
Evolving Acquired Vemurafenib Resistance in a BRAF V600E Mutant Melanoma PDTX Model to Reveal New Potential Targets
 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. PDTX Generation
2.3. BRAF Genotyping
2.4. Animal Model of Vemurafenib Treatment
2.5. RNA Isolation and RNAseq of Whole Genome mRNA
2.6. Bioinformatic Analysis of RNAseq Data
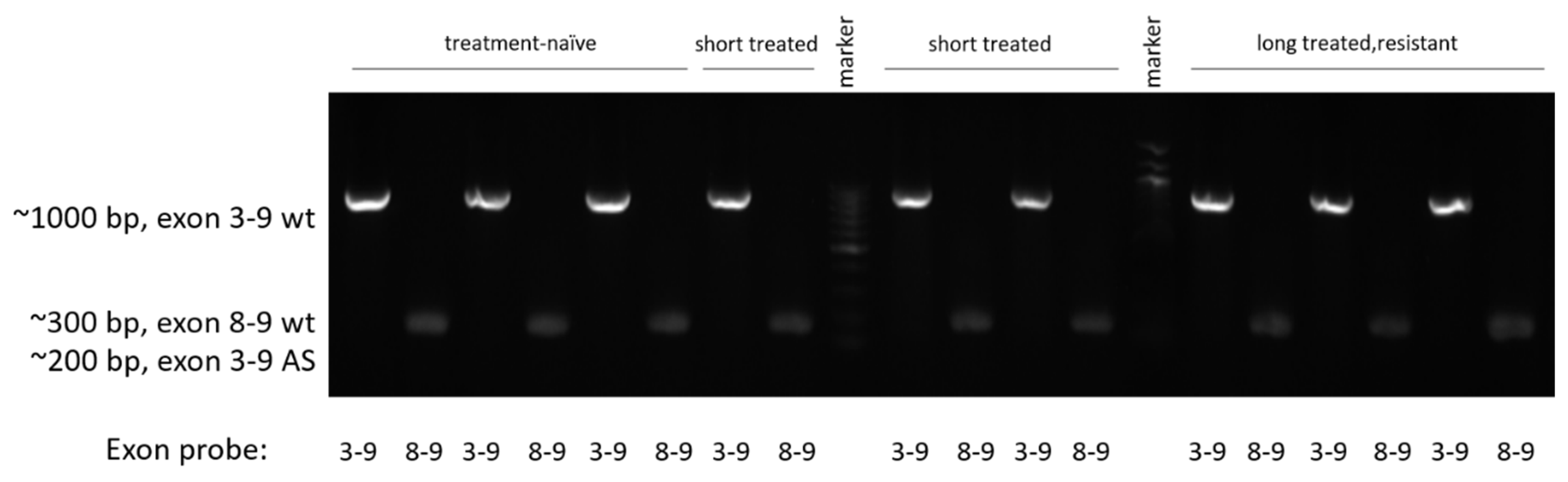
2.7. Alternative Splicing Detection
2.8. Western Blot
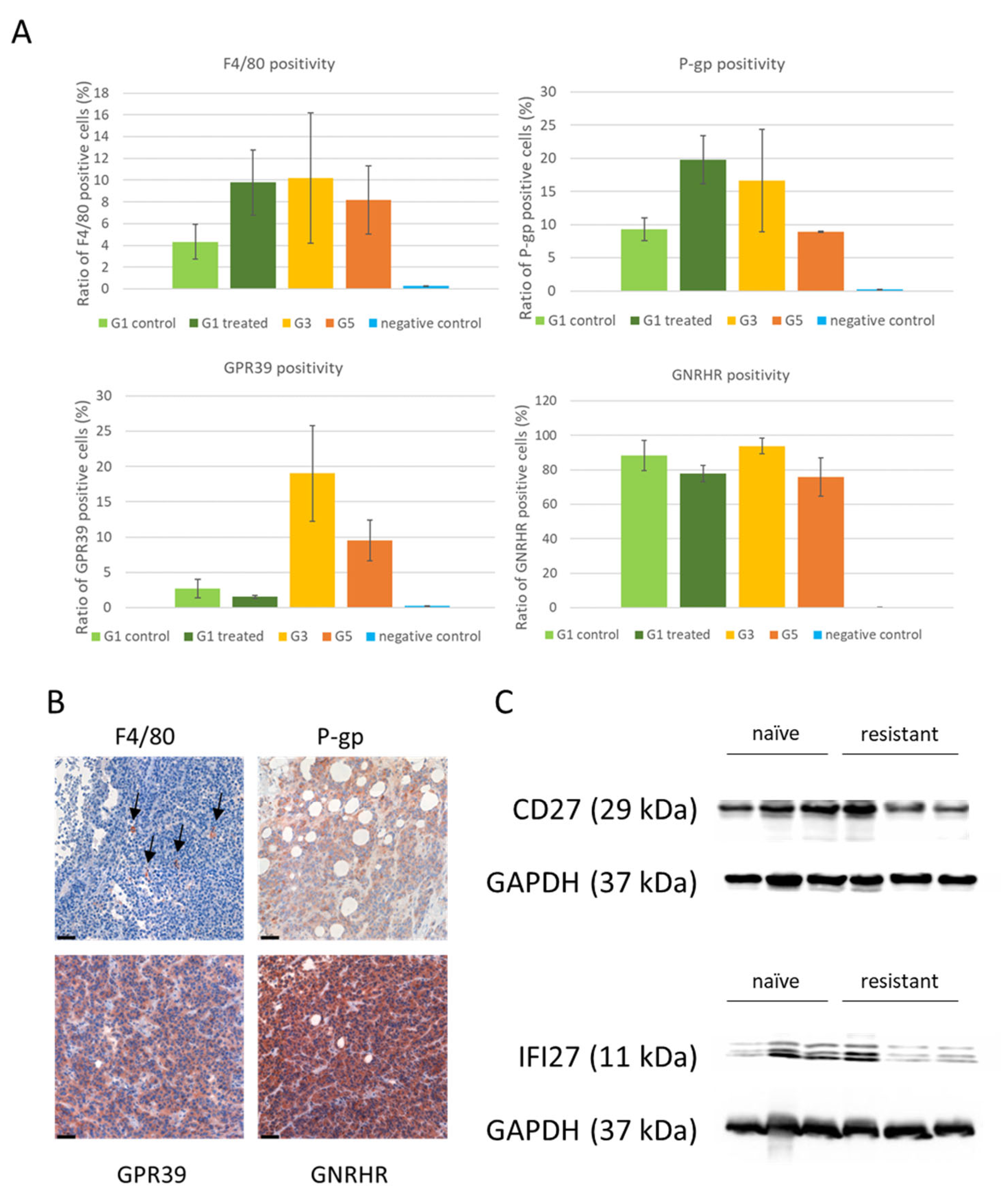
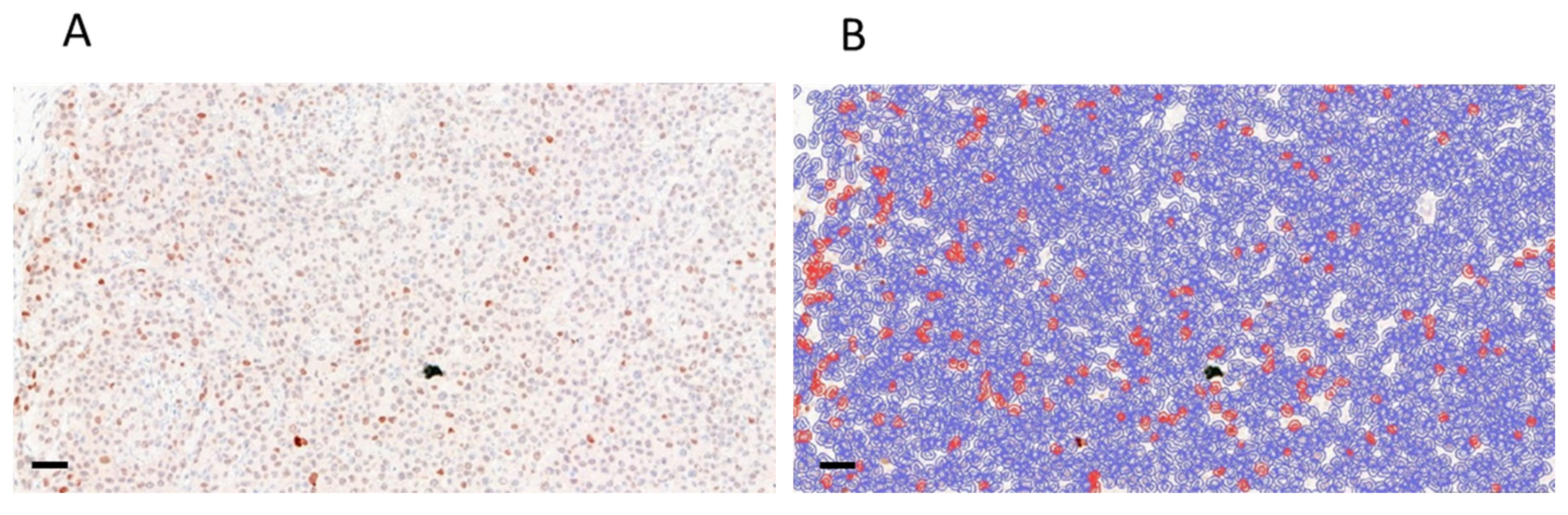
2.9. Immunohistochemistry
2.10. Data Processing and Statistical Analyses
3. Results
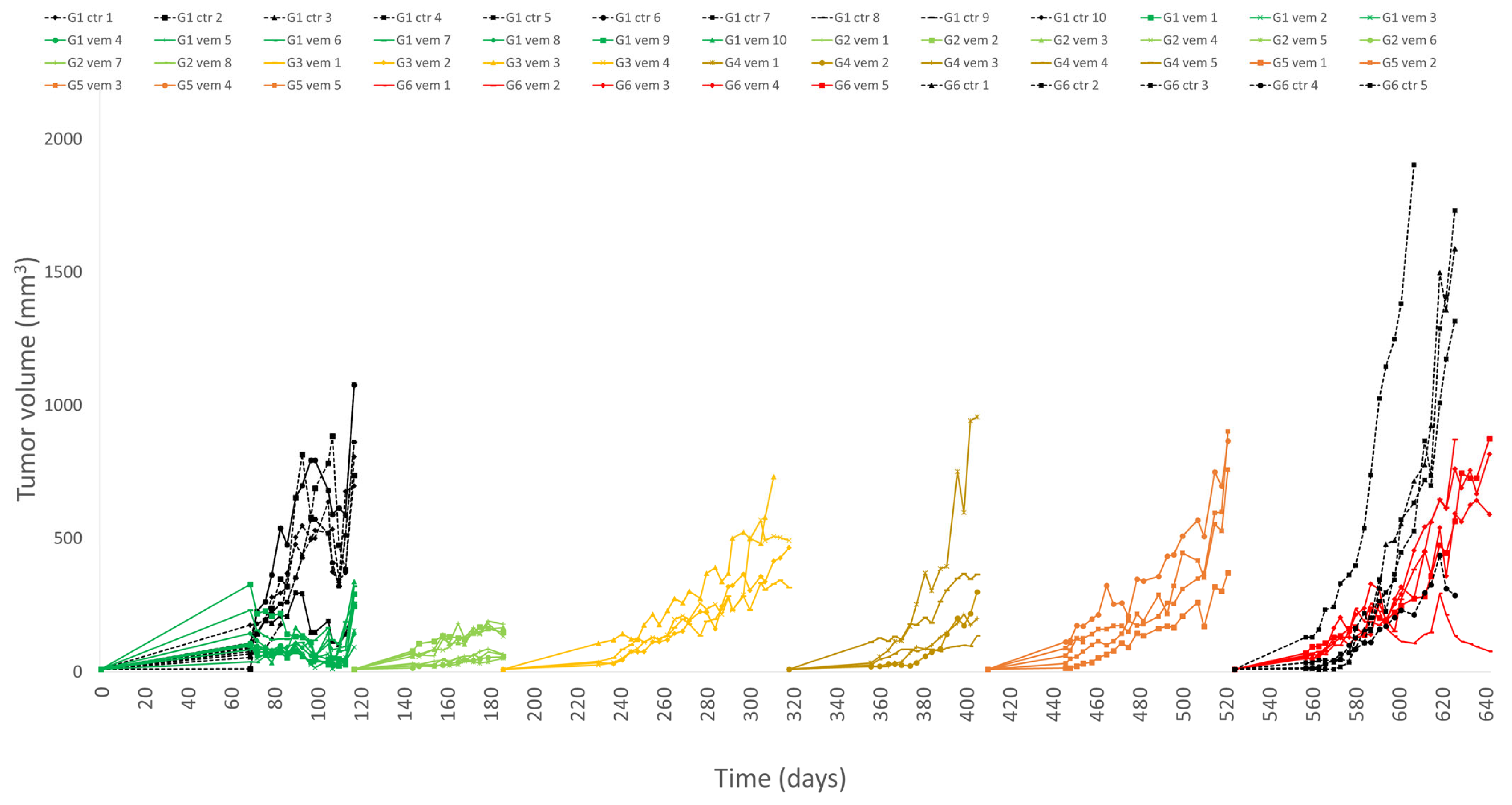
3.1. PDTX Growth and Vemurafenib Resistance In Vivo
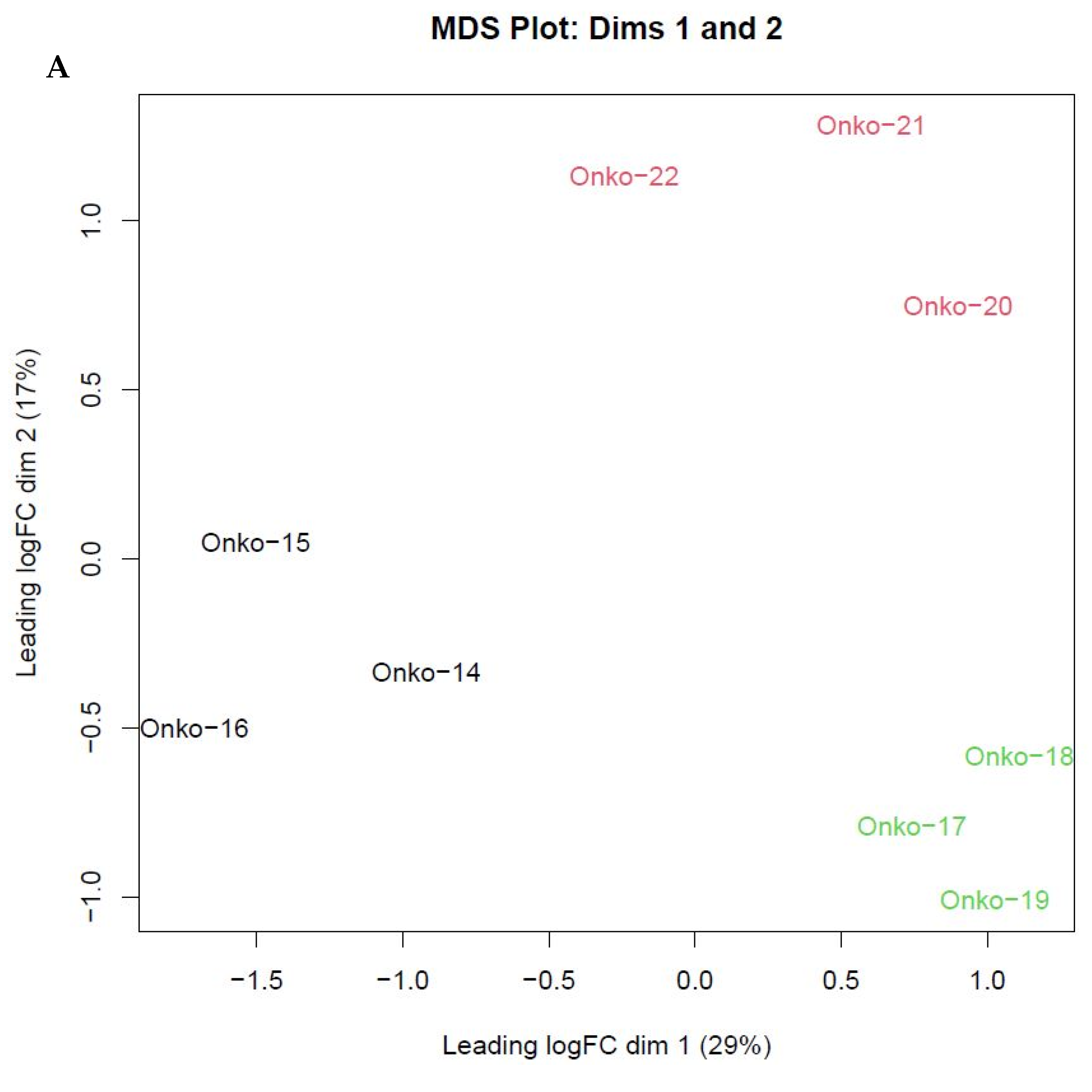
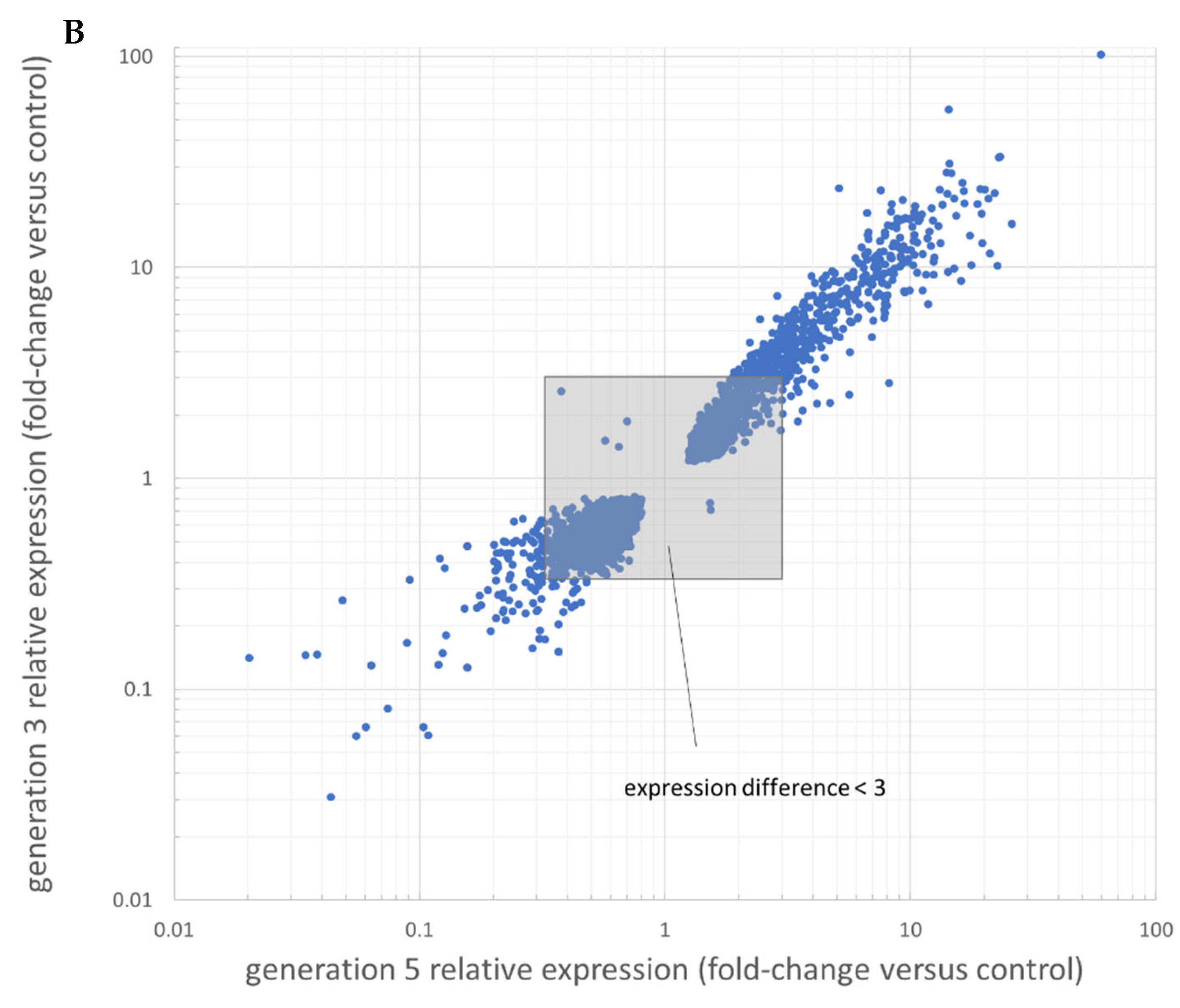
3.2. RNAseq Results Reveal Differentially Expressed Genes over Vemurafenib Treatment
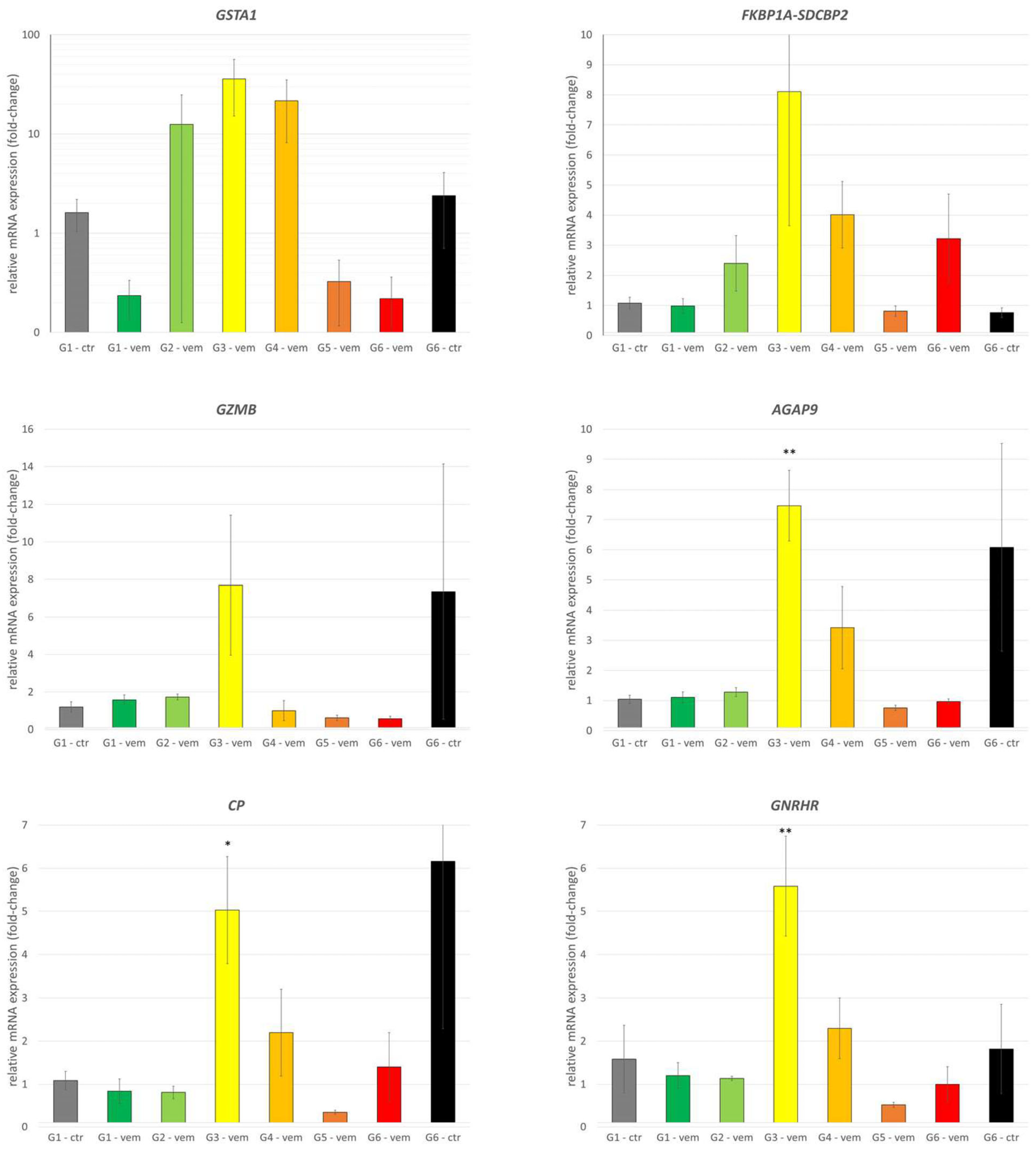
3.3. Detailed Analysis of Highlighted Genes of Interest by qPCR
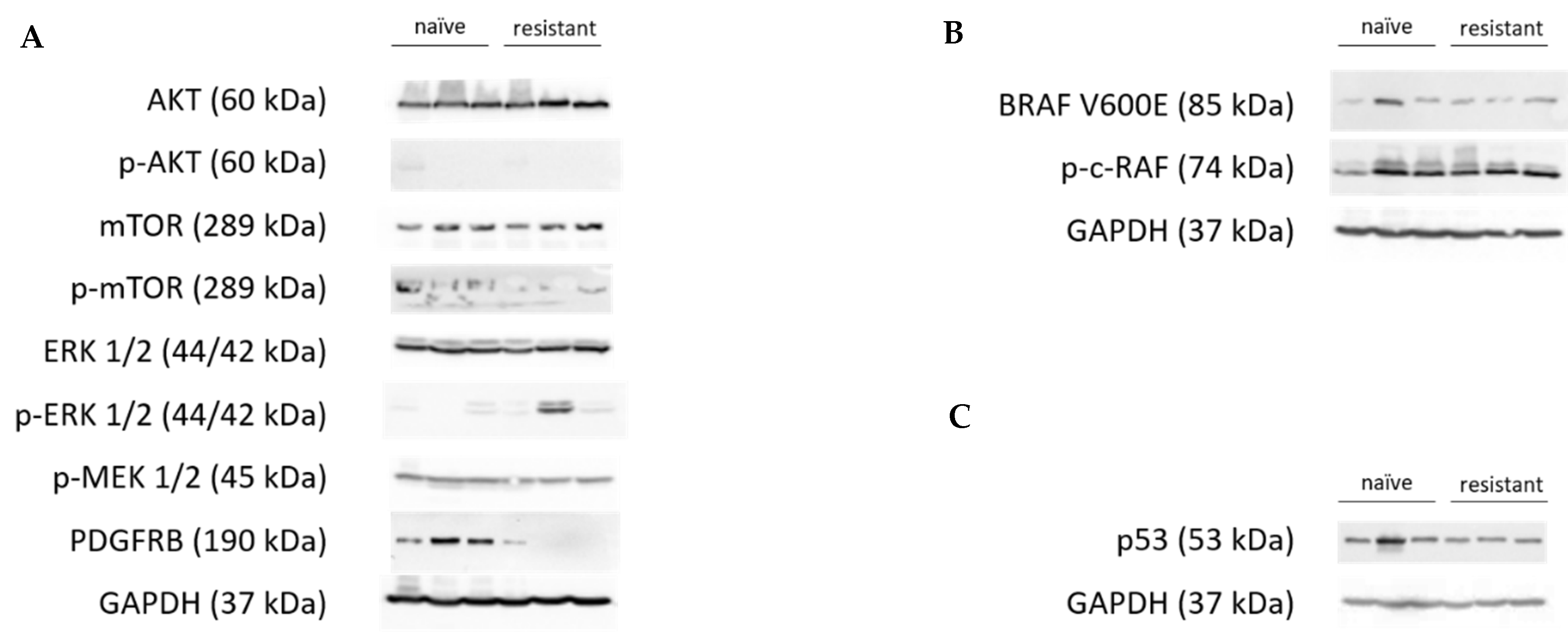
3.4. Protein Expression of Differentially Expressed Genes
3.5. Analysis of Known Resistance Mechanisms in the Present Resistance Model
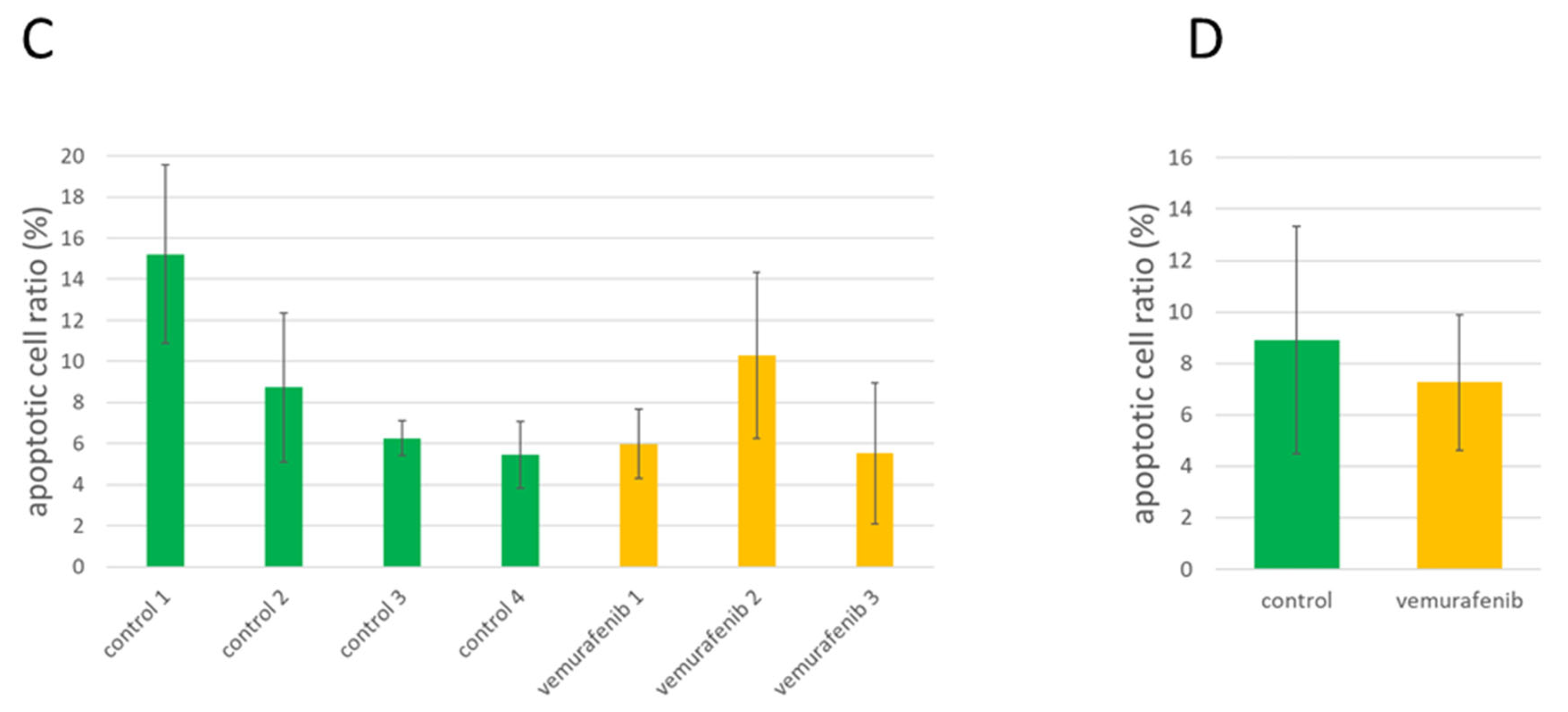
3.6. Analysis of Apoptotic Activity in Control and Treated PDTX Tumors
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer Statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef]
- Malignant Melanoma: Practice Essentials, Background, Etiology. Available online: https://emedicine.medscape.com/article/280245-overview (accessed on 25 April 2023).
- Sassolas, B.; Leccia, M.T.; Godard, C.; Benmahamed, L.; Flinois, A.; Levy-Bachelot, L.; Bédane, C. Real-World Treatment Patterns and Clinical Outcomes in Advanced Cutaneous Melanoma Patients in France. J. Eur. Acad. Dermatol. Venereol. 2018, 32, 587–594. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, K.T.; Yasothan, U.; Kirkpatrick, P. Vemurafenib. Nat. Rev. Drug Discov. 2011, 10, 811–812. [Google Scholar] [CrossRef] [PubMed]
- Subbiah, V.; Baik, C.; Kirkwood, J.M. Clinical Development of BRAF plus MEK Inhibitor Combinations. Trends Cancer 2020, 6, 797–810. [Google Scholar] [CrossRef]
- Proietti, I.; Skroza, N.; Bernardini, N.; Tolino, E.; Balduzzi, V.; Marchesiello, A.; Michelini, S.; Volpe, S.; Mambrin, A.; Mangino, G.; et al. Mechanisms of Acquired BRAF Inhibitor Resistance in Melanoma: A Systematic Review. Cancers 2020, 12, 2801. [Google Scholar] [CrossRef]
- Shi, H.; Hugo, W.; Kong, X.; Hong, A.; Koya, R.C.; Moriceau, G.; Chodon, T.; Guo, R.; Johnson, D.B.; Dahlman, K.B.; et al. Acquired Resistance and Clonal Evolution in Melanoma during BRAF Inhibitor Therapy. Cancer Discov. 2014, 4, 80–93. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y.; Guo, W. A Review of the Molecular Pathways Involved in Resistance to BRAF Inhibitors in Patients with Advanced-Stage Melanoma. Med. Sci. Monit. 2020, 26, e920957-1. [Google Scholar] [CrossRef]
- Torres-Collado, A.X.; Knott, J.; Jazirehi, A.R. Reversal of Resistance in Targeted Therapy of Metastatic Melanoma: Lessons Learned from Vemurafenib (BRAFV600E-Specific Inhibitor). Cancers 2018, 10, 157. [Google Scholar] [CrossRef] [Green Version]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.J.; et al. Combined BRAF and MEK Inhibition versus BRAF Inhibition Alone in Melanoma. N. Engl. J. Med. 2014, 371, 1877–1888. [Google Scholar] [CrossRef] [Green Version]
- Robert, C.; Karaszewska, B.; Schachter, J.; Rutkowski, P.; Mackiewicz, A.; Stroiakovski, D.; Lichinitser, M.; Dummer, R.; Grange, F.; Mortier, L.; et al. Improved Overall Survival in Melanoma with Combined Dabrafenib and Trametinib. N. Engl. J. Med. 2015, 372, 30–39. [Google Scholar] [CrossRef] [Green Version]
- Niu, N.; Wang, L. In Vitro Human Cell Line Models to Predict Clinical Response to Anticancer Drugs. Pharmacogenomics 2015, 16, 273–285. [Google Scholar] [CrossRef] [Green Version]
- Kemper, K.; Krijgsman, O.; Kong, X.; Cornelissen-Steijger, P.; Shahrabi, A.; Weeber, F.; van der Velden, D.L.; Bleijerveld, O.B.; Kuilman, T.; Kluin, R.J.C.; et al. BRAF(V600E) Kinase Domain Duplication Identified in Therapy-Refractory Melanoma Patient-Derived Xenografts. Cell Rep. 2016, 16, 263–277. [Google Scholar] [CrossRef] [Green Version]
- Monsma, D.J.; Cherba, D.M.; Eugster, E.E.; Dylewski, D.L.; Davidson, P.T.; Peterson, C.A.; Borgman, A.S.; Winn, M.E.; Dykema, K.J.; Webb, C.P.; et al. Melanoma Patient Derived Xenografts Acquire Distinct Vemurafenib Resistance Mechanisms. Am. J. Cancer Res. 2015, 5, 1507–1518. [Google Scholar]
- Van Allen, E.M.; Wagle, N.; Sucker, A.; Treacy, D.J.; Johannessen, C.M.; Goetz, E.M.; Place, C.S.; Taylor-Weiner, A.; Whittaker, S.; Kryukov, G.V.; et al. The Genetic Landscape of Clinical Resistance to RAF Inhibition in Metastatic Melanoma. Cancer Discov. 2014, 4, 94–109. [Google Scholar] [CrossRef] [Green Version]
- Lito, P.; Pratilas, C.A.; Joseph, E.W.; Tadi, M.; Halilovic, E.; Zubrowski, M.; Huang, A.; Wong, W.L.; Callahan, M.K.; Merghoub, T.; et al. Relief of Profound Feedback Inhibition of Mitogenic Signaling by Raf Inhibitors Attenuates Their Activity in Brafv600e Melanomas. Cancer Cell 2012, 22, 668–682. [Google Scholar] [CrossRef] [Green Version]
- Arozarena, I.; Wellbrock, C. Overcoming Resistance to BRAF Inhibitors. Ann. Transl. Med. 2017, 5, 387. [Google Scholar] [CrossRef] [Green Version]
- Smith, M.P.; Brunton, H.; Rowling, E.J.; Ferguson, J.; Arozarena, I.; Miskolczi, Z.; Lee, J.L.; Girotti, M.R.; Marais, R.; Levesque, M.P.; et al. Inhibiting Drivers of Non-Mutational Drug Tolerance Is a Salvage Strategy for Targeted Melanoma Therapy. Cancer Cell 2016, 29, 270–284. [Google Scholar] [CrossRef] [Green Version]
- Martin, M. Cutadapt Removes Adapter Sequences from High-Throughput Sequencing Reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-Based Genome Alignment and Genotyping with HISAT2 and HISAT-Genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve Years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- Kovaka, S.; Zimin, A.V.; Pertea, G.M.; Razaghi, R.; Salzberg, S.L.; Pertea, M. Transcriptome Assembly from Long-Read RNA-Seq Alignments with StringTie2. Genome Biol. 2019, 20, 278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2021; Available online: Https://Www.R-Project.Org/ (accessed on 15 April 2023).
- Law, C.W.; Chen, Y.; Shi, W.; Smyth, G.K. Voom: Precision Weights Unlock Linear Model Analysis Tools for RNA-Seq Read Counts. Genome Biol. 2014, 15, R29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, R.; Holik, A.Z.; Su, S.; Jansz, N.; Chen, K.; Leong, H.S.; Blewitt, M.E.; Asselin-Labat, M.-L.; Smyth, G.K.; Ritchie, M.E. Why Weight? Modelling Sample and Observational Level Variability Improves Power in RNA-Seq Analyses. Nucleic Acids Res. 2015, 43, e97. [Google Scholar] [CrossRef] [PubMed]
- Smyth, G.K. Limma: Linear Models for Microarray Data. In Bioinformatics and Computational Biology Solutions Using R and Bioconductor; Gentleman, R., Carey, V.J., Huber, W., Irizarry, R.A., Dudoit, S., Eds.; Statistics for Biology and Health; Springer: New York, NY, USA, 2005; pp. 397–420. ISBN 978-0-387-29362-2. [Google Scholar]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Statist. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Bokharaie, H.; Kolch, W.; Krstic, A. Analysis of Alternative MRNA Splicing in Vemurafenib-Resistant Melanoma Cells. Biomolecules 2022, 12, 993. [Google Scholar] [CrossRef]
- Cserepes, M.; Türk, D.; Tóth, S.; Pape, V.F.S.; Gaál, A.; Gera, M.; Szabó, J.E.; Kucsma, N.; Várady, G.; Vértessy, B.G.; et al. Unshielding Multidrug Resistant Cancer through Selective Iron Depletion of P-Glycoprotein-Expressing Cells. Cancer Res. 2020, 80, 663–674. [Google Scholar] [CrossRef]
- Ladányi, A.; Somlai, B.; Gilde, K.; Fejös, Z.; Gaudi, I.; Tímár, J. T-Cell Activation Marker Expression on Tumor-Infiltrating Lymphocytes As Prognostic Factor in Cutaneous Malignant Melanoma. Clin. Cancer Res. 2004, 10, 521–530. [Google Scholar] [CrossRef] [Green Version]
- Bankhead, P.; Loughrey, M.B.; Fernández, J.A.; Dombrowski, Y.; McArt, D.G.; Dunne, P.D.; McQuaid, S.; Gray, R.T.; Murray, L.J.; Coleman, H.G.; et al. QuPath: Open Source Software for Digital Pathology Image Analysis. Sci. Rep. 2017, 7, 16878. [Google Scholar] [CrossRef] [Green Version]
- Wilson, M.A.; Schuchter, L.M. Chemotherapy for Melanoma. In Melanoma; Kaufman, H.L., Mehnert, J.M., Eds.; Cancer Treatment and Research; Springer International Publishing: Cham, Switzerland, 2016; pp. 209–229. ISBN 978-3-319-22539-5. [Google Scholar]
- Teixido, C.; Castillo, P.; Martinez-Vila, C.; Arance, A.; Alos, L. Molecular Markers and Targets in Melanoma. Cells 2021, 10, 2320. [Google Scholar] [CrossRef]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.-J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2019, 381, 1535–1546. [Google Scholar] [CrossRef] [Green Version]
- Ascierto, P.A.; Kirkwood, J.M.; Grob, J.-J.; Simeone, E.; Grimaldi, A.M.; Maio, M.; Palmieri, G.; Testori, A.; Marincola, F.M.; Mozzillo, N. The Role of BRAF V600 Mutation in Melanoma. J. Transl. Med. 2012, 10, 85. [Google Scholar] [CrossRef] [Green Version]
- Holderfield, M.; Deuker, M.M.; McCormick, F.; McMahon, M. Targeting RAF Kinases for Cancer Therapy: BRAF-Mutated Melanoma and Beyond. Nat. Rev. Cancer 2014, 14, 455–467. [Google Scholar] [CrossRef] [Green Version]
- McArthur, G.A. Combination Therapies to Inhibit the RAF/MEK/ERK Pathway in Melanoma: We Are Not Done Yet. Front. Oncol. 2015, 5, 161. [Google Scholar] [CrossRef] [Green Version]
- Facchinetti, F.; Lacroix, L.; Mezquita, L.; Scoazec, J.-Y.; Loriot, Y.; Tselikas, L.; Gazzah, A.; Rouleau, E.; Adam, J.; Michiels, S.; et al. Molecular Mechanisms of Resistance to BRAF and MEK Inhibitors in BRAFV600E Non-Small Cell Lung Cancer. Eur. J. Cancer 2020, 132, 211–223. [Google Scholar] [CrossRef]
- Wu, C.-P.; Ambudkar, S.V. The Pharmacological Impact of ATP-Binding Cassette Drug Transporters on Vemurafenib-Based Therapy. Acta Pharm. Sin. B 2014, 4, 105–111. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Dou, Y.; Xu, H.; Jiang, Z. The Expression and Clinical Significance of GPR39 in Colon Cancer. Ir. J. Med. Sci. 2022, 191, 1577–1585. [Google Scholar] [CrossRef]
- Miao, M.; Masengere, H.; Yu, G.; Shan, F. Reevaluation of NOD/SCID Mice as NK Cell-Deficient Models. Biomed. Res. Int. 2021, 2021, 8851986. [Google Scholar] [CrossRef]
- Song, F.; Yi, Y.; Li, C.; Hu, Y.; Wang, J.; Smith, D.E.; Jiang, H. Regulation and Biological Role of the Peptide/Histidine Transporter SLC15A3 in Toll-like Receptor-Mediated Inflammatory Responses in Macrophage. Cell Death Dis. 2018, 9, 770. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Chen, C.; Liu, Z.; Guo, H.; Lu, W.; Hu, W.; Lin, Z. PABPC1-Induced Stabilization of IFI27 MRNA Promotes Angiogenesis and Malignant Progression in Esophageal Squamous Cell Carcinoma through Exosomal MiRNA-21-5p. J. Exp. Clin. Cancer Res. 2022, 41, 111. [Google Scholar] [CrossRef]
- Szekely, B.; Bossuyt, V.; Li, X.; Wali, V.B.; Patwardhan, G.A.; Frederick, C.; Silber, A.; Park, T.; Harigopal, M.; Pelekanou, V.; et al. Immunological Differences between Primary and Metastatic Breast Cancer. Ann. Oncol. 2018, 29, 2232–2239. [Google Scholar] [CrossRef] [PubMed]
- Rebecca, V.W.; Wood, E.; Fedorenko, I.V.; Paraiso, K.H.T.; Haarberg, H.E.; Chen, Y.; Xiang, Y.; Sarnaik, A.; Gibney, G.T.; Sondak, V.K.; et al. Evaluating Melanoma Drug Response and Therapeutic Escape with Quantitative Proteomics. Mol. Cell Proteom. 2014, 13, 1844–1854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szakács, G.; Paterson, J.K.; Ludwig, J.A.; Booth-Genthe, C.; Gottesman, M.M. Targeting Multidrug Resistance in Cancer. Nat. Rev. Drug Discov. 2006, 5, 219–234. [Google Scholar] [CrossRef] [PubMed]
- Durmus, S.; Sparidans, R.W.; Wagenaar, E.; Beijnen, J.H.; Schinkel, A.H. Oral Availability and Brain Penetration of the B-RAFV600E Inhibitor Vemurafenib Can Be Enhanced by the P-Glycoprotein (ABCB1) and Breast Cancer Resistance Protein (ABCG2) Inhibitor Elacridar. Mol. Pharm. 2012, 9, 3236–3245. [Google Scholar] [CrossRef] [PubMed]
- Tamaki, A.; Ierano, C.; Szakacs, G.; Robey, R.W.; Bates, S.E. The Controversial Role of ABC Transporters in Clinical Oncology. Essays Biochem. 2011, 50, 209–232. [Google Scholar] [CrossRef]
- Qiu, J.-G.; Zhang, Y.-J.; Li, Y.; Zhao, J.-M.; Zhang, W.-J.; Jiang, Q.-W.; Mei, X.-L.; Xue, Y.-Q.; Qin, W.-M.; Yang, Y.; et al. Trametinib Modulates Cancer Multidrug Resistance by Targeting ABCB1 Transporter. Oncotarget 2015, 6, 15494–15509. [Google Scholar] [CrossRef] [Green Version]
- Das Thakur, M.; Salangsang, F.; Landman, A.S.; Sellers, W.R.; Pryer, N.K.; Levesque, M.P.; Dummer, R.; McMahon, M.; Stuart, D.D. Modelling Vemurafenib Resistance in Melanoma Reveals a Strategy to Forestall Drug Resistance. Nature 2013, 494, 251–255. [Google Scholar] [CrossRef] [Green Version]
- Fofaria, N.M.; Frederick, D.T.; Sullivan, R.J.; Flaherty, K.T.; Srivastava, S.K. Overexpression of Mcl-1 Confers Resistance to BRAFV600E Inhibitors Alone and in Combination with MEK1/2 Inhibitors in Melanoma. Oncotarget 2015, 6, 40535–40556. [Google Scholar] [CrossRef]
- Singh, M.K.; Altameemi, S.; Lares, M.; Newton, M.A.; Setaluri, V. Role of Dual Specificity Phosphatases (DUSPs) in Melanoma Cellular Plasticity and Drug Resistance. Sci. Rep. 2022, 12, 14395. [Google Scholar] [CrossRef]
- Shen, S.; Faouzi, S.; Souquere, S.; Roy, S.; Routier, E.; Libenciuc, C.; André, F.; Pierron, G.; Scoazec, J.-Y.; Robert, C. Melanoma Persister Cells Are Tolerant to BRAF/MEK Inhibitors via ACOX1-Mediated Fatty Acid Oxidation. Cell Rep. 2020, 33, 108421. [Google Scholar] [CrossRef]
- Romano, E.; Pradervand, S.; Paillusson, A.; Weber, J.; Harshman, K.; Muehlethaler, K.; Speiser, D.; Peters, S.; Rimoldi, D.; Michielin, O. Identification of Multiple Mechanisms of Resistance to Vemurafenib in a Patient with BRAFV600E-Mutated Cutaneous Melanoma Successfully Rechallenged after Progression. Clin. Cancer Res. 2013, 19, 5749–5757. [Google Scholar] [CrossRef] [Green Version]
- Wilmott, J.S.; Tembe, V.; Howle, J.R.; Sharma, R.; Thompson, J.F.; Rizos, H.; Lo, R.S.; Kefford, R.F.; Scolyer, R.A.; Long, G.V. Intratumoral Molecular Heterogeneity in a BRAF-Mutant, BRAF Inhibitor-Resistant Melanoma: A Case Illustrating the Challenges for Personalized Medicine. Mol. Cancer Ther. 2012, 11, 2704–2708. [Google Scholar] [CrossRef] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer | Reverse Primer |
|---|---|---|
| AGAP9 | CCACCACTGATGAGGACCTG | ATGACGTCCACCCCGTCA |
| GZMB | CCCTGGGAAAACACTCACACA | GCACAACTCAATGGTACTGTCG |
| GNRHR | TGTCTGGAAAGATCCGAGTGA | AGGTTGGCTAAGGTCAGATGTT |
| CD27 | TCAGCAACTGGGCACAGAAA | GGATCACACTGAGCAGCCTT |
| CP | GGGCCATCTACCCTGATAACA | TTAAAGGTCCGATGAGTCCTGA |
| IFI27 | TGCTCTCACCTCATCAGCAGT | CACAACTCCTCCAATCACAACT |
| GPR39 | TGTCCCCGAGTTTGAGGTG | GAAGGCCCATCACGAAGATGA |
| GSTA1 | CTGCCCGTATGTCCACCTG | AGCTCCTCGACGTAGTAGAGA |
| SLC15A3 | TGGCGTTTATTCAGCAGAACA | TCTCTGGCCGAGTGTCGTT |
| FKBP1A-SDCBP2 | CACTACCCTGCACTGAGCTG | CATGACGTCCACCCCGTCAG |
| PDGFA | GCAAGACCAGGACGGTCATTT | GGCACTTGACACTGCTCGT |
| ABCB1 | TTGCTGCTTACATTCAGGTTTCA | AGCCTATCTCCTGTCGCATTA |
| RPLP0 | AGCCCAGAACACTGGTCTC | ACTCAGGATTTCAATGGTGCC |
| BRAF exon 3 | AGCAAGCTAGATGCACTCCA | --- |
| BRAF exon 8 | CCAAATTCTCACCAGTCCGT | --- |
| BRAF exon 9 | --- | ACCACGAAATCCTTGGTCTC |
| Upregulated genes | Fold-change: generation 5/control | Fold-change: generation 3/control | Positive prognostic value (ProteinAtlas) | Negative prognostic value (ProteinAtlas) |
| AGAP9 | 59.54 | 102.30 | UC | RCC; CRC |
| FKBP1A-SDCBP2 | 25.83 | 16.11 | --- | HCC; RCC |
| GZMB | 8.21 | 9.49 | EC; BC | RCC |
| CP | 9.49 | 7.60 | --- | RCC |
| GNRHR | 6.41 | 8.94 | --- | --- |
| ACBD7-DCLRE1CP1 | 7.80 | 5.77 | HNSCC; OC | RCC |
| CD27 | 3.04 | 2.86 | Melanoma malignum; RCC | HNSCC; CC; EC |
| Downregulated genes | Fold-change: control/generation 5 | Fold-change: control/generation 3 | Positive prognostic value (ProteinAtlas) | Negative prognostic value (ProteinAtlas) |
| GPR39 | 22.97 | 32.52 | --- | Pancreas |
| GSTA1 | 18.17 | 16.61 | NSCLC | RCC |
| TNNT3 | 16.55 | 15.13 | CC | --- |
| SLC15A3 | 15.78 | 7.71 | CC | --- |
| IFI27 | 49.45 | 7.11 | OC | --- |
| ZNF141 | 29.13 | 6.88 | UC | --- |
| PDGFA | 5.13 | 5.31 | --- | glioma, HNSCC, UC |
| MDR genes | Fold-change: generation 5/control | Fold-change: generation 3/control | Positive prognostic value (ProteinAtlas) | Negative prognostic value (ProteinAtlas) |
| ABCB1 | n.s. | n.s. | RCC; Pancreas | --- |
| ABCG2 | n.s. | n.s. | --- | --- |
| ABCC1 | n.s. | n.s. | --- | RCC; HCC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tóvári, J.; Vári-Mező, D.; Surguta, S.E.; Ladányi, A.; Kigyós, A.; Cserepes, M. Evolving Acquired Vemurafenib Resistance in a BRAF V600E Mutant Melanoma PDTX Model to Reveal New Potential Targets. Cells 2023, 12, 1919. https://doi.org/10.3390/cells12141919
Tóvári J, Vári-Mező D, Surguta SE, Ladányi A, Kigyós A, Cserepes M. Evolving Acquired Vemurafenib Resistance in a BRAF V600E Mutant Melanoma PDTX Model to Reveal New Potential Targets. Cells. 2023; 12(14):1919. https://doi.org/10.3390/cells12141919
Chicago/Turabian StyleTóvári, József, Diána Vári-Mező, Sára Eszter Surguta, Andrea Ladányi, Attila Kigyós, and Mihály Cserepes. 2023. "Evolving Acquired Vemurafenib Resistance in a BRAF V600E Mutant Melanoma PDTX Model to Reveal New Potential Targets" Cells 12, no. 14: 1919. https://doi.org/10.3390/cells12141919