

Deficiency of Adipose Aryl Hydrocarbon Receptor Protects against Diet-Induced Metabolic Dysfunction through Sexually Dimorphic Mechanisms


Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals and Experimental Timeline
2.2. Activity Behavior Monitoring
2.3. Metabolic Chamber Measurements
2.4. Glucose Tolerance Test (GTT)
2.5. Histological Analysis and Immunohistochemistry
2.6. Immunofluorescence
2.7. q-PCR
2.8. Western Blot
2.9. ELISA
2.10. Serum Triglyceride Assay
2.11. Statistical Analysis
3. Results
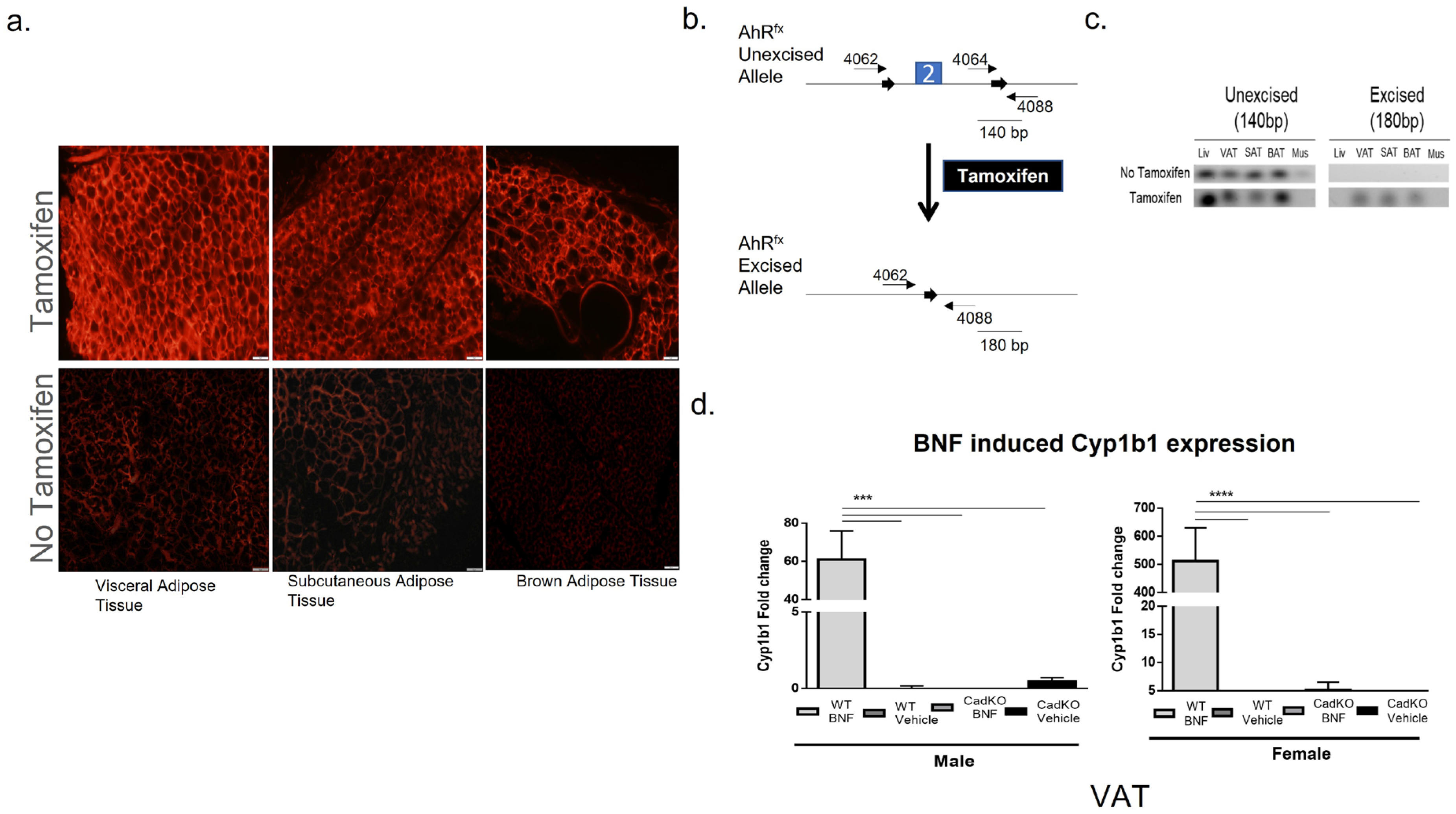
3.1. Conditional Deletion of AhR from Adipose Tissue
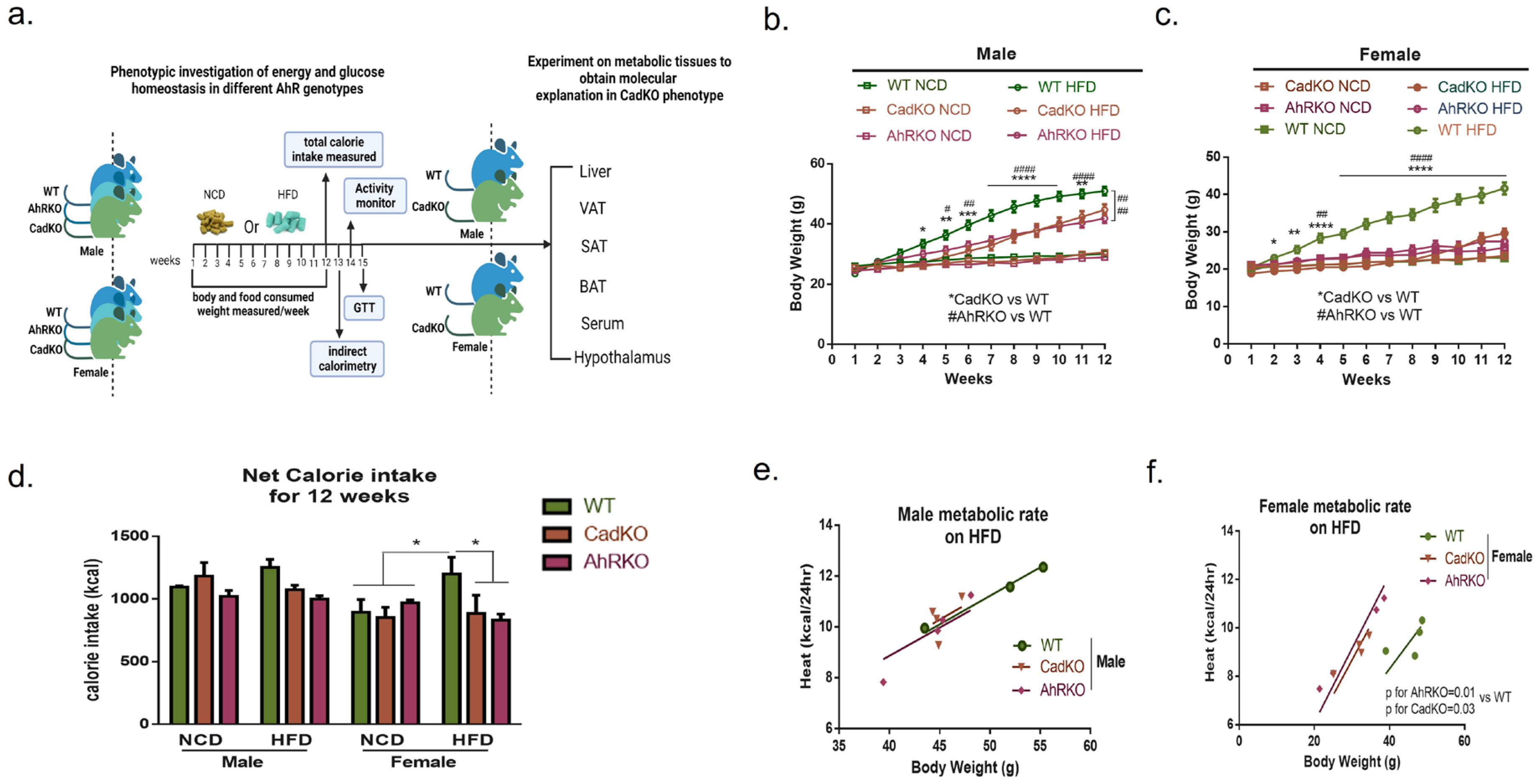
3.2. Sex-Specific Effects of Global and Adipose-Specific AhR Deletion on HFD-Induced Weight Gain and Metabolic Rate
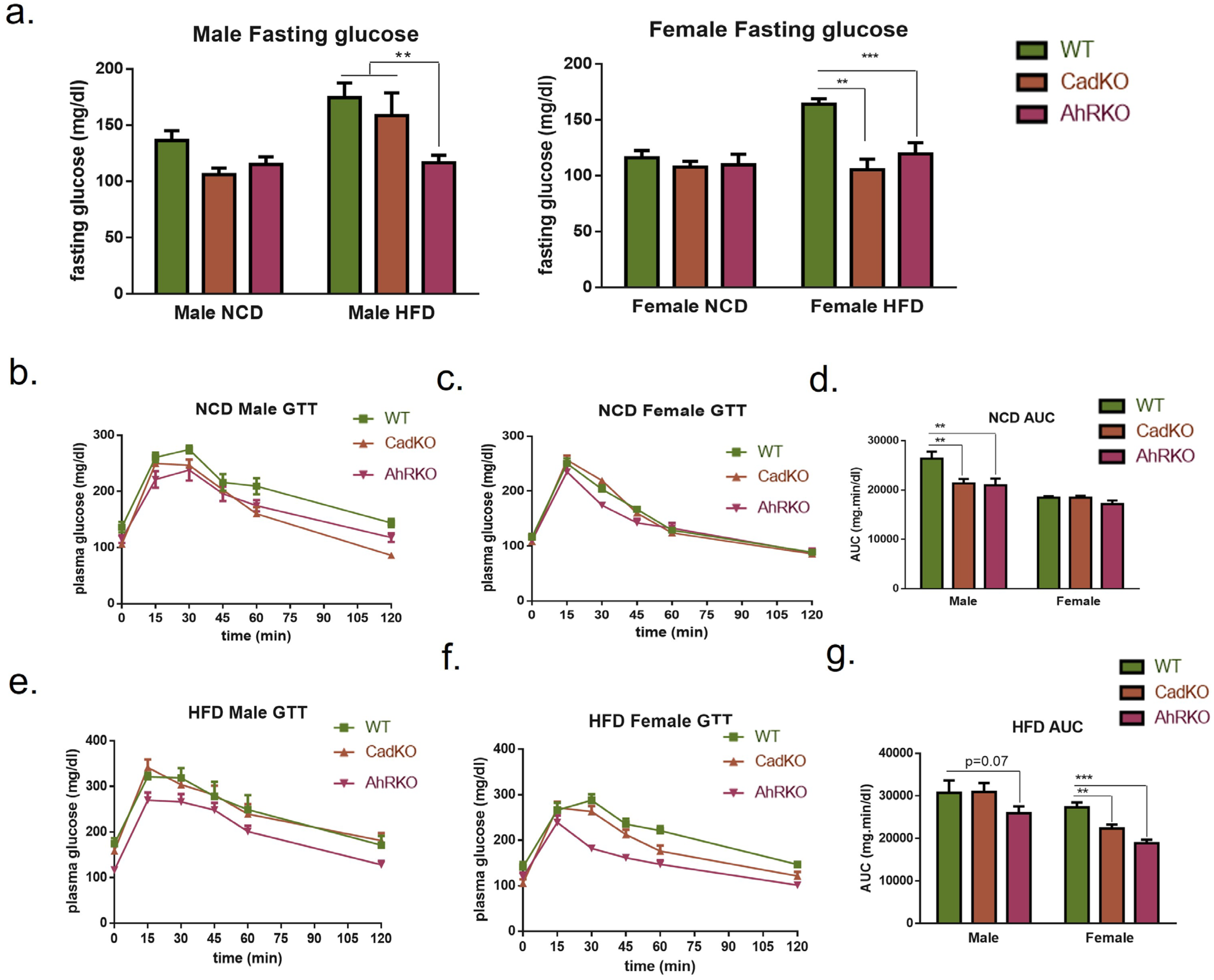
3.3. CadKO Improves Systemic Glucose Homeostasis and Protects against HFD-Induced Insulin Resistance in Females
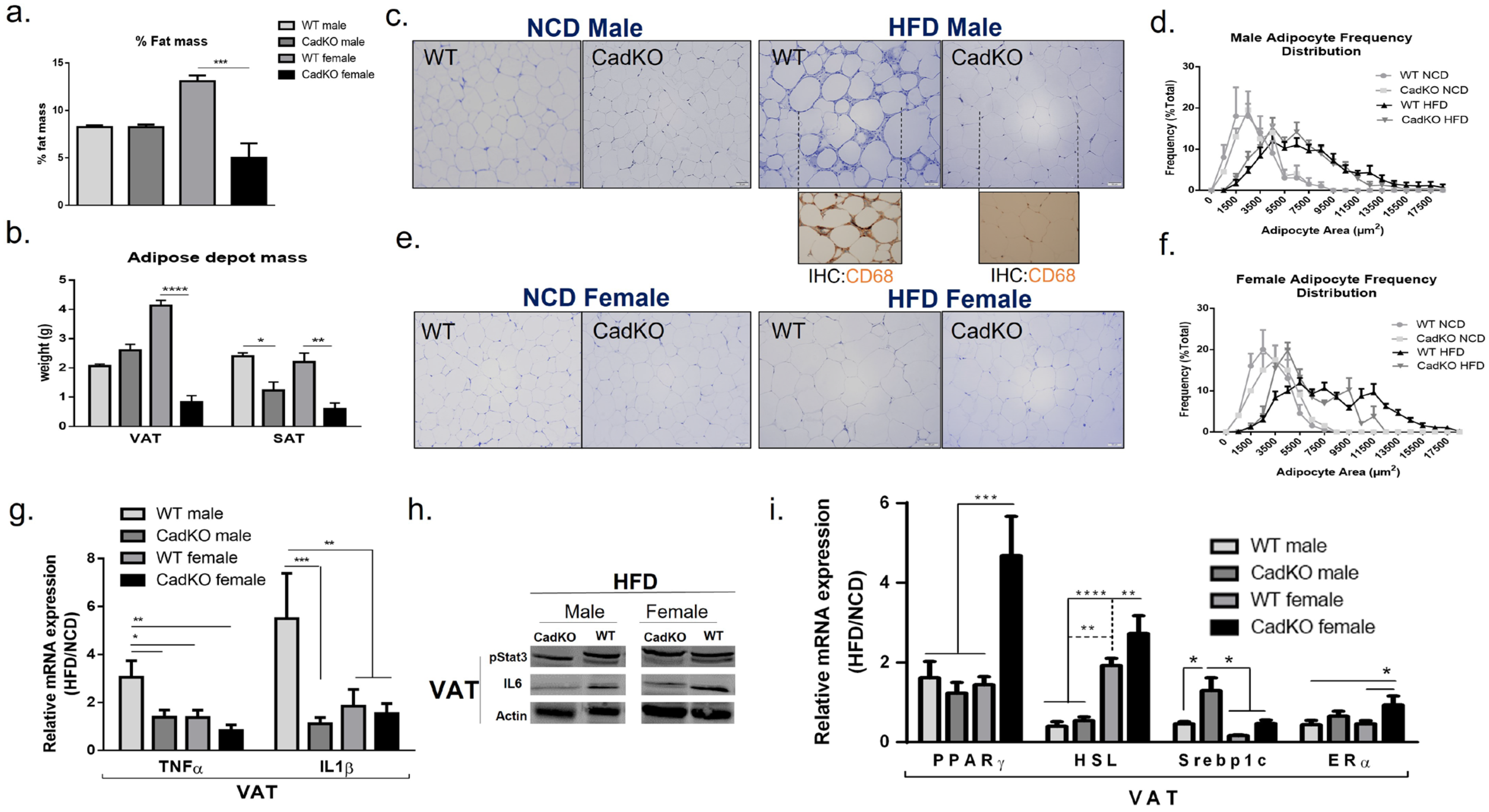
3.4. CadKO Protects from HFD-Induced Adiposity in Females and Inflammation in Males
3.5. Adipose-Specific Deletion of AhR Protects Endocrine Function of White Adipocytes in Females
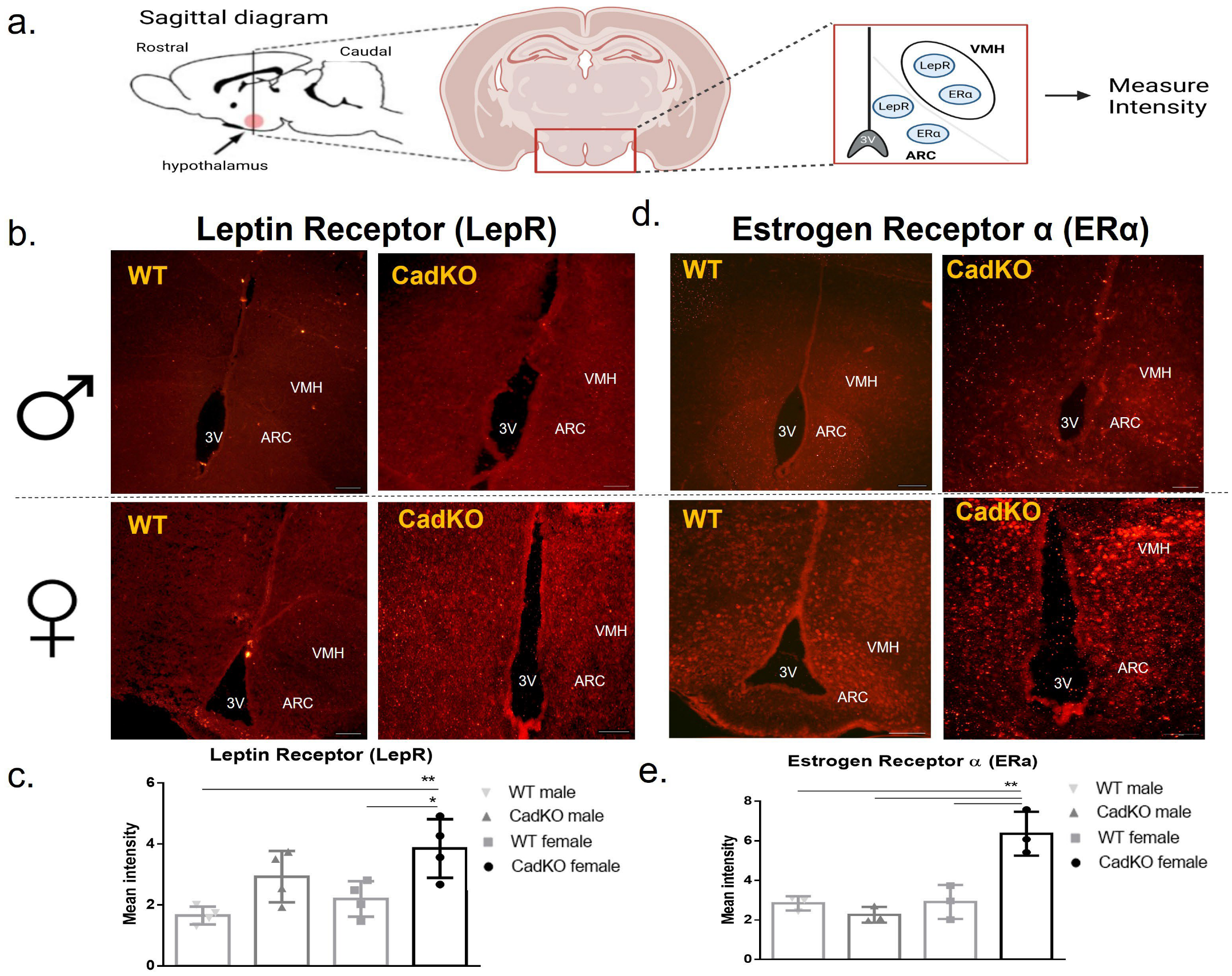
3.6. HFD-Fed Female CadKO Mice Have Increased Leptin and Estrogen Receptors
3.7. CadKO Protection against Liver Pathogenesis Is Sexually Dimorphic
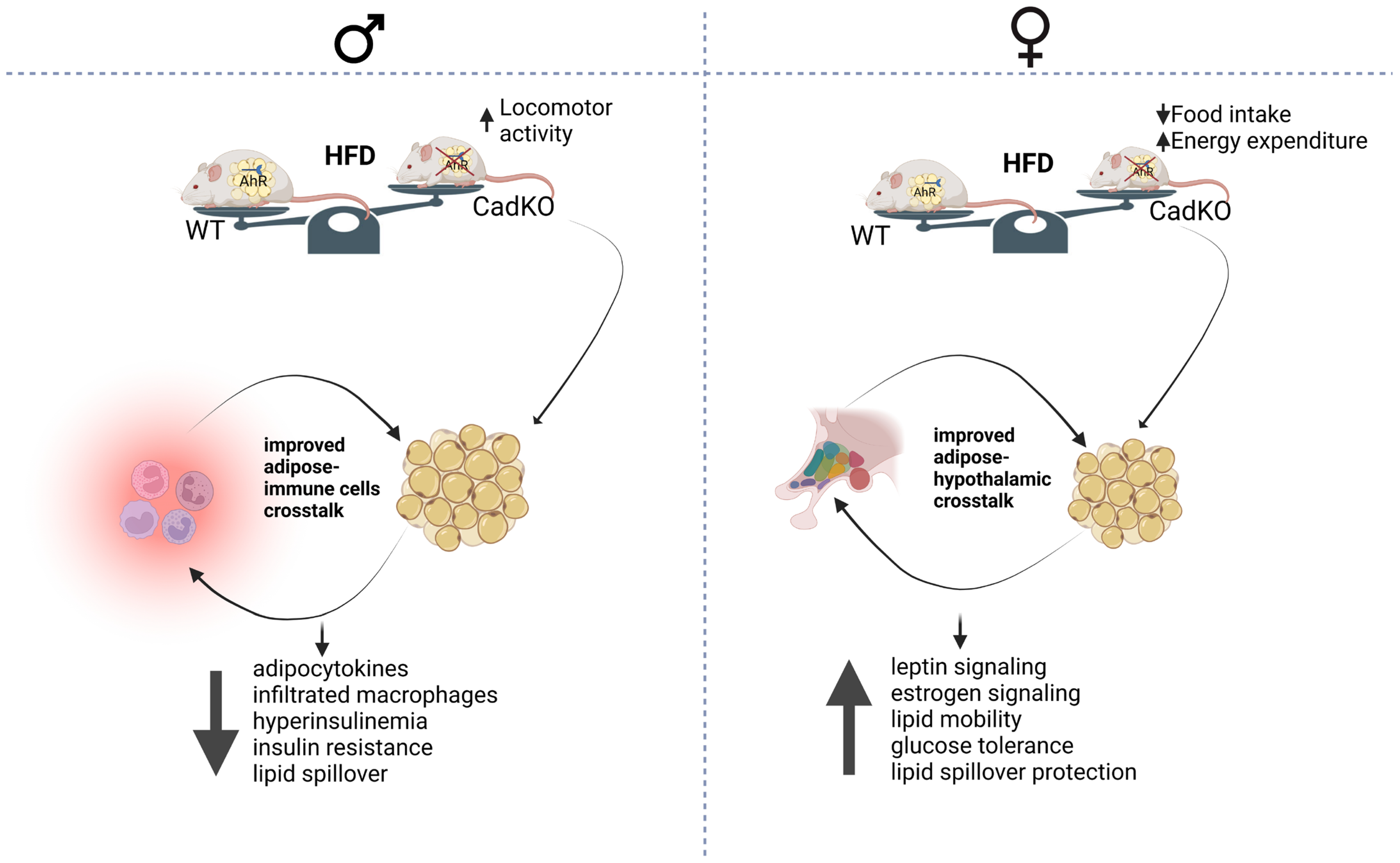
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Frühbeck, G. Overview of Adipose Tissue and Its Role in Obesity and Metabolic Disorders. Methods Mol. Biol. 2008, 456, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Kershaw, E.E.; Flier, J.S. Adipose Tissue as an Endocrine Organ. J. Clin. Endocrinol. Metab. 2004, 89, 2548–2556. [Google Scholar] [CrossRef] [PubMed]
- Sandoval, D.; Cota, D.; Seeley, R.J. The integrative role of CNS fuel-sensing mechanisms in energy balance and glucose regu-lation. Annu. Rev. Physiol. 2008, 70, 513–535. [Google Scholar] [CrossRef]
- Haque, N.; Tischkau, S.A. Sexual Dimorphism in Adipose-Hypothalamic Crosstalk and the Contribution of Aryl Hydrocarbon Receptor to Regulate Energy Homeostasis. Int. J. Mol. Sci. 2022, 23, 7679. [Google Scholar] [CrossRef] [PubMed]
- Considine, R.V.; Sinha, M.K.; Heiman, M.L.; Kriauciunas, A.; Stephens, T.W.; Nyce, M.R.; Ohannesian, J.P.; Marco, C.C.; McKee, L.J.; Bauer, T.L.; et al. Serum Immunoreactive-Leptin Concentrations in Normal-Weight and Obese Humans. N. Engl. J. Med. 1996, 334, 292–295. [Google Scholar] [CrossRef] [PubMed]
- Caro, J.F.; Kolaczynski, J.W.; Nyce, M.R.; Ohannesian, J.P.; Opentanova, I.; Goldman, W.H.; Lynn, R.B.; Zhang, P.L.; Sinha, M.K.; Considine, R.V. Decreased cerebrospi-nal-fluid/serum leptin ratio in obesity: A possible mechanism for leptin resistance. Lancet 1996, 348, 159–161. [Google Scholar] [CrossRef]
- Friedman, J.M. Leptin and the endocrine control of energy balance. Nat. Metab. 2019, 1, 754–764. [Google Scholar] [CrossRef]
- Cone, R.D. Anatomy and regulation of the central melanocortin system. Nat. Neurosci. 2005, 8, 571–578. [Google Scholar] [CrossRef]
- Dietze-Schroeder, D.; Sell, H.; Uhlig, M.; Koenen, M.; Eckel, J. Autocrine Action of Adiponectin on Human Fat Cells Prevents the Release of Insulin Resistance-Inducing Factors. Diabetes 2005, 54, 2003–2011. [Google Scholar] [CrossRef] [Green Version]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W., Jr. Obesity is associated with macro-phage accumulation in adipose tissue. J. Clin. Investig. 2003, 112, 1796–1808. [Google Scholar] [CrossRef]
- Hotamisligil, G.S.; Shargill, N.S.; Spiegelman, B.M. Adipose expression of tumor necrosis factor-alpha: Direct role in obesi-ty-linked insulin resistance. Science 1993, 259, 87–91. [Google Scholar] [CrossRef]
- Kanda, H.; Tateya, S.; Tamori, Y.; Kotani, K.; Hiasa, K.; Kitazawa, R.; Kitazawa, S.; Miyachi, H.; Maeda, S.; Egashira, K.; et al. MCP-1 contributes to macrophage infiltration into adi-pose tissue, insulin resistance, and hepatic steatosis in obesity. J. Clin. Investig. 2006, 116, 1494–1505. [Google Scholar] [CrossRef] [PubMed]
- Ema, M.; Sogawa, K.; Watanabe, N.; Chujoh, Y.; Matsushita, N.; Gotoh, O.; Funae, Y.; Fujii-Kuriyama, Y. cDNA cloning and structure of mouse putative Ah receptor. Biochem. Biophys. Res. Commun. 1992, 184, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Burbach, K.M.; Poland, A.; Bradfield, C.A. Cloning of the Ah-receptor cDNA reveals a distinctive ligand-activated transcrip-tion factor. Proc. Natl. Acad. Sci. USA 1992, 89, 8185–8189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fierens, S.; Mairesse, H.; Heilier, J.-F.; De Burbure, C.; Focant, J.-F.; Eppe, G.; De Pauw, E.; Bernard, A. Dioxin/polychlorinated biphenyl body burden, diabetes and endometriosis: Findings in a population-based study in Belgium. Biomarkers 2003, 8, 529–534. [Google Scholar] [CrossRef]
- Henriksen, G.L.; Ketchum, N.S.; Michalek, J.E.; Swaby, J.A. Serum Dioxin and Diabetes Mellitus in Veterans of Operation Ranch Hand. Epidemiology 1997, 8, 252–258. [Google Scholar]
- Lee, D.H.; Lee, I.K.; Song, K.; Steffes, M.; Toscano, W.; Baker, B.A.; Jacobs, D.R., Jr. A strong dose-response relation between serum con-centrations of persistent organic pollutants and diabetes: Results from the National Health and Examination Survey 1999–2002. Diabetes Care 2006, 29, 1638–1644. [Google Scholar] [CrossRef] [Green Version]
- Magliano, D.J.; Rancière, F.; Slama, R.; Roussel, R.; Kiviranta, H.; Coumoul, X.; Balkau, B.; Botton, J. Exposure to persistent organic pollutants and the risk of type 2 diabetes: A case-cohort study. Diabetes Metab. 2021, 47, 101234. [Google Scholar] [CrossRef]
- Turyk, M.; Anderson, H.A.; Knobeloch, L.; Imm, P.; Persky, V.W. Prevalence of diabetes and body burdens of polychlorinated biphenyls, polybrominated diphenyl ethers, and p,p′-diphenyldichloroethene in Great Lakes sport fish consumers. Chemosphere 2009, 75, 674–679. [Google Scholar] [CrossRef]
- Wang, S.L.; Tsai, P.C.; Yang, C.Y.; Guo, Y.L. Increased risk of diabetes and polychlorinated biphenyls and dioxins: A 24-year follow-up study of the Yucheng cohort. Diabetes Care 2008, 31, 1574–1579. [Google Scholar] [CrossRef] [Green Version]
- McMillan, B.J.; Bradfield, C.A. The Aryl hydrocarbon receptor is activated by modified low-density lipoprotein. Proc. Natl. Acad. Sci. USA 2007, 104, 1412–1417. [Google Scholar] [CrossRef] [Green Version]
- Seefeld, M.D.; Corbett, S.W.; Keesey, R.E.; Peterson, R.E. Characterization of the wasting syndrome in rats treated with 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicol. Appl. Pharmacol. 1984, 73, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Mezrich, J.D.; Fechner, J.H.; Zhang, X.; Johnson, B.P.; Burlingham, W.J.; Bradfield, C.A. An Interaction between Kynurenine and the Aryl Hydrocarbon Receptor Can Generate Regulatory T Cells. J. Immunol. 2010, 185, 3190–3198. [Google Scholar] [CrossRef] [Green Version]
- Manzella, C.; Singhal, M.; Alrefai, W.A.; Saksena, S.; Dudeja, P.K.; Gill, R.K. Serotonin is an endogenous regulator of intesti-nal CYP1A1 via AhR. Sci. Rep. 2018, 8, 6103. [Google Scholar] [CrossRef] [Green Version]
- Shimba, S.; Todoroki, K.; Aoyagi, T.; Tezuka, M. Depletion of Arylhydrocarbon Receptor during Adipose Differentiation in 3T3-L1 Cells. Biochem. Biophys. Res. Commun. 1998, 249, 131–137. [Google Scholar] [CrossRef]
- Shimba, S.; Hayashi, M.; Ohno, T.; Tezuka, M. Transcriptional Regulation of the AhR Gene during Adipose Differentiation. Biol. Pharm. Bull. 2003, 26, 1266–1271. [Google Scholar] [CrossRef] [Green Version]
- Baker, N.A.; Karounos, M.; English, V.; Fang, J.; Wei, Y.; Stromberg, A.; Sunkara, M.; Morris, A.J.; Swanson, H.I.; Cassis, L.A. Coplanar polychlorinated biphenyls impair glu-cose homeostasis in lean C57BL/6 mice and mitigate beneficial effects of weight loss on glucose homeostasis in obese mice. Environ. Health Perspect. 2013, 121, 105–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khazaal, A.Q.; Haque, N.; Krager, C.R.; Krager, S.L.; Chambers, C.; Wilber, A.; Tischkau, S.A. Aryl hydrocarbon receptor affects circa-dian-regulated lipolysis through an E-Box-dependent mechanism. Mol. Cell. Endocrinol. 2023, 559, 111809. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.X.; Wang, C.; Zhang, Z.M.; Jaeger, C.D.; Krager, S.L.; Bottum, K.M.; Liu, J.; Liao, D.F.; Tischkau, S.A. Aryl hydrocarbon receptor deficiency pro-tects mice from diet-induced adiposity and metabolic disorders through increased energy expenditure. Int. J. Obes. (Lond.) 2015, 39, 1300–1309. [Google Scholar] [CrossRef] [Green Version]
- Kerley-Hamilton, J.S.; Trask, H.W.; Ridley, C.J.; DuFour, E.; Ringelberg, C.S.; Nurinova, N.; Wong, D.; Moodie, K.L.; Shipman, S.L.; Moore, J.H.; et al. Obesity Is Mediated by Differential Aryl Hydrocarbon Receptor Signaling in Mice Fed a Western Diet. Environ. Health Perspect. 2012, 120, 1252–1259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moyer, B.J.; Rojas, I.Y.; Kerley-Hamilton, J.S.; Hazlett, H.F.; Nemani, K.V.; Trask, H.W.; West, R.J.; Lupien, L.E.; Collins, A.J.; Ringelberg, C.S.; et al. Inhibition of the aryl hydrocar-bon receptor prevents Western diet-induced obesity. Model for AHR activation by kynurenine via oxidized-LDL, TLR2/4, TGFβ, and IDO1. Toxicol. Appl. Pharmacol. 2016, 300, 13–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wada, T.; Sunaga, H.; Miyata, K.; Shirasaki, H.; Uchiyama, Y.; Shimba, S. Aryl Hydrocarbon Receptor Plays Protective Roles against High Fat Diet (HFD)-induced Hepatic Steatosis and the Subsequent Lipotoxicity via Direct Transcriptional Regulation of Socs3 Gene Expression. J. Biol. Chem. 2016, 291, 7004–7016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girer, N.G.; Carter, D.; Bhattarai, N.; Mustafa, M.; Denner, L.; Porter, C.; Elferink, C.J. Inducible Loss of the Aryl Hydrocarbon Re-ceptor Activates Perigonadal White Fat Respiration and Brown Fat Thermogenesis via Fibroblast Growth Factor 21. Int. J. Mol. Sci. 2019, 20, 950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walisser, J.A.; Glover, E.; Pande, K.; Liss, A.L.; Bradfield, C.A. Aryl hydrocarbon receptor-dependent liver development and hepatotoxicity are mediated by different cell types. Proc. Natl. Acad. Sci. USA 2005, 102, 17858–17863. [Google Scholar] [CrossRef] [Green Version]
- Jaeger, C.; Khazaal, A.Q.; Xu, C.; Sun, M.; Krager, S.L.; Tischkau, S.A. Aryl Hydrocarbon Receptor Deficiency Alters Circa-dian and Metabolic Rhythmicity. J. Biol. Rhythms. 2017, 32, 109–120. [Google Scholar] [CrossRef] [Green Version]
- Huang, T.; Song, J.; Gao, J.; Cheng, J.; Xie, H.; Zhang, L.; Wang, Y.-H.; Gao, Z.; Wang, Y.; Wang, X.; et al. Adipocyte-derived kynurenine promotes obesity and insulin resistance by activating the AhR/STAT3/IL-6 signaling. Nat. Commun. 2022, 13, 3489. [Google Scholar] [CrossRef]
- Shi, H.; Strader, A.D.; Woods, S.C.; Seeley, R.J. Sexually dimorphic responses to fat loss after caloric restriction or surgical lipectomy. Am. J. Physiol. Metab. 2007, 293, E316–E326. [Google Scholar] [CrossRef] [Green Version]
- Palmer, B.F.; Clegg, D.J. The sexual dimorphism of obesity. Mol. Cell. Endocrinol. 2015, 402, 113–119. [Google Scholar] [CrossRef] [Green Version]
- Karastergiou, K.; Smith, S.R.; Greenberg, A.S.; Fried, S.K. Sex differences in human adipose tissues—The biology of pear shape. Biol. Sex Differ. 2012, 3, 13. [Google Scholar] [CrossRef] [Green Version]
- Kautzky-Willer, A.; Harreiter, J.; Pacini, G. Sex and Gender Differences in Risk, Pathophysiology and Complications of Type 2 Diabetes Mellitus. Endocr. Rev. 2016, 37, 278–316. [Google Scholar] [CrossRef] [Green Version]
- Ye, R.; Wang, Q.A.; Tao, C.; Vishvanath, L.; Shao, M.; McDonald, J.G.; Gupta, R.K.; Scherer, P.E. Impact of tamoxifen on adipocyte lineage trac-ing: Inducer of adipogenesis and prolonged nuclear translocation of Cre recombinase. Mol. Metab. 2015, 4, 771–778. [Google Scholar] [CrossRef]
- Moyer, B.J.; Rojas, I.Y.; Kerley-Hamilton, J.S.; Nemani, K.V.; Trask, H.W.; Ringelberg, C.S.; Gimi, B.; Demidenko, E.; Tomlinson, C.R. Obesity and fatty liver are prevented by inhibition of the aryl hydrocarbon receptor in both female and male mice. Nutr. Res. 2017, 44, 38–50. [Google Scholar] [CrossRef] [PubMed]
- Baker, N.A.; Shoemaker, R.; English, V.; Larian, N.; Sunkara, M.; Morris, A.J.; Walker, M.; Yiannikouris, F.; Cassis, L.A. Effects of Adipocyte Aryl Hydrocarbon Receptor Deficiency on PCB-Induced Disruption of Glucose Homeostasis in Lean and Obese Mice. Environ. Health Perspect. 2015, 123, 944–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gourronc, F.A.; Markan, K.R.; Kulhankova, K.; Zhu, Z.; Sheehy, R.; Quelle, D.E.; Zingman, L.V.; Kurago, Z.B.; Ankrum, J.A.; Klingelhutz, A.J. Pdgfrα-Cre mediated knockout of the aryl hydrocarbon receptor protects mice from high-fat diet induced obesity and hepatic steatosis. PLoS ONE 2020, 15, e0236741. [Google Scholar] [CrossRef] [PubMed]
- Preitner, F.; Mody, N.; Graham, T.E.; Peroni, O.D.; Kahn, B.B. Long-term Fenretinide treatment prevents high-fat di-et-induced obesity, insulin resistance, and hepatic steatosis. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E1420–E1429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- West, D.B.; Boozer, C.N.; Moody, D.L.; Atkinson, R.L. Dietary obesity in nine inbred mouse strains. Am. J. Physiol. Integr. Comp. Physiol. 1992, 262, R1025–R1032. [Google Scholar] [CrossRef]
- Jardí, F.; Laurent, M.R.; Kim, N.; Khalil, R.; De Bundel, D.; Van Eeckhaut, A.; Van Helleputte, L.; Deboel, L.; Dubois, V.; Schollaert, D.; et al. Testosterone boosts physical activity in male mice via dopaminergic pathways. Sci. Rep. 2018, 8, 957. [Google Scholar] [CrossRef] [Green Version]
- Zhao, R.-X.; He, Q.; Sha, S.; Song, J.; Qin, J.; Liu, P.; Sun, Y.-J.; Sun, L.; Hou, X.; Chen, L. Increased AHR Transcripts Correlate With Pro-inflammatory T-Helper Lymphocytes Polarization in Both Metabolically Healthy Obesity and Type 2 Diabetic Patients. Front. Immunol. 2020, 11, 1644. [Google Scholar] [CrossRef]
- Cranmer, M.; Louie, S.; Kennedy, R.H.; Kern, P.A.; Fonseca, V.A. Exposure to 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) Is Associated with Hyperinsulinemia and Insulin Resistance. Toxicol. Sci. 2000, 56, 431–436. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Zhang, L.; Allman, E.L.; Hubbard, T.D.; Murray, I.A.; Hao, F.; Tian, Y.; Gui, W.; Nichols, R.G.; Smith, P.B.; et al. The aryl hydrocarbon receptor activates ceramide biosynthesis in mice contributing to hepatic lipogenesis. Toxicology 2021, 458, 152831. [Google Scholar] [CrossRef]
- Dou, H.; Duan, Y.; Zhang, X.; Yu, Q.; Di, Q.; Song, Y.; Li, P.; Gong, Y. Aryl hydrocarbon receptor (AhR) regulates adipocyte differentiation by assembling CRL4B ubiquitin ligase to target PPARγ for proteasomal degradation. J. Biol. Chem. 2019, 294, 18504–18515. [Google Scholar] [CrossRef]
- Matthews, J.; Gustafsson, J.-A. Estrogen receptor and aryl hydrocarbon receptor signaling pathways. Nucl. Recept. Signal. 2006, 4, e016. [Google Scholar] [CrossRef]
- Heine, P.A.; Taylor, J.A.; Iwamoto, G.A.; Lubahn, D.B.; Cooke, P.S. Increased adipose tissue in male and female estrogen re-ceptor-alpha knockout mice. Proc. Natl. Acad. Sci. USA 2000, 97, 12729–12734. [Google Scholar] [CrossRef] [Green Version]
- Pedersen, S.B.; Børglum, J.D.; Møller-Pedersen, T.; Richelsen, B. Effects of in vivo estrogen treatment on adipose tissue metab-olism and nuclear estrogen receptor binding in isolated rat adipocytes. Mol. Cell. Endocrinol. 1992, 85, 13–19. [Google Scholar] [CrossRef]
- Arsenescu, V.; Arsenescu, R.I.; King, V.; Swanson, H.; Cassis, L.A. Polychlorinated biphenyl-77 induces adipocyte differentia-tion and proinflammatory adipokines and promotes obesity and atherosclerosis. Environ. Health Perspect. 2008, 116, 761–768. [Google Scholar] [CrossRef] [Green Version]
- Nishiumi, S.; Yoshida, M.; Azuma, T.; Yoshida, K.-I.; Ashida, H. 2,3,7,8-Tetrachlorodibenzo-p-Dioxin Impairs an Insulin Signaling Pathway through the Induction of Tumor Necrosis Factor-α in Adipocytes. Toxicol. Sci. 2010, 115, 482–491. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.J.; Pelloux, V.; Guyot, E.; Tordjman, J.; Bui, L.-C.; Chevallier, A.; Forest, C.; Benelli, C.; Clément, K.; Barouki, R. Inflammatory Pathway Genes Belong to Major Targets of Persistent Organic Pollutants in Adipose Cells. Environ. Health Perspect. 2012, 120, 508–514. [Google Scholar] [CrossRef] [Green Version]
- Martins, A.B.; Brownlow, M.L.; Araújo, B.B.; Garnica-Siqueira, M.C.; Zaia, D.A.M.; Leite, C.M.; Zaia, C.T.B.V.; Uchoa, E.T. Arcuate nucleus of the hypothalamus contributes to the hypophagic effect and plasma metabolic changes induced by vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide. Neurochem. Int. 2022, 155, 105300. [Google Scholar] [CrossRef]
- Butera, P.C.; Beikirch, R.J. Central implants of diluted estradiol: Independent effects on ingestive and reproductive behaviors of ovariectomized rats. Brain Res. 1989, 491, 266–273. [Google Scholar] [CrossRef]
- Palmer, K.; Gray, J.M. Central vs. peripheral effects of estrogen on food intake and lipoprotein lipase activity in ovariecto-mized rats. Physiol. Behav. 1986, 37, 187–189. [Google Scholar] [CrossRef]
- Xu, Y.; Nedungadi, T.P.; Zhu, L.; Sobhani, N.; Irani, B.G.; Davis, K.E.; Zhang, X.; Zou, F.; Gent, L.M.; Hahner, L.D.; et al. Distinct Hypothalamic Neurons Mediate Estrogenic Effects on Energy Homeostasis and Reproduction. Cell Metab. 2011, 14, 453–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Civitarese, A.E.; Ukropcova, B.; Carling, S.; Hulver, M.; DeFronzo, R.A.; Mandarino, L.; Ravussin, E.; Smith, S.R. Role of adiponectin in human skeletal muscle bioenergetics. Cell Metab. 2006, 4, 75–87. [Google Scholar] [CrossRef] [Green Version]
- Yoon, M.J.; Lee, G.Y.; Chung, J.J.; Ahn, Y.H.; Hong, S.H.; Kim, J.B. Adiponectin increases fatty acid oxidation in skeletal mus-cle cells by sequential activation of AMP-activated protein kinase, p38 mitogen-activated protein kinase, and peroxisome pro-liferator-activated receptor alpha. Diabetes 2006, 55, 2562–2570. [Google Scholar] [CrossRef] [Green Version]
- Yamauchi, T.; Kamon, J.; Minokoshi, Y.; Ito, Y.; Waki, H.; Uchida, S.; Yamashita, S.; Noda, M.; Kita, S.; Ueki, K.; et al. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat. Med. 2002, 8, 1288–1295. [Google Scholar] [CrossRef] [PubMed]
- Clegg, D.J.; Riedy, C.A.; Smith, K.A.B.; Benoit, S.C.; Woods, S.C. Differential Sensitivity to Central Leptin and Insulin in Male and Female Rats. Diabetes 2003, 52, 682–687. [Google Scholar] [CrossRef] [Green Version]
- Rosenbaum, M.; Nicolson, M.; Hirsch, J.; Heymsfield, S.B.; Gallagher, D.; Chu, F.; Leibel, R.L. Effects of gender, body composition, and menopause on plasma concentrations of leptin. J. Clin. Endocrinol. Metab. 1996, 81, 3424–3427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saad, M.F.; Damani, S.; Gingerich, R.L.; Riad-Gabriel, M.G.; Khan, A.; Boyadjian, R.; Jinagouda, S.D.; El-Tawil, K.; Rude, R.K.; Kamdar, V. Sexual Dimorphism in Plasma Leptin Concentration1. J. Clin. Endocrinol. Metab. 1997, 82, 579–584. [Google Scholar] [CrossRef] [Green Version]
- Fried, S.K.; Lee, M.-J.; Karastergiou, K. Shaping fat distribution: New insights into the molecular determinants of depot- and sex-dependent adipose biology. Obesity 2015, 23, 1345–1352. [Google Scholar] [CrossRef] [Green Version]
- Fujii, H.; Kawada, N.; Japan Study Group of NAFLD (JSG-NAFLD). The Role of Insulin Resistance and Diabetes in Nonalcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2020, 21, 3863. [Google Scholar] [CrossRef]
- Lee, Y.; Wang, M.Y.; Kakuma, T.; Wang, Z.W.; Babcock, E.; McCorkle, K.; Higa, M.; Zhou, Y.T.; Unger, R.H. Liporegulation in diet-induced obesity. The an-tisteatotic role of hyperleptinemia. J. Biol. Chem. 2001, 276, 5629–5635. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Hatzakis, E.; Nichols, R.G.; Hao, R.; Correll, J.; Smith, P.B.; Chiaro, C.R.; Perdew, G.H.; Patterson, A.D. Metabolomics Reveals that Aryl Hydrocarbon Re-ceptor Activation by Environmental Chemicals Induces Systemic Metabolic Dysfunction in Mice. Environ. Sci. Technol. 2015, 49, 8067–8077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gatselis, N.K.; Ntaios, G.; Makaritsis, K.; Dalekos, G.N. Adiponectin: A key playmaker adipocytokine in non-alcoholic fatty liver disease. Clin. Exp. Med. 2013, 14, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Wada, T.; Febbraio, M.; He, J.; Matsubara, T.; Lee, M.J.; Gonzalez, F.J.; Xie, W. A novel role for the dioxin receptor in fatty acid me-tabolism and hepatic steatosis. Gastroenterology 2010, 139, 653–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NCD | HFD | |||||||
|---|---|---|---|---|---|---|---|---|
| CadKO Male | WT Male | CadKO Female | WT Female | CadKO Male | WT Male | CadKO Female | WT Female | |
| Insulin (ng/mL) | 0.34 ± 0.22 # | 0.81 ± 0.30 # | 0.46 ± 0.34 | 0.63 ± 0.34 | 1.59 ± 0.46 $ | 6.46 ± 5.14 $ | 0.49 ± 0.34 | 0.88 ± 0.54 |
| Leptin (ng/mL) | 7.76 ± 2.64 # | 5.27 ± 3.88 # | 4.77 ± 2.57 # | 5.56 ± 1.01 # | 40.40 ± 2.76 $ | 43.47 ± 2.40 | 28.81 ± 11.19 | 38.67 ± 3.19 |
| Adiponectin (mg/mL) | 48.52 ± 10.38 | 35.85 ± 6.25 $,# | 75.75 ± 18.9 | 71.52 ± 31 # | 71.42 ± 9.64 | 73.90 ± 7.96 # | 78.15 ± 24.22 | 106.85 ± 26.6 |
| 17β-estradiol (pg/mL) | 155.10 ± 1.99 | 155.73 ± 12.4 $,# | 161.39 ± 13.4 | 178.8 ± 16.63 | 144.76 ± 24.92 | 125.74 ± 13 $ | 161.48 ± 11.5 | 160.13 ± 6.57 |
| Triglycerides (mg/dL) | n/a | n/a | n/a | n/a | 195.88 ± 26 $ | 181.74 ± 6.23 | 155.33 ± 17.78 | 173.20 ± 5.68 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haque, N.; Ojo, E.S.; Krager, S.L.; Tischkau, S.A. Deficiency of Adipose Aryl Hydrocarbon Receptor Protects against Diet-Induced Metabolic Dysfunction through Sexually Dimorphic Mechanisms. Cells 2023, 12, 1748. https://doi.org/10.3390/cells12131748
Haque N, Ojo ES, Krager SL, Tischkau SA. Deficiency of Adipose Aryl Hydrocarbon Receptor Protects against Diet-Induced Metabolic Dysfunction through Sexually Dimorphic Mechanisms. Cells. 2023; 12(13):1748. https://doi.org/10.3390/cells12131748
Chicago/Turabian StyleHaque, Nazmul, Emmanuel S. Ojo, Stacey L. Krager, and Shelley A. Tischkau. 2023. "Deficiency of Adipose Aryl Hydrocarbon Receptor Protects against Diet-Induced Metabolic Dysfunction through Sexually Dimorphic Mechanisms" Cells 12, no. 13: 1748. https://doi.org/10.3390/cells12131748