, 151–174. [Google Scholar] [CrossRef] [Green Version]
- Johnstone, I.M. On the Distribution of the Largest Eigenvalue in Principal Components Analysis. Ann. Stat. 2001, 29, 295–327. [Google Scholar] [CrossRef]
- Patterson, N.; Price, A.L.; Reich, D. Population Structure and Eigenanalysis. PLoS Genet. 2006, 2, e190. [Google Scholar] [CrossRef] [PubMed]
- Longo, S.K.; Guo, M.G.; Ji, A.L.; Khavari, P.A. Integrating Single-Cell and Spatial Transcriptomics to Elucidate Intercellular Tissue Dynamics. Nat. Rev. Genet. 2021, 22, 627–644. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Hyeon, D.Y.; Hwang, D. Single-Cell Multiomics: Technologies and Data Analysis Methods. Exp. Mol. Med. 2020, 52, 1428–1442. [Google Scholar] [CrossRef] [PubMed]
- Miao, Z.; Humphreys, B.D.; McMahon, A.P.; Kim, J. Multi-Omics Integration in the Age of Million Single-Cell Data. Nat. Rev. Nephrol. 2021, 17, 710–724. [Google Scholar] [CrossRef]
- Stuart, T.; Butler, A.; Hoffman, P.; Hafemeister, C.; Papalexi, E.; III, W.M.M.; Hao, Y.; Stoeckius, M.; Smibert, P.; Satija, R. Comprehensive Integration of Single-Cell Data. Cell 2019, 177, 1888–1902. [Google Scholar] [CrossRef]
- Hie, B.; Bryson, B.; Berger, B. Efficient Integration of Heterogeneous Single-Cell Transcriptomes Using Scanorama. Nat. Biotechnol. 2019, 37, 685–691. [Google Scholar] [CrossRef]
- Korsunsky, I.; Millard, N.; Fan, J.; Slowikowski, K.; Zhang, F.; Wei, K.; Baglaenko, Y.; Brenner, M.; Loh, P.; Raychaudhuri, S. Fast, Sensitive, and Accurate Integration of Single Cell Data with Harmony. Nat. Methods 2019, 16, 1289–1296. [Google Scholar] [CrossRef]
- Haghverdi, L.; Lun, A.T.; Morgan, M.D.; Marioni, J.C. Batch Effects in Single-Cell RNA-Sequencing Data Are Corrected by Matching Mutual Nearest Neighbors. Nat. Biotechnol. 2018. [Google Scholar] [CrossRef]
- Abdelaal, T.; Mourragui, S.; Mahfouz, A.; Reinders, M.J.T. SpaGE: Spatial Gene Enhancement Using ScRNA-Seq. Nucleic Acids Res. 2020, 48, e107. [Google Scholar] [CrossRef] [PubMed]
- Welch, J.D.; Kozareva, V.; Ferreira, A.; Vanderburg, C.; Martin, C.; Macosko, E.Z. Single-Cell Multi-Omic Integration Compares and Contrasts Features of Brain Cell Identity. Cell 2019, 177, 1873–1887.e17. [Google Scholar] [CrossRef] [PubMed]
- Lopez, R.; Regier, J.; Cole, M.B.; Jordan, M.I.; Yosef, N. Deep Generative Modeling for Single-Cell Transcriptomics. Nat. Methods 2018, 15, 1053–1058. [Google Scholar] [CrossRef]
- Biancalani, T.; Scalia, G.; Buffoni, L.; Avasthi, R.; Lu, Z.; Sanger, A.; Tokcan, N.; Vanderburg, C.R.; Segerstolpe, Å.; Zhang, M.; et al. Deep Learning and Alignment of Spatially Resolved Single-Cell Transcriptomes with Tangram. Nat. Methods 2021, 18, 1352–1362. [Google Scholar] [CrossRef]
- Gong, B.; Zhou, Y.; Purdom, E. Cobolt: Integrative Analysis of Multimodal Single-Cell Sequencing Data. Genome Biol. 2021, 22, 351. [Google Scholar] [CrossRef] [PubMed]
- Ashuach, T.; Gabitto, M.I.; Koodli, R.V.; Saldi, G.-A.; Jordan, M.I.; Yosef, N. MultiVI: Deep Generative Model for the Integration of Multimodal Data. Nat. Methods 2023. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Stuart, T.; Kowalski, M.H.; Choudhary, S.; Hoffman, P.; Hartman, A.; Srivastava, A.; Molla, G.; Madad, S.; Fernandez-Granda, C. Dictionary Learning for Integrative, Multimodal and Scalable Single-Cell Analysis. Nat. Biotechnol. 2023, 1–12. [Google Scholar] [CrossRef]
- Avila Cobos, F.; Alquicira-Hernandez, J.; Powell, J.E.; Mestdagh, P.; De Preter, K. Benchmarking of Cell Type Deconvolution Pipelines for Transcriptomics Data. Nat. Commun. 2020, 11, 5650. [Google Scholar] [CrossRef]
- Dong, R.; Yuan, G.-C. SpatialDWLS: Accurate Deconvolution of Spatial Transcriptomic Data. Genome Biol. 2021, 22, 145. [Google Scholar] [CrossRef] [PubMed]
- Cable, D.M.; Murray, E.; Zou, L.S.; Goeva, A.; Macosko, E.Z.; Chen, F.; Irizarry, R.A. Robust Decomposition of Cell Type Mixtures in Spatial Transcriptomics. Nat. Biotechnol. 2022, 40, 517–526. [Google Scholar] [CrossRef] [PubMed]
- Tsoucas, D.; Dong, R.; Chen, H.; Zhu, Q.; Guo, G.; Yuan, G.-C. Accurate Estimation of Cell-Type Composition from Gene Expression Data. Nat. Commun. 2019, 10, 2975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elosua-Bayes, M.; Nieto, P.; Mereu, E.; Gut, I.; Heyn, H. SPOTlight: Seeded NMF Regression to Deconvolute Spatial Transcriptomics Spots with Single-Cell Transcriptomes. Nucleic Acids Res. 2021, 49, e50. [Google Scholar] [CrossRef]
- Argelaguet, R.; Arnol, D.; Bredikhin, D.; Deloro, Y.; Velten, B.; Marioni, J.C.; Stegle, O. MOFA+: A Statistical Framework for Comprehensive Integration of Multi-Modal Single-Cell Data. Genome Biol. 2020, 21, 1–17. [Google Scholar] [CrossRef]
- Argelaguet, R.; Velten, B.; Arnol, D.; Dietrich, S.; Zenz, T.; Marioni, J.C.; Buettner, F.; Huber, W.; Stegle, O. Multi-Omics Factor Analysis—a Framework for Unsupervised Integration of Multi-omics Data Sets. Mol. Syst. Biol. 2018, 14, e8124. [Google Scholar] [CrossRef]
- Gayoso, A.; Steier, Z.; Lopez, R.; Regier, J.; Nazor, K.L.; Streets, A.; Yosef, N. Joint Probabilistic Modeling of Single-Cell Multi-Omic Data with TotalVI. Nat. Methods 2021, 18, 272–282. [Google Scholar] [CrossRef]
- Hao, Y.; Hao, S.; Andersen-Nissen, E.; Mauck, W.M.; Zheng, S.; Butler, A.; Lee, M.J.; Wilk, A.J.; Darby, C.; Zager, M. Integrated Analysis of Multimodal Single-Cell Data. Cell 2021, 184, 3573–3587.e29. [Google Scholar] [CrossRef]
- Gayoso, A.; Lopez, R.; Xing, G.; Boyeau, P.; Valiollah Pour Amiri, V.; Hong, J.; Wu, K.; Jayasuriya, M.; Mehlman, E.; Langevin, M. A Python Library for Probabilistic Analysis of Single-Cell Omics Data. Nat. Biotechnol. 2022, 40, 163–166. [Google Scholar] [CrossRef]
- Tran, H.T.N.; Ang, K.S.; Chevrier, M.; Zhang, X.; Lee, N.Y.S.; Goh, M.; Chen, J. A Benchmark of Batch-Effect Correction Methods for Single-Cell RNA Sequencing Data. Genome Biol. 2020, 21, 1–32. [Google Scholar] [CrossRef] [Green Version]
- Luecken, M.D.; Büttner, M.; Chaichoompu, K.; Danese, A.; Interlandi, M.; Müller, M.F.; Strobl, D.C.; Zappia, L.; Dugas, M.; Colomé-Tatché, M. Benchmarking Atlas-Level Data Integration in Single-Cell Genomics. Nat. Methods 2022, 19, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Lotfollahi, M.; Wolf, F.A.; Theis, F.J. ScGen Predicts Single-Cell Perturbation Responses. Nat. Methods 2019, 16, 715–721. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Zhang, W.; Guo, C.; Xu, H.; Li, L.; Fang, M.; Hu, Y.; Zhang, X.; Yao, X.; Tang, M. Benchmarking Spatial and Single-Cell Transcriptomics Integration Methods for Transcript Distribution Prediction and Cell Type Deconvolution. Nat. Methods 2022, 19, 662–670. [Google Scholar] [CrossRef]
- Baron, M.; Veres, A.; Wolock, S.L.; Faust, A.L.; Gaujoux, R.; Vetere, A.; Ryu, J.H.; Wagner, B.K.; Shen-Orr, S.S.; Klein, A.M.; et al. A Single-Cell Transcriptomic Map of the Human and Mouse Pancreas Reveals Inter- and Intra-Cell Population Structure. Cell Syst. 2016, 3, 346–360.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masuda, T.; Sankowski, R.; Staszewski, O.; Böttcher, C.; Amann, L.; Sagar; Scheiwe, C.; Nessler, S.; Kunz, P.; van Loo, G.; et al. Spatial and Temporal Heterogeneity of Mouse and Human Microglia at Single-Cell Resolution. Nature 2019, 566, 388–392. [Google Scholar] [CrossRef] [PubMed]
- Emont, M.P.; Jacobs, C.; Essene, A.L.; Pant, D.; Tenen, D.; Colleluori, G.; Di Vincenzo, A.; Jørgensen, A.M.; Dashti, H.; Stefek, A.; et al. A Single-Cell Atlas of Human and Mouse White Adipose Tissue. Nature 2022, 603, 926–933. [Google Scholar] [CrossRef]
- Ni, Z.; Prasad, A.; Chen, S.; Halberg, R.B.; Arkin, L.M.; Drolet, B.A.; Newton, M.A.; Kendziorski, C. SpotClean Adjusts for Spot Swapping in Spatial Transcriptomics Data. Nat. Commun. 2022, 13, 2971. [Google Scholar] [CrossRef]
- Shan, Y.; Zhang, Q.; Guo, W.; Wu, Y.; Miao, Y.; Xin, H.; Lian, Q.; Gu, J. TIST: Transcriptome and Histopathological Image Integrative Analysis for Spatial Transcriptomics. Genom. Proteom. Bioinform. 2022, 20, 974–988. [Google Scholar] [CrossRef]
- Chen, W.; Zhang, S.; Williams, J.; Ju, B.; Shaner, B.; Easton, J.; Wu, G.; Chen, X. A Comparison of Methods Accounting for Batch Effects in Differential Expression Analysis of UMI Count Based Single Cell RNA Sequencing. Comput. Struct. Biotechnol. J. 2020, 18, 861–873. [Google Scholar] [CrossRef]
- Smith, J.M.; Szathmary, E. The Major Transitions in Evolution; OUP Oxford: Oxford, UK, 1997; ISBN 0-19-158600-5. [Google Scholar]
- Calcott, B.; Sterelny, K. The Major Transitions in Evolution Revisited; MIT Press: Cambridge, MA, USA, 2011; ISBN 0-262-29453-2. [Google Scholar]
- Gilbert, S.F. Developmental Biology. 6th Editio. Sunderland MA Sinauer Assoc. 2000. [Google Scholar]
- Valls, P.O.; Esposito, A. Signalling Dynamics, Cell Decisions, and Homeostatic Control in Health and Disease. Curr. Opin. Cell Biol. 2022, 75, 102066. [Google Scholar] [CrossRef] [PubMed]
- Altan-Bonnet, G.; Mukherjee, R. Cytokine-Mediated Communication: A Quantitative Appraisal of Immune Complexity. Nat. Rev. Immunol. 2019, 19, 205–217. [Google Scholar] [CrossRef]
- Graeber, T.G.; Eisenberg, D. Bioinformatic Identification of Potential Autocrine Signaling Loops in Cancers from Gene Expression Profiles. Nat. Genet. 2001, 29, 295–300. [Google Scholar] [CrossRef]
- Hu, Z.; Yuan, J.; Long, M.; Jiang, J.; Zhang, Y.; Zhang, T.; Xu, M.; Fan, Y.; Tanyi, J.L.; Montone, K.T. The Cancer Surfaceome Atlas Integrates Genomic, Functional and Drug Response Data to Identify Actionable Targets. Nat. Cancer 2021, 2, 1406–1422. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [Green Version]
- Toda, S.; Frankel, N.W.; Lim, W.A. Engineering Cell–Cell Communication Networks: Programming Multicellular Behaviors. Curr. Opin. Chem. Biol. 2019, 52, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Lim, W.; Mayer, B.; Pawson, T. Cell Signaling; Taylor & Francis: Abingdon, UK, 2014; ISBN 1-317-57362-5. [Google Scholar]
- Monte, I.; Ishida, S.; Zamarreño, A.M.; Hamberg, M.; Franco-Zorrilla, J.M.; García-Casado, G.; Gouhier-Darimont, C.; Reymond, P.; Takahashi, K.; García-Mina, J.M. Ligand-Receptor Co-Evolution Shaped the Jasmonate Pathway in Land Plants. Nat. Chem. Biol. 2018, 14, 480–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moyle, W.R.; Campbell, R.K.; Myers, R.V.; Bernard, M.P.; Han, Y.; Wang, X. Co-Evolution of Ligand-Receptor Pairs. Nature 1994, 368, 251–255. [Google Scholar] [CrossRef]
- Armingol, E.; Officer, A.; Harismendy, O.; Lewis, N.E. Deciphering Cell–Cell Interactions and Communication from Gene Expression. Nat. Rev. Genet. 2021, 22, 71–88. [Google Scholar] [CrossRef]
- Efremova, M.; Vento-Tormo, M.; Teichmann, S.A.; Vento-Tormo, R. CellPhoneDB: Inferring Cell–Cell Communication from Combined Expression of Multi-Subunit Ligand–Receptor Complexes. Nat. Protoc. 2020, 15, 1484–1506. [Google Scholar] [CrossRef]
- Garcia-Alonso, L.; Handfield, L.-F.; Roberts, K.; Nikolakopoulou, K.; Fernando, R.C.; Gardner, L.; Woodhams, B.; Arutyunyan, A.; Polanski, K.; Hoo, R. Mapping the Temporal and Spatial Dynamics of the Human Endometrium in Vivo and in Vitro. Nat. Genet. 2021, 53, 1698–1711. [Google Scholar] [CrossRef]
- Jin, S.; Guerrero-Juarez, C.F.; Zhang, L.; Chang, I.; Ramos, R.; Kuan, C.-H.; Myung, P.; Plikus, M.V.; Nie, Q. Inference and Analysis of Cell-Cell Communication Using CellChat. Nat. Commun. 2021, 12, 1088. [Google Scholar] [CrossRef]
- Browaeys, R.; Saelens, W.; Saeys, Y. NicheNet: Modeling Intercellular Communication by Linking Ligands to Target Genes. Nat. Methods 2020, 17, 159–162. [Google Scholar] [CrossRef]
- Cillo, A.R.; Kürten, C.H.; Tabib, T.; Qi, Z.; Onkar, S.; Wang, T.; Liu, A.; Duvvuri, U.; Kim, S.; Soose, R.J. Immune Landscape of Viral-and Carcinogen-Driven Head and Neck Cancer. Immunity 2020, 52, 183–199.e9. [Google Scholar] [CrossRef] [PubMed]
- Dries, R.; Zhu, Q.; Dong, R.; Eng, C.-H.L.; Li, H.; Liu, K.; Fu, Y.; Zhao, T.; Sarkar, A.; Bao, F. Giotto: A Toolbox for Integrative Analysis and Visualization of Spatial Expression Data. Genome Biol. 2021, 22, 1–31. [Google Scholar] [CrossRef] [PubMed]
- Cabello-Aguilar, S.; Alame, M.; Kon-Sun-Tack, F.; Fau, C.; Lacroix, M.; Colinge, J. SingleCellSignalR: Inference of Intercellular Networks from Single-Cell Transcriptomics. Nucleic Acids Res. 2020, 48, e55. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, R.; Zhang, S.; Song, S.; Jiang, C.; Han, G.; Wang, M.; Ajani, J.; Futreal, A.; Wang, L. ITALK: An R Package to Characterize and Illustrate Intercellular Communication. bioRxiv 2019. [Google Scholar] [CrossRef] [Green Version]
- Cang, Z.; Nie, Q. Inferring Spatial and Signaling Relationships between Cells from Single Cell Transcriptomic Data. Nat. Commun. 2020, 11, 2084. [Google Scholar] [CrossRef]
- Choi, H.; Sheng, J.; Gao, D.; Li, F.; Durrans, A.; Ryu, S.; Lee, S.B.; Narula, N.; Rafii, S.; Elemento, O. Transcriptome Analysis of Individual Stromal Cell Populations Identifies Stroma-Tumor Crosstalk in Mouse Lung Cancer Model. Cell Rep. 2015, 10, 1187–1201. [Google Scholar] [CrossRef] [Green Version]
- Pham, D.; Tan, X.; Xu, J.; Grice, L.F.; Lam, P.Y.; Raghubar, A.; Vukovic, J.; Ruitenberg, M.J.; Nguyen, Q. StLearn: Integrating Spatial Location, Tissue Morphology and Gene Expression to Find Cell Types, Cell-Cell Interactions and Spatial Trajectories within Undissociated Tissues. bioRxiv 2020. [Google Scholar] [CrossRef]
- Wang, S.; Karikomi, M.; MacLean, A.L.; Nie, Q. Cell Lineage and Communication Network Inference via Optimization for Single-Cell Transcriptomics. Nucleic Acids Res. 2019, 47, e66. [Google Scholar] [CrossRef] [Green Version]
- Hou, R.; Denisenko, E.; Ong, H.T.; Ramilowski, J.A.; Forrest, A.R. Predicting Cell-to-Cell Communication Networks Using NATMI. Nat. Commun. 2020, 11, 5011. [Google Scholar] [CrossRef]
- Noël, F.; Massenet-Regad, L.; Carmi-Levy, I.; Cappuccio, A.; Grandclaudon, M.; Trichot, C.; Kieffer, Y.; Mechta-Grigoriou, F.; Soumelis, V. Dissection of Intercellular Communication Using the Transcriptome-Based Framework ICELLNET. Nat. Commun. 2021, 12, 1089. [Google Scholar] [CrossRef]
- Dimitrov, D.; Türei, D.; Garrido-Rodriguez, M.; Burmedi, P.L.; Nagai, J.S.; Boys, C.; Ramirez Flores, R.O.; Kim, H.; Szalai, B.; Costa, I.G. Comparison of Methods and Resources for Cell-Cell Communication Inference from Single-Cell RNA-Seq Data. Nat. Commun. 2022, 13, 3224. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Zhang, J.; Wu, Z.; Sun, X. Inferring Microenvironmental Regulation of Gene Expression from Single-Cell RNA Sequencing Data Using ScMLnet with an Application to COVID-19. Brief. Bioinform. 2021, 22, 988–1005. [Google Scholar] [CrossRef] [PubMed]
- Tyler, S.R.; Rotti, P.G.; Sun, X.; Yi, Y.; Xie, W.; Winter, M.C.; Flamme-Wiese, M.J.; Tucker, B.A.; Mullins, R.F.; Norris, A.W. PyMINEr Finds Gene and Autocrine-Paracrine Networks from Human Islet ScRNA-Seq. Cell Rep. 2019, 26, 1951–1964.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raredon, M.S.B.; Yang, J.; Garritano, J.; Wang, M.; Kushnir, D.; Schupp, J.C.; Adams, T.S.; Greaney, A.M.; Leiby, K.L.; Kaminski, N. Computation and Visualization of Cell–Cell Signaling Topologies in Single-Cell Systems Data Using Connectome. Sci. Rep. 2022, 12, 4187. [Google Scholar] [CrossRef]
- Tsuyuzaki, K.; Ishii, M.; Nikaido, I. Uncovering Hypergraphs of Cell-Cell Interaction from Single Cell RNA-Sequencing Data. bioRxiv 2019. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Peng, T.; Gao, L.; Tan, K. CytoTalk: De Novo Construction of Signal Transduction Networks Using Single-Cell Transcriptomic Data. Sci. Adv. 2021, 7, eabf1356. [Google Scholar] [CrossRef]
- Armingol, E.; Baghdassarian, H.M.; Martino, C.; Perez-Lopez, A.; Aamodt, C.; Knight, R.; Lewis, N.E. Context-Aware Deconvolution of Cell–Cell Communication with Tensor-Cell2cell. Nat. Commun. 2022, 13, 3665. [Google Scholar] [CrossRef] [PubMed]
- Shao, X.; Li, C.; Yang, H.; Lu, X.; Liao, J.; Qian, J.; Wang, K.; Cheng, J.; Yang, P.; Chen, H. Knowledge-Graph-Based Cell-Cell Communication Inference for Spatially Resolved Transcriptomic Data with SpaTalk. Nat. Commun. 2022, 13, 4429. [Google Scholar] [CrossRef] [PubMed]
- Jakobsson, J.E.; Spjuth, O.; Lagerström, M.C. ScConnect: A Method for Exploratory Analysis of Cell–Cell Communication Based on Single-Cell RNA-Sequencing Data. Bioinformatics 2021, 37, 3501–3508. [Google Scholar] [CrossRef]
- Cang, Z.; Zhao, Y.; Almet, A.A.; Stabell, A.; Ramos, R.; Plikus, M.V.; Atwood, S.X.; Nie, Q. Screening Cell–Cell Communication in Spatial Transcriptomics via Collective Optimal Transport. Nat. Methods 2023, 20, 218–228. [Google Scholar] [CrossRef] [PubMed]
- Cherry, C.; Maestas, D.R.; Han, J.; Andorko, J.I.; Cahan, P.; Fertig, E.J.; Garmire, L.X.; Elisseeff, J.H. Intercellular Signaling Dynamics from a Single Cell Atlas of the Biomaterials Response. bioRxiv 2020. [Google Scholar] [CrossRef]
- Baruzzo, G.; Cesaro, G.; Di Camillo, B. Identify, Quantify and Characterize Cellular Communication from Single-Cell RNA Sequencing Data with ScSeqComm. Bioinformatics 2022, 38, 1920–1929. [Google Scholar] [CrossRef]
- Wilk, A.J.; Shalek, A.K.; Holmes, S.; Blish, C.A. Comparative Analysis of Cell–Cell Communication at Single-Cell Resolution. Nat. Biotechnol. 2023, 1–14. [Google Scholar] [CrossRef]
- Tang, Z.; Zhang, T.; Yang, B.; Su, J.; Song, Q. SpaCI: Deciphering Spatial Cellular Communications through Adaptive Graph Model. Brief Bioinform. 2023, 24, bbac563. [Google Scholar] [CrossRef]
- Li, Z.; Wang, T.; Liu, P.; Huang, Y. SpatialDM: Rapid Identification of Spatially Co-Expressed Ligand-Receptor Reveals Cell-Cell Communication Patterns. bioRxiv 2022. [Google Scholar] [CrossRef]
- Liu, Q.; Hsu, C.-Y.; Li, J.; Shyr, Y. Dysregulated Ligand–Receptor Interactions from Single-Cell Transcriptomics. Bioinformatics 2022, 38, 3216–3221. [Google Scholar] [CrossRef]
- Xin, Y.; Lyu, P.; Jiang, J.; Zhou, F.; Wang, J.; Blackshaw, S.; Qian, J. LRLoop: A Method to Predict Feedback Loops in Cell–Cell Communication. Bioinformatics 2022, 38, 4117–4126. [Google Scholar] [CrossRef]
- Yang, Y.; Li, G.; Zhong, Y.; Xu, Q.; Lin, Y.-T.; Roman-Vicharra, C.; Chapkin, R.S.; Cai, J.J. ScTenifoldXct: A Semi-Supervised Method for Predicting Cell-Cell Interactions and Mapping Cellular Communication Graphs. Cell Syst. 2023. [Google Scholar] [CrossRef]
- Vahid, M.R.; Kurlovs, A.H.; Andreani, T.; Augé, F.; Olfati-Saber, R.; de Rinaldis, E.; Rapaport, F.; Savova, V. DiSiR: Fast and Robust Method to Identify Ligand–Receptor Interactions at Subunit Level from Single-Cell RNA-Sequencing Data. NAR Genomics Bioinforma. 2023, 5, lqad030. [Google Scholar] [CrossRef]
- Rao, N.; Pai, R.; Mishra, A.; Ginhoux, F.; Chan, J.; Sharma, A.; Zafar, H. Charting Spatial Ligand-Target Activity Using Renoir. bioRxiv 2023. [Google Scholar] [CrossRef]
- Subedi, S.; Park, Y.P. Single-Cell Pair-Wise Relationships Untangled by Composite Embedding Model. iScience 2023, 26. [Google Scholar] [CrossRef]
- Liu, S.; Zhang, Y.; Peng, J.; Shang, X. An Improved Hierarchical Variational Autoencoder for Cell–Cell Communication Estimation Using Single-Cell RNA-Seq Data. Brief. Funct. Genomics 2023, elac056. [Google Scholar] [CrossRef]
- Burdziak, C.; Alonso-Curbelo, D.; Walle, T.; Reyes, J.; Barriga, F.M.; Haviv, D.; Xie, Y.; Zhao, Z.; Zhao, C.J.; Chen, H.-A. Epigenetic Plasticity Cooperates with Cell-Cell Interactions to Direct Pancreatic Tumorigenesis. Science 2023, 380, eadd5327. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Feng, X.; Wu, Z.; Kajigaya, S.; Young, N.S. CellCallEXT: Analysis of Ligand–Receptor and Transcription Factor Activities in Cell–Cell Communication of Tumor Immune Microenvironment. Cancers 2022, 14, 4957. [Google Scholar] [CrossRef] [PubMed]
- Bridges, K.; Miller-Jensen, K. Mapping and Validation of ScRNA-Seq-Derived Cell-Cell Communication Networks in the Tumor Microenvironment. Front. Immunol. 2022, 13, 885267. [Google Scholar] [CrossRef] [PubMed]
- Song, D.; Yang, D.; Powell, C.A.; Wang, X. Cell–Cell Communication: Old Mystery and New Opportunity. Cell Biol. Toxicol. 2019, 35, 89–93. [Google Scholar] [CrossRef] [Green Version]
- Peng, L.; Wang, F.; Wang, Z.; Tan, J.; Huang, L.; Tian, X.; Liu, G.; Zhou, L. Cell–Cell Communication Inference and Analysis in the Tumour Microenvironments from Single-Cell Transcriptomics: Data Resources and Computational Strategies. Brief. Bioinform. 2022, 23, bbac234. [Google Scholar] [CrossRef]
- Buccitelli, C.; Selbach, M. MRNAs, Proteins and the Emerging Principles of Gene Expression Control. Nat. Rev. Genet. 2020, 21, 630–644. [Google Scholar] [CrossRef]
- Maier, T.; Güell, M.; Serrano, L. Correlation of MRNA and Protein in Complex Biological Samples. FEBS Lett. 2009, 583, 3966–3973. [Google Scholar] [CrossRef] [Green Version]
- Ramilowski, J.A.; Goldberg, T.; Harshbarger, J.; Kloppmann, E.; Lizio, M.; Satagopam, V.P.; Itoh, M.; Kawaji, H.; Carninci, P.; Rost, B. A Draft Network of Ligand–Receptor-Mediated Multicellular Signalling in Human. Nat. Commun. 2015, 6, 7866. [Google Scholar] [CrossRef] [Green Version]
- Shao, X.; Liao, J.; Li, C.; Lu, X.; Cheng, J.; Fan, X. CellTalkDB: A Manually Curated Database of Ligand–Receptor Interactions in Humans and Mice. Brief. Bioinform. 2021, 22, bbaa269. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, T.; Wang, J.; Zou, B.; Li, L.; Yao, L.; Chen, K.; Ning, L.; Wu, B.; Zhao, X. Cellinker: A Platform of Ligand–Receptor Interactions for Intercellular Communication Analysis. Bioinformatics 2021, 37, 2025–2032. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, T.; Hu, X.; Wang, M.; Wang, J.; Zou, B.; Tan, P.; Cui, T.; Dou, Y.; Ning, L. CellCall: Integrating Paired Ligand–Receptor and Transcription Factor Activities for Cell–Cell Communication. Nucleic Acids Res. 2021, 49, 8520–8534. [Google Scholar] [CrossRef]
- Deng, M.; Wang, Y.; Yan, Y. Mining Cell–Cell Signaling in Single-Cell Transcriptomics Atlases. Curr. Opin. Cell Biol. 2022, 76, 102101. [Google Scholar] [CrossRef]
- Liu, Y.; Li, J.S.S.; Rodiger, J.; Comjean, A.; Attrill, H.; Antonazzo, G.; Brown, N.H.; Hu, Y.; Perrimon, N. FlyPhoneDB: An Integrated Web-Based Resource for Cell–Cell Communication Prediction in Drosophila. Genetics 2022, 220, iyab235. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Ma, D.; Ding, Q.; Zhou, Y.; Zheng, H.-L. PlantPhoneDB: A Manually Curated Pan-plant Database of Ligand-receptor Pairs Infers Cell–Cell Communication. Plant Biotechnol. J. 2022, 20, 2123–2134. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.P.; Du, J.; Lagoudas, G.; Jiao, Y.; Sawyer, A.; Drummond, D.C.; Lauffenburger, D.A.; Raue, A. Analysis of Single-Cell RNA-Seq Identifies Cell-Cell Communication Associated with Tumor Characteristics. Cell Rep. 2018, 25, 1458–1468.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Sun, D.; Wang, C. Evaluation of Cell-Cell Interaction Methods by Integrating Single-Cell RNA Sequencing Data with Spatial Information. Genome Biol. 2022, 23, 1–38. [Google Scholar] [CrossRef] [PubMed]
- Almet, A.A.; Cang, Z.; Jin, S.; Nie, Q. The Landscape of Cell–Cell Communication through Single-Cell Transcriptomics. Curr. Opin. Syst. Biol. 2021, 26, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Shao, X.; Lu, X.; Liao, J.; Chen, H.; Fan, X. New Avenues for Systematically Inferring Cell-Cell Communication: Through Single-Cell Transcriptomics Data. Protein Cell 2020, 11, 866–880. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.; Zhang, S.; Song, L.; Wang, B.; Wei, L.; Zhang, F. Applications and Analytical Tools of Cell Communication Based on Ligand-Receptor Interactions at Single Cell Level. Cell Biosci. 2021, 11, 121. [Google Scholar] [CrossRef]
- Hill, A.V. The Possible Effects of the Aggregation of the Molecules of Hemoglobin on Its Dissociation Curves. J. Physiol. 1910, 40, iv–vii. [Google Scholar]
- Langmuir, I. The Adsorption of Gases on Plane Surfaces of Glass, Mica and Platinum. J. Am. Chem. Soc. 1918, 40, 1361–1403. [Google Scholar] [CrossRef] [Green Version]
- Pavličev, M.; Wagner, G.P.; Chavan, A.R.; Owens, K.; Maziarz, J.; Dunn-Fletcher, C.; Kallapur, S.G.; Muglia, L.; Jones, H. Single-Cell Transcriptomics of the Human Placenta: Inferring the Cell Communication Network of the Maternal-Fetal Interface. Genome Res. 2017, 27, 349–361. [Google Scholar] [CrossRef] [Green Version]
- Sheikh, B.N.; Bondareva, O.; Guhathakurta, S.; Tsang, T.H.; Sikora, K.; Aizarani, N.; Holz, H.; Grün, D.; Hein, L.; Akhtar, A. Systematic Identification of Cell-Cell Communication Networks in the Developing Brain. iScience 2019, 21, 273–287. [Google Scholar] [CrossRef] [Green Version]
- Camp, J.G.; Sekine, K.; Gerber, T.; Loeffler-Wirth, H.; Binder, H.; Gac, M.; Kanton, S.; Kageyama, J.; Damm, G.; Seehofer, D. Multilineage Communication Regulates Human Liver Bud Development from Pluripotency. Nature 2017, 546, 533–538. [Google Scholar] [CrossRef]
- Wang, L.I.; Yu, P.; Zhou, B.; Song, J.; Li, Z.; Zhang, M.; Guo, G.; Wang, Y.; Chen, X.; Han, L. Single-Cell Reconstruction of the Adult Human Heart during Heart Failure and Recovery Reveals the Cellular Landscape Underlying Cardiac Function. Nat. Cell Biol. 2020, 22, 108–119. [Google Scholar] [CrossRef]
- Raredon, M.S.B.; Adams, T.S.; Suhail, Y.; Schupp, J.C.; Poli, S.; Neumark, N.; Leiby, K.L.; Greaney, A.M.; Yuan, Y.; Horien, C. Single-Cell Connectomic Analysis of Adult Mammalian Lungs. Sci. Adv. 2019, 5, eaaw3851. [Google Scholar] [CrossRef] [Green Version]
- Rieckmann, J.C.; Geiger, R.; Hornburg, D.; Wolf, T.; Kveler, K.; Jarrossay, D.; Sallusto, F.; Shen-Orr, S.S.; Lanzavecchia, A.; Mann, M. Social Network Architecture of Human Immune Cells Unveiled by Quantitative Proteomics. Nat. Immunol. 2017, 18, 583–593. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Xu, L.; Liu, J.; Huang, P.; Tan, Y.; Chen, S. Cell–Cell Communication Alterations via Intercellular Signaling Pathways in Substantia Nigra of Parkinson’s Disease. Front. Aging Neurosci. 2022, 14. [Google Scholar] [CrossRef]
- Devkota, P.; Wuchty, S. Controllability Analysis of Molecular Pathways Points to Proteins That Control the Entire Interaction Network. Sci. Rep. 2020, 10, 2943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thurley, K.; Wu, L.F.; Altschuler, S.J. Modeling Cell-to-Cell Communication Networks Using Response-Time Distributions. Cell Syst. 2018, 6, 355–367.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francis, K.; Palsson, B.O. Effective Intercellular Communication Distances Are Determined by the Relative Time Constants for Cyto/Chemokine Secretion and Diffusion. Proc. Natl. Acad. Sci. USA 1997, 94, 12258–12262. [Google Scholar] [CrossRef]
- Gupta, P.B.; Fillmore, C.M.; Jiang, G.; Shapira, S.D.; Tao, K.; Kuperwasser, C.; Lander, E.S. Stochastic State Transitions Give Rise to Phenotypic Equilibrium in Populations of Cancer Cells. Cell 2011, 146, 633–644. [Google Scholar] [CrossRef] [Green Version]
- Baccin, C.; Al-Sabah, J.; Velten, L.; Helbling, P.M.; Grünschläger, F.; Hernández-Malmierca, P.; Nombela-Arrieta, C.; Steinmetz, L.M.; Trumpp, A.; Haas, S. Combined Single-Cell and Spatial Transcriptomics Reveal the Molecular, Cellular and Spatial Bone Marrow Niche Organization. Nat. Cell Biol. 2020, 22, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Fawkner-Corbett, D.; Antanaviciute, A.; Parikh, K.; Jagielowicz, M.; Gerós, A.S.; Gupta, T.; Ashley, N.; Khamis, D.; Fowler, D.; Morrissey, E. Spatiotemporal Analysis of Human Intestinal Development at Single-Cell Resolution. Cell 2021, 184, 810–826.e23. [Google Scholar] [CrossRef]
- Arnol, D.; Schapiro, D.; Bodenmiller, B.; Saez-Rodriguez, J.; Stegle, O. Modeling Cell-Cell Interactions from Spatial Molecular Data with Spatial Variance Component Analysis. Cell Rep. 2019, 29, 202–211.e6. [Google Scholar] [CrossRef] [Green Version]
- Tanevski, J.; Flores, R.O.R.; Gabor, A.; Schapiro, D.; Saez-Rodriguez, J. Explainable Multi-View Framework for Dissecting Intercellular Signaling from Highly Multiplexed Spatial Data. bioRxiv 2020. [Google Scholar] [CrossRef]
- Vandereyken, K.; Sifrim, A.; Thienpont, B.; Voet, T. Methods and Applications for Single-Cell and Spatial Multi-Omics. Nat. Rev. Genet. 2023, 24, 494–515. [Google Scholar] [CrossRef]
- Mantri, M.; Scuderi, G.J.; Abedini-Nassab, R.; Wang, M.F.Z.; McKellar, D.; Shi, H.; Grodner, B.; Butcher, J.T.; De Vlaminck, I. Spatiotemporal Single-Cell RNA Sequencing of Developing Chicken Hearts Identifies Interplay between Cellular Differentiation and Morphogenesis. Nat. Commun. 2021, 12, 1771. [Google Scholar] [CrossRef]
- Chen, A.; Liao, S.; Cheng, M.; Ma, K.; Wu, L.; Lai, Y.; Qiu, X.; Yang, J.; Xu, J.; Hao, S.; et al. Spatiotemporal Transcriptomic Atlas of Mouse Organogenesis Using DNA Nanoball-Patterned Arrays. Cell 2022, 185, 1777–1792.e21. [Google Scholar] [CrossRef] [PubMed]
- Karras, P.; Bordeu, I.; Pozniak, J.; Nowosad, A.; Pazzi, C.; Van Raemdonck, N.; Landeloos, E.; Van Herck, Y.; Pedri, D.; Bervoets, G.; et al. A Cellular Hierarchy in Melanoma Uncouples Growth and Metastasis. Nature 2022, 610, 190–198. [Google Scholar] [CrossRef]
- Lim, W.A.; Lee, C.M.; Tang, C. Design Principles of Regulatory Networks: Searching for the Molecular Algorithms of the Cell. Mol. Cell 2013, 49, 202–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
| Platform | Modality | Brief Description |
|---|---|---|
| Single-Cell Transcriptomics | ||
| scRNA-seq | Gene Expression | These include platforms using UMI counts such as 10x Chromium [12] and raw read counts such as SMART-seq [13]/SMART-seq2 [14]. |
| snRNA-seq | Gene Expression | This method only measures the nuclear transcripts. However, nuclei dissociation may be more applicable when whole-cell dissociation is challenging. In the research we reviewed, scRNA-seq is commonly used as an umbrella term for various single-cell RNA sequencing techniques, including snRNA-seq. |
| Spatial Transcriptomics | ||
| High-Plex RNA Imaging (HPRI) | Gene Expression with Spatial Information | The resolution is at single-cell or subcellular level. Techniques include in situ sequencing [15], smFISH [16], STARmap [17], MERFISH [18], and seqFISH [19]/seqFISH+ [20]. |
| Spatial Barcoding | Gene Expression with Spatial Information | Align a tissue to a plate with spots encoding the spatial position, and measure transcripts of each spot. Each spot often contains multiple cells. Techniques include spatial transcriptomics [3], 10× Visium (https://www.10xgenomics.com/products/spatial-gene-expression accessed on 25 May 2023), HDST [21], and Slide-seq [22]/Slide-seq2 [23]. |
| Single-Cell Epigenomics | ||
| scATAC-seq | Chromatin Accessibility | Techniques include plate- or array-based methods such as Fluidigm C1 [24], droplet-based methods such as 10x Chromium [25], or split-pooling based methods such as sciATAC-seq [26]. |
| Single-Cell Methylation | DNA Methylation | Techniques include scRRBS [27], snmC-seq2 [28], and scCGI-seq [29]. |
| Single-Cell Multimodality | ||
| 10x Multiome (https://www.10xgenomics.com/products/single-cell-multiome-atac-plus-gene-expression accessed on 25 May 2023), SHARE-seq [30] | Gene Expression + Chromatin Accessibility | Simultaneous high-throughput ATAC and RNA expression. |
| CITE-seq [31] | Gene Expression + Protein Levels | CITE-seq can measure both the transcriptome and cell surface protein. |
| ASAP-seq [32] | Chromatin Accessibility + Protein Levels | This technique pairs scATAC-seq with detection of the cell surface and intracellular protein markers. |
| Method | Description |
|---|---|
| CTA (Cell-Type Activity) | Sum of the weighted expression |
| Ucell | Mann–Whitney U statistic |
| AUCell | Area under the curve |
| ssGSEA | Rank-based enrichment score |
| SCSS | Sum of UMI, normalized by library size |
| GSVA | Kernel density estimation |
| Singscore | Normalized mean percentile rank |
| ScType | Cluster summary enrichment score |
| JASMINE | Approximate mean of gene ranks and the enrichment of the signatures |
| AddModuleScore (Seurat) | Average expression level |
| scCATCH | Evidence-based scoring |
| Method | Description | Reference |
|---|---|---|
| DE | Differentially expressed genes | [88] |
| DD | Differentially distributed genes by Kolmogorov–Smirnov test | |
| DV | Differentially variable genes by Bartlett’s test | |
| BD | Bimodally distributed by bimodality index | [94] |
| DP | Differentially proportioned genes by chi-squared test | |
| M3Drop | Dropout-based feature selection | [96] |
| E-test | Entropy-based feature selection | [91] |
| FEAST | Unsupervised consensus clustering followed by F-test for ranking features | [95] |
| Tool | Year | Reference Database | Algorithm | Ref. |
|---|---|---|---|---|
| SingleR | 2019(**) | Built-in celldex (transcriptome of pure cell types) | Spearman | [63] |
| scmap-cell | 2018(**) | Annotated transcriptome | K-nearest neighbor (KNN) | [110] |
| Garnett | 2019(**) | Marker genes | Elastic net regression | [101] |
| CellAssign | 2019(**) | Marker genes | Probabilistic Bayesian model | [108] |
| scPred | 2019(**) | Annotated transcriptome | Support vector machines (SVM) | [98] |
| singleCellNet | 2019(*) | Annotated transcriptome | Random Forest | [99] |
| CHETAH | 2019(*) | Annotated transcriptome | Spearman and confidence | [109] |
| cellTypist | 2022(*) | Annotated transcriptome | L2-regularized logistic regression | [103] |
| CellBlast | 2020 | Annotated transcriptome | Neural network-based generative model | [105] |
| sciBET | 2020 | Annotated transcriptome | Multinomial-distribution model | [91] |
| scClassify | 2020 | Annotated transcriptome | Weighted KNN | [97] |
| scDeepSort | 2021 | Annotated transcriptome | Weighted graph neural network | [104] |
| scBERT | 2022 | Annotated transcriptome | BERT | [106] |
| scAnotate | 2023 | Annotated transcriptome | Random Forest | [100] |
| TOSICA | 2023 | Annotated transcriptome | Transformer | [111] |
| Tool | Input Data Demonstrated | scRNA-seq Data Preprocessing | Methods/Algorithms | Application/Output |
|---|---|---|---|---|
| Mapping | ||||
| scVI | scRNA-seq <-> scRNA-seq | Raw count matrix | Probabilistic modeling, neural networks, variational inference | scRNA-seq cell level and gene level batch correction scRNA-seq mapping |
| scANVI | scRNA-seq <-> scRNA-seq | Raw count matrix for UMI counts, gene length normalized count for read counts | Probabilistic modeling, neural networks, variational inference | scRNA-seq cell level and gene level batch correction scRNA-seq mapping annotation of single cells from annotated reference cells |
| MNN/fastMNN | scRNA-seq <-> scRNA-seq | Normalized with the library size, log transformed | Randomized SVD, MNN, weighted average of correction vectors | scRNA-seq cell level and gene level batch correction scRNA-seq mapping |
| Scanorama | scRNA-seq <-> scRNA-seq | L2-normalized for each cell | Randomized SVD, MNN, weighted average of correction vectors | scRNA-seq cell level and gene level batch correction scRNA-seq mapping |
| Seurat V3 | scRNA-seq <-> scRNA-seq scRNA-seq <-> HPRI scRNA-seq <-> CITE-seq scRNA-seq <-> scATAC-seq | Normalized with the library size, log transformed, gene scaled | CCA, MNN, anchor scoring and weighting | scRNA-seq cell level and gene level batch correction scRNA-seq mapping Multimodal data mapping |
| Harmony | scRNA-seq <-> scRNA-seq scRNA-seq <-> HPRI | Normalized with the library size, log transformed, gene scaled, PCs from PCA | Maximum batch diversity soft k-means clustering, linear mixture model correction | scRNA-seq cell level batch correction scRNA-seq mapping Multimodal data mapping |
| LIGER | scRNA-seq <-> scRNA-seq scRNA-seq <-> HPRI scRNA-seq <-> single cell DNA methylation | Normalized with the library size, gene scaled but not centered | Integrative non-negative matrix factorization, shared factor neighborhood clustering | scRNA-seq cell level batch correction scRNA-seq mapping Multimodal data mapping |
| SpaGE | scRNA-seq <-> HPRI | Normalized with the library size, log transformed, gene scaled | SVD on the cosine similarity matrix of PCs from each modality | Multimodal data mapping |
| gimVI | scRNA-seq <-> HPRI | Raw count matrix | Probabilistic modeling, neural networks, variational inference | Multimodal data mapping (for gene imputation) |
| Tangram | scRNA-seq <-> spatial barcoding and HPRI | Normalized with the library size | Direct minimization of a Kullback–Leibler divergence and cosine distances | Multimodal data mapping |
| Cobolt | Unimodal data sets and multimodal data set | Raw count matrix | Probabilistic modeling, neural networks, variational inference | Multimodal data mapping |
| MultiVI | Unimodal data sets and multimodal data set | Raw count matrix | Probabilistic modeling, neural networks, variational inference | Multimodal data mapping |
| Seurat V5 | Unimodal data sets and multimodal data sets | Depends on the mapping method in the first mapping step | Dictionary learning, Laplacian eigen-decomposition, sketching | Multimodal data mapping |
| Deconvolution | ||||
| Cell2location | scRNA-seq <-> spatial barcoding data | Raw count matrix | Bayesian negative binomial models, approximate variational inference | Estimate the absolute cell type abundance for each spot of spatial data |
| RCTD | scRNA-seq <-> spatial barcoding data | Raw count matrix | Poisson–lognormal models | Estimate the proportion of cell types for each spot of spatial data |
| stereoscope | scRNA-seq <-> spatial barcoding data | Raw count matrix | Negative binomial models | Estimate the proportion of cell types for each spot of spatial data |
| SpatialDWLS | scRNA-seq <-> spatial barcoding data | Normalized with the library size, log transformed | Enrichment analysis, dampened weighted least squares | Estimate the proportion of cell types for each spot of spatial data |
| SPOTlight | scRNA-seq <-> spatial barcoding data | Gene scaled | A seeded non-negative matrix factorization (NMF) regression and non-negative least squares | Estimate the proportion of cell types for each spot of spatial data |
| DestVI | scRNA-seq <-> spatial barcoding data | Raw count matrix | Probabilistic modeling, negative binomial models, neural networks, variational inference | Estimate both the proportion of cell types and the variations within each cell type for each spot of spatial data |
| Multimodality Fusion | ||||
| MOFA+ | scNMT-seq, single cell DNA methylation | Normalized with the library size, log transformed | Multi-omics factor analysis | Low-dimension visualization and clustering |
| totalVI | CITE-seq | Raw count matrix | Joint probabilistic modeling of genes and proteins, neural networks, variational inference | Low-dimension visualization and clustering Multimodality mapping Protein background decoupling, batch correction Protein level imputation Differential expression |
| Seurat V4 | CITE-seq, SHARE-seq, ASAP-seq | Normalized with the library size, log transformed, gene scaled | Supervised PCA, weighted similarity measure from different modalities | Low-dimension visualization and clustering Multimodality mapping |
| Tools | Year | Description | Ref. |
|---|---|---|---|
| CellPhoneDB | 2020 (**) | Infer CCC from combined expression of multi-subunit LRs, updated with spatial information integration. | [175,176] |
| CellChat | 2021 (**) | Quantify CCC by a mass action-based model. | [177] |
| NicheNet | 2020 (**) | Infer CCC by incorporating signaling network information. | [178] |
| CellTalker | 2020 (*) | Infer CCC by differentially expressed LRs. | [179] |
| Giotto | 2021 (*) | Identify CCC by incorporating spatial information. | [180] |
| SingleCellSignalR | 2020 (*) | Quantify CCC by regularized ligand–receptor expression product. | [181] |
| iTALK | 2019 (*) | Comparative and longitudinal CCC analysis. | [182] |
| SpaOTsc | 2020 (*) | Infer CCC using an optimal transport model, incorporating downstream expression information. | [183] |
| CCCExplorer | 2015 (*) | Infer CCC by incorporating downstream TF expression. | [184] |
| stLearn | 2020 (*) | Integrating spatial transcriptomics for CCC inference. | [185] |
| SoptSC | 2019 (*) | Score CCC by integrating downstream signaling measurements. | [186] |
| NATMI | 2020 | Extensive cell–cell communication network analysis. | [187] |
| ICELLNET | 2021 | Integrate LR information for CCC inference. | [188] |
| LIANA | 2022 | Infer CCC by a consensus combination of ligand–receptor methods and resources. | [189] |
| scMLnet | 2021 | Infer CCC by multilayer network method, which incorporates gene regulatory networks information. | [190] |
| PyMINEr | 2019 | Quantify CCC by differentially expressed LRs. | [191] |
| Connectome | 2022 | Differential and comparative CCC analysis. | [192] |
| scTensor | 2019 | Represent LR data as a tensor and then take decomposition. | [193] |
| CytoTalk | 2021 | Use crosstalk score to rank CCC signal. | [194] |
| Tensor-cell2cell | 2022 | Using tensor decomposition to characterize context dependent CCC. | [195] |
| SpaTalk | 2023 | Integrate spatial information to constrain CCC inference. | [196] |
| scConnect | 2021 | Inferring CCC by incorporating interactions in a multi-directional graph. | [197] |
| COMMOT | 2023 | Incorporate spatial information and biochemical reaction information to CCC reconstruction. | [198] |
| Domino | 2022 | Infer CCC by correlating expression of receptor to expression pattern in gene regulatory network. | [199] |
| scSeqComm | 2022 | Identify and quantify CCC through the evidence of ongoing intercellular and intracellular signaling. | [200] |
| Scriabin | 2023 | Infer CCC using a binning approach, without cell aggregation or down sampling. | [201] |
| spaCI | 2023 | Use adaptive graph model with attention mechanisms to incorporate both spatial locations and gene expression profiles of cells for CCC inference. | [202] |
| SpatialDM | 2022 | Spatial association/co-expression of LRs. | [203] |
| scLR | 2022 | Examine dysregulated ligand–receptor interactions between two conditions. | [204] |
| LRLoop | 2023 | Infer feedback loops in CCC. | [205] |
| scTenifoldXct | 2023 | Use manifold alignment for CCC inference, with comparative interaction analysis included. | [206] |
| DiSiR | 2023 | Considering interactions that are not listed in reference LR databases. | [207] |
| Renoir | 2023 | Infer CCC across a spatial topology and delineate spatial-specific communication niches. | [208] |
| SPRUCE | 2023 | Systematically infer common CCC patterns embedded in single-cell RNA-seq data. | [209] |
| HiVAE | 2023 | Quantify CCC by transfer entropy with hierarchical variational autoencoder. | [210] |
| Calligraphy | 2023 | Utilizes LR modularity to robustly infer the CCC. | [211] |
| CellCallEXT | 2022 | Identify LR that alter the expression of downstream genes between two conditions. | [212] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, C.; Chen, W.; Jin, H.; Chen, X. A Review of Single-Cell RNA-Seq Annotation, Integration, and Cell–Cell Communication. Cells 2023, 12, 1970. https://doi.org/10.3390/cells12151970
Cheng C, Chen W, Jin H, Chen X. A Review of Single-Cell RNA-Seq Annotation, Integration, and Cell–Cell Communication. Cells. 2023; 12(15):1970. https://doi.org/10.3390/cells12151970
Chicago/Turabian StyleCheng, Changde, Wenan Chen, Hongjian Jin, and Xiang Chen. 2023. "A Review of Single-Cell RNA-Seq Annotation, Integration, and Cell–Cell Communication" Cells 12, no. 15: 1970. https://doi.org/10.3390/cells12151970