

Starvation Protects Hepatocytes from Inflammatory Damage through Paradoxical mTORC1 Signaling

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Antibodies
2.3. Cell Culture, Cell Stimulations and Treatments
2.4. Cell Death Assay and Quantification
2.5. Real-Time Glo Necrosis/Apoptosis Assay
2.6. siRNA Transfections
2.7. Western Blot and Immune Detection
2.8. Protein Synthesis Determination
2.9. Immunocytochemistry
2.10. Statistical Analysis
3. Results
3.1. Starvation Protects Immortalized Hepatocytes against Pro-Inflammatory Cytokine/PAMP-Induced Cell Damage
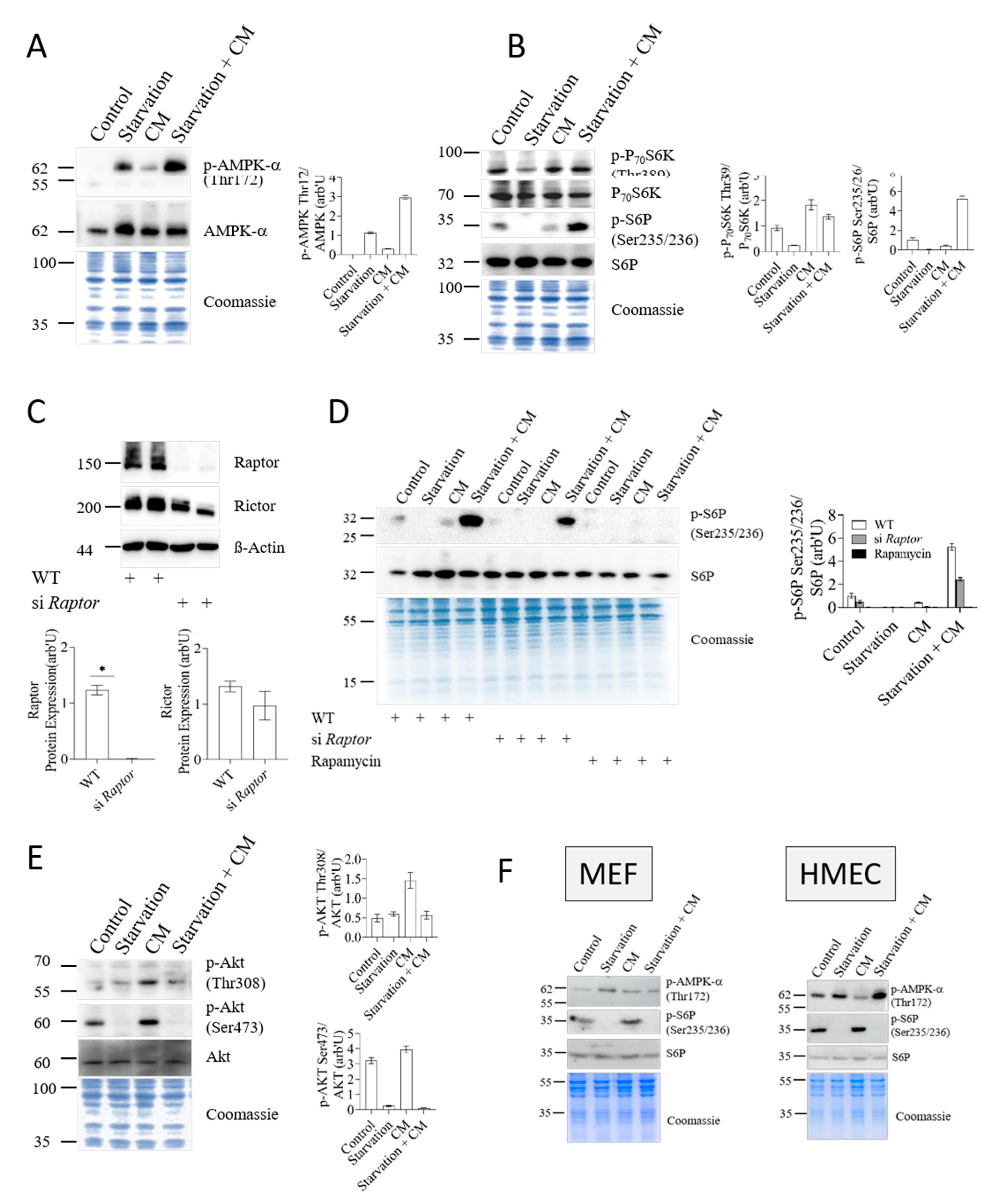
3.2. Nutrient- and Energy-Sensing Pathways Respond to Starvation in Hepatocytes
3.3. AMPK and mTORC1, but Not mTORC2, Mediate Starvation-Induced Protection against Inflammatory Stress
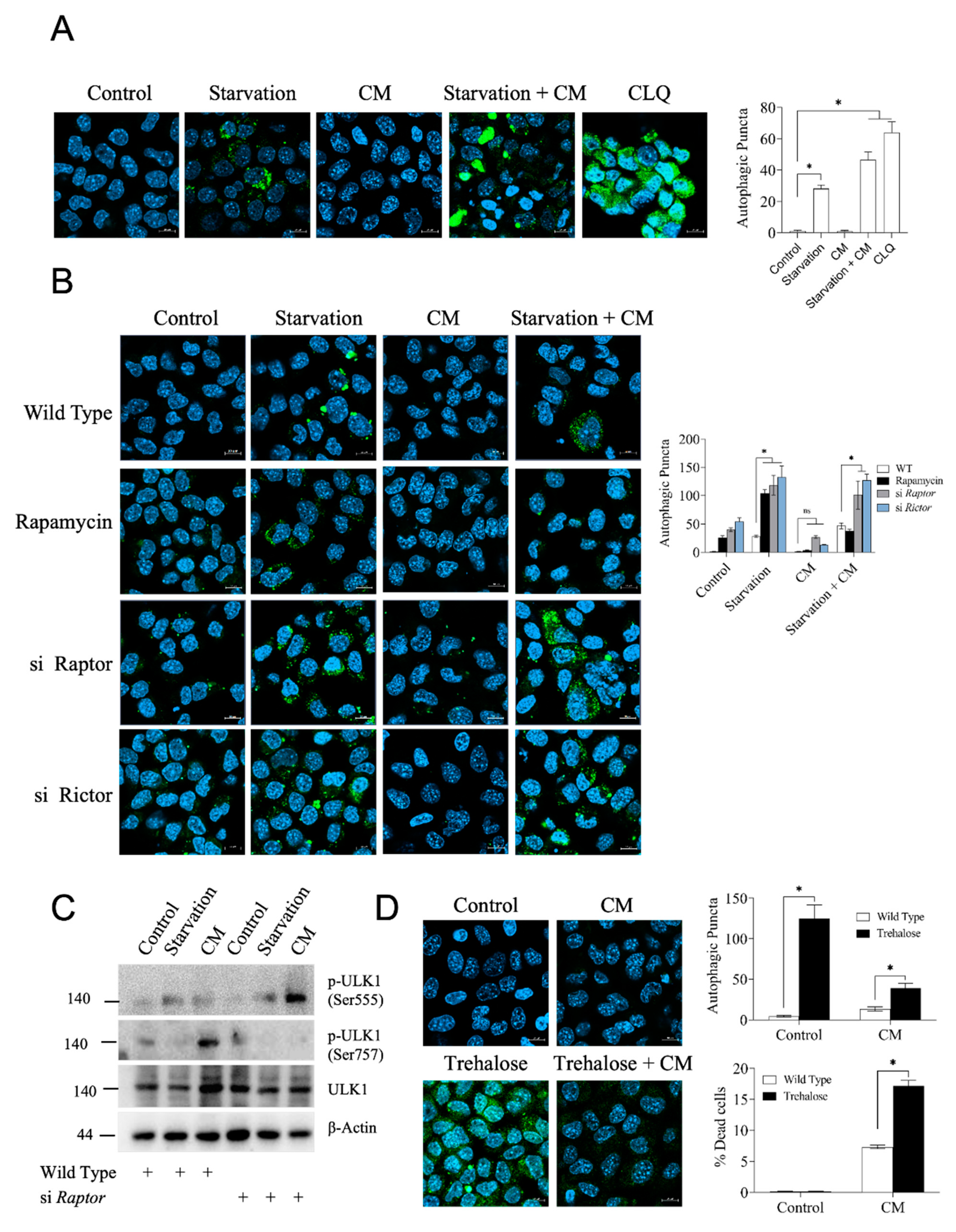
3.4. High Autophagy Rate Does Not Protect Hepatocytes from Inflammatory Damage
3.5. Protein Synthesis Rate Correlates with Cell Viability and Is Strongly Dependent on mTORC1
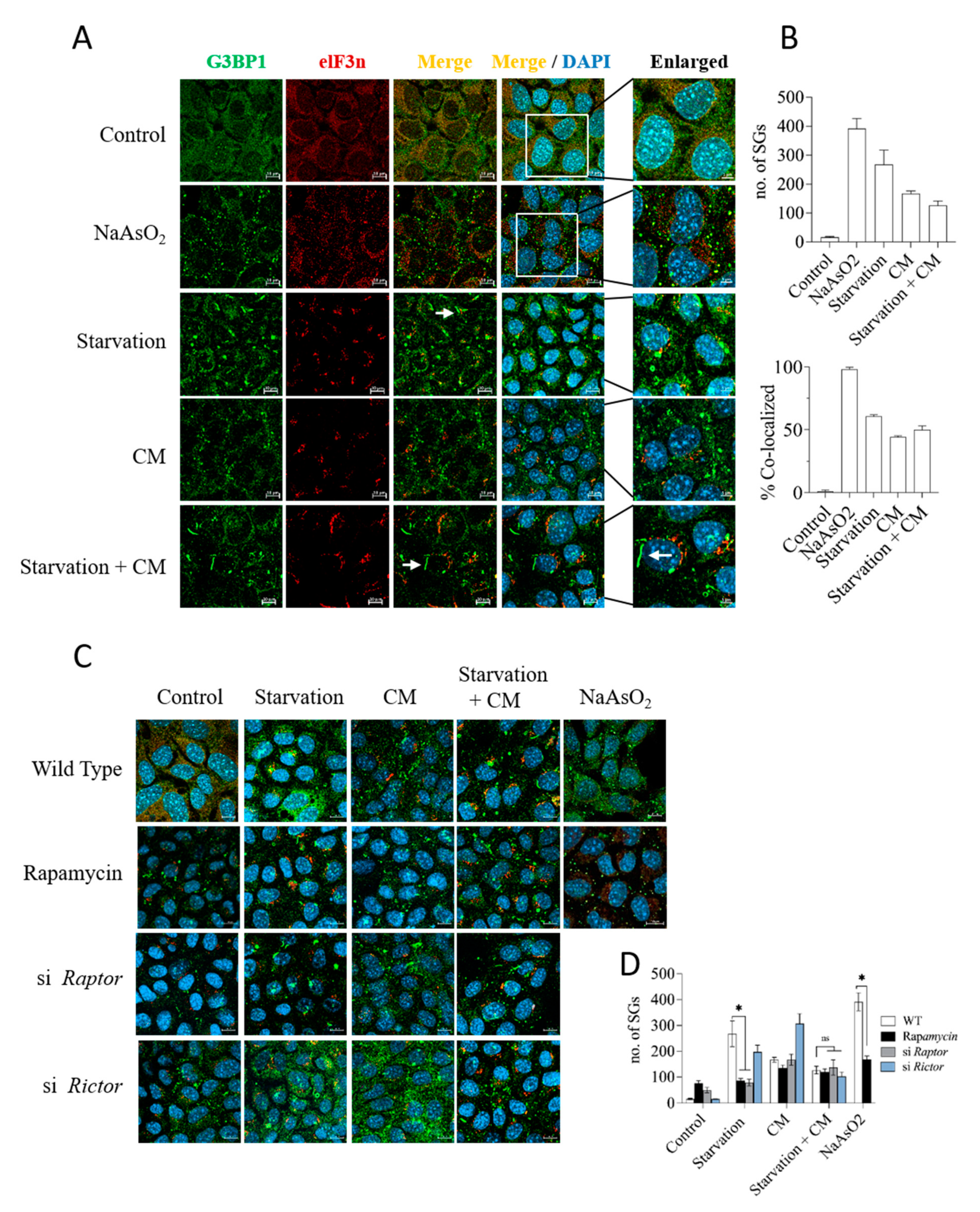
3.6. Starvation Induces Stress Granule Formation in Dependency on mTORC1, but Stress Granules Do Not Accumulate in Protected Cells
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.D.; Coopersmith, C.M.; et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 2016, 315, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Hotchkiss, R.S.; Monneret, G.; Payen, D. Sepsis-induced immunosuppression: From cellular dysfunctions to immunotherapy. Nat. Rev. Immunol. 2013, 13, 862–874. [Google Scholar] [PubMed] [Green Version]
- Rubio, I.; Osuchowski, M.F.; Shankar-Hari, M.; Skirecki, T.; Winkler, M.S.; Lachmann, G.; La Rosee, P.; Monneret, G.; Venet, F.; Bauer, M.; et al. Current gaps in sepsis immunology: New opportunities for translational research. Lancet Infect. Dis. 2019, 19, e422–e436. [Google Scholar] [CrossRef]
- Sun, J.; Zhang, J.; Wang, X.; Ji, F.; Ronco, C.; Tian, J.; Yin, Y. Gut-liver crosstalk in sepsis-induced liver injury. Crit. Care 2020, 24, 614. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, H.; Hashimoto, K.; Yuan, S.; Zhang, J. The gut-liver axis in sepsis: Interaction mechanisms and therapeutic potential. Crit. Care 2022, 26, 213. [Google Scholar] [CrossRef]
- Strnad, P.; Tacke, F.; Koch, A.; Trautwein, C. Liver—Guardian, modifier and target of sepsis. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 55–66. [Google Scholar] [CrossRef]
- Weis, S.; Carlos, A.R.; Moita, M.R.; Singh, S.; Blankenhaus, B.; Cardoso, S.; Larsen, R.; Rebelo, S.; Schauble, S.; Del Barrio, L.; et al. Metabolic Adaptation Establishes Disease Tolerance to Sepsis. Cell 2017, 169, 1263–1275.e14. [Google Scholar] [CrossRef] [Green Version]
- Wang, A.; Huen, S.C.; Luan, H.H.; Yu, S.; Zhang, C.; Gallezot, J.D.; Booth, C.J.; Medzhitov, R. Opposing Effects of Fasting Metabolism on Tissue Tolerance in Bacterial and Viral Inflammation. Cell 2016, 166, 1512–1525.e12. [Google Scholar] [CrossRef] [Green Version]
- Grau, T.; Bonet, A.; Rubio, M.; Mateo, D.; Farre, M.; Acosta, J.A.; Blesa, A.; Montejo, J.C.; de Lorenzo, A.G.; Mesejo, A.; et al. Liver dysfunction associated with artificial nutrition in critically ill patients. Crit. Care 2007, 11, R10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davidson, M.D.; Khetani, S.R. Intermittent Starvation Extends the Functional Lifetime of Primary Human Hepatocyte Cultures. Toxicol. Sci. 2020, 174, 266–277. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, G.R.; Carling, D. AMP-activated protein kinase: The current landscape for drug development. Nat. Rev. Drug. Discov. 2019, 18, 527–551. [Google Scholar] [CrossRef] [PubMed]
- Viollet, B.; Guigas, B.; Leclerc, J.; Hebrard, S.; Lantier, L.; Mounier, R.; Andreelli, F.; Foretz, M. AMP-activated protein kinase in the regulation of hepatic energy metabolism: From physiology to therapeutic perspectives. Acta Physiol. 2009, 196, 81–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardie, D.G. Sensing of energy and nutrients by AMP-activated protein kinase. Am. J. Clin. Nutr. 2011, 93, 891S–896S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angin, Y.; Beauloye, C.; Horman, S.; Bertrand, L. Regulation of Carbohydrate Metabolism, Lipid Metabolism, and Protein Metabolism by AMPK. Exp. Suppl. 2016, 107, 23–43. [Google Scholar] [PubMed]
- Kikuchi, S.; Piraino, G.; O’Connor, M.; Wolfe, V.; Ridings, K.; Lahni, P.; Zingarelli, B. Hepatocyte-Specific Deletion of AMPKalpha1 Results in Worse Outcomes in Mice Subjected to Sepsis in a Sex-Specific Manner. Front. Immunol. 2020, 11, 210. [Google Scholar] [CrossRef]
- Inata, Y.; Kikuchi, S.; Samraj, R.S.; Hake, P.W.; O’Connor, M.; Ledford, J.R.; O’Connor, J.; Lahni, P.; Wolfe, V.; Piraino, G.; et al. Autophagy and mitochondrial biogenesis impairment contribute to age-dependent liver injury in experimental sepsis: Dysregulation of AMP-activated protein kinase pathway. FASEB J. 2018, 32, 728–741. [Google Scholar] [CrossRef] [Green Version]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [Green Version]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Zhang, G.; Xu, X.; Su, W.; Yu, B. mTOR-rictor is the Ser473 kinase for AKT1 in mouse one-cell stage embryos. Mol. Cell. Biochem. 2012, 361, 249–257. [Google Scholar] [CrossRef]
- Byrnes, K.; Blessinger, S.; Bailey, N.T.; Scaife, R.; Liu, G.; Khambu, B. Therapeutic regulation of autophagy in hepatic metabolism. Acta Pharm. Sin. B 2022, 12, 33–49. [Google Scholar] [CrossRef]
- Byles, V.; Cormerais, Y.; Kalafut, K.; Barrera, V.; Hughes Hallett, J.E.; Sui, S.H.; Asara, J.M.; Adams, C.M.; Hoxhaj, G.; Ben-Sahra, I.; et al. Hepatic mTORC1 signaling activates ATF4 as part of its metabolic response to feeding and insulin. Mol. Metab. 2021, 53, 101309. [Google Scholar] [CrossRef] [PubMed]
- Hindupur, S.K.; Gonzalez, A.; Hall, M.N. The opposing actions of target of rapamycin and AMP-activated protein kinase in cell growth control. Cold Spring Harb. Perspect. Biol. 2015, 7, a019141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tee, A.R.; Manning, B.D.; Roux, P.P.; Cantley, L.C.; Blenis, J. Tuberous sclerosis complex gene products, Tuberin and Hamartin, control mTOR signaling by acting as a GTPase-activating protein complex toward Rheb. Curr. Biol. 2003, 13, 1259–1268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Manning, B.D. The TSC1-TSC2 complex: A molecular switchboard controlling cell growth. Biochem. J. 2008, 412, 179–190. [Google Scholar] [CrossRef] [Green Version]
- Corradetti, M.N.; Inoki, K.; Bardeesy, N.; DePinho, R.A.; Guan, K.L. Regulation of the TSC pathway by LKB1: Evidence of a molecular link between tuberous sclerosis complex and Peutz-Jeghers syndrome. Genes. Dev. 2004, 18, 1533–1538. [Google Scholar] [CrossRef] [Green Version]
- Marquardt, J.U.; Edlich, F. Predisposition to Apoptosis in Hepatocellular Carcinoma: From Mechanistic Insights to Therapeutic Strategies. Front. Oncol. 2019, 9, 1421. [Google Scholar] [CrossRef] [Green Version]
- Fabregat, I. Dysregulation of apoptosis in hepatocellular carcinoma cells. World J. Gastroenterol. 2009, 15, 513–520. [Google Scholar] [CrossRef]
- Lai, J.H.; Tan, T.H. CD28 signaling causes a sustained down-regulation of I kappa B alpha which can be prevented by the immunosuppressant rapamycin. J. Biol. Chem. 1994, 269, 30077–30080. [Google Scholar] [CrossRef] [PubMed]
- Jundt, F.; Raetzel, N.; Muller, C.; Calkhoven, C.F.; Kley, K.; Mathas, S.; Lietz, A.; Leutz, A.; Dorken, B. A rapamycin derivative (everolimus) controls proliferation through down-regulation of truncated CCAAT enhancer binding protein {beta} and NF-{kappa}B activity in Hodgkin and anaplastic large cell lymphomas. Blood 2005, 106, 1801–1807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reiter, A.K.; Bolster, D.R.; Crozier, S.J.; Kimball, S.R.; Jefferson, L.S. Repression of protein synthesis and mTOR signaling in rat liver mediated by the AMPK activator aminoimidazole carboxamide ribonucleoside. Am. J. Physiol. Endocrinol. Metab. 2005, 288, E980–E988. [Google Scholar] [CrossRef]
- Cheung, P.C.; Salt, I.P.; Davies, S.P.; Hardie, D.G.; Carling, D. Characterization of AMP-activated protein kinase gamma-subunit isoforms and their role in AMP binding. Biochem. J. 2000, 346 Pt 3, 659–669. [Google Scholar] [CrossRef]
- Figueiredo, N.; Chora, A.; Raquel, H.; Pejanovic, N.; Pereira, P.; Hartleben, B.; Neves-Costa, A.; Moita, C.; Pedroso, D.; Pinto, A.; et al. Anthracyclines induce DNA damage response-mediated protection against severe sepsis. Immunity 2013, 39, 874–884. [Google Scholar] [CrossRef] [Green Version]
- Hofius, D.; Munch, D.; Bressendorff, S.; Mundy, J.; Petersen, M. Role of autophagy in disease resistance and hypersensitive response-associated cell death. Cell Death Differ. 2011, 18, 1257–1262. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.C.; Guan, K.L. mTOR: A pharmacologic target for autophagy regulation. J. Clin. Investig. 2015, 125, 25–32. [Google Scholar] [CrossRef] [Green Version]
- Nazio, F.; Strappazzon, F.; Antonioli, M.; Bielli, P.; Cianfanelli, V.; Bordi, M.; Gretzmeier, C.; Dengjel, J.; Piacentini, M.; Fimia, G.M.; et al. mTOR inhibits autophagy by controlling ULK1 ubiquitylation, self-association and function through AMBRA1 and TRAF6. Nat. Cell Biol. 2013, 15, 406–416. [Google Scholar] [CrossRef] [PubMed]
- Dunlop, E.A.; Tee, A.R. The kinase triad, AMPK, mTORC1 and ULK1, maintains energy and nutrient homoeostasis. Biochem. Soc. Trans. 2013, 41, 939–943. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.H.; Jun, C.B.; Ro, S.H.; Kim, Y.M.; Otto, N.M.; Cao, J.; Kundu, M.; Kim, D.H. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol. Biol. Cell 2009, 20, 1992–2003. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, S.; Davies, J.E.; Huang, Z.; Tunnacliffe, A.; Rubinsztein, D.C. Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and alpha-synuclein. J. Biol. Chem. 2007, 282, 5641–5652. [Google Scholar] [CrossRef] [Green Version]
- Holz, M.K.; Ballif, B.A.; Gygi, S.P.; Blenis, J. mTOR and S6K1 mediate assembly of the translation preinitiation complex through dynamic protein interchange and ordered phosphorylation events. Cell 2005, 123, 569–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, X.; Lin, Y.; Ortiz-Vega, S.; Yonezawa, K.; Avruch, J. Rheb binds and regulates the mTOR kinase. Curr. Biol. 2005, 15, 702–713. [Google Scholar] [CrossRef] [Green Version]
- Wek, R.C.; Jiang, H.Y.; Anthony, T.G. Coping with stress: eIF2 kinases and translational control. Biochem. Soc. Trans. 2006, 34 Pt 1, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Holcik, M.; Sonenberg, N. Translational control in stress and apoptosis. Nat. Rev. Mol. Cell Biol. 2005, 6, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Gallinetti, J.; Harputlugil, E.; Mitchell, J.R. Amino acid sensing in dietary-restriction-mediated longevity: Roles of signal-transducing kinases GCN2 and TOR. Biochem. J. 2013, 449, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Li, Z.; Xu, R.; Zhang, J.; Zhou, Q.; Gao, R.; Lu, H.; Lan, Y.; Zhao, K.; He, H.; et al. The PERK/PKR-eIF2alpha Pathway Negatively Regulates Porcine Hemagglutinating Encephalomyelitis Virus Replication by Attenuating Global Protein Translation and Facilitating Stress Granule Formation. J. Virol. 2022, 96, e0169521. [Google Scholar] [CrossRef]
- Onomoto, K.; Yoneyama, M.; Fung, G.; Kato, H.; Fujita, T. Antiviral innate immunity and stress granule responses. Trends Immunol. 2014, 35, 420–428. [Google Scholar] [CrossRef]
- Protter, D.S.W.; Parker, R. Principles and Properties of Stress Granules. Trends Cell Biol. 2016, 26, 668–679. [Google Scholar] [CrossRef] [Green Version]
- Sfakianos, A.P.; Mellor, L.E.; Pang, Y.F.; Kritsiligkou, P.; Needs, H.; Abou-Hamdan, H.; Desaubry, L.; Poulin, G.B.; Ashe, M.P.; Whitmarsh, A.J. The mTOR-S6 kinase pathway promotes stress granule assembly. Cell Death Differ. 2018, 25, 1766–1780. [Google Scholar] [CrossRef] [Green Version]
- Thedieck, K.; Holzwarth, B.; Prentzell, M.T.; Boehlke, C.; Klasener, K.; Ruf, S.; Sonntag, A.G.; Maerz, L.; Grellscheid, S.N.; Kremmer, E.; et al. Inhibition of mTORC1 by astrin and stress granules prevents apoptosis in cancer cells. Cell 2013, 154, 859–874. [Google Scholar] [CrossRef] [Green Version]
- Wippich, F.; Bodenmiller, B.; Trajkovska, M.G.; Wanka, S.; Aebersold, R.; Pelkmans, L. Dual specificity kinase DYRK3 couples stress granule condensation/dissolution to mTORC1 signaling. Cell 2013, 152, 791–805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, Y.; Jin, J.; Li, J.; Ye, M.; Jin, X. The roles of G3BP1 in human diseases (review). Gene 2022, 821, 146294. [Google Scholar] [CrossRef] [PubMed]
- Yao, R.Q.; Ren, C.; Xia, Z.F.; Yao, Y.M. Organelle-specific autophagy in inflammatory diseases: A potential therapeutic target underlying the quality control of multiple organelles. Autophagy 2021, 17, 385–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medzhitov, R.; Schneider, D.S.; Soares, M.P. Disease tolerance as a defense strategy. Science 2012, 335, 936–941. [Google Scholar] [CrossRef] [Green Version]
- Sun, D.; Muthukumar, A.R.; Lawrence, R.A.; Fernandes, G. Effects of calorie restriction on polymicrobial peritonitis induced by cecum ligation and puncture in young C57BL/6 mice. Clin. Diagn. Lab. Immunol. 2001, 8, 1003–1011. [Google Scholar] [CrossRef] [Green Version]
- Hasegawa, A.; Iwasaka, H.; Hagiwara, S.; Asai, N.; Nishida, T.; Noguchi, T. Alternate day calorie restriction improves systemic inflammation in a mouse model of sepsis induced by cecal ligation and puncture. J. Surg. Res. 2012, 174, 136–141. [Google Scholar] [CrossRef]
- Liu, L.J.; Xu, M.; Zhu, J.; Li, N.; Zhao, X.Z.; Gao, H.M. Adiponectin alleviates liver injury in sepsis rats through AMPK/MTOR pathway. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 10745–10752. [Google Scholar]
- Li, Z.; Zhang, J.; Mulholland, M.; Zhang, W. mTOR activation protects liver from ischemia/reperfusion-induced injury through NF-kappaB pathway. FASEB J. 2017, 31, 3018–3026. [Google Scholar] [CrossRef]
- Aki, T.; Unuma, K.; Uemura, K. Emerging roles of mitochondria and autophagy in liver injury during sepsis. Cell Stress 2017, 1, 79–89. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.C.; Wang, J.L.; Ren, H.Z.; Shi, X.L. Autophagy plays a double-edged sword role in liver diseases. J. Physiol. Biochem. 2022, 78, 9–17. [Google Scholar] [CrossRef]
- Crowell, K.T.; Soybel, D.I.; Lang, C.H. Restorative Mechanisms Regulating Protein Balance in Skeletal Muscle During Recovery From Sepsis. Shock 2017, 47, 463–473. [Google Scholar] [CrossRef] [Green Version]
- Abraham, E.; Singer, M. Mechanisms of sepsis-induced organ dysfunction. Crit. Care Med. 2007, 35, 2408–2416. [Google Scholar] [CrossRef]
- Yin, X.M.; Ding, W.X.; Gao, W. Autophagy in the liver. Hepatology 2008, 47, 1773–1785. [Google Scholar] [CrossRef]
- Deter, R.L.; De Duve, C. Influence of glucagon, an inducer of cellular autophagy, on some physical properties of rat liver lysosomes. J. Cell Biol. 1967, 33, 437–449. [Google Scholar] [CrossRef] [Green Version]
- Ueno, T.; Komatsu, M. Autophagy in the liver: Functions in health and disease. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 170–184. [Google Scholar] [CrossRef] [PubMed]
- Meldau, S.; Erb, M.; Baldwin, I.T. Defence on demand: Mechanisms behind optimal defence patterns. Ann. Bot. 2012, 110, 1503–1514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Advani, V.M.; Ivanov, P. Stress granule subtypes: An emerging link to neurodegeneration. Cell. Mol. Life Sci. 2020, 77, 4827–4845. [Google Scholar] [CrossRef]
- Lavalee, M.; Curdy, N.; Laurent, C.; Fournie, J.J.; Franchini, D.M. Cancer cell adaptability: Turning ribonucleoprotein granules into targets. Trends Cancer 2021, 7, 902–915. [Google Scholar] [CrossRef]
- McCormick, C.; Khaperskyy, D.A. Translation inhibition and stress granules in the antiviral immune response. Nat. Rev. Immunol. 2017, 17, 647–660. [Google Scholar] [CrossRef] [PubMed]
- Biskup, C.; Rubio, I. Real-time visualization and quantification of native Ras activation in single living cells. Methods Mol. Biol. 2014, 1120, 285–305. [Google Scholar]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hussain, I.; Sureshkumar, H.K.; Bauer, M.; Rubio, I. Starvation Protects Hepatocytes from Inflammatory Damage through Paradoxical mTORC1 Signaling. Cells 2023, 12, 1668. https://doi.org/10.3390/cells12121668
Hussain I, Sureshkumar HK, Bauer M, Rubio I. Starvation Protects Hepatocytes from Inflammatory Damage through Paradoxical mTORC1 Signaling. Cells. 2023; 12(12):1668. https://doi.org/10.3390/cells12121668
Chicago/Turabian StyleHussain, Iqra, Harini K. Sureshkumar, Michael Bauer, and Ignacio Rubio. 2023. "Starvation Protects Hepatocytes from Inflammatory Damage through Paradoxical mTORC1 Signaling" Cells 12, no. 12: 1668. https://doi.org/10.3390/cells12121668