
Role of a Novel Heparanase Inhibitor on the Balance between Apoptosis and Autophagy in U87 Human Glioblastoma Cells
 , ,
, ,  , , and
, , and
Abstract
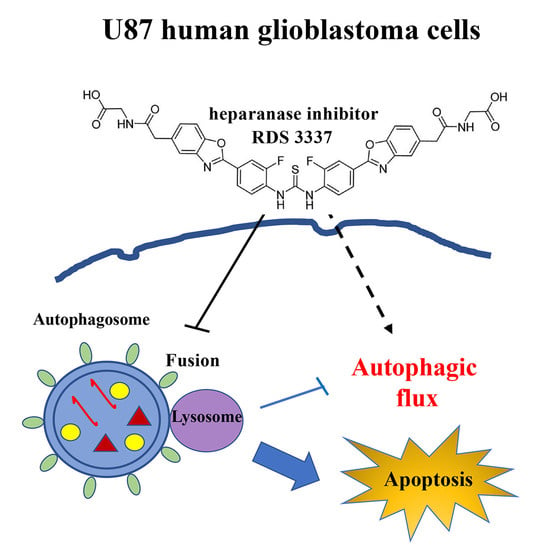
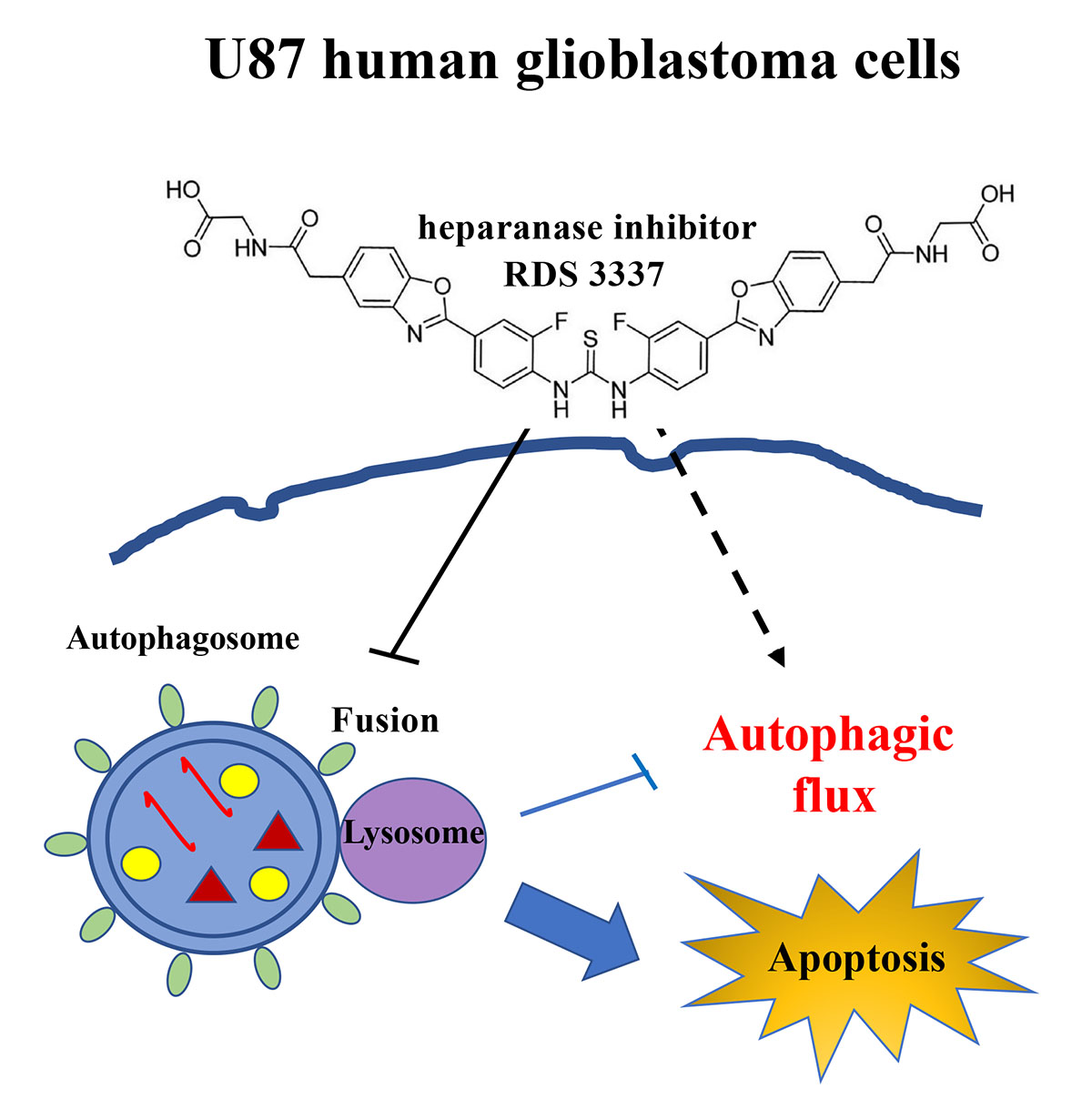
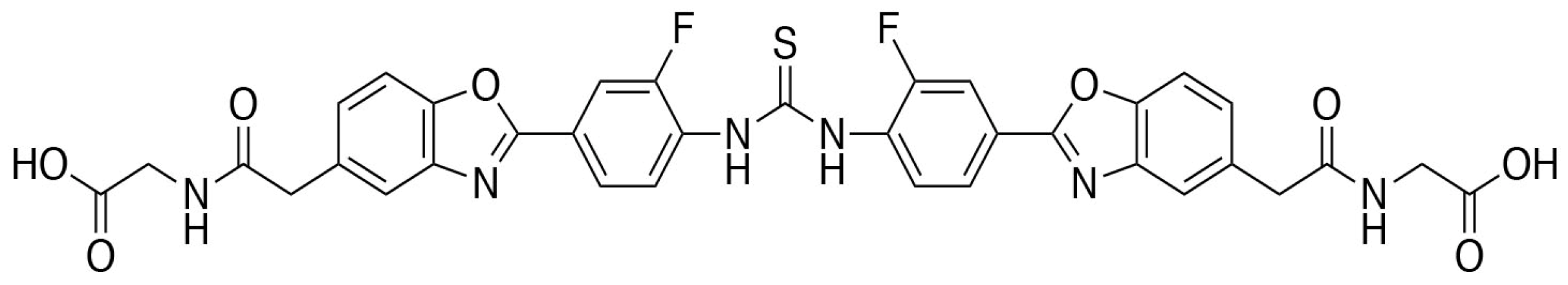
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Cultures and Treatments
2.2. In Vitro Treatment with HPSE Inhibitors
2.3. Cell Viability Assays
2.3.1. Trypan Blue Assay
2.3.2. WST-1 Cell Viability Assay
2.3.3. BrdU Cell Proliferation Assay
2.4. Preparation of Cell Extracts
2.5. Western Blot Analysis for Autophagy and Apoptosis-Related Proteins
2.6. Autophagy Evaluation by Flow Cytometry
2.7. Apoptosis Evaluation by Flow Cytometry
2.7.1. Propidium Iodide Staining
2.7.2. AnnexinV/Propidium Iodide (PI)
2.8. Statistical Analysis
3. Results
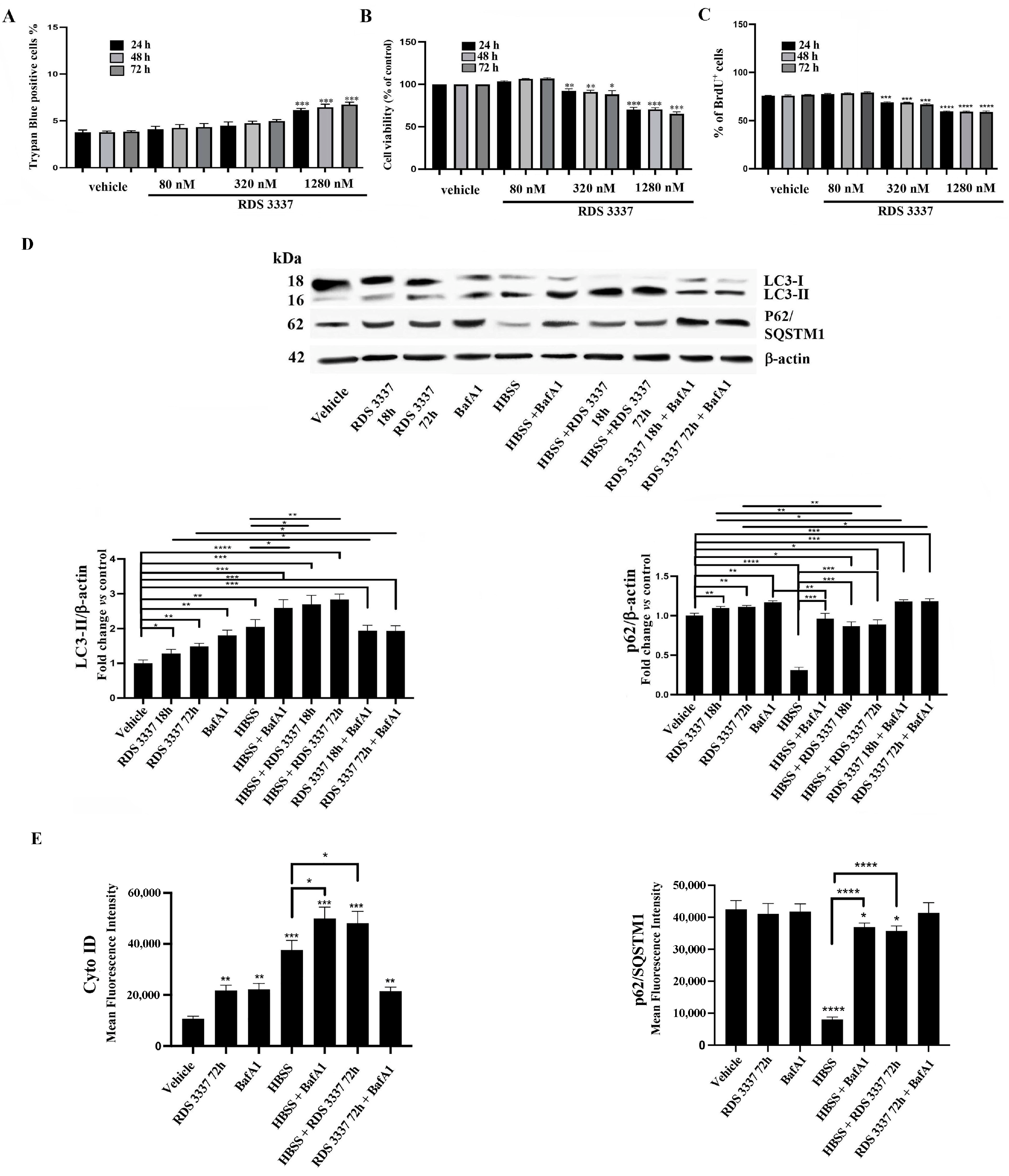
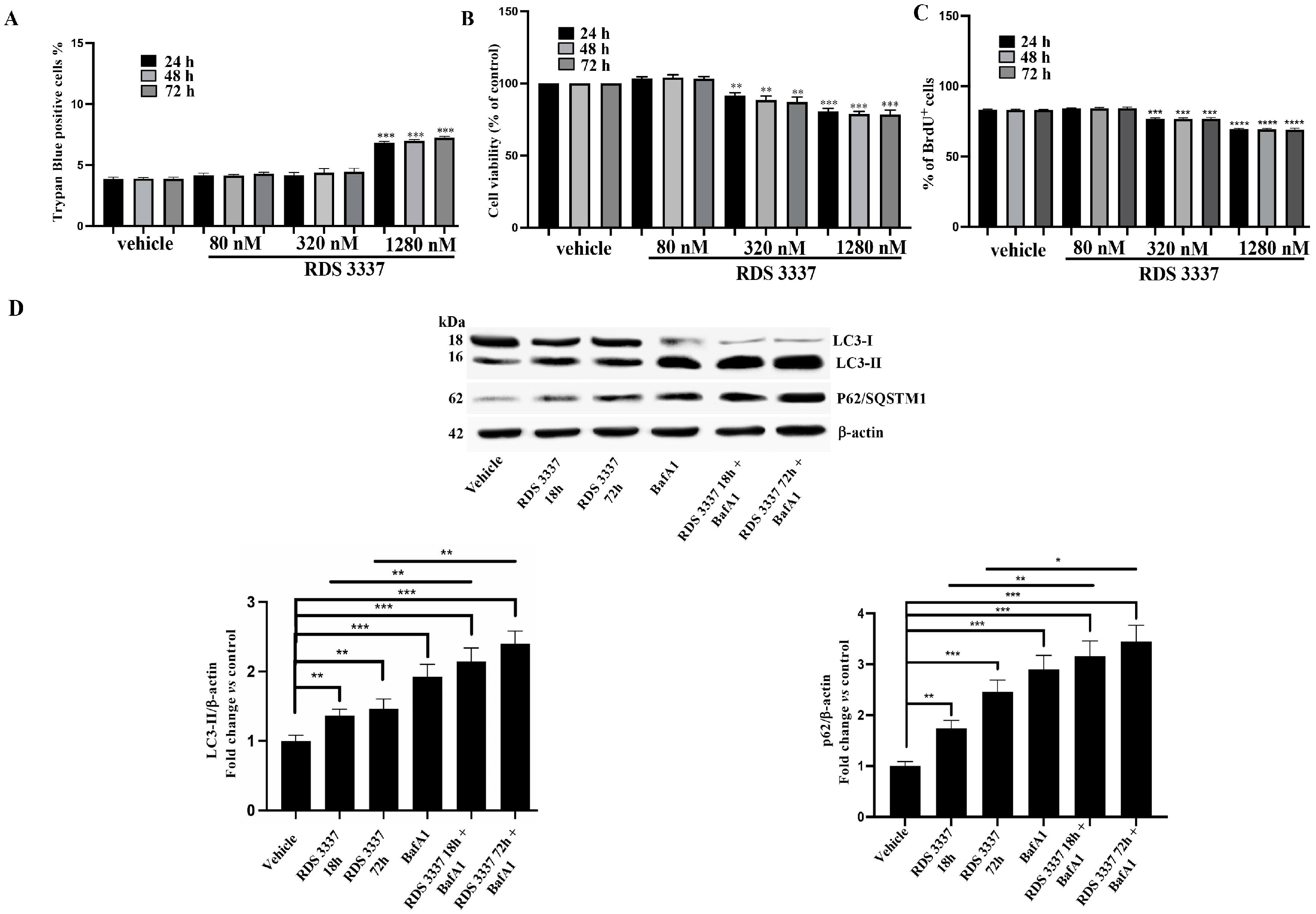
3.1. HPSE Inhibitor RDS 3337 Promotes LC3-II and p62 Accumulation in RPE-1 Cells
3.2. HPSE Inhibitor RDS 3337 Blocks Autophagic Flux in U87 Human Glioblastoma Cells
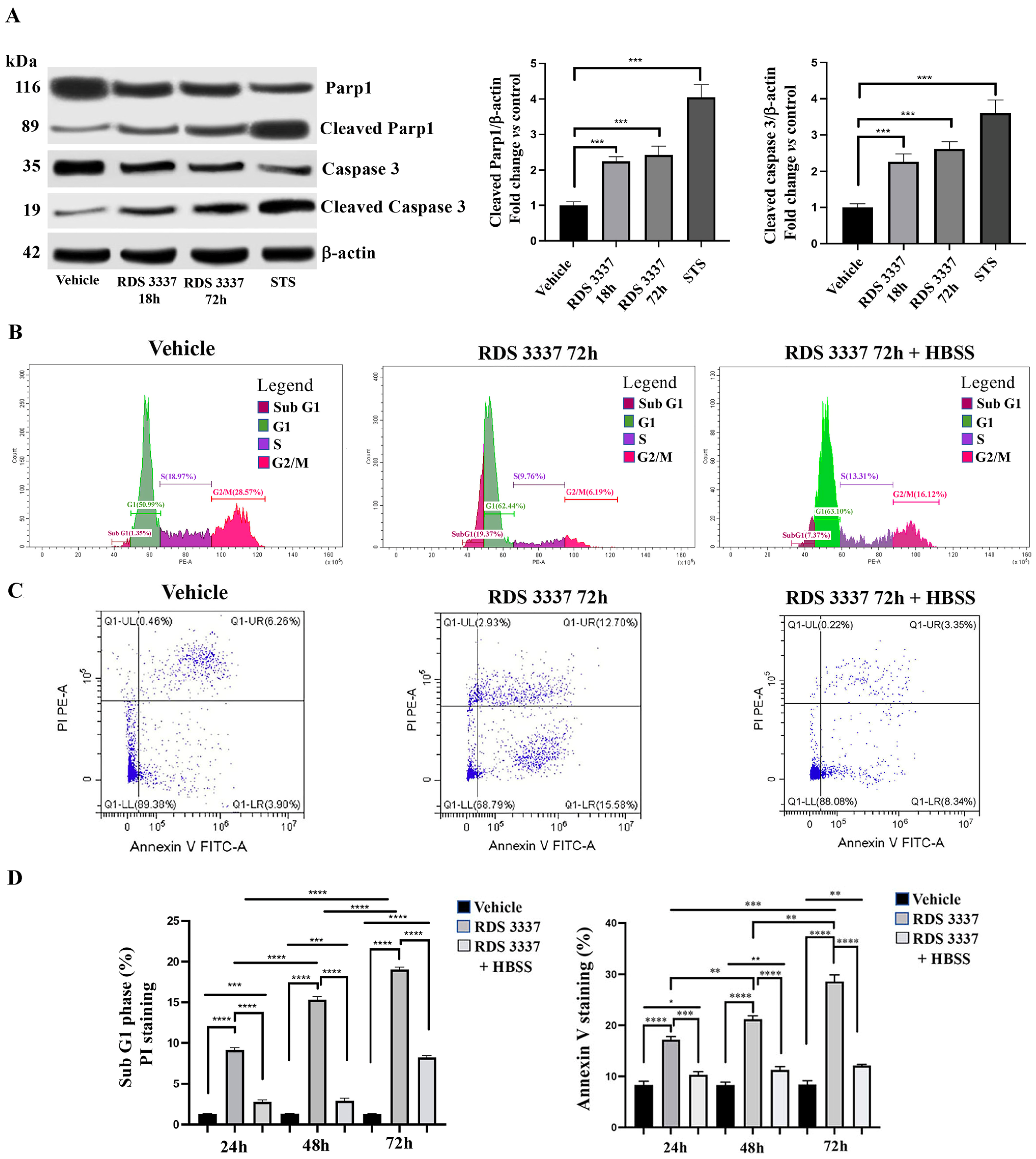
3.3. RDS 3337 Inhibitor Sensitizes U87 Human Glioblastoma Cells to Apoptosis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vlodavsky, I.; Singh, P.; Boyango, I.; Gutter-Kapon, L.; Elkin, M.; Sanderson, R.D.; Ilan, N. Heparanase: From basic research to therapeutic applications in cancer and inflammation. Drug Resist. Update 2016, 29, 54–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, V.M.; Wade, A.; McKinney, A.; Chen, K.; Lindberg, O.R.; Engler, J.R.; Persson, A.I.; Phillips, J.J. Heparan Sulfate Glycosaminoglycans in Glioblastoma Promote Tumor Invasion. Mol. Cancer Res. 2017, 15, 1623–1633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.J.; Shi, L.; Wang, X.M.; Yang, G.W. Heparanase is a novel biomarker for immune infiltration and prognosis in breast cancer. Aging 2021, 13, 20836–20852. [Google Scholar] [CrossRef]
- Hammond, E.; Khurana, A.; Shridhar, V.; Dredge, K. The Role of Heparanase and Sulfatases in the Modification of Heparan Sulfate Proteoglycans within the Tumor Microenvironment and Opportunities for Novel Cancer Therapeutics. Front. Oncol. 2014, 4, 195. [Google Scholar] [CrossRef] [PubMed]
- Gingis-Velitski, S.; Zetser, A.; Kaplan, V.; Ben-Zaken, O.; Cohen, E.; Levy-Adam, F.; Bashenko, Y.; Flugelman, M.Y.; Vlodavsky, I.; Ilan, N. Heparanase uptake is mediated by cell membrane heparan sulfate proteoglycans. J. Biol. Chem. 2004, 279, 44084–444092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shteingauz, A.; Ilan, N.; Vlodavsky, I. Processing of heparanase is mediated by syndecan-1 cytoplasmic domain and involves syntenin and α-actinin. Cell. Mol. Life Sci. 2014, 71, 4457–4470. [Google Scholar] [CrossRef] [Green Version]
- Nadav, L.; Eldor, A.; Yacoby-Zeevi, O.; Zamir, E.; Pecker, I.; Ilan, N.; Geiger, B.; Vlodavsky, I.; Katz, B.Z. Activation, processing and trafficking of extracellular heparanase by primary human fibroblasts. J. Cell Sci. 2002, 115, 2179–2187. [Google Scholar] [CrossRef]
- Zetser, A.; Levy-Adam, F.; Kaplan, V.; Gingis-Velitski, S.; Bashenko, Y.; Schubert, S.; Flugelman, M.Y.; Vlodavsky, I.; Ilan, N. Processing and activation of latent heparanase occurs in lysosomes. J. Cell Sci. 2004, 117, 2249–2258. [Google Scholar] [CrossRef] [Green Version]
- Abboud-Jarrous, G.; Rangini-Guetta, Z.; Aingorn, H.; Atzmon, R.; Elgavish, S.; Peretz, T.; Vlodavsky, I. Site-directed mutagenesis, proteolytic cleavage, and activation of human proheparanase. J. Biol. Chem. 2005, 280, 13568–13575. [Google Scholar] [CrossRef] [Green Version]
- Abboud-Jarrous, G.; Atzmon, R.; Peretz, T.; Palermo, C.; Gadea, B.B.; Joyce, J.A.; Vlodavsky, I. Cathepsin L is responsible for processing and activation of proheparanase through multiple cleavages of a linker segment. J. Biol. Chem. 2008, 283, 18167–18176. [Google Scholar] [CrossRef] [Green Version]
- Sanderson, R.D.; Bandari, S.K.; Vlodavsky, I. Proteases and glycosidases on the surface of exosomes: Newly discovered mechanisms for extracellular remodeling. Matrix Biol. 2019, 75–76, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Shteingauz, A.; Boyango, I.; Naroditsky, I.; Hammond, E.; Gruber, M.; Doweck, I.; Ilan, N.; Vlodavsky, I. Heparanase Enhances Tumor Growth and Chemoresistance by Promoting Autophagy. Cancer Res. 2015, 75, 3946–3957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, M.D.; Sanderson, R.D. Heparan sulfate in the nucleus and its control of cellular functions. Matrix Biol. 2014, 35, 56–59. [Google Scholar] [CrossRef] [PubMed]
- Fux, L.; Ilan, N.; Sanderson, R.D.; Vlodavsky, I. Heparanase: Busy at the cell surface. Trends Biochem. Sci. 2009, 34, 511–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shafat, I.; Ben-Arush, M.W.; Issakov, J.; Meller, I.; Naroditsky, I.; Tortoreto, M.; Cassinelli, G.; Lanzi, C.; Pisano, C.; Ilan, N.; et al. Pre-clinical and clinical significance of heparanase in Ewing’s sarcoma. J. Cell. Mol. Med. 2011, 15, 1857–1864. [Google Scholar] [CrossRef]
- Vlodavsky, I.; Beckhove, P.; Lerner, I.; Pisano, C.; Meirovitz, A.; Ilan, N.; Elkin, M. Significance of heparanase in cancer and inflammation. Cancer Microenviron. 2012, 5, 115–132. [Google Scholar] [CrossRef] [Green Version]
- Ilan, N.; Shteingauz, A.; Vlodavsky, I. Function from within: Autophagy induction by HPSE/heparanase--new possibilities for intervention. Autophagy 2015, 11, 2387–2399. [Google Scholar] [CrossRef] [Green Version]
- Masola, V.; Bellin, G.; Gambaro, G.; Onisto, M. Heparanase: A Multitasking Protein Involved in Extracellular Matrix (ECM) Remodeling and Intracellular Events. Cells 2018, 28, 236. [Google Scholar] [CrossRef] [Green Version]
- He, C.; Klionsky, D.J. Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 2009, 43, 67–93. [Google Scholar] [CrossRef] [Green Version]
- Matarrese, P.; Garofalo, T.; Manganelli, V.; Gambardella, L.; Marconi, M.; Grasso, M.; Tinari, A.; Misasi, R.; Malorni, W.; Sorice, M. Evidence for the involvement of GD3 ganglioside in autophagosome formation and maturation. Autophagy 2014, 10, 750–765. [Google Scholar] [CrossRef]
- Garofalo, T.; Matarrese, P.; Manganelli, V.; Marconi, M.; Tinari, A.; Gambardella, L.; Faggioni, A.; Misasi, R.; Sorice, M.; Malorni, W. Evidence for the involvement of lipid rafts localized at the ER-mitochondria associated membranes in autophagosome formation. Autophagy 2016, 12, 917–935. [Google Scholar] [CrossRef] [Green Version]
- Manganelli, V.; Matarrese, P.; Antonioli, M.; Gambardella, L.; Vescovo, T.; Gretzmeier, C.; Longo, A.; Capozzi, A.; Recalchi, S.; Riitano, G.; et al. Raft-like lipid microdomains drive autophagy initiation via AMBRA1-ERLIN1 molecular association within MAMs. Autophagy 2021, 17, 2528–2548. [Google Scholar] [CrossRef] [PubMed]
- Murrow, L.; Debnath, J. Autophagy as a stress-response and quality-control mechanism: Implications for cell injury and human disease. Annu. Rev. Pathol. 2013, 8, 105–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marinković, M.; Šprung, M.; Buljubašić, M.; Novak, I. Autophagy Modulation in Cancer: Current Knowledge on Action and Therapy. Oxid. Med. Cell. Longev. 2018, 31, 8023821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; He, Z.; Simon, H.U. Protective role of autophagy and autophagy-related protein 5 in early tumorigenesis. J. Mol. Med. 2015, 93, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Chavez-Dominguez, R.; Perez-Medina, M.; Lopez-Gonzalez, J.S.; Galicia-Velasco, M.; Aguilar-Cazares, D. The Double-Edge Sword of Autophagy in Cancer: From Tumor Suppression to Pro-tumor Activity. Front. Oncol. 2020, 10, 578418. [Google Scholar] [CrossRef]
- Heyman, B.; Yang, Y. Mechanisms of heparanase inhibitors in cancer therapy. Exp. Hematol. 2016, 44, 1002–1012. [Google Scholar] [CrossRef] [Green Version]
- Mohan, C.D.; Hari, S.; Preetham, H.D.; Rangappa, S.; Barash, U.; Ilan, N.; Nayak, S.C.; Gupta, V.K.; Basappa; Vlodavsky, I.; et al. Targeting Heparanase in Cancer: Inhibition by Synthetic, Chemically Modified, and Natural Compounds. iScience 2019, 31, 360–390. [Google Scholar] [CrossRef] [Green Version]
- Weissmann, M.; Arvatz, G.; Horowitz, N.; Feld, S.; Naroditsky, I.; Zhang, Y.; Ng, M.; Hammond, E.; Nevo, E.; Vlodavsky, I.; et al. Heparanase-neutralizing antibodies attenuate lymphoma tumor growth and metastasis. Proc. Natl. Acad. Sci. USA 2016, 113, 704–709. [Google Scholar] [CrossRef]
- Chhabra, M. Chapter 6—Biological therapeutic modalities. In Translational Biotechnology; Academic Press: Cambridge, MA, USA, 2021; pp. 137–164. [Google Scholar] [CrossRef]
- Messore, A.; Madia, V.N.; Pescatori, L.; Saccoliti, F.; Tudino, V.; De Leo, A.; Bortolami, M.; De Vita, D.; Scipione, L.; Pepi, F.; et al. Novel Symmetrical Benzazolyl Derivatives Endowed with Potent Anti-Heparanase Activity. J. Med. Chem. 2018, 61, 10834–10859. [Google Scholar] [CrossRef] [Green Version]
- Madia, V.N.; Messore, A.; Pescatori, L.; Saccoliti, F.; Tudino, V.; De Leo, A.; Bortolami, M.; Scipione, L.; Costi, R.; Rivara, S.; et al. Novel Benzazole Derivatives Endowed with Potent Antiheparanase Activity. J. Med. Chem. 2018, 61, 6918–6936. [Google Scholar] [CrossRef] [PubMed]
- Capozzi, A.; Riitano, G.; Recalchi, S.; Manganelli, V.; Costi, R.; Saccoliti, F.; Pulcinelli, F.; Garofalo, T.; Misasi, R.; Longo, A.; et al. Effect of heparanase inhibitor on tissue factor overexpression in platelets and endothelial cells induced by anti-β2-GPI antibodies. J. Thromb. Haemost. 2021, 19, 2302–2313. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition)1. Autophagy 2021, 17, 1–382. [Google Scholar] [CrossRef] [PubMed]
- Mitrakas, A.G.; Kalamida, D.; Giatromanolaki, A.; Pouliliou, S.; Tsolou, A.; Kyranas, R.; Koukourakis, M.I. Autophagic flux response and glioblastoma sensitivity to radiation. Cancer Biol. Med. 2018, 15, 260–274. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.L.; Gutter-Kapon, L.; Ilan, N.; Batool, T.; Singh, K.; Digre, A.; Luo, Z.; Sandler, S.; Shaked, Y.; Sanderson, R.D.; et al. Significance of host heparanase in promoting tumor growth and metastasis. Matrix Biol. 2020, 93, 25–42. [Google Scholar] [CrossRef]
- Koganti, R.; Suryawanshi, R.; Shukla, D. Heparanase, cell signaling, and viral infections. Cell. Mol. Life Sci. 2020, 77, 5059–5077. [Google Scholar] [CrossRef]
- Wei, R.R.; Sun, D.N.; Yang, H.; Yan, J.; Zhang, X.; Zheng, X.L.; Fu, X.H.; Geng, M.Y.; Huang, X.; Ding, J. CTC clusters induced by heparanase enhance breast cancer metastasis. Acta Pharmacol. Sin. 2018, 39, 1326–1337. [Google Scholar] [CrossRef] [Green Version]
- Gingis-Velitski, S.; Zetser, A.; Flugelman, M.Y.; Vlodavsky, I.; Ilan, N. Heparanase induces endothelial cell migration via protein kinase B/Akt activation. J. Biol. Chem. 2004, 279, 23536–23541. [Google Scholar] [CrossRef] [Green Version]
- Riaz, A.; Ilan, N.; Vlodavsky, I.; Li, J.P.; Johansson, S. Characterization of heparanase-induced phosphatidylinositol 3-kinase-AKT activation and its integrin dependence. J. Biol. Chem. 2013, 288, 12366–12375. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.; Zhang, L.; Jin, J.; Zhu, W.; Xu, Y.; Wu, Y.; Wang, Y.; Chen, H.; Webster, K.A.; Chen, H.; et al. Heparanase released from mesenchymal stem cells activates integrin beta1/HIF-2alpha/Flk-1 signaling and promotes endothelial cell migration and angiogenesis. Stem Cells 2015, 33, 1850–1862. [Google Scholar] [CrossRef]
- Cohen-Kaplan, V.; Naroditsky, I.; Zetser, A.; Ilan, N.; Vlodavsky, I.; Doweck, I. Heparanase induces VEGF C and facilitates tumor lymphangiogenesis. Int. J. Cancer 2008, 123, 2566–2573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen-Kaplan, V.; Jrbashyan, J.; Yanir, Y.; Naroditsky, I.; Ben-Izhak, O.; Ilan, N.; Doweck, I.; Vlodavsky, I. Heparanase induces signal transducer and activator of transcription (STAT) protein phosphorylation: Pre-clinical and clinical significance in head and neck cancer. J. Biol. Chem. 2012, 287, 6668–6678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, M.; Tang, B.; Wang, S.; Tang, L.; Wen, D.; Vlodavsky, I.; Yang, S.M. Non-enzymatic heparanase enhances gastric tumor proliferation via TFEB-dependent autophagy. Oncogenesis 2022, 11, 49. [Google Scholar] [CrossRef] [PubMed]
- Tanida, I.; Minematsu-Ikeguchi, N.; Ueno, T.; Kominami, E. Lysosomal turnover, but not a cellular level, of endogenous LC3 is a marker for autophagy. Autophagy 2005, 1, 84–91. [Google Scholar] [CrossRef] [Green Version]
- Dunlop, E.A.; Tee, A.R. mTOR and autophagy: A dynamic relationship governed by nutrients and energy. Semin. Cell Dev. Biol. 2014, 36, 121–129. [Google Scholar] [CrossRef]
- Matarrese, P.; Manganelli, V.; Garofalo, T.; Tinari, A.; Gambardella, L.; Ndebele, K.; Khosravi-Far, R.; Sorice, M.; Esposti, M.D.; Malorni, W. Endosomal compartment contributes to the propagation of CD95/Fas-mediated signals in type II cells. Biochem. J. 2008, 413, 467–478. [Google Scholar] [CrossRef]
- Sun, W.L.; Chen, J.; Wang, Y.P.; Zheng, H. Autophagy protects breast cancer cells from epirubicin-induced apoptosis and facilitates epirubicin-resistance development. Autophagy 2011, 7, 1035–1044. [Google Scholar] [CrossRef] [Green Version]
- Mariño, G.; Niso-Santano, M.; Baehrecke, E.H.; Kroemer, G. Self-consumption: The interplay of autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2014, 15, 81–94. [Google Scholar] [CrossRef] [Green Version]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9–14. [Google Scholar] [CrossRef]
- Das, S.; Shukla, N.; Singh, S.S.; Kushwaha, S.; Shrivastava, R. Mechanism of interaction between autophagy and apoptosis in cancer. Apoptosis 2021, 26, 512–533. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manganelli, V.; Misasi, R.; Riitano, G.; Capozzi, A.; Mattei, V.; Caglar, T.R.; Ialongo, D.; Madia, V.N.; Messore, A.; Costi, R.; et al. Role of a Novel Heparanase Inhibitor on the Balance between Apoptosis and Autophagy in U87 Human Glioblastoma Cells. Cells 2023, 12, 1891. https://doi.org/10.3390/cells12141891
Manganelli V, Misasi R, Riitano G, Capozzi A, Mattei V, Caglar TR, Ialongo D, Madia VN, Messore A, Costi R, et al. Role of a Novel Heparanase Inhibitor on the Balance between Apoptosis and Autophagy in U87 Human Glioblastoma Cells. Cells. 2023; 12(14):1891. https://doi.org/10.3390/cells12141891
Chicago/Turabian StyleManganelli, Valeria, Roberta Misasi, Gloria Riitano, Antonella Capozzi, Vincenzo Mattei, Tuba Rana Caglar, Davide Ialongo, Valentina Noemi Madia, Antonella Messore, Roberta Costi, and et al. 2023. "Role of a Novel Heparanase Inhibitor on the Balance between Apoptosis and Autophagy in U87 Human Glioblastoma Cells" Cells 12, no. 14: 1891. https://doi.org/10.3390/cells12141891