
Flow Cytometry: The Next Revolution
 ,
, 
 and
and
Abstract
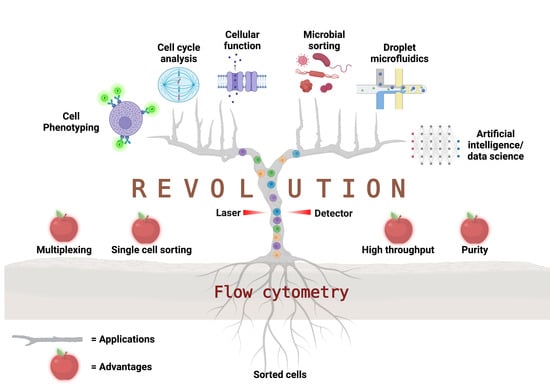
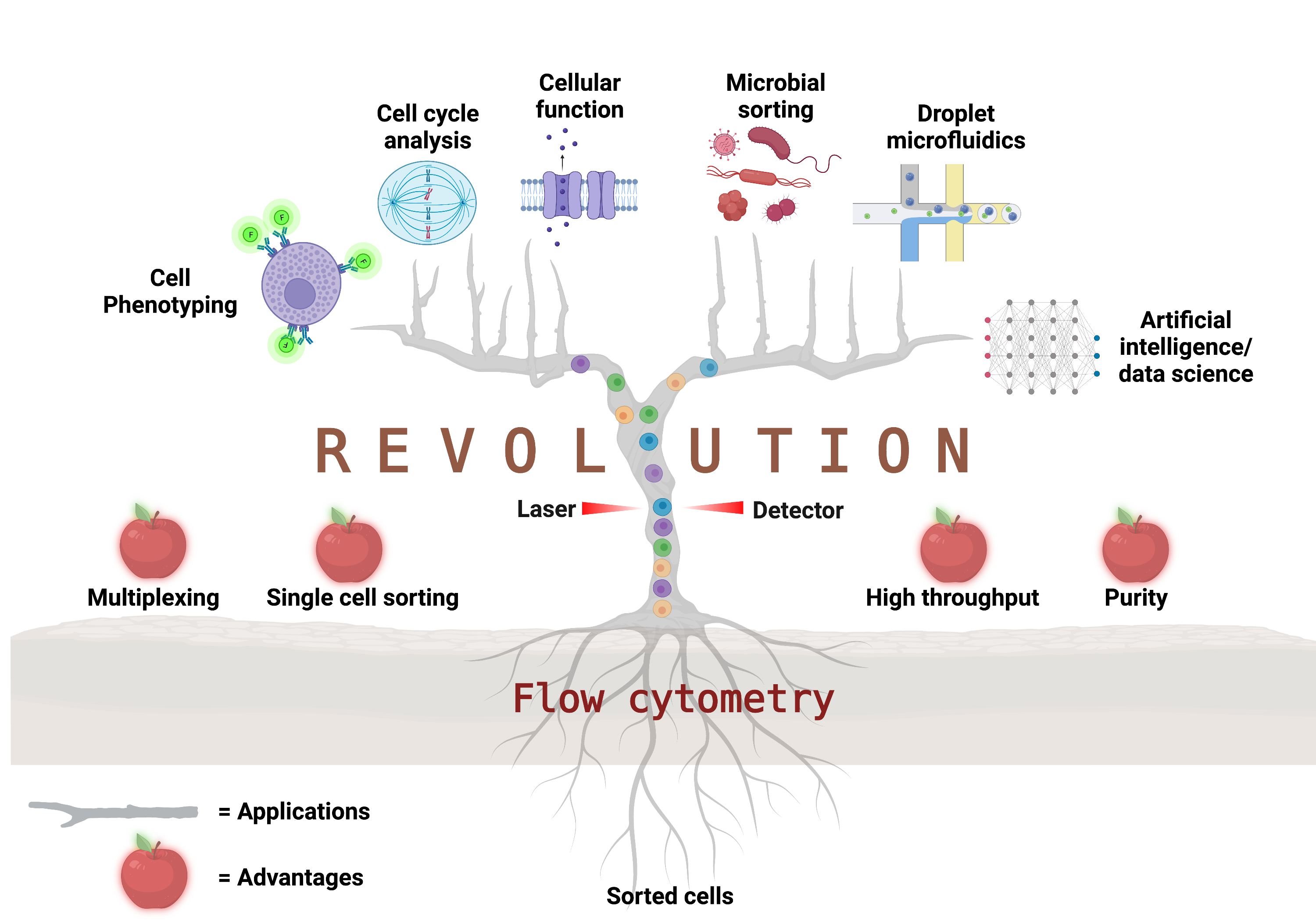
:
1. Background
2. Fluorescence Detection Principles
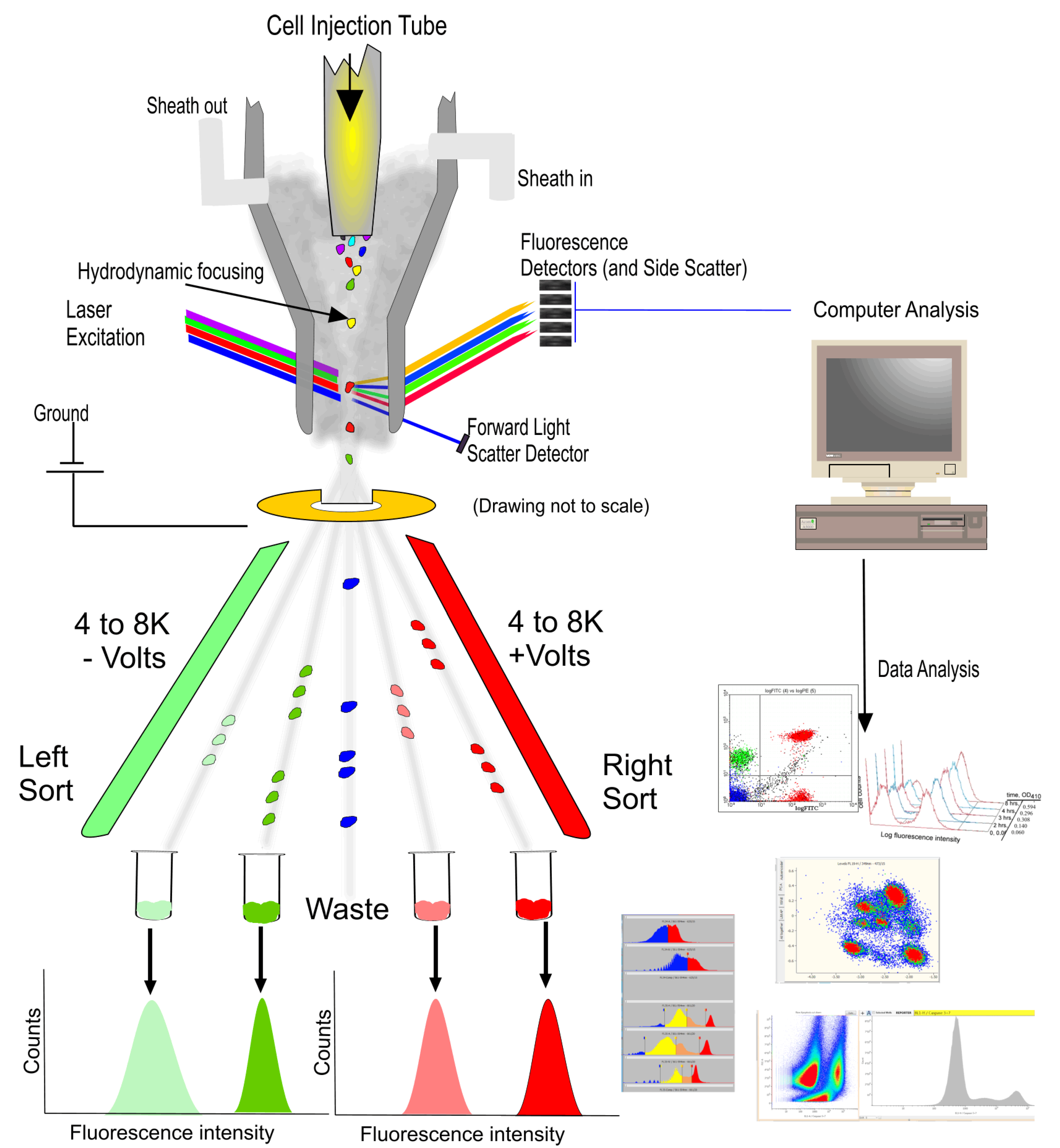
3. The Principles of Flow Cytometry
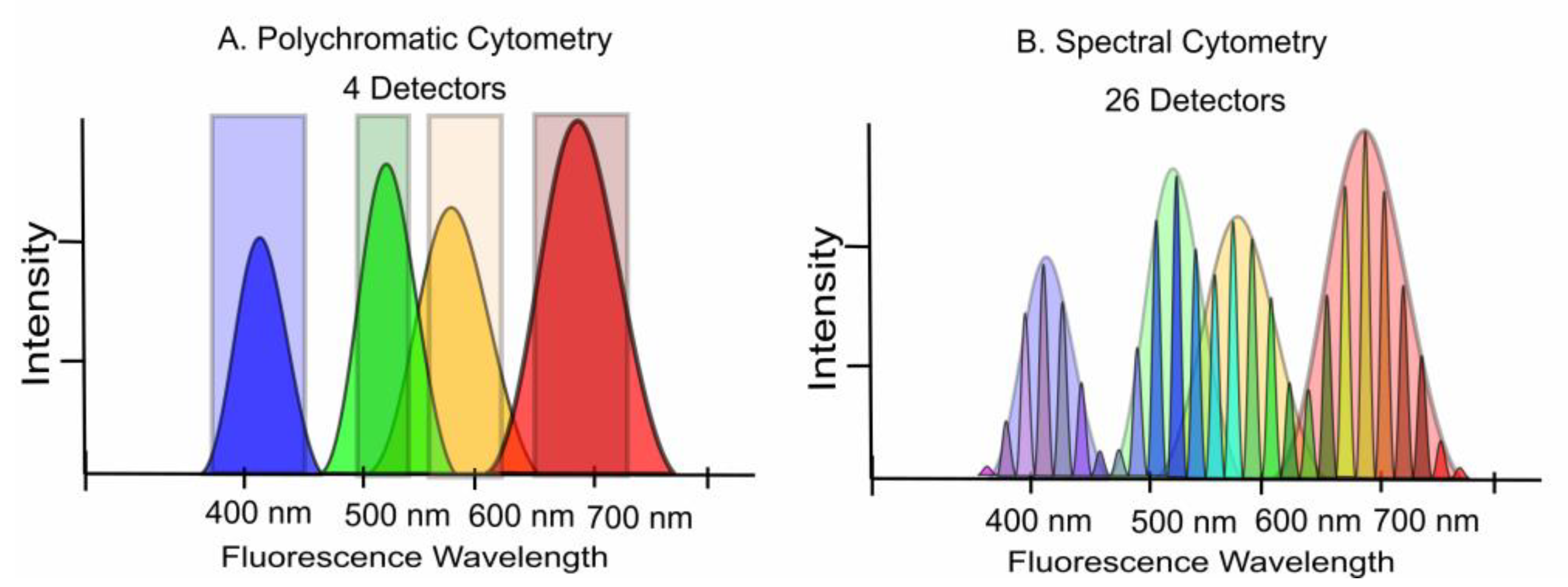
4. Polychromatic vs. Spectral Cytometry
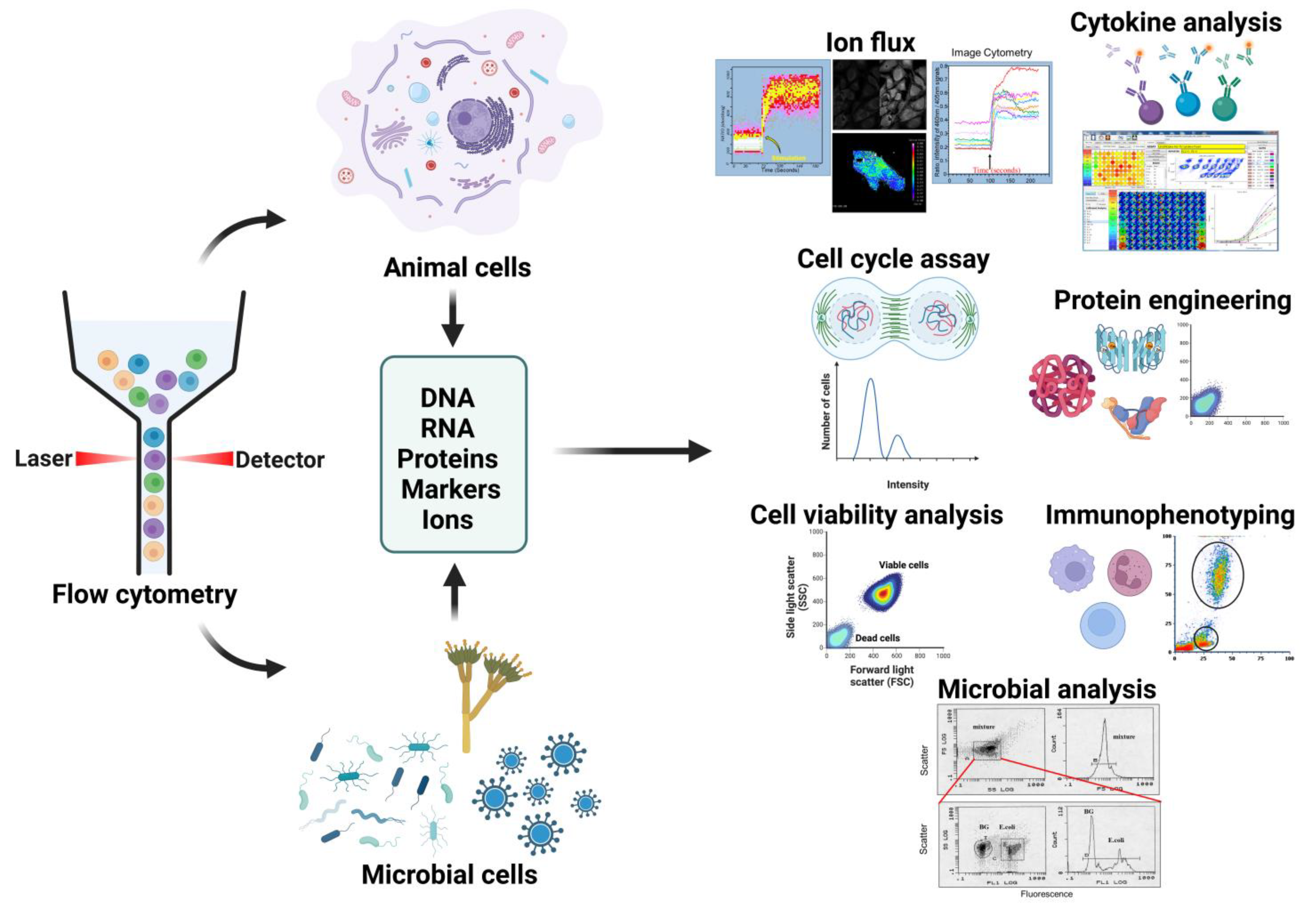
5. Applications of Flow Cytometry
5.1. Cell Phenotyping
5.2. Viability Assays
5.3. Cell Cycle Analysis
5.4. Ion Flux Assays
5.5. Cellular Function Measurement
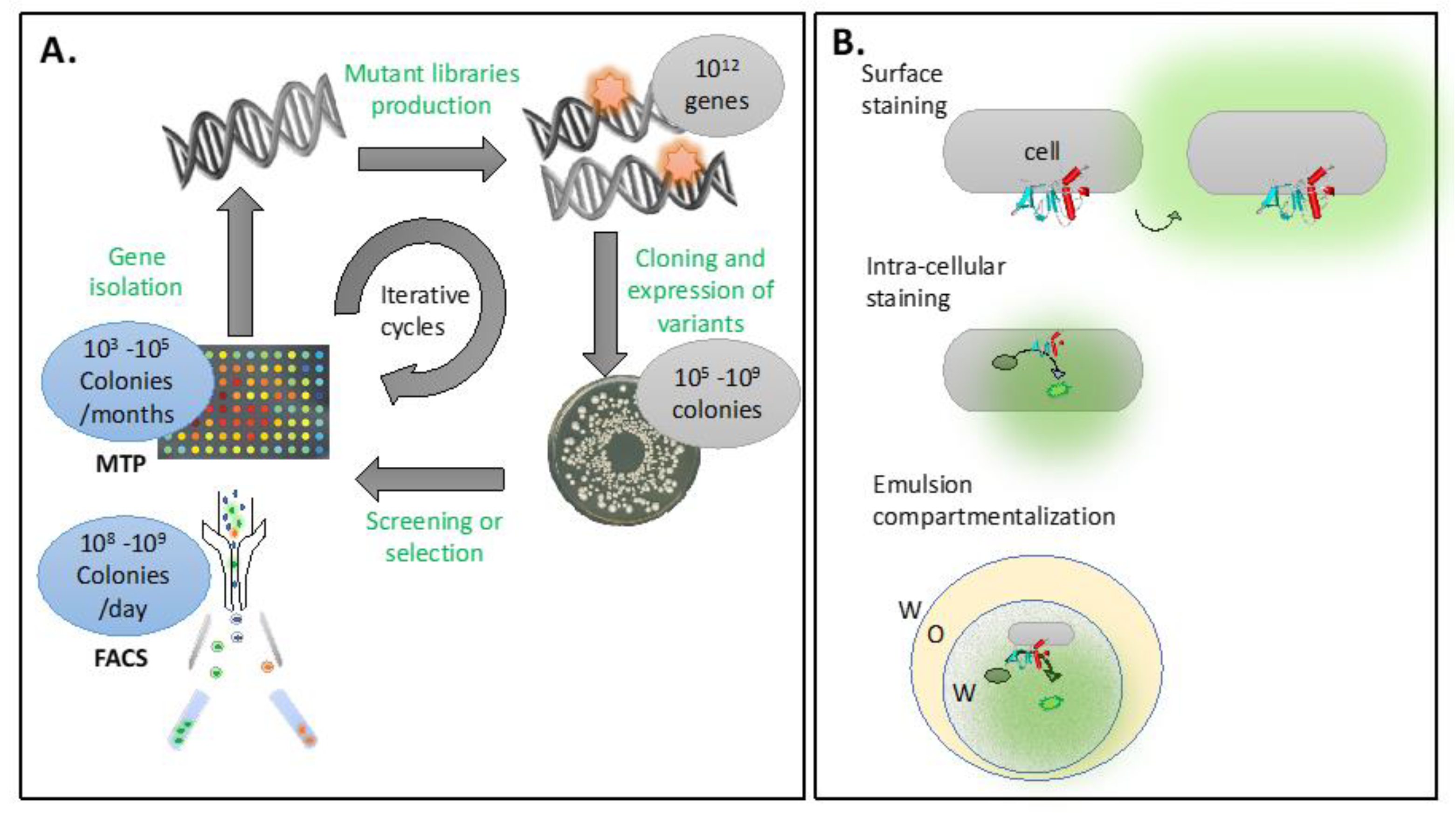
5.6. Protein Engineering
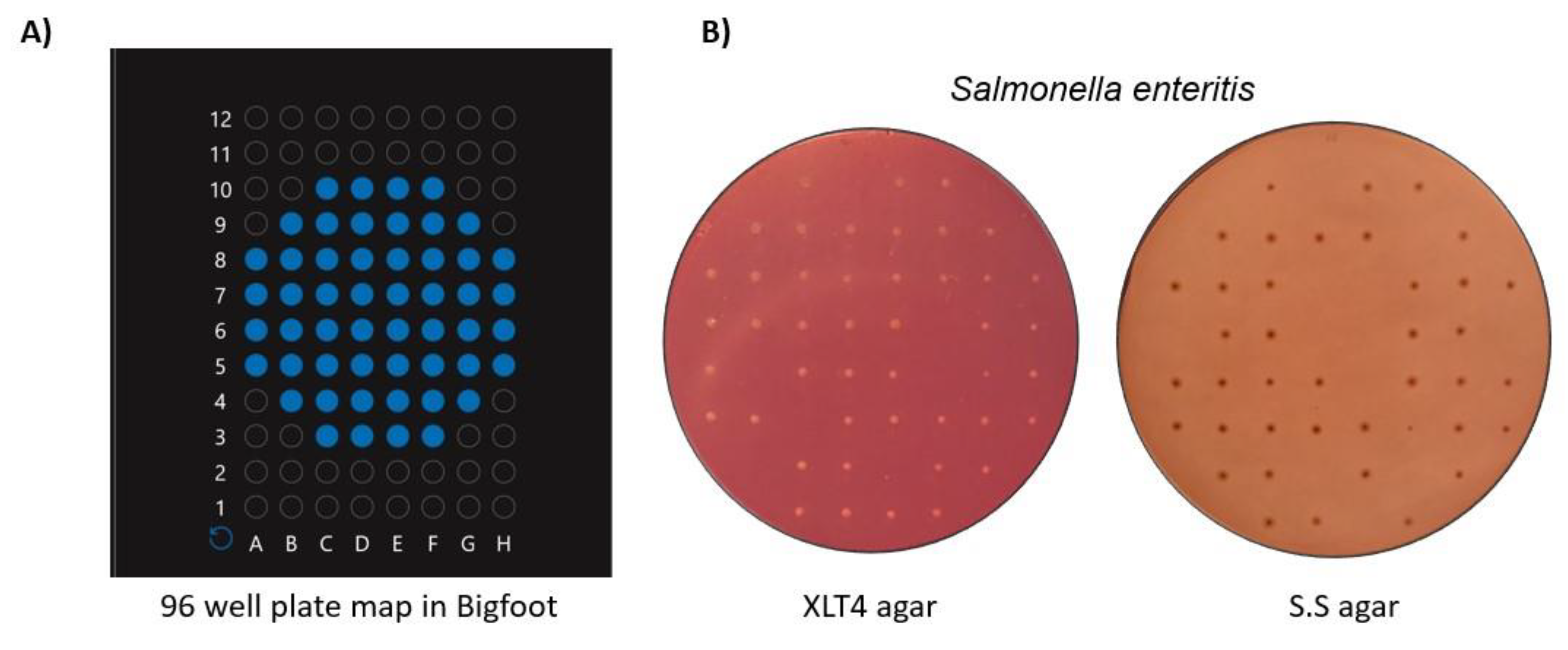
5.7. Bacterial Cell Sorting
5.8. Droplet Microfluidics
5.9. Next-Generation Bioengineered Biologics
6. Flow Cytometry Data Science and Informatics
6.1. Spectral Unmixing and Compensation
6.2. Data Pre-processing and Curation
6.3. Data Visualization
6.4. Automated Gating and Cell Population Identification
6.5. Biomarker Discovery and Predictive Modeling
6.6. Omics Integration
7. Cytometry Market Opportunities
8. Next-Generation Instruments
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kohler, A. Mikrophotographische Untersuchungen Mit Ultraviolettem Licht; Arthur H. Thomas Co: Chadds Ford Township, PA, USA, 1904. [Google Scholar]
- Feulgen, R.; Rossenback, H. Mikroskopisch-chemischer Nachweis einer Nucleinsäure vom Typus der Thymonucleinsäure und die darauf beruhende elektive Färbungvon Zellkernen in mikroskopischen Präparaten. Hoppe-Seyler’s Z. Physiol. Chem. 1924, 135, 203–248. [Google Scholar] [CrossRef]
- Casperson, T. Uber den chemischen Aufbau der Strukturen des Zellkernes. Scand Arch. Physiol. 1936, 73, 1–158. [Google Scholar] [CrossRef]
- Casperson, O. Quantitative cytochemical Studies on normal, malignant premalignant and atypical cell populations from the himan uterine cervix. Acta Cytol. 1964, 8, 45. [Google Scholar]
- Papanicolaou, G.N.; Traut, H.F. The Diagnostic Value of Vaginal Smears in Carcinoma of the Uterus. Am. J. Obstet. Gynecol. 1941, 42, 193–206. [Google Scholar] [CrossRef]
- Mellors, R.C.; Keane, J.F., Jr.; Papanicolaou, G.N. Nucleic acid contents of the squamous cancer cell. Science 1952, 116, 265–269. [Google Scholar] [CrossRef]
- Mendelsohn, M.L. The two-wavelength method of microspectrophotometry. J. Biophys. Biochem. Cytol. 1958, 4, 425–431. [Google Scholar] [CrossRef]
- Moldavan, A. Photo-electric technique for the counting of microscopical cells. Science 1934, 80, 188–189. [Google Scholar] [CrossRef]
- Coulter, W.H. Apparatus for Treatment of Fluids Requiring Sterilization or Pasteurization. U.S. Patent 2,799,216, 16 July 1957. [Google Scholar]
- Parker, J.C.; Horst, W.R. Device for Automatically Counting Blood Cells. U.S. Patent 2,807,416, 13 July 1953. [Google Scholar]
- Coulter, W.H. High speed automatic blood cell counter and cell size analyzer. Proc. Natl. Electron. Conf. 1956, 12, 1034–1042. [Google Scholar]
- Crosland-Taylor, P.J. A device for counting small particles suspended in fluid through a tube. Nature 1953, 171, 37–38. [Google Scholar] [CrossRef]
- Stokes, G.G. On the change of refrangibility of light. Philos. Trans. R. Soc. 1852, 142, 463–562. [Google Scholar] [CrossRef] [Green Version]
- Jablonski, A. Efficiency of anti-Stokes fluorescence in dyes. Nature 1933, 131, 839–840. [Google Scholar] [CrossRef]
- Hedley, D.W. Method for analysis of cellular DNA content of paraffin-embedded pathological material using flow cytometry. J. Histochem. Cytochem. 1983, 31, 1333–1335. [Google Scholar] [CrossRef]
- Robinson, J.P. Multispectral cytometry: The next generation. In Biophotonics International; Laurin Publishing: Pittsfield, MA, USA, 2004; pp. 36–40. [Google Scholar]
- Robinson, J.P.; Rajwa, B.; Grégori, G.; Jones, J.; Patsekin, V. Collection hardware for high speed multispectral single particle analysis. Cytometry 2004, 59, 12. [Google Scholar]
- Robinson, J.P.; Rajwa, B.; Grégori, G.; Jones, J.; Patsekin, V. Multispectral cytometry of single bio-particles using a 32-channel detector. In Advanced Biomedical and Clinical Diagnostic Systems III; Society of Photo Optical: Bellingham, WA, USA, 2005; pp. 359–365. [Google Scholar]
- Bendall, S.C.; Simonds, E.F.; Qiu, P.; Amir, E.A.D.; Krutzik, P.O.; Finck, R.; Bruggner, R.V.; Melamed, R.; Trejo, A.; Ornatsky, O.I.; et al. Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science 2011, 332, 687–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schasfoort, R.B.M.; Abali, F.; Stojanovic, I.; Vidarsson, G.; Terstappen, L. Trends in SPR Cytometry: Advances in Label-Free Detection of Cell Parameters. Biosensors 2018, 8, 102. [Google Scholar] [CrossRef] [Green Version]
- Watson, D.A.; Brown, L.O.; Gaskill, D.F.; Naivar, M.; Graves, S.W.; Doorn, S.K.; Nolan, J.P. A flow cytometer for the measurement of Raman spectra. Cytom. Part A 2008, 73, 119–128. [Google Scholar] [CrossRef]
- Nolan, J.P.; Duggan, E.; Liu, E.; Condello, D.; Dave, I.; Stoner, S.A. Single cell analysis using surface enhanced Raman scattering (SERS) tags. Methods 2012, 57, 272–279. [Google Scholar] [CrossRef] [Green Version]
- Cheung, K.; Gawad, S.; Renaud, P. Impedance spectroscopy flow cytometry: On-chip label-free cell differentiation. Cytom. Part A 2005, 65, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Liu, Y.S.; Irimia, D.; Demirci, U.; Yang, L.; Zamir, L.; Rodriguez, W.R.; Toner, M.; Bashir, R. Cell detection and counting through cell lysate impedance spectroscopy in microfluidic devices. Lab Chip 2007, 7, 746–755. [Google Scholar] [CrossRef] [Green Version]
- Schade-Kampmann, G.; Huwiler, A.; Hebeisen, M.; Hessler, T.; Di, B.M. On-chip non-invasive and label-free cell discrimination by impedance spectroscopy. Cell Prolif. 2008, 41, 830–840. [Google Scholar] [CrossRef]
- Robinson, J.P.; Patsekin, V.; Grégori, G.; Rajwa, B.; Jones, J. Multispectral flow cytometry: Next generation tools for automated classification. Microsc. Microanal. 2005, 11, 2–3. [Google Scholar] [CrossRef] [Green Version]
- Grégori, G.; Patsekin, V.; Rajwa, B.; Jones, J.; Ragheb, K.; Holdman, C.; Robinson, J.P. Hyperspectral cytometry at the single-cell level using a 32-channel photodetector. Cytom. Part A 2012, 81, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Nolan, J.P. The evolution of spectral flow cytometry. Cytometry Part A 2022, 101, 812–817. [Google Scholar] [CrossRef] [PubMed]
- Nolan, J.P.; Condello, D. Spectral flow cytometry. Curr. Protoc. Cytom. 2013, 63, 1–27. [Google Scholar] [CrossRef] [Green Version]
- Robinson, J.P. Flow cytometry: Past and future. Biotechniques 2022, 72, 159–169. [Google Scholar] [CrossRef]
- Novo, D. A comparison of spectral unmixing to conventional compensation for the calculation of fluorochrome abundances from flow cytometric data. Cytom. Part A 2022, 101, 885–891. [Google Scholar] [CrossRef]
- Novo, D.; Grégori, G.; Rajwa, B. Generalized unmixing model for multispectral flow cytometry utilizing nonsquare compensation matrices. Cytom. Part A 2013, 83, 508–520. [Google Scholar] [CrossRef] [Green Version]
- Roederer, M. Spectral compensation for flow cytometry: Visualization artifacts, limitations, and caveats. Cytometry 2001, 45, 194–205. [Google Scholar] [CrossRef]
- Kohler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495–497. [Google Scholar] [CrossRef]
- Kung, P.; Goldstein, G.; Reinherz, E.L.; Schlossman, S.F. Monoclonal antibodies defining distinctive human T cell surface antigens. Science 1979, 206, 347–349. [Google Scholar] [CrossRef]
- Hulett, H.R.; Bonner, W.A.; Barrett, J.; Herzenberg, L.A. Cell sorting: Automated separation of mammalian cells as a function of intracellular fluorescence. Science 1969, 166, 747–749. [Google Scholar] [CrossRef] [PubMed]
- Bonner, W.A.; Hulett, H.R.; Sweet, R.G.; Herzenberg, L.A. Fluorescence activated cell sorting. Rev. Sci. Instrum. 1972, 43, 404–409. [Google Scholar] [CrossRef]
- Hulett, H.R.; Bonner, W.A.; Sweet, R.G.; Herzenberg, L.A. Development and application of a rapid cell sorter. Clin. Chem. 1973, 19, 813–816. [Google Scholar] [CrossRef] [PubMed]
- Julius, M.H.; Sweet, R.G.; Fathman, C.G.; Herzenberg, L.A. Fluorescence-activated cell sorting and its applications. In Mammalian Cells: Probes and Problems; Richmond, C.R., Ed.; US Energy Research and Development Administration: Oak Ridge, TN, USA, 1975; pp. 107–121. [Google Scholar]
- Loken, M.R.; Parks, D.R.; Herzenberg, L.A. Two-color immunofluorescence using a fluorescence-activated cell sorter. J. Histochem. Cytochem. 1977, 25, 899–907. [Google Scholar] [CrossRef] [Green Version]
- Parks, D.R.; Bryan, V.M.; Oi, V.T.; Herzenberg, L.A. Antigen-specific identification and cloning of hybridomas with a fluorescence-activated cell sorter. Proc. Natl. Acad. Sci. USA 1979, 76, 1962–1966. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Woffendin, C.; Bahner, I.; Ranga, U.; Xu, L.; Yang, Z.Y.; King, S.R.; Kohn, D.B.; Nabel, G.J. Gene transfer into human umbilical cord blood-derived CD34+ cells by particle-mediated gene transfer. Gene Ther. 1998, 5, 692–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, G.B.; McMullen, M.; Turner, S.; Olszak, I.T.; Scadden, D.T.; McClure, M.O.; Poznansky, M.C. Isolation and transduction of CD34+ cells from small quantities of peripheral blood from HIV-1-infected patients not treated with hemopoietic growth factors. J. Acquir. Immune Defic. Syndr. 1999, 21, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Bozzi, F.; Conca, E.; Manenti, G.; Negri, T.; Brich, S.; Gronchi, A.; Pierotti, M.A.; Tamborini, E.; Pilotti, S. High CD133 expression levels in gastrointestinal stromal tumors. Cytom. Part B Clin. Cytom. 2011, 80, 238–247. [Google Scholar] [CrossRef]
- Bartolovic, K.; Balabanov, S.; Berner, B.; Buhring, H.J.; Komor, M.; Becker, S.; Hoelzer, D.; Kanz, L.; Hofmann, W.K.; Brummendorf, T.H. Clonal heterogeneity in growth kinetics of CD34+. Stem Cells 2005, 23, 946–957. [Google Scholar]
- Verfaillie, C.M.; Almeida-Porada, G.; Wissink, S.; Zanjani, E.D. Kinetics of engraftment of CD34(−) and CD34(+) cells from mobilized blood differs from that of CD34(−) and CD34(+) cells from bone marrow. Exp. Hematol. 2000, 28, 1071–1079. [Google Scholar] [CrossRef] [Green Version]
- Boye, E.; Steen, H.B.; Skarstad, K. Flow cytometry of bacteria: A promising tool in experimental and clinical microbiology. J. Gen. Microbiol. 1983, 129, 973–980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Betz, J.W.; Aretz, W.; Hartel, W. Use of flow cytometry in industrial microbiology for strain improvement programs. Cytometry 1984, 5, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Tyndall, R.L.; Hand, R.E., Jr.; Mann, R.C.; Evans, C.; Jernigan, R. Application of flow cytometry to detection and characterization of Legionella spp. Appl. Environ. Microbiol. 1985, 49, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Davey, H.M.; Davey, C.L.; Kell, D.B. On the determination of the size of microbial cells using flow cytometry. In Flow Cytometry in Microbiology; Lloyd, D., Ed.; Springer: New York, NY, USA, 1993; pp. 49–66. [Google Scholar]
- Hamori, E.; Arndt-Jovin, D.J.; Grimwade, B.G.; Jovin, T.M. Selection of viable cells with known DNA content. Cytometry 1980, 1, 132–135. [Google Scholar] [CrossRef] [PubMed]
- Suga, H.; Matsumoto, D.; Inoue, K.; Shigeura, T.; Eto, H.; Aoi, N.; Kato, H.; Abe, H.; Yoshimura, K. Numerical measurement of viable and nonviable adipocytes and other cellular components in aspirated fat tissue. Plast. Reconstr. Surg. 2008, 122, 103–114. [Google Scholar] [CrossRef] [Green Version]
- Mozdziak, P.E.; Pulvermacher, P.M.; Schultz, E.; Schell, K. Hoechst fluorescence intensity can be used to separate viable bromodeoxyuridine-labeled cells from viable non-bromodeoxyuridine-labeled cells. Cytometry 2000, 41, 89–95. [Google Scholar] [CrossRef]
- Keij, J.F.; Bell-Prince, C.; Steinkamp, J.A. Simultaneous analysis of relative DNA and glutathione content in viable cells by phase-resolved flow cytometry. Cytometry 1999, 35, 48–54. [Google Scholar] [CrossRef]
- Dauber, K.; Becker, D.; Odendahl, M.; Seifried, E.; Bonig, H.; Tonn, T. Enumeration of viable CD34(+) cells by flow cytometry in blood, bone marrow and cord blood: Results of a study of the novel BD stem cell enumeration kit. Cytotherapy 2011, 13, 449–458. [Google Scholar] [CrossRef]
- Perfetto, S.P.; Chattopadhyay, P.K.; Lamoreaux, L.; Nguyen, R.; Ambrozak, D.; Koup, R.A.; Roederer, M. Amine reactive dyes: An effective tool to discriminate live and dead cells in polychromatic flow cytometry. J. Immunol. Methods 2006, 313, 199–208. [Google Scholar] [CrossRef]
- Dachary-Prigent, J.; Freyssinet, J.M.; Pasquet, J.M.; Carron, J.C.; Nurden, A.T. Annexin V as a probe of aminophospholipid exposure and platelet membrane vesiculation: A flow cytometry study showing a role for free sulfhydryl groups. Blood 1993, 81, 2554–2565. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.; Gurtu, V.; Kain, S.R.; Yan, G. Early detection of apoptosis using a fluorescent conjugate of annexin V. BioTechniques 1997, 23, 525–531. [Google Scholar] [CrossRef]
- Van Heerde, W.L.; Robert-Offerman, S.; Dumont, E.; Hofstra, L.; Doevendans, P.A.; Smits, J.F.; Daemen, M.J.; Reutelingsperger, C.P. Markers of apoptosis in cardiovascular tissues: Focus on Annexin V. Cardiovasc. Res. 2000, 45, 549–559. [Google Scholar] [CrossRef] [Green Version]
- Pinto, L.A.; Trivett, M.T.; Wallace, D.; Higgins, J.; Baseler, M.; Terabe, M.; Belyakov, I.M.; Berzofsky, J.A.; Hildesheim, A. Fixation and cryopreservation of whole blood and isolated mononuclear cells: Influence of different procedures on lymphocyte subset analysis by flow cytometry. Cytom. Part B Clin. Cytom. 2005, 63, 47–55. [Google Scholar] [CrossRef]
- Stewart, J.C.; Villasmil, M.L.; Frampton, M.W. Changes in fluorescence intensity of selected leukocyte surface markers following fixation. Cytom. Part A 2007, 71, 379–385. [Google Scholar] [CrossRef]
- Otali, D.; Stockard, C.R.; Oelschlager, D.K.; Wan, W.; Manne, U.; Watts, S.A.; Grizzle, W.E. Combined effects of formalin fixation and tissue processing on immunorecognition. Biotech. Histochem. 2009, 84, 223–247. [Google Scholar] [CrossRef] [PubMed]
- Kaplow, L.S.; Dauber, H.; Lerner, E. Assessment of monocyte esterase activity by flow cytometry. J. Histochem. Cytochem. 1976, 24, 363–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valet, G.K.; Raffael, A. Cytostatic Drug Testing on Patient Tumor Cells. 1 Determination of Intracellular pH and Esterase Activity in Vital Cells by Flow-Cytometry; Paesel GmbH: Rheinberg, Germany, 1989; pp. 3–21. [Google Scholar]
- Breeuwer, P.; Drocourt, J.L.; Bunschoten, N.; Zwietering, M.H.; Rombouts, F.M.; Abee, T. Characterization of uptake and hydrolysis of fluorescein diacetate and carboxyfluorescein diacetate by intracellular esterases in Saccharomyces cerevisiae, which result in accumulation of fluorescent product. Appl. Environ. Microbiol. 1995, 61, 1614–1619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishan, A. Flow cytofluorometric analysis of mammalian cell cycle by propidium iodide staining. J. Cell Biol. 1975, 66, 188–193. [Google Scholar] [CrossRef]
- Darzynkiewicz, Z.; Traganos, F.; Sharpless, T.K.; Melamed, M.R. Cell cycle-related changes in nuclear chromatin of stimulated lymphocytes as measured by flow cytometry. Cancer Res. 1977, 37, 4635–4640. [Google Scholar]
- Darzynkiewicz, Z.; Sharpless, T.; Staiano-Coico, L.; Melamed, M.R. Subcompartments of the G 1 phase of cell cycle detected by flow cytometry. Proc. Natl. Acad. Sci. USA 1980, 77, 6696–6699. [Google Scholar] [CrossRef]
- Simonetti, S.; Natalini, A.; Peruzzi, G.; Nicosia, A.; Folgori, A.; Capone, S.; Santoni, A.; Di Rosa, F. A DNA/Ki67-Based Flow Cytometry Assay for Cell Cycle Analysis of Antigen-Specific CD8 T Cells in Vaccinated Mice. J. Vis. Exp. 2021, 167, e61867. [Google Scholar] [CrossRef]
- Al Zouabi, N.N.; Roberts, C.M.; Lin, Z.P.; Ratner, E.S. Flow Cytometric Analyses of p53-Mediated Cell Cycle Arrest and Apoptosis in Cancer Cells. Methods Mol. Biol. 2021, 2255, 43–53. [Google Scholar] [CrossRef]
- Zhu, T.; Wang, Y.; Ding, W.; Xu, J.; Chen, R.; Xie, J.; Zhu, W.; Jia, L.; Ma, T. Anticancer activity and DNA-binding investigations of the Cu(II) and Ni(II) complexes with coumarin derivative. Chem. Biol. Drug Des. 2015, 85, 385–393. [Google Scholar] [CrossRef]
- Zribi, N.; Chakroun, N.F.; Elleuch, H.; Abdallah, F.B.; Ben Hamida, A.S.; Gargouri, J.; Fakhfakh, F.; Keskes, L.A. Sperm DNA fragmentation and oxidation are independent of malondialdheyde. Reprod. Biol. Endocrinol. 2011, 9, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Wu, X.; Li, J.; Sun, Y.; Gao, P.; Zhang, C.; Zhang, H.; Zhou, G. MDR1 (multidrug resistence 1) can regulate GCS (glucosylceramide synthase) in breast cancer cells. J. Surg. Oncol. 2011, 104, 466–471. [Google Scholar] [CrossRef]
- Ligasová, A.; Frydrych, I.; Koberna, K. Basic Methods of Cell Cycle Analysis. Int. J. Mol. Sci. 2023, 24, 3674. [Google Scholar] [CrossRef]
- Christo, S.N.; Diener, K.R.; Nordon, R.E.; Brown, M.P.; Griesser, H.J.; Vasilev, K.; Christo, F.C.; Hayball, J.D. Scrutinizing calcium flux oscillations in T lymphocytes to deduce the strength of stimulus. Sci. Rep. 2015, 5, 7760. [Google Scholar] [CrossRef] [Green Version]
- Scharenberg, A.M.; Humphries, L.A.; Rawlings, D.J. Calcium signalling and cell-fate choice in B cells. Nat. Rev. Immunol. 2007, 7, 778–789. [Google Scholar] [CrossRef] [Green Version]
- Theorell, J.; Bryceson, Y.T. Analysis of Intracellular Ca2+ Mobilization in Human NK Cell Subsets by Flow Cytometry. In Natural Killer Cells; Somanchi, S.S., Ed.; Springer: New York, NY, USA, 2016; Volume 1441, pp. 117–130. [Google Scholar]
- Ngkelo, A.; Richart, A.; Kirk, J.A.; Bonnin, P.; Vilar, J.; Lemitre, M.; Marck, P.; Branchereau, M.; Le Gall, S.; Renault, N.; et al. Mast cells regulate myofilament calcium sensitization and heart function after myocardial infarction. J. Exp. Med. 2016, 213, 1353–1374. [Google Scholar] [CrossRef] [PubMed]
- Brini, M.; Calì, T.; Ottolini, D.; Carafoli, E. Neuronal calcium signaling: Function and dysfunction. Cell. Mol. Life Sci. 2014, 71, 2787–2814. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Stadler, B.M.; Heiner, D.C.; Skvaril, F.; De Weck, A.L. Flow cytometric analysis of human basophil degranulation: Degranulation induced by anti-IgE, anti-IgG4 and the calcium ionophore A23187. Clin. Allergy 1981, 11, 21–30. [Google Scholar] [CrossRef]
- Valet, G.K.; Raffael, A.; Russmann, L. Determination of intracellular calcium in vital cells by flow cytometry. Naturwissenschaften 1985, 72, 600–602. [Google Scholar] [CrossRef]
- Vandenberghe, P.A.; Ceuppens, J.L. Flow cytometric measurement of cytoplasmic free calcium in human peripheral blood T lymphocytes with fluo-3, a new fluorescent calcium indicator. J. Immunol. Methods 1990, 127, 197–205. [Google Scholar] [CrossRef]
- Do Ceu, M.M.; Sansonetty, F.; Goncalves, M.J.; O’Connor, J.E. Flow cytometric kinetic assay of calcium mobilization in whole blood platelets using Fluo-3 and CD41. Cytometry 1999, 35, 302–310. [Google Scholar]
- Valet, G.; Raffael, A. The flow-cytometric determination of intracellular calcium in vital cells with the indo-1 dye. Int. Symp. Clin. Cytom. Histometry 1986, 141, 53–63. [Google Scholar]
- Jennings, L.K.; Dockter, M.E.; Wall, C.D.; Fox, C.F.; Kennedy, D.M. Calcium mobilization in human platelets using indo-1 and flow cytometry. Blood 1989, 74, 2674–2680. [Google Scholar] [CrossRef] [Green Version]
- Vines, A.; McBean, G.J.; Blanco-Fernández, A. A flow-cytometric method for continuous measurement of intracellular Ca2+ concentration. Cytom. Part A 2010, 77, 1091–1097. [Google Scholar] [CrossRef]
- Cappadone, C.; Malucelli, E.; Zini, M.; Farruggia, G.; Picone, G.; Gianoncelli, A.; Notargiacomo, A.; Fratini, M.; Pignatti, C.; Iotti, S.; et al. Assessment and Imaging of Intracellular Magnesium in SaOS-2 Osteosarcoma Cells and Its Role in Proliferation. Nutrients 2021, 13, 1376. [Google Scholar] [CrossRef] [PubMed]
- Aliotta, A.; Bertaggia Calderara, D.; Alberio, L. Flow Cytometric Monitoring of Dynamic Cytosolic Calcium, Sodium, and Potassium Fluxes Following Platelet Activation. Cytom. Part A 2020, 97, 933–944. [Google Scholar] [CrossRef]
- Chow, S.; Hedley, D. Flow Cytometric Measurement of Intracellular pH. Curr. Protoc. Cytom. 1997. [Google Scholar] [CrossRef]
- Malin-Berdel, J.; Valet, G. Flow cytometric determination of esterase and phosphatase activities and kinetics in hematopoietic cells with fluorogenic substrates. Cytometry 1980, 1, 222–228. [Google Scholar] [CrossRef]
- Rothe, G.; Valet, G. Flow cytometric analysis of respiratory burst activity in phagocytes with hydroethidine and 2′,7′-dichlorofluorescin. J. Leukoc. Biol. 1990, 47, 440–448. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.P.; Bruner, L.H.; Ward, P.A.; Phan, S.H. Measurement of intracellular fluorescence of human monocytes indicative of oxidative metabolism. Proc. Int. Congr. Immunol. 1986, 6, 304–310. [Google Scholar]
- Wolber, R.A.; Duque, R.E.; Robinson, J.P.; Oberman, H.A. Oxidative product formation in irradiated neutrophils. A flow cytometric analysis. Transfusion 1987, 27, 167–170. [Google Scholar]
- Loesche, W.J.; Robinson, J.P.; Flynn, M.; Hudson, J.L.; Duque, R.E. Reduced oxidative function in gingival crevicualar neutrophisl in periodontal disease. Infect. Immun. 1988, 56, 156–160. [Google Scholar] [CrossRef]
- Dunn, P.A.; Tyrer, H.W. Quantitation of neutrophil phagocytosis, using fluorescent latex beads. Correlation of microscopy and flow cytometry. J. Lab. Clin. Med. 1981, 98, 374–381. [Google Scholar]
- Steinkamp, J.A.; Wilson, J.S.; Saunders, G.C.; Stewart, C.C. Phagocytosis: Flow cytometric quantitation with fluorescent microspheres. Science 1982, 215, 64–66. [Google Scholar] [CrossRef]
- Bassoe, C.F.; Laerum, O.D.; Glette, J.; Hopen, G.; Haneberg, B.; Solberg, C.O. Simultaneous measurement of phagocytosis and phagosomal pH by flow cytometry: Role of polymorphonuclear neutrophilic leukocyte granules in phagosome acidification. Cytometry 1983, 4, 254–262. [Google Scholar] [CrossRef] [PubMed]
- Bassoe, C.F.; Laerum, O.D.; Solberg, C.O.; Haneberg, B. Phagocytosis of bacteria by human leukocytes measured by flow cytometry. Proc. Soc. Exp. Biol. Med. 1983, 174, 182–186. [Google Scholar] [CrossRef]
- Bjerknes, R.; Bassoe, C.F. Human leukocyte phagocytosis of zymosan particles measured by flow cytometry. Acta Pathol. Microbiol. Immunol. Scand. Sect. C Immunol. 1983, 91, 341–348. [Google Scholar]
- Bratosin, D.; Tissier, J.P.; Lapillonne, H.; Hermine, O.; de Villemeur, T.B.; Cotoraci, C.; Montreuil, J.; Mignot, C. A cytometric study of the red blood cells in Gaucher disease reveals their abnormal shape that may be involved in increased erythrophagocytosis. Cytom. B Clin.Cytom. 2011, 80, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Overland, H.S.; Pettersen, E.F.; Ronneseth, A.; Wergeland, H.I. Phagocytosis by B-cells and neutrophils in Atlantic salmon (Salmo salar L.) and Atlantic cod (Gadus morhua L.). Fish Shellfish. Immunol. 2010, 28, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Vander Top, E.A.; Perry, G.A.; Gentry-Nielsen, M.J. A novel flow cytometric assay for measurement of in vivo pulmonary neutrophil phagocytosis. BMC Microbiol. 2006, 6, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiezer, M.J.; Meijer, C.; Wallast-Groenewoud, H.P.; Tool, A.T.; Prins, H.A.; Houdijk, A.P.; Beelen, R.H.; Meijer, S.; Hack, C.E.; van Leeuwen, P.A. Impaired leukocyte phagocytosis in patients undergoing hemihepatectomy for liver metastases. Liver Transpl. Surg. 1999, 5, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Ruff, A.J.; Dennig, A.; Wirtz, G.; Blanusa, M.; Schwaneberg, U. Flow Cytometer-Based High-Throughput Screening System for Accelerated Directed Evolution of P450 Monooxygenases. ACS Catal. 2012, 2, 2724–2728. [Google Scholar] [CrossRef]
- Ostafe, R.; Prodanovic, R.; Nazor, J.; Fischer, R. Ultra-High-Throughput Screening Method for the Directed Evolution of Glucose Oxidase. Chem. Biol. 2014, 21, 414–421. [Google Scholar] [CrossRef]
- Menghiu, G.; Ostafe, V.; Prodanović, R.; Fischer, R.; Ostafe, R. A High-Throughput Screening System Based on Fluorescence-Activated Cell Sorting for the Directed Evolution of Chitinase A. Int. J. Mol. Sci. 2021, 22, 3041. [Google Scholar] [CrossRef]
- Ostafe, R.; Prodanovic, R.; Commandeur, U.; Fischer, R. Flow cytometry-based ultra-high-throughput screening assay for cellulase activity. Anal. Biochem. 2013, 435, 93–98. [Google Scholar] [CrossRef]
- Körfer, G.; Pitzler, C.; Vojcic, L.; Martinez, R.; Schwaneberg, U. In vitro flow cytometry-based screening platform for cellulase engineering. Sci. Rep. 2016, 6, 26128. [Google Scholar] [CrossRef] [Green Version]
- Ilić Đurđić, K.; Ece, S.; Ostafe, R.; Vogel, S.; Schillberg, S.; Fischer, R.; Prodanović, R. Improvement in oxidative stability of versatile peroxidase by flow cytometry-based high-throughput screening system. Biochem. Eng. J. 2020, 157, 107555. [Google Scholar] [CrossRef]
- Lipovšek, D.; Antipov, E.; Armstrong, K.A.; Olsen, M.J.; Klibanov, A.M.; Tidor, B.; Wittrup, K.D. Selection of Horseradish Peroxidase Variants with Enhanced Enantioselectivity by Yeast Surface Display. Chem. Biol. 2007, 14, 1176–1185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cummins, I.; Steel, P.G.; Edwards, R. Identification of a carboxylesterase expressed in protoplasts using fluorescence-activated cell sorting. Plant Biotechnol. J. 2007, 5, 354–359. [Google Scholar] [CrossRef] [PubMed]
- Aharoni, A.; Thieme, K.; Chiu, C.P.C.; Buchini, S.; Lairson, L.L.; Chen, H.; Strynadka, N.C.J.; Wakarchuk, W.W.; Withers, S.G. High-throughput screening methodology for the directed evolution of glycosyltransferases. Nat. Methods 2006, 3, 609–614. [Google Scholar] [CrossRef] [PubMed]
- Mastrobattista, E.; Taly, V.; Chanudet, E.; Treacy, P.; Kelly, B.T.; Griffiths, A.D. High-Throughput Screening of Enzyme Libraries: In Vitro Evolution of a β-Galactosidase by Fluorescence-Activated Sorting of Double Emulsions. Chem. Biol. 2005, 12, 1291–1300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aharoni, A.; Amitai, G.; Bernath, K.; Magdassi, S.; Tawfik, D.S. High-Throughput Screening of Enzyme Libraries: Thiolactonases Evolved by Fluorescence-Activated Sorting of Single Cells in Emulsion Compartments. Chem. Biol. 2005, 12, 1281–1289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prodanovic, R.; Ostafe, R.; Scacioc, A.; Schwaneberg, U. Ultrahigh Throughput Screening System for Directed Glucose Oxidase Evolution in Yeast Cells. Comb. Chem. High Throughput Screen. 2011, 14, 55–60. [Google Scholar] [CrossRef]
- Popovic, M.; Prodanovic, R.; Ostafe, R.; Schillberg, S.; Fischer, R.; Gavrovic-Jankulovic, M. Yeast surface display is a novel tool for the rapid immunological characterization of plant-derived food allergens. Immunol. Res. 2015, 61, 230–239. [Google Scholar] [CrossRef]
- Griswold, K.E.; Kawarasaki, Y.; Ghoneim, N.; Benkovic, S.J.; Iverson, B.L.; Georgiou, G. Evolution of highly active enzymes by homology-independent recombination. Proc. Natl. Acad. Sci. USA 2005, 102, 10082–10087. [Google Scholar] [CrossRef]
- Liu, L.; Li, Y.; Liotta, D.; Lutz, S. Directed evolution of an orthogonal nucleoside analog kinase via fluorescence-activated cell sorting. Nucleic Acids Res. 2009, 37, 4472–4481. [Google Scholar] [CrossRef] [Green Version]
- Prodanovic, R.; Ostafe, R.; Blanusa, M.; Schwaneberg, U. Vanadium bromoperoxidase-coupled fluorescent assay for flow cytometry sorting of glucose oxidase gene libraries in double emulsions. Anal. Bioanal. Chem. 2012, 404, 1439–1447. [Google Scholar] [CrossRef]
- Shapiro, H.M. Microbial analysis at the single-cell level: Tasks and techniques. J. Microbiol. Methods 2000, 42, 3–16. [Google Scholar] [CrossRef]
- Ambriz-Avina, V.; Contreras-Garduno, J.A.; Pedraza-Reyes, M. Applications of flow cytometry to characterize bacterial physiological responses. BioMed Res. Int. 2014, 2014, 461941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, S.; Nebe-von-Caron, G. Functional single-cell analyses: Flow cytometry and cell sorting of microbial populations and communities. FEMS Microbiol. Rev. 2010, 34, 554–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapinusova, G.; Lopez Marin, M.A.; Uhlik, O. Reaching unreachables: Obstacles and successes of microbial cultivation and their reasons. Front. Microbiol. 2023, 14, 1089630. [Google Scholar] [CrossRef] [PubMed]
- Biorad. Sample Preparation Protocol. Available online: https://www.bio-rad-antibodies.com/flow-cytometry-sample-preparation.html#:~:text=Single%20cells%20must%20be%20suspended,2%2C000%E2%80%9320%2C000%20cells%2Fsecond. (accessed on 3 June 2023).
- Narayana Iyengar, S.; Kumar, T.; Martensson, G.; Russom, A. High resolution and rapid separation of bacteria from blood using elasto-inertial microfluidics. Electrophoresis 2021, 42, 2538–2551. [Google Scholar] [CrossRef]
- Pitt, W.G.; Alizadeh, M.; Husseini, G.A.; McClellan, D.S.; Buchanan, C.M.; Bledsoe, C.G.; Robison, R.A.; Blanco, R.; Roeder, B.L.; Melville, M.; et al. Rapid separation of bacteria from blood-review and outlook. Biotechnol. Prog. 2016, 32, 823–839. [Google Scholar] [CrossRef] [Green Version]
- McHugh, I.O.L.; Tucker, A.L. Flow cytometry for the rapid detection of bacteria in cell culture production medium. Cytom. Part A 2007, 71, 1019–1026. [Google Scholar] [CrossRef]
- Wallner, G.; Fuchs, B.; Spring, S.; Beisker, W.; Amann, R. Flow sorting of microorganisms for molecular analysis. Appl. Environ. Microbiol. 1997, 63, 4223–4231. [Google Scholar] [CrossRef] [Green Version]
- Karlsson, R.; Gonzales-Siles, L.; Gomila, M.; Busquets, A.; Salva-Serra, F.; Jaen-Luchoro, D.; Jakobsson, H.E.; Karlsson, A.; Boulund, F.; Kristiansson, E.; et al. Proteotyping bacteria: Characterization, differentiation and identification of pneumococcus and other species within the Mitis Group of the genus Streptococcus by tandem mass spectrometry proteomics. PLoS ONE 2018, 13, e0208804. [Google Scholar] [CrossRef] [Green Version]
- Karlsson, R.; Thorsell, A.; Gomila, M.; Salva-Serra, F.; Jakobsson, H.E.; Gonzales-Siles, L.; Jaen-Luchoro, D.; Skovbjerg, S.; Fuchs, J.; Karlsson, A.; et al. Discovery of Species-unique Peptide Biomarkers of Bacterial Pathogens by Tandem Mass Spectrometry-based Proteotyping. Mol. Cell. Proteom. 2020, 19, 518–528. [Google Scholar] [CrossRef]
- Robinson, J.P. Spectral Flow Cytometry—Quo Vadimus? Cytom. Part A 2019, 95, 823–824. [Google Scholar] [CrossRef] [Green Version]
- FluoroFinder. Cytometers—Analyzers, and Cell Sorters. Available online: https://fluorofinder.com/cytometers/ (accessed on 3 June 2023).
- Schmid, I.; Lambert, C.; Ambrozak, D.; Perfetto, S.P. Standard safety practices for sorting of unfixed cells. Curr. Protoc. Cytom. 2007. [Google Scholar] [CrossRef] [PubMed]
- ThermoFisher. Bigfoot Spectral Cell Sorter Features. Available online: https://www.thermofisher.com/us/en/home/life-science/cell-analysis/flow-cytometry/flow-cytometers/bigfoot-spectral-cell-sorter/features.html?gclid=CjwKCAjwrdmhBhBBEiwA4Hx5g49IaWVVv0liQQ49wa1iTpQSQ_omgjWlw4NjDh9F95_iCUbL_JaHsBoCyD4QAvD_BwE&ef_id=CjwKCAjwrdmhBhBBEiwA4Hx5g49IaWVVv0liQQ49wa1iTpQSQ_omgjWlw4NjDh9F95_iCUbL_JaHsBoCyD4QAvD_BwE:G:s&s_kwcid=AL!3652!3!610458580017!!!g!!!12667906722!123084934209&cid=bid_pca_fsi_r01_co_cp1359_pjt0000_bid00000_0se_gaw_dy_lgn_ins (accessed on 3 June 2023).
- Read, S.C.; Irwin, R.J.; Poppe, C.; Harris, J. A comparison of two methods for isolation of Salmonella from poultry litter samples. Poult. Sci. 1994, 73, 1617–1621. [Google Scholar] [CrossRef] [PubMed]
- Andrews, W.H.; Poelma, P.L.; Wilson, C.R. Comparative efficiency of brilliant green, bismuth sulfite, Salmonella-Shigella, hektoen enteric, and xylose lysine desoxycholate agars for the recovery of Salmonella from foods: Collaborative study. J. Assoc. Off. Anal. Chem. 1981, 64, 899–928. [Google Scholar] [CrossRef]
- Amselem, G.; Guermonprez, C.; Drogue, B.; Michelin, S.; Baroud, C.N. Universal microfluidic platform for bioassays in anchored droplets. Lab Chip 2016, 16, 4200–4211. [Google Scholar] [CrossRef]
- Garstecki, P.; Fuerstman, M.J.; Stone, H.A.; Whitesides, G.M. Formation of droplets and bubbles in a microfluidic T-junction—Scaling and mechanism of break-up. Lab Chip 2006, 6, 437–446. [Google Scholar] [CrossRef]
- Nisisako, T.; Torii, T.; Higuchi, T. Droplet formation in a microchannel network. Lab Chip 2002, 2, 24–26. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.H.; Li, S.W.; Tan, J.; Luo, G. Correlations of droplet formation in T-junction microfluidic devices: From squeezing to dripping. Microfluid. Nanofluidics 2008, 5, 711–717. [Google Scholar] [CrossRef]
- Garstecki, P.; Gitlin, I.; DiLuzio, W.; Whitesides, G.M.; Kumacheva, E.; Stone, H.A. Formation of monodisperse bubbles in a microfluidic flow-focusing device. Appl. Phys. Lett. 2004, 85, 2649–2651. [Google Scholar] [CrossRef] [Green Version]
- Dixon, A.J.; Rickel, J.M.R.; Shin, B.D.; Klibanov, A.L.; Hossack, J.A. In Vitro Sonothrombolysis Enhancement by Transiently Stable Microbubbles Produced by a Flow-Focusing Microfluidic Device. Ann. Biomed. Eng. 2018, 46, 222–232. [Google Scholar] [CrossRef]
- Mu, K.; Si, T.; Li, E.; Xu, R.X.; Ding, H. Numerical study on droplet generation in axisymmetric flow focusing upon actuation. Phys. Fluids 2018, 30, 012111. [Google Scholar] [CrossRef]
- Hayat, Z.; El Abed, A.I. High-Throughput Optofluidic Acquisition of Microdroplets in Microfluidic Systems. Micromachines 2018, 9, 183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, S.; Nie, Z.; Seo, M.; Lewis, P.; Kumacheva, E.; Stone, H.A.; Garstecki, P.; Weibel, D.B.; Gitlin, I.; Whitesides, G.M. Generation of Monodisperse Particles by Using Microfluidics: Control over Size, Shape, and Composition. Angew. Chem. Int. Ed. 2005, 44, 724–728. [Google Scholar] [CrossRef] [PubMed]
- Dunkel, P.; Hayat, Z.; Barosi, A.; Bchellaoui, N.; Dhimane, H.; Dalko, P.I.; El Abed, A.I. Photolysis-driven merging of microdroplets in microfluidic chambers. Lab Chip 2016, 16, 1484–1491. [Google Scholar] [CrossRef]
- Gu, H.; Duits, M.H.G.; Mugele, F. Droplets Formation and Merging in Two-Phase Flow Microfluidics. Int. J. Mol. Sci. 2011, 12, 2572–2597. [Google Scholar] [CrossRef] [Green Version]
- Varma, V.B.; Ray, A.; Wang, Z.M.; Wang, Z.P.; Ramanujan, R.V. Droplet Merging on a Lab-on-a-Chip Platform by Uniform Magnetic Fields. Sci. Rep. 2016, 6, 37671. [Google Scholar] [CrossRef] [Green Version]
- Berry, S.B.; Lee, J.J.; Berthier, J.; Berthier, E.; Theberge, A.B. Droplet incubation and splitting in open microfluidic channels. Anal. Methods 2019, 11, 4528–4536. [Google Scholar] [CrossRef] [PubMed]
- Agnihotri, S.N.; Raveshi, M.R.; Bhardwaj, R.; Neild, A. Microfluidic Valves for Selective on-Chip Droplet Splitting at Multiple Sites. Langmuir 2020, 36, 1138–1146. [Google Scholar] [CrossRef]
- Fallah, K.; Fattahi, E. Splitting of droplet with different sizes inside a symmetric T-junction microchannel using an electric field. Sci. Rep. 2022, 12, 3226. [Google Scholar] [CrossRef]
- Mazutis, L.; Gilbert, J.; Ung, W.L.; Weitz, D.A.; Griffiths, A.D.; Heyman, J.A. Single-cell analysis and sorting using droplet-based microfluidics. Nat. Protoc. 2013, 8, 870–891. [Google Scholar] [CrossRef]
- Huang, C.; Jiang, Y.; Li, Y.; Zhang, H. Droplet Detection and Sorting System in Microfluidics: A Review. Micromachines 2023, 14, 103. [Google Scholar] [CrossRef] [PubMed]
- Marchand, G.; Dubois, P.; Delattre, C.; Vinet, F.; Blanchard-Desce, M.; Vaultier, M. Organic Synthesis in Soft Wall-Free Microreactors: Real-Time Monitoring of Fluorogenic Reactions. Anal. Chem. 2008, 80, 6051–6055. [Google Scholar] [CrossRef]
- Moragues, T.; Mitchell, S.; Faust Akl, D.; Pérez-Ramírez, J.; deMello, A. Droplet-Based Microfluidics Platform for the Synthesis of Single-Atom Heterogeneous Catalysts. Small Struct. 2023, 4, 2200284. [Google Scholar] [CrossRef]
- Nielsen, J.B.; Hanson, R.L.; Almughamsi, H.M.; Pang, C.; Fish, T.R.; Woolley, A.T. Microfluidics: Innovations in Materials and Their Fabrication and Functionalization. Anal. Chem. 2020, 92, 150–168. [Google Scholar] [CrossRef]
- Utech, S.; Prodanovic, R.; Mao, A.S.; Ostafe, R.; Mooney, D.J.; Weitz, D.A. Microfluidic Generation of Monodisperse, Structurally Homogeneous Alginate Microgels for Cell Encapsulation and 3D Cell Culture. Adv. Healthc. Mater. 2015, 4, 1628–1633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, C.; Chi, J.; Shang, L.; Fan, Q.; Ye, F. Droplet microfluidics-based biomedical microcarriers. Acta Biomater. 2022, 138, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Sun, M.; Zhang, J.; Hu, R.; Fu, W.; Xuanyuan, T.; Liu, W. Single-cell droplet microfluidics for biomedical applications. Analyst 2022, 147, 2294–2316. [Google Scholar] [CrossRef]
- Abate, A.R.; Hung, T.; Sperling, R.A.; Mary, P.; Rotem, A.; Agresti, J.J.; Weiner, M.A.; Weitz, D.A. DNA sequence analysis with droplet-based microfluidics. Lab Chip 2013, 13, 4864–4869. [Google Scholar] [CrossRef] [Green Version]
- Sukovich, D.J.; Lance, S.T.; Abate, A.R. Sequence specific sorting of DNA molecules with FACS using 3dPCR. Sci. Rep. 2017, 7, 39385. [Google Scholar] [CrossRef] [Green Version]
- Bjork, S.M.; Joensson, H.N. Microfluidics for cell factory and bioprocess development. Anal. Biotechnol. 2019, 55, 95–102. [Google Scholar] [CrossRef]
- Ho, C.M.B.; Sun, Q.; Teo, A.J.T.; Wibowo, D.; Gao, Y.; Zhou, J.; Huang, Y.; Tan, S.H.; Zhao, C.-X. Development of a Microfluidic Droplet-Based Microbioreactor for Microbial Cultivation. ACS Biomater. Sci. Eng. 2020, 6, 3630–3637. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Zhang, Y.; Xu, Q.; Sun, X.; Meng, F. Recent Advances on Sorting Methods of High-Throughput Droplet-Based Microfluidics in Enzyme Directed Evolution. Front. Chem. 2021, 9, 666867. [Google Scholar] [CrossRef] [PubMed]
- Chiu, F.W.Y.; Stavrakis, S. High-throughput droplet-based microfluidics for directed evolution of enzymes. Electrophoresis 2019, 40, 2860–2872. [Google Scholar] [CrossRef]
- Ostafe, R.; Prodanovic, R.; Lloyd Ung, W.; Weitz, D.A.; Fischer, R. A high-throughput cellulase screening system based on droplet microfluidics. Biomicrofluidics 2014, 8, 041102. [Google Scholar] [CrossRef] [Green Version]
- Prodanović, R.; Ung, W.L.; Ilić Đurđić, K.; Fischer, R.; Weitz, D.A.; Ostafe, R. A High-Throughput Screening System Based on Droplet Microfluidics for Glucose Oxidase Gene Libraries. Molecules 2020, 25, 2418. [Google Scholar] [CrossRef] [PubMed]
- Obexer, R.; Godina, A.; Garrabou, X.; Mittl, P.R.E.; Baker, D.; Griffiths, A.D.; Hilvert, D. Emergence of a catalytic tetrad during evolution of a highly active artificial aldolase. Nat. Chem. 2017, 9, 50–56. [Google Scholar] [CrossRef]
- Ma, C.; Tan, Z.L.; Lin, Y.; Han, S.; Xing, X.; Zhang, C. Gel microdroplet–based high-throughput screening for directed evolution of xylanase-producing Pichia pastoris. J. Biosci. Bioeng. 2019, 128, 662–668. [Google Scholar] [CrossRef] [PubMed]
- Vallejo, D.; Nikoomanzar, A.; Paegel, B.M.; Chaput, J.C. Fluorescence-Activated Droplet Sorting for Single-Cell Directed Evolution. ACS Synth. Biol. 2019, 8, 1430–1440. [Google Scholar] [CrossRef]
- Holland-Moritz, D.A.; Wismer, M.K.; Mann, B.F.; Farasat, I.; Devine, P.; Guetschow, E.D.; Mangion, I.; Welch, C.J.; Moore, J.C.; Sun, S.; et al. Mass Activated Droplet Sorting (MADS) Enables High-Throughput Screening of Enzymatic Reactions at Nanoliter Scale. Angew. Chem. Int. Ed. 2020, 59, 4470–4477. [Google Scholar] [CrossRef]
- Gielen, F.; Hours, R.; Emond, S.; Fischlechner, M.; Schell, U.; Hollfelder, F. Ultrahigh-throughput–directed enzyme evolution by absorbance-activated droplet sorting (AADS). Proc. Natl. Acad. Sci. USA 2016, 113, E7383–E7389. [Google Scholar] [CrossRef]
- Kempa, E.E.; Smith, C.A.; Li, X.; Bellina, B.; Richardson, K.; Pringle, S.; Galman, J.L.; Turner, N.J.; Barran, P.E. Coupling Droplet Microfluidics with Mass Spectrometry for Ultrahigh-Throughput Analysis of Complex Mixtures up to and above 30 Hz. Anal. Chem. 2020, 92, 12605–12612. [Google Scholar] [CrossRef]
- Goto, H.; Kanai, Y.; Yotsui, A.; Shimokihara, S.; Shitara, S.; Oyobiki, R.; Fujiwara, K.; Watanabe, T.; Einaga, Y.; Matsumoto, Y.; et al. Microfluidic screening system based on boron-doped diamond electrodes and dielectrophoretic sorting for directed evolution of NAD(P)-dependent oxidoreductases. Lab Chip 2020, 20, 852–861. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ren, L.; Su, Y.; Ji, Y.; Liu, Y.; Li, C.; Li, X.; Zhang, Y.; Wang, W.; Hu, Q.; et al. Raman-Activated Droplet Sorting (RADS) for Label-Free High-Throughput Screening of Microalgal Single-Cells. Anal. Chem. 2017, 89, 12569–12577. [Google Scholar] [CrossRef] [PubMed]
- Hale, W.; Rossetto, G.; Greenhalgh, R.; Finch, G.; Utz, M. High-resolution nuclear magnetic resonance spectroscopy in microfluidic droplets. Lab Chip 2018, 18, 3018–3024. [Google Scholar] [CrossRef] [Green Version]
- Yan, J.; Bauer, W.-A.C.; Fischlechner, M.; Hollfelder, F.; Kaminski, C.F.; Huck, W.T.S. Monodisperse Water-in-Oil-in-Water (W/O/W) Double Emulsion Droplets as Uniform Compartments for High-Throughput Analysis via Flow Cytometry. Micromachines 2013, 4, 402–413. [Google Scholar] [CrossRef] [Green Version]
- Brower, K.K.; Carswell-Crumpton, C.; Klemm, S.; Cruz, B.; Kim, G.; Calhoun, S.G.K.; Nichols, L.; Fordyce, P.M. Double emulsion flow cytometry with high-throughput single droplet isolation and nucleic acid recovery. Lab Chip 2020, 20, 2062–2074. [Google Scholar] [CrossRef] [PubMed]
- Report, B.M. Global Markets for Bioengineered Protein Drugs; BCC Publishing: Wellesley, MA, USA, 2023. [Google Scholar]
- Nelson, P.N.; Reynolds, G.M.; Waldron, E.E.; Ward, E.; Giannopoulos, K.; Murray, P.G. Monoclonal antibodies. Mol. Pathol. 2000, 53, 111–117. [Google Scholar] [CrossRef]
- Mullard, A. FDA approves 100th monoclonal antibody product. Nat. Rev. Drug Discov. 2021, 20, 491–495. [Google Scholar] [CrossRef]
- Kaplon, H.; Crescioli, S.; Chenoweth, A.; Visweswaraiah, J.; Reichert, J.M. Antibodies to watch in 2023. MAbs 2023, 15, 2153410. [Google Scholar] [CrossRef]
- Pedrioli, A.; Oxenius, A. Single B cell technologies for monoclonal antibody discovery. Trends Immunol. 2021, 42, 1143–1158. [Google Scholar] [CrossRef]
- Georgiou, G.; Ippolito, G.C.; Beausang, J.; Busse, C.E.; Wardemann, H.; Quake, S.R. The promise and challenge of high-throughput sequencing of the antibody repertoire. Nat. Biotechnol. 2014, 32, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Parola, C.; Neumeier, D.; Reddy, S.T. Integrating high-throughput screening and sequencing for monoclonal antibody discovery and engineering. Immunology 2018, 153, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Abul-Husn, N.S.; Kenny, E.E. Personalized Medicine and the Power of Electronic Health Records. Cell 2019, 177, 58–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, K.C.; Boyd, A.W.; Fisher, D.C.; Slaughenhoupt, B.; Groopman, J.E.; O’Hara, C.J.; Daley, J.F.; Schlossman, S.F.; Nadler, L.M. Isolation and functional analysis of human B cell populations. I. Characterization of the B1+B2+ and B1+B2- subsets. J. Immunol. 1985, 134, 820–827. [Google Scholar] [CrossRef] [PubMed]
- El Debs, B.; Utharala, R.; Balyasnikova, I.V.; Griffiths, A.D.; Merten, C.A. Functional single-cell hybridoma screening using droplet-based microfluidics. Proc. Natl. Acad. Sci. USA 2012, 109, 11570–11575. [Google Scholar] [CrossRef]
- Garraud, O.; Borhis, G.; Badr, G.; Degrelle, S.; Pozzetto, B.; Cognasse, F.; Richard, Y. Revisiting the B-cell compartment in mouse and humans: More than one B-cell subset exists in the marginal zone and beyond. BMC Immunol. 2012, 13, 63. [Google Scholar] [CrossRef]
- Konforte, D.; Simard, N.; Paige, C.J. IL-21: An Executor of B Cell Fate1. J. Immunol. 2009, 182, 1781–1787. [Google Scholar] [CrossRef] [Green Version]
- Lanzavecchia, A. Antigen-specific interaction between T and B cells. Nature 1985, 314, 537–539. [Google Scholar] [CrossRef]
- Bernasconi, N.L.; Traggiai, E.; Lanzavecchia, A. Maintenance of serological memory by polyclonal activation of human memory B cells. Science 2002, 298, 2199–2202. [Google Scholar] [CrossRef]
- Tiller, T.; Meffre, E.; Yurasov, S.; Tsuiji, M.; Nussenzweig, M.C.; Wardemann, H. Efficient generation of monoclonal antibodies from single human B cells by single cell RT-PCR and expression vector cloning. J. Immunol. Methods 2008, 329, 112–124. [Google Scholar] [CrossRef] [Green Version]
- Kang, S.; Yang, M.; Hong, Z.; Zhang, L.; Huang, Z.; Chen, X.; He, S.; Zhou, Z.; Zhou, Z.; Chen, Q.; et al. Crystal structure of SARS-CoV-2 nucleocapsid protein RNA binding domain reveals potential unique drug targeting sites. Acta Pharm. Sin. B 2020, 10, 1228–1238. [Google Scholar] [CrossRef] [PubMed]
- Ogunniyi, A.O.; Story, C.M.; Papa, E.; Guillen, E.; Love, J.C. Screening individual hybridomas by microengraving to discover monoclonal antibodies. Nat. Protoc. 2009, 4, 767–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greener, J.G.; Kandathil, S.M.; Moffat, L.; Jones, D.T. A guide to machine learning for biologists. Nat. Rev. Mol. Cell Biol. 2022, 23, 40–55. [Google Scholar] [CrossRef]
- Shin, H.C.; Roth, H.R.; Gao, M.; Lu, L.; Xu, Z.; Nogues, I.; Yao, J.; Mollura, D.; Summers, R.M. Deep Convolutional Neural Networks for Computer-Aided Detection: CNN Architectures, Dataset Characteristics and Transfer Learning. IEEE Trans. Med. Imaging 2016, 35, 1285–1298. [Google Scholar] [CrossRef] [Green Version]
- O’Neill, K.; Aghaeepour, N.; Špidlen, J.; Brinkman, R. Flow Cytometry Bioinformatics. PLoS Comput. Biol. 2013, 9, e1003365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liechti, T.; Weber, L.M.; Ashhurst, T.M.; Stanley, N.; Prlic, M.; Van Gassen, S.; Mair, F. An updated guide for the perplexed: Cytometry in the high-dimensional era. Nat. Immunol. 2021, 22, 1190–1197. [Google Scholar] [CrossRef]
- Rahim, A.; Meskas, J.; Drissler, S.; Yue, A.; Lorenc, A.; Laing, A.; Saran, N.; White, J.; Abeler-Dörner, L.; Hayday, A.; et al. High throughput automated analysis of big flow cytometry data. Methods 2018, 134–135, 164–176. [Google Scholar] [CrossRef] [PubMed]
- Saeys, Y.; Van Gassen, S.; Lambrecht, B.N. Computational flow cytometry: Helping to make sense of high-dimensional immunology data. Nat. Rev. Immunol. 2016, 16, 449–462. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Bhattacharya, S.; Butte, A.J. Application of Machine Learning for Cytometry Data. Front. Immunol. 2022, 12, 787574. [Google Scholar] [CrossRef]
- Liu, P.; Liu, S.; Fang, Y.; Xue, X.; Zou, J.; Tseng, G.; Konnikova, L. Recent Advances in Computer-Assisted Algorithms for Cell Subtype Identification of Cytometry Data. Front. Cell Dev. Biol. 2020, 8, 234. [Google Scholar] [CrossRef]
- Keyes, T.J.; Domizi, P.; Lo, Y.-C.; Nolan, G.P.; Davis, K.L. A Cancer Biologist’s Primer on Machine Learning Applications in High-Dimensional Cytometry. Cytom. Part A 2020, 97, 782–799. [Google Scholar] [CrossRef]
- Lucchesi, S.; Furini, S.; Medaglini, D.; Ciabattini, A. From Bivariate to Multivariate Analysis of Cytometric Data: Overview of Computational Methods and Their Application in Vaccination Studies. Vaccines 2020, 8, 138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nolan, J.P.; Condello, D.; Duggan, E.; Naivar, M.; Novo, D. Visible and near infrared fluorescence spectral flow cytometry. Cytom. Part A 2013, 83, 253–264. [Google Scholar] [CrossRef] [Green Version]
- Ferrer-Font, L.; Kraker, G.; Hally, K.E.; Price, K.M. Ensuring Full Spectrum Flow Cytometry Data Quality for High-Dimensional Data Analysis. Curr. Protoc. 2023, 3, e657. [Google Scholar] [CrossRef]
- Bagwell, C.B.; Adams, E.G. Fluorescence Spectral Overlap Compensation for Any Number of Flow Cytometry Parameters. Ann. N. Y. Acad. Sci. 1993, 677, 167–184. [Google Scholar] [CrossRef]
- Keshava, N.; Mustard, J.F. Spectral unmixing. IEEE Signal Process. Mag. 2002, 19, 44–57. [Google Scholar] [CrossRef]
- Wang, R.; Lemus, A.A.; Henneberry, C.M.; Ying, Y.; Feng, Y.; Valm, A.M. Unmixing biological fluorescence image data with sparse and low-rank Poisson regression. Bioinformatics 2023, 39, btad159. [Google Scholar] [CrossRef] [PubMed]
- Van Gassen, S.; Gaudilliere, B.; Angst, M.S.; Saeys, Y.; Aghaeepour, N. CytoNorm: A Normalization Algorithm for Cytometry Data. Cytom. Part A 2020, 97, 268–278. [Google Scholar] [CrossRef] [Green Version]
- Hahne, F.; Khodabakhshi, A.H.; Bashashati, A.; Wong, C.-J.; Gascoyne, R.D.; Weng, A.P.; Seyfert-Margolis, V.; Bourcier, K.; Asare, A.; Lumley, T.; et al. Per-channel basis normalization methods for flow cytometry data. Cytom. Part A 2010, 77, 121–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finak, G.; Jiang, W.; Krouse, K.; Wei, C.; Sanz, I.; Phippard, D.; Asare, A.; De Rosa, S.C.; Self, S.; Gottardo, R. High-throughput flow cytometry data normalization for clinical trials. Cytom. Part A 2014, 85, 277–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fletez-Brant, K.; Špidlen, J.; Brinkman, R.R.; Roederer, M.; Chattopadhyay, P.K. flowClean: Automated identification and removal of fluorescence anomalies in flow cytometry data. Cytom. Part A 2016, 89, 461–471. [Google Scholar] [CrossRef] [Green Version]
- Ashhurst, T.M.; Marsh-Wakefield, F.; Putri, G.H.; Spiteri, A.G.; Shinko, D.; Read, M.N.; Smith, A.L.; King, N.J.C. Integration, exploration, and analysis of high-dimensional single-cell cytometry data using Spectre. Cytom. Part A 2022, 101, 237–253. [Google Scholar] [CrossRef]
- Quintelier, K.; Couckuyt, A.; Emmaneel, A.; Aerts, J.; Saeys, Y.; Van Gassen, S. Analyzing high-dimensional cytometry data using FlowSOM. Nat. Protoc. 2021, 16, 3775–3801. [Google Scholar] [CrossRef] [PubMed]
- Qiu, P.; Simonds, E.F.; Bendall, S.C.; Gibbs, K.D.; Bruggner, R.V.; Linderman, M.D.; Sachs, K.; Nolan, G.P.; Plevritis, S.K. Extracting a cellular hierarchy from high-dimensional cytometry data with SPADE. Nat. Biotechnol. 2011, 29, 886–891. [Google Scholar] [CrossRef] [Green Version]
- Amir, E.-a.D.; Davis, K.L.; Tadmor, M.D.; Simonds, E.F.; Levine, J.H.; Bendall, S.C.; Shenfeld, D.K.; Krishnaswamy, S.; Nolan, G.P.; Pe’er, D. viSNE enables visualization of high dimensional single-cell data and reveals phenotypic heterogeneity of leukemia. Nat. Biotechnol. 2013, 31, 545–552. [Google Scholar] [CrossRef] [Green Version]
- Becht, E.; McInnes, L.; Healy, J.; Dutertre, C.-A.; Kwok, I.W.H.; Ng, L.G.; Ginhoux, F.; Newell, E.W. Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol. 2019, 37, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Moon, K.R.; van Dijk, D.; Wang, Z.; Gigante, S.; Burkhardt, D.B.; Chen, W.S.; Yim, K.; Elzen, A.v.d.; Hirn, M.J.; Coifman, R.R.; et al. Visualizing structure and transitions in high-dimensional biological data. Nat. Biotechnol. 2019, 37, 1482–1492. [Google Scholar] [CrossRef] [PubMed]
- Mair, F.; Hartmann, F.J.; Mrdjen, D.; Tosevski, V.; Krieg, C.; Becher, B. The end of gating? An introduction to automated analysis of high dimensional cytometry data. Eur. J. Immunol. 2016, 46, 34–43. [Google Scholar] [CrossRef]
- Aghaeepour, N.; Nikolic, R.; Hoos, H.H.; Brinkman, R.R. Rapid cell population identification in flow cytometry data. Cytom. Part A 2011, 79, 6–13. [Google Scholar] [CrossRef] [Green Version]
- Weber, L.M.; Robinson, M.D. Comparison of clustering methods for high-dimensional single-cell flow and mass cytometry data. Cytom. Part A 2016, 89, 1084–1096. [Google Scholar] [CrossRef] [Green Version]
- Finak, G.; Bashashati, A.; Brinkman, R.; Gottardo, R. Merging Mixture Components for Cell Population Identification in Flow Cytometry. Adv. Bioinform. 2009, 2009, 247646. [Google Scholar] [CrossRef] [Green Version]
- Dorfman, D.M.; LaPlante, C.D.; Li, B. FLOCK cluster analysis of plasma cell flow cytometry data predicts bone marrow involvement by plasma cell neoplasia. Leuk. Res. 2016, 48, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Qiu, P. Automatically generate two-dimensional gating hierarchy from clustered cytometry data. Cytom. Part A 2018, 93, 1039–1050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Commenges, D.; Alkhassim, C.; Gottardo, R.; Hejblum, B.; Thiébaut, R. cytometree: A binary tree algorithm for automatic gating in cytometry analysis. Cytom. Part A 2018, 93, 1132–1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aghaeepour, N.; Finak, G.; Hoos, H.; Mosmann, T.R.; Brinkman, R.; Gottardo, R.; Scheuermann, R.H.; FlowCAP Consortium; Dream Consortium. Critical assessment of automated flow cytometry data analysis techniques. Nat. Methods 2013, 10, 228–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo, K.; Brinkman, R.R.; Gottardo, R. Automated gating of flow cytometry data via robust model-based clustering. Cytom. Part A 2008, 73, 321–332. [Google Scholar] [CrossRef]
- Dundar, M.; Akova, F.; Yerebakan, H.Z.; Rajwa, B. A non-parametric Bayesian model for joint cell clustering and cluster matching: Identification of anomalous sample phenotypes with random effects. BMC Bioinform. 2014, 15, 314. [Google Scholar] [CrossRef]
- Azad, A.; Rajwa, B.; Pothen, A. Immunophenotype Discovery, Hierarchical Organization, and Template-Based Classification of Flow Cytometry Samples. Front. Oncol. 2016, 6, 188. [Google Scholar] [CrossRef] [Green Version]
- Béné, M.C.; Lacombe, F.; Porwit, A. Unsupervised flow cytometry analysis in hematological malignancies: A new paradigm. Int. J. Lab. Hematol. 2021, 43, 54–64. [Google Scholar] [CrossRef]
- Hu, Z.; Tang, A.; Singh, J.; Bhattacharya, S.; Butte, A.J. A robust and interpretable end-to-end deep learning model for cytometry data. Proc. Natl. Acad. Sci. USA 2020, 117, 21373–21380. [Google Scholar] [CrossRef]
- Rajwa, B.; Wallace, P.K.; Griffiths, E.A.; Dundar, M. Automated assessment of disease progression in acute myeloid leukemia by probabilistic analysis of flow cytometry data. IEEE Trans. Biomed. Eng. 2017, 64, 1089–1098. [Google Scholar] [CrossRef]
- Rajwa, B.; Venkatapathi, M.; Ragheb, K.; Banada, P.P.; Hirleman, E.D.; Lary, T.; Robinson, J.P. Automated classification of bacterial particles in flow by multiangle scatter measurement and support vector machine classifier. Cytom. Part A 2008, 73, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Aghaeepour, N.; Jalali, A.; O’Neill, K.; Chattopadhyay, P.K.; Roederer, M.; Hoos, H.H.; Brinkman, R.R. RchyOptimyx: Cellular hierarchy optimization for flow cytometry. Cytom. Part A 2012, 81, 1022–1030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Neill, K.; Jalali, A.; Aghaeepour, N.; Hoos, H.; Brinkman, R.R. Enhanced flowType/RchyOptimyx: A Bioconductor pipeline for discovery in high-dimensional cytometry data. Bioinformatics 2014, 30, 1329–1330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malek, M.; Taghiyar, M.J.; Chong, L.; Finak, G.; Gottardo, R.; Brinkman, R.R. flowDensity: Reproducing manual gating of flow cytometry data by automated density-based cell population identification. Bioinformatics 2014, 31, 606–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greene, E.; Finak, G.; D’Amico, L.A.; Bhardwaj, N.; Church, C.D.; Morishima, C.; Ramchurren, N.; Taube, J.M.; Nghiem, P.T.; Cheever, M.A.; et al. New interpretable machine-learning method for single-cell data reveals correlates of clinical response to cancer immunotherapy. Patterns 2021, 2, 100372. [Google Scholar] [CrossRef]
- Nicora, G.; Vitali, F.; Dagliati, A.; Geifman, N.; Bellazzi, R. Integrated Multi-Omics Analyses in Oncology: A Review of Machine Learning Methods and Tools. Front. Oncol. 2020, 10, 1030. [Google Scholar] [CrossRef]
- Subramanian, I.; Verma, S.; Kumar, S.; Jere, A.; Anamika, K. Multi-omics Data Integration, Interpretation, and Its Application. Bioinform. Biol. Insights 2020, 14, 1177932219899051. [Google Scholar] [CrossRef] [Green Version]
- Whetton, A.D.; Preston, G.W.; Abubeker, S.; Geifman, N. Proteomics and Informatics for Understanding Phases and Identifying Biomarkers in COVID-19 Disease. J. Proteome Res. 2020, 19, 4219–4232. [Google Scholar] [CrossRef]
- Franco, J.; Rajwa, B.; Ferreira, C.R.; Sundberg, J.P.; HogenEsch, H. Lipidomic profiling of the epidermis in a mouse model of dermatitis reveals sexual dimorphism and changes in lipid composition before the onset of clinical disease. Metabolites 2020, 10, 299. [Google Scholar] [CrossRef]
- Hira, Z.M.; Gillies, D.F. A Review of Feature Selection and Feature Extraction Methods Applied on Microarray Data. Adv. Bioinform. 2015, 2015, 198363. [Google Scholar] [CrossRef] [PubMed]
- Bruggner, R.V.; Bodenmiller, B.; Dill, D.L.; Tibshirani, R.J.; Nolan, G.P. Automated identification of stratifying signatures in cellular subpopulations. Proc. Natl. Acad. Sci. USA 2014, 111, E2770–E2777. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Wen, B.; Gao, Q.; Zhang, B. Feature Selection Methods for Protein Biomarker Discovery from Proteomics or Multiomics Data. Mol. Cell. Proteom. 2021, 20, 100083. [Google Scholar] [CrossRef] [PubMed]
- Mamoshina, P.; Vieira, A.; Putin, E.; Zhavoronkov, A. Applications of Deep Learning in Biomedicine. Mol. Pharm. 2016, 13, 1445–1454. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.D.; Wang, D. Multiview learning for understanding functional multiomics. PLoS Comput. Biol. 2020, 16, e1007677. [Google Scholar] [CrossRef]
- Cossarizza, A.; Chang, H.D.; Radbruch, A.; Acs, A.; Adam, D.; Adam-Klages, S.; Agace, W.W.; Aghaeepour, N.; Akdis, M.; Allez, M.; et al. Guidelines for the use of flow cytometry and cell sorting in immunological studies (second edition). Eur. J. Immunol. 2019, 49, 1457–1973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, M.; Robinson, J.P. Quantum approach for nanoparticle fluorescence by sub-ns photon detection. Cytom. Part A 2020, 99, 145–151. [Google Scholar] [CrossRef]
- Yamamoto, M.; Hernandez, K.; Robinson, J.P. Photon Spectroscopy by Picoseconds Differential Geiger-Mode Si Photomultiplier; SPIE: San Francisco, CA, USA, 2018; Volume 10500, pp. 10501–10513. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fluorochrome | Excitation Spectrum | Emission Spectrum |
|---|---|---|
| Hoechst | 350 nm | 450 nm (blue) |
| DAPI | 350 nm | 450 nm (blue) |
| Pacific Blue | 405 nm | 450 nm (blue) |
| Brilliant Violet | 405 nm | Numerous Lines |
| FITC | 488 nm (blue) | 530 nm (green) |
| PE | 488–530 nm (green) | 575 nm (orange-red) |
| PerCP | 488 nm (blue) | 675 nm (orange) |
| APC | 633 nm (red) | 660 nm (far-red) |
| Alexa Fluor 488 | 488 nm (blue) | 519 nm (green) |
| Alexa Fluor 647 | 633 nm (red) | 668 nm (far-red) |
| Alexa Fluor 700 | 633 nm (red) | 719 nm (far-red) |
| Pacific Blue | 405 nm (violet) | 455 nm (blue) |
| BV421 | 405 nm (violet) | 421 nm (blue) |
| BV510 | 405 nm (violet) | 510 nm (green) |
| BV605 | 405 nm (violet) | 605 nm (orange) |
| BV650 | 405 nm (violet) | 650 nm (red) |
| BV711 | 405 nm (violet) | 711 nm (far-red) |
| PE-Cy7 | 633 nm (red) | 780 nm (near-infrared) |
| APC-Cy7 | 633 nm (red) | 785 nm (near-infrared) |
| PerCP-Cy5.5 | 488 nm (blue) | 695 nm (red) |
| Qdot 605 | 405 nm (violet) | 605 nm (orange) |
| Qdot 655 | 405 nm (violet) | 655 nm (red) |
| Qdot 705 | 405 nm (violet) | 705 nm (far-red) |
| Qdot 800 | 405 nm (violet) | 800 nm (near-infrared) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Robinson, J.P.; Ostafe, R.; Iyengar, S.N.; Rajwa, B.; Fischer, R. Flow Cytometry: The Next Revolution. Cells 2023, 12, 1875. https://doi.org/10.3390/cells12141875
Robinson JP, Ostafe R, Iyengar SN, Rajwa B, Fischer R. Flow Cytometry: The Next Revolution. Cells. 2023; 12(14):1875. https://doi.org/10.3390/cells12141875
Chicago/Turabian StyleRobinson, J. Paul, Raluca Ostafe, Sharath Narayana Iyengar, Bartek Rajwa, and Rainer Fischer. 2023. "Flow Cytometry: The Next Revolution" Cells 12, no. 14: 1875. https://doi.org/10.3390/cells12141875