
Tumor Microenvironment Role in Pancreatic Cancer Stem Cells
 , , and
, , and 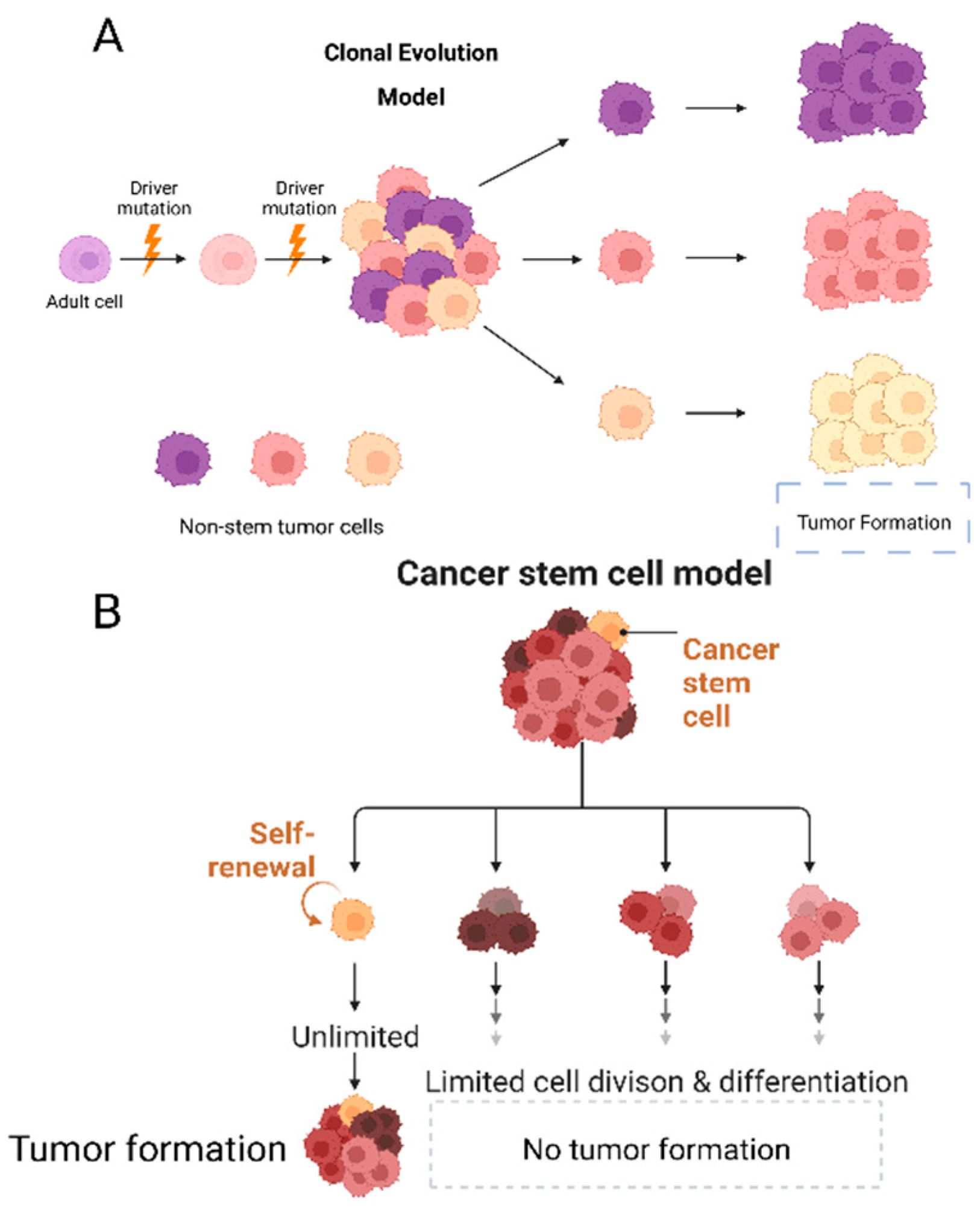{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Clonal Evolution of Tumoral Populations
3. Cancer Stem Cells (CSCs)
4. Pancreatic Cancer Stem Cells
5. Pancreatic Cancer Stem Cells Origin
6. Tumor Microenvironment (TME)
6.1. Hypoxic Niche
6.2. Acidic Niche
6.3. Mechanical Microenvironment
6.4. Innervated Niche
6.5. Metabolic Microenvironment
6.6. Exosomes
7. TME in PDAC
Exosomes in PDAC TME
8. Therapeutic Approaches against PDAC
9. Looking for a Better CSC Characterization and Isolating System
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Barman, S.; Fatima, I.; Singh, A.B.; Dhawan, P. Pancreatic Cancer and Therapy: Role and Regulation of Cancer Stem Cells. Int. J. Mol. Sci. 2021, 22, 4765. [Google Scholar] [CrossRef]
- Hermann, P.C.; Huber, S.L.; Herrler, T.; Aicher, A.; Ellwart, J.W.; Guba, M.; Bruns, C.J.; Heeschen, C. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell 2007, 1, 313–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [Green Version]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef]
- Balachandran, V.P.; Beatty, G.L.; Dougan, S.K. Broadening the Impact of Immunotherapy to Pancreatic Cancer: Challenges and Opportunities. Gastroenterology 2019, 156, 2056–2072. [Google Scholar] [CrossRef]
- Kleeff, J.; Korc, M.; Apte, M.; La Vecchia, C.; Johnson, C.D.; Biankin, A.V.; Neale, R.E.; Tempero, M.; Tuveson, D.A.; Hruban, R.H.; et al. Pancreatic cancer. Nat. Rev. Dis. Prim. 2016, 2, 16022. [Google Scholar] [CrossRef]
- He, J.; Ahuja, N.; Makary, M.A.; Cameron, J.L.; Eckhauser, F.E.; Choti, M.A.; Hruban, R.H.; Pawlik, T.M.; Wolfgang, C.L. 2564 resected periampullary adenocarcinomas at a single institution: Trends over three decades. HPB 2014, 16, 83–90. [Google Scholar] [CrossRef] [Green Version]
- Muraro, M.J.; Dharmadhikari, G.; Grün, D.; Groen, N.; Dielen, T.; Jansen, E.; van Gurp, L.; Engelse, M.A.; Carlotti, F.; de Koning, E.J.; et al. A Single-Cell Transcriptome Atlas of the Human Pancreas. Cell Syst. 2016, 3, 385–394.e383. [Google Scholar] [CrossRef] [Green Version]
- Kopp, J.L.; von Figura, G.; Mayes, E.; Liu, F.F.; Dubois, C.L.; Morris, J.P.; Pan, F.C.; Akiyama, H.; Wright, C.V.; Jensen, K.; et al. Identification of Sox9-dependent acinar-to-ductal reprogramming as the principal mechanism for initiation of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 22, 737–750. [Google Scholar] [CrossRef] [Green Version]
- Gidekel Friedlander, S.Y.; Chu, G.C.; Snyder, E.L.; Girnius, N.; Dibelius, G.; Crowley, D.; Vasile, E.; DePinho, R.A.; Jacks, T. Context-dependent transformation of adult pancreatic cells by oncogenic K-Ras. Cancer Cell 2009, 16, 379–389. [Google Scholar] [CrossRef] [Green Version]
- Nowell, P.C. The clonal evolution of tumor cell populations. Science 1976, 194, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Clara, J.A.; Monge, C.; Yang, Y.; Takebe, N. Targeting signalling pathways and the immune microenvironment of cancer stem cells-a clinical update. Nat. Rev. Clin. Oncol. 2020, 17, 204–232. [Google Scholar] [CrossRef] [PubMed]
- Walcher, L.; Kistenmacher, A.K.; Suo, H.; Kitte, R.; Dluczek, S.; Strauß, A.; Blaudszun, A.R.; Yevsa, T.; Fricke, S.; Kossatz-Boehlert, U. Cancer Stem Cells-Origins and Biomarkers: Perspectives for Targeted Personalized Therapies. Front. Immunol. 2020, 11, 1280. [Google Scholar] [CrossRef]
- Kesh, K.; Gupta, V.K.; Durden, B.; Garrido, V.; Mateo-Victoriano, B.; Lavania, S.P.; Banerjee, S. Therapy Resistance, Cancer Stem Cells and ECM in Cancer: The Matrix Reloaded. Cancers 2020, 12, 3067. [Google Scholar] [CrossRef] [PubMed]
- Bajaj, J.; Diaz, E.; Reya, T. Stem cells in cancer initiation and progression. J. Cell Biol. 2020, 219, e201911053. [Google Scholar] [CrossRef]
- Nguyen, K.; McConnell, E.; Edwards, O.; Collins-Burow, B.M.; Burow, M.E. GD2+ cancer stem cells in triple-negative breast cancer: Mechanisms of resistance to breast cancer therapies. Cancer Drug Resist. 2022, 5, 721–726. [Google Scholar] [CrossRef]
- Talukdar, S.; Bhoopathi, P.; Emdad, L.; Das, S.; Sarkar, D.; Fisher, P.B. Dormancy and cancer stem cells: An enigma for cancer therapeutic targeting. Adv. Cancer Res. 2019, 141, 43–84. [Google Scholar] [CrossRef]
- Cho, Y.; Kim, Y.K. Cancer Stem Cells as a Potential Target to Overcome Multidrug Resistance. Front. Oncol. 2020, 10, 764. [Google Scholar] [CrossRef]
- Phi, L.T.H.; Sari, I.N.; Yang, Y.G.; Lee, S.H.; Jun, N.; Kim, K.S.; Lee, Y.K.; Kwon, H.Y. Cancer Stem Cells (CSCs) in Drug Resistance and their Therapeutic Implications in Cancer Treatment. Stem Cells Int. 2018, 2018, 5416923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.H.; Reed-Newman, T.; Anant, S.; Ramasamy, T.S. Regulatory Role of Quiescence in the Biological Function of Cancer Stem Cells. Stem Cell Rev. Rep. 2020, 16, 1185–1207. [Google Scholar] [CrossRef] [PubMed]
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994, 367, 645–648. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Li, B.; Liu, F.; Zhang, M.; Wang, Q.; Liu, Y.; Yao, Y.; Li, D. The epithelial to mesenchymal transition (EMT) and cancer stem cells: Implication for treatment resistance in pancreatic cancer. Mol. Cancer 2017, 16, 52. [Google Scholar] [CrossRef] [Green Version]
- Brunner, T.B.; Kunz-Schughart, L.A.; Grosse-Gehling, P.; Baumann, M. Cancer stem cells as a predictive factor in radiotherapy. Semin. Radiat. Oncol. 2012, 22, 151–174. [Google Scholar] [CrossRef]
- Dalerba, P.; Cho, R.W.; Clarke, M.F. Cancer stem cells: Models and concepts. Annu. Rev. Med. 2007, 58, 267–284. [Google Scholar] [CrossRef] [Green Version]
- López-Lázaro, M. Stem cell division theory of cancer. Cell Cycle 2015, 14, 2547–2548. [Google Scholar] [CrossRef] [Green Version]
- Rubio, D.; Garcia-Castro, J.; Martín, M.C.; de la Fuente, R.; Cigudosa, J.C.; Lloyd, A.C.; Bernad, A. Spontaneous human adult stem cell transformation. Cancer Res. 2005, 65, 3035–3039. [Google Scholar] [CrossRef] [Green Version]
- Baccelli, I.; Trumpp, A. The evolving concept of cancer and metastasis stem cells. J. Cell Biol. 2012, 198, 281–293. [Google Scholar] [CrossRef] [Green Version]
- Barker, N.; Ridgway, R.A.; van Es, J.H.; van de Wetering, M.; Begthel, H.; van den Born, M.; Danenberg, E.; Clarke, A.R.; Sansom, O.J.; Clevers, H. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature 2009, 457, 608–611. [Google Scholar] [CrossRef]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef]
- Burns, J.S.; Abdallah, B.M.; Guldberg, P.; Rygaard, J.; Schrøder, H.D.; Kassem, M. Tumorigenic heterogeneity in cancer stem cells evolved from long-term cultures of telomerase-immortalized human mesenchymal stem cells. Cancer Res. 2005, 65, 3126–3135. [Google Scholar] [CrossRef] [Green Version]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Heidt, D.G.; Dalerba, P.; Burant, C.F.; Zhang, L.; Adsay, V.; Wicha, M.; Clarke, M.F.; Simeone, D.M. Identification of pancreatic cancer stem cells. Cancer Res. 2007, 67, 1030–1037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Wu, J.J.; Hynes, M.; Dosch, J.; Sarkar, B.; Welling, T.H.; Pasca di Magliano, M.; Simeone, D.M. c-Met is a marker of pancreatic cancer stem cells and therapeutic target. Gastroenterology 2011, 141, 2218–2227.e2215. [Google Scholar] [CrossRef] [PubMed]
- Ishiwata, T.; Matsuda, Y.; Yoshimura, H.; Sasaki, N.; Ishiwata, S.; Ishikawa, N.; Takubo, K.; Arai, T.; Aida, J. Pancreatic cancer stem cells: Features and detection methods. Pathol. Oncol. Res. 2018, 24, 797–805. [Google Scholar] [CrossRef]
- Gu, G.; Dubauskaite, J.; Melton, D.A. Direct evidence for the pancreatic lineage: NGN3+ cells are islet progenitors and are distinct from duct progenitors. Development 2002, 129, 2447–2457. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Law, A.C.; Rajagopal, J.; Anderson, W.J.; Gray, P.A.; Melton, D.A. A multipotent progenitor domain guides pancreatic organogenesis. Dev. Cell 2007, 13, 103–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanger, B.Z.; Tanaka, A.J.; Melton, D.A. Organ size is limited by the number of embryonic progenitor cells in the pancreas but not the liver. Nature 2007, 445, 886–891. [Google Scholar] [CrossRef]
- Byrnes, L.E.; Wong, D.M.; Subramaniam, M.; Meyer, N.P.; Gilchrist, C.L.; Knox, S.M.; Tward, A.D.; Ye, C.J.; Sneddon, J.B. Lineage dynamics of murine pancreatic development at single-cell resolution. Nat. Commun. 2018, 9, 3922. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.X.; Qiu, W.L.; Yang, L.; Zhang, Y.; He, M.Y.; Li, L.C.; Xu, C.R. Defining multistep cell fate decision pathways during pancreatic development at single-cell resolution. EMBO J. 2019, 38, e100164. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, J.; Liss, A.S.; Sontheimer, A.; Mino-Kenudson, M.; Castillo, C.F.; Warshaw, A.L.; Thayer, S.P. Pancreatic duct glands (PDGs) are a progenitor compartment responsible for pancreatic ductal epithelial repair. Stem Cell Res. 2015, 15, 190–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Storz, P. Acinar cell plasticity and development of pancreatic ductal adenocarcinoma. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 296–304. [Google Scholar] [CrossRef]
- Westphalen, C.B.; Takemoto, Y.; Tanaka, T.; Macchini, M.; Jiang, Z.; Renz, B.W.; Chen, X.; Ormanns, S.; Nagar, K.; Tailor, Y.; et al. Dclk1 Defines Quiescent Pancreatic Progenitors that Promote Injury-Induced Regeneration and Tumorigenesis. Cell Stem Cell 2016, 18, 441–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, J.; Sun, B.F.; Chen, C.Y.; Zhou, J.Y.; Chen, Y.S.; Chen, H.; Liu, L.; Huang, D.; Jiang, J.; Cui, G.S.; et al. Single-cell RNA-seq highlights intra-tumoral heterogeneity and malignant progression in pancreatic ductal adenocarcinoma. Cell Res. 2019, 29, 725–738. [Google Scholar] [CrossRef]
- Roy, N.; Malik, S.; Villanueva, K.E.; Urano, A.; Lu, X.; Von Figura, G.; Seeley, E.S.; Dawson, D.W.; Collisson, E.A.; Hebrok, M. Brg1 promotes both tumor-suppressive and oncogenic activities at distinct stages of pancreatic cancer formation. Genes Dev. 2015, 29, 658–671. [Google Scholar] [CrossRef] [Green Version]
- Balkwill, F.R.; Capasso, M.; Hagemann, T. The tumor microenvironment at a glance. J. Cell Sci. 2012, 125, 5591–5596. [Google Scholar] [CrossRef] [Green Version]
- Fridman, W.H.; Pagès, F.; Sautès-Fridman, C.; Galon, J. The immune contexture in human tumours: Impact on clinical outcome. Nat. Rev. Cancer 2012, 12, 298–306. [Google Scholar] [CrossRef]
- Hinshaw, D.C.; Shevde, L.A. The Tumor Microenvironment Innately Modulates Cancer Progression. Cancer Res. 2019, 79, 4557–4566. [Google Scholar] [CrossRef] [Green Version]
- Baghban, R.; Roshangar, L.; Jahanban-Esfahlan, R.; Seidi, K.; Ebrahimi-Kalan, A.; Jaymand, M.; Kolahian, S.; Javaheri, T.; Zare, P. Tumor microenvironment complexity and therapeutic implications at a glance. Cell Commun. Signal. 2020, 18, 59. [Google Scholar] [CrossRef] [Green Version]
- Jin, M.Z.; Jin, W.L. The updated landscape of tumor microenvironment and drug repurposing. Signal Transduct. Target. Ther. 2020, 5, 166. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Yu, D. Tumor microenvironment as a therapeutic target in cancer. Pharmacol. Ther. 2021, 221, 107753. [Google Scholar] [CrossRef] [PubMed]
- Xiao, M.; He, J.; Yin, L.; Chen, X.; Zu, X.; Shen, Y. Tumor-Associated Macrophages: Critical Players in Drug Resistance of Breast Cancer. Front. Immunol. 2021, 12, 799428. [Google Scholar] [CrossRef] [PubMed]
- Denisenko, T.V.; Budkevich, I.N.; Zhivotovsky, B. Cell death-based treatment of lung adenocarcinoma. Cell Death Dis. 2018, 9, 117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schito, L.; Semenza, G.L. Hypoxia-Inducible Factors: Master Regulators of Cancer Progression. Trends Cancer 2016, 2, 758–770. [Google Scholar] [CrossRef] [Green Version]
- Heddleston, J.M.; Li, Z.; Lathia, J.D.; Bao, S.; Hjelmeland, A.B.; Rich, J.N. Hypoxia inducible factors in cancer stem cells. Br. J. Cancer 2010, 102, 789–795. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Han, Z.; Zhu, Y.; Chen, J.; Li, W. Role of hypoxia inducible factor-1 in cancer stem cells (Review). Mol. Med. Rep. 2021, 23, 17. [Google Scholar] [CrossRef] [PubMed]
- Otrock, Z.K.; Hatoum, H.A.; Awada, A.H.; Ishak, R.S.; Shamseddine, A.I. Hypoxia-inducible factor in cancer angiogenesis: Structure, regulation and clinical perspectives. Crit. Rev. Oncol. Hematol. 2009, 70, 93–102. [Google Scholar] [CrossRef]
- Zhang, W.; Shi, X.; Peng, Y.; Wu, M.; Zhang, P.; Xie, R.; Wu, Y.; Yan, Q.; Liu, S.; Wang, J. HIF-1α Promotes Epithelial-Mesenchymal Transition and Metastasis through Direct Regulation of ZEB1 in Colorectal Cancer. PLoS ONE 2015, 10, e0129603. [Google Scholar] [CrossRef]
- WARBURG, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sattler, U.G.; Meyer, S.S.; Quennet, V.; Hoerner, C.; Knoerzer, H.; Fabian, C.; Yaromina, A.; Zips, D.; Walenta, S.; Baumann, M.; et al. Glycolytic metabolism and tumour response to fractionated irradiation. Radiother. Oncol. 2010, 94, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Paolini, L.; Adam, C.; Beauvillain, C.; Preisser, L.; Blanchard, S.; Pignon, P.; Seegers, V.; Chevalier, L.M.; Campone, M.; Wernert, R.; et al. Lactic Acidosis Together with GM-CSF and M-CSF Induces Human Macrophages toward an Inflammatory Protumor Phenotype. Cancer Immunol. Res. 2020, 8, 383–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colegio, O.R.; Chu, N.Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014, 513, 559–563. [Google Scholar] [CrossRef] [Green Version]
- Hunt, T.K.; Aslam, R.S.; Beckert, S.; Wagner, S.; Ghani, Q.P.; Hussain, M.Z.; Roy, S.; Sen, C.K. Aerobically derived lactate stimulates revascularization and tissue repair via redox mechanisms. Antioxid. Redox Signal. 2007, 9, 1115–1124. [Google Scholar] [CrossRef] [Green Version]
- Angelin, A.; Gil-de-Gómez, L.; Dahiya, S.; Jiao, J.; Guo, L.; Levine, M.H.; Wang, Z.; Quinn, W.J.; Kopinski, P.K.; Wang, L.; et al. Foxp3 Reprograms T Cell Metabolism to Function in Low-Glucose, High-Lactate Environments. Cell Metab. 2017, 25, 1282–1293.e1287. [Google Scholar] [CrossRef] [Green Version]
- Ayad, N.M.E.; Weaver, V.M. Tension in tumour cells keeps metabolism high. Nature 2020, 578, 517–518. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Lv, J.; Liang, X.; Yin, X.; Zhang, L.; Chen, D.; Jin, X.; Fiskesund, R.; Tang, K.; Ma, J.; et al. Fibrin Stiffness Mediates Dormancy of Tumor-Repopulating Cells via a Cdc42-Driven Tet2 Epigenetic Program. Cancer Res. 2018, 78, 3926–3937. [Google Scholar] [CrossRef] [Green Version]
- Nagelkerke, A.; Bussink, J.; Rowan, A.E.; Span, P.N. The mechanical microenvironment in cancer: How physics affects tumours. Semin. Cancer Biol. 2015, 35, 62–70. [Google Scholar] [CrossRef]
- Calvo, F.; Ege, N.; Grande-Garcia, A.; Hooper, S.; Jenkins, R.P.; Chaudhry, S.I.; Harrington, K.; Williamson, P.; Moeendarbary, E.; Charras, G.; et al. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat. Cell Biol. 2013, 15, 637–646. [Google Scholar] [CrossRef]
- Xiong, G.; Chen, J.; Zhang, G.; Wang, S.; Kawasaki, K.; Zhu, J.; Zhang, Y.; Nagata, K.; Li, Z.; Zhou, B.P.; et al. Hsp47 promotes cancer metastasis by enhancing collagen-dependent cancer cell-platelet interaction. Proc. Natl. Acad. Sci. USA 2020, 117, 3748–3758. [Google Scholar] [CrossRef] [Green Version]
- Magnon, C.; Hall, S.J.; Lin, J.; Xue, X.; Gerber, L.; Freedland, S.J.; Frenette, P.S. Autonomic nerve development contributes to prostate cancer progression. Science 2013, 341, 1236361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayala, G.E.; Dai, H.; Powell, M.; Li, R.; Ding, Y.; Wheeler, T.M.; Shine, D.; Kadmon, D.; Thompson, T.; Miles, B.J.; et al. Cancer-related axonogenesis and neurogenesis in prostate cancer. Clin. Cancer Res. 2008, 14, 7593–7603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jobling, P.; Pundavela, J.; Oliveira, S.M.; Roselli, S.; Walker, M.M.; Hondermarck, H. Nerve-Cancer Cell Cross-talk: A Novel Promoter of Tumor Progression. Cancer Res. 2015, 75, 1777–1781. [Google Scholar] [CrossRef] [Green Version]
- Liebig, C.; Ayala, G.; Wilks, J.A.; Berger, D.H.; Albo, D. Perineural invasion in cancer: A review of the literature. Cancer 2009, 115, 3379–3391. [Google Scholar] [CrossRef] [PubMed]
- Mauffrey, P.; Tchitchek, N.; Barroca, V.; Bemelmans, A.P.; Firlej, V.; Allory, Y.; Roméo, P.H.; Magnon, C. Progenitors from the central nervous system drive neurogenesis in cancer. Nature 2019, 569, 672–678. [Google Scholar] [CrossRef] [PubMed]
- Cervantes-Villagrana, R.D.; Albores-García, D.; Cervantes-Villagrana, A.R.; García-Acevez, S.J. Tumor-induced neurogenesis and immune evasion as targets of innovative anti-cancer therapies. Signal Transduct. Target. Ther. 2020, 5, 99. [Google Scholar] [CrossRef]
- Zahalka, A.H.; Frenette, P.S. Nerves in cancer. Nat. Rev. Cancer 2020, 20, 143–157. [Google Scholar] [CrossRef]
- Faulkner, S.; Jobling, P.; March, B.; Jiang, C.C.; Hondermarck, H. Tumor Neurobiology and the War of Nerves in Cancer. Cancer Discov. 2019, 9, 702–710. [Google Scholar] [CrossRef] [Green Version]
- Schulz, A.; Büttner, R.; Hagel, C.; Baader, S.L.; Kluwe, L.; Salamon, J.; Mautner, V.F.; Mindos, T.; Parkinson, D.B.; Gehlhausen, J.R.; et al. The importance of nerve microenvironment for schwannoma development. Acta Neuropathol. 2016, 132, 289–307. [Google Scholar] [CrossRef] [Green Version]
- Osswald, M.; Jung, E.; Sahm, F.; Solecki, G.; Venkataramani, V.; Blaes, J.; Weil, S.; Horstmann, H.; Wiestler, B.; Syed, M.; et al. Brain tumour cells interconnect to a functional and resistant network. Nature 2015, 528, 93–98. [Google Scholar] [CrossRef]
- Zeng, Q.; Michael, I.P.; Zhang, P.; Saghafinia, S.; Knott, G.; Jiao, W.; McCabe, B.D.; Galván, J.A.; Robinson, H.P.C.; Zlobec, I.; et al. Synaptic proximity enables NMDAR signalling to promote brain metastasis. Nature 2019, 573, 526–531. [Google Scholar] [CrossRef] [PubMed]
- Borden, P.; Houtz, J.; Leach, S.D.; Kuruvilla, R. Sympathetic innervation during development is necessary for pancreatic islet architecture and functional maturation. Cell Rep. 2013, 4, 287–301. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, F.; Hamanaka, R.; Wheaton, W.W.; Weinberg, S.; Joseph, J.; Lopez, M.; Kalyanaraman, B.; Mutlu, G.M.; Budinger, G.R.; Chandel, N.S. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. USA 2010, 107, 8788–8793. [Google Scholar] [CrossRef] [Green Version]
- Mao, X.Y.; Jin, M.Z.; Chen, J.F.; Zhou, H.H.; Jin, W.L. Live or let die: Neuroprotective and anti-cancer effects of nutraceutical antioxidants. Pharmacol. Ther. 2018, 183, 137–151. [Google Scholar] [CrossRef]
- Qu, P.; Boelte, K.C.; Lin, P.C. Negative regulation of myeloid-derived suppressor cells in cancer. Immunol. Investig. 2012, 41, 562–580. [Google Scholar] [CrossRef]
- Sena, L.A.; Li, S.; Jairaman, A.; Prakriya, M.; Ezponda, T.; Hildeman, D.A.; Wang, C.R.; Schumacker, P.T.; Licht, J.D.; Perlman, H.; et al. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity 2013, 38, 225–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, A.; Scholer-Dahirel, A.; Mechta-Grigoriou, F. The role of reactive oxygen species and metabolism on cancer cells and their microenvironment. Semin. Cancer Biol. 2014, 25, 23–32. [Google Scholar] [CrossRef]
- Ailuno, G.; Baldassari, S.; Lai, F.; Florio, T.; Caviglioli, G. Exosomes and Extracellular Vesicles as Emerging Theranostic Platforms in Cancer Research. Cells 2020, 9, 2569. [Google Scholar] [CrossRef]
- Jayasinghe, M.K.; Tan, M.; Peng, B.; Yang, Y.; Sethi, G.; Pirisinu, M.; Le, M.T.N. New approaches in extracellular vesicle engineering for improving the efficacy of anti-cancer therapies. Semin. Cancer Biol. 2021, 74, 62–78. [Google Scholar] [CrossRef]
- Cocucci, E.; Racchetti, G.; Meldolesi, J. Shedding microvesicles: Artefacts no more. Trends Cell Biol. 2009, 19, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Ashrafizaveh, S.; Ashrafizadeh, M.; Zarrabi, A.; Husmandi, K.; Zabolian, A.; Shahinozzaman, M.; Aref, A.R.; Hamblin, M.R.; Nabavi, N.; Crea, F.; et al. Long non-coding RNAs in the doxorubicin resistance of cancer cells. Cancer Lett. 2021, 508, 104–114. [Google Scholar] [CrossRef] [PubMed]
- Mathivanan, S.; Ji, H.; Simpson, R.J. Exosomes: Extracellular organelles important in intercellular communication. J. Proteom. 2010, 73, 1907–1920. [Google Scholar] [CrossRef] [PubMed]
- D’Asti, E.; Garnier, D.; Lee, T.H.; Montermini, L.; Meehan, B.; Rak, J. Oncogenic extracellular vesicles in brain tumor progression. Front. Physiol. 2012, 3, 294. [Google Scholar] [CrossRef] [Green Version]
- Vlassov, A.V.; Magdaleno, S.; Setterquist, R.; Conrad, R. Exosomes: Current knowledge of their composition, biological functions, and diagnostic and therapeutic potentials. Biochim. Biophys. Acta 2012, 1820, 940–948. [Google Scholar] [CrossRef] [PubMed]
- Wee, I.; Syn, N.; Sethi, G.; Goh, B.C.; Wang, L. Role of tumor-derived exosomes in cancer metastasis. Biochim. Biophys. Acta Rev. Cancer 2019, 1871, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Denzer, K.; Kleijmeer, M.J.; Heijnen, H.F.; Stoorvogel, W.; Geuze, H.J. Exosome: From internal vesicle of the multivesicular body to intercellular signaling device. J. Cell Sci. 2000, 113 Pt 19, 3365–3374. [Google Scholar] [CrossRef]
- Colombo, M.; Raposo, G.; Théry, C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu. Rev. Cell Dev. Biol. 2014, 30, 255–289. [Google Scholar] [CrossRef]
- Hu, T.; Wolfram, J.; Srivastava, S. Extracellular Vesicles in Cancer Detection: Hopes and Hypes. Trends Cancer 2021, 7, 122–133. [Google Scholar] [CrossRef]
- Huyan, T.; Li, H.; Peng, H.; Chen, J.; Yang, R.; Zhang, W.; Li, Q. Extracellular Vesicles-Advanced Nanocarriers in Cancer Therapy: Progress and Achievements. Int. J. Nanomed. 2020, 15, 6485–6502. [Google Scholar] [CrossRef]
- EL Andaloussi, S.; Mäger, I.; Breakefield, X.O.; Wood, M.J. Extracellular vesicles: Biology and emerging therapeutic opportunities. Nat. Rev. Drug Discov. 2013, 12, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Azmi, A.S.; Bao, B.; Sarkar, F.H. Exosomes in cancer development, metastasis, and drug resistance: A comprehensive review. Cancer Metastasis Rev. 2013, 32, 623–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bebelman, M.P.; Smit, M.J.; Pegtel, D.M.; Baglio, S.R. Biogenesis and function of extracellular vesicles in cancer. Pharmacol. Ther. 2018, 188, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Guo, Z.; Chen, W.; Wang, X.; Cao, M.; Han, X.; Zhang, K.; Teng, B.; Cao, J.; Wu, W.; et al. M2 Macrophage-Derived Exosomes Promote Angiogenesis and Growth of Pancreatic Ductal Adenocarcinoma by Targeting E2F2. Mol. Ther. 2021, 29, 1226–1238. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhang, J.; Zhang, Z.; Gao, Z.; Qi, Y.; Qiu, W.; Pan, Z.; Guo, Q.; Li, B.; Zhao, S.; et al. Hypoxic glioma-derived exosomes promote M2-like macrophage polarization by enhancing autophagy induction. Cell Death Dis. 2021, 12, 373. [Google Scholar] [CrossRef]
- Parolini, I.; Federici, C.; Raggi, C.; Lugini, L.; Palleschi, S.; De Milito, A.; Coscia, C.; Iessi, E.; Logozzi, M.; Molinari, A.; et al. Microenvironmental pH is a key factor for exosome traffic in tumor cells. J. Biol. Chem. 2009, 284, 34211–34222. [Google Scholar] [CrossRef] [Green Version]
- Hsu, Y.L.; Hung, J.Y.; Chang, W.A.; Lin, Y.S.; Pan, Y.C.; Tsai, P.H.; Wu, C.Y.; Kuo, P.L. Hypoxic lung cancer-secreted exosomal miR-23a increased angiogenesis and vascular permeability by targeting prolyl hydroxylase and tight junction protein ZO-1. Oncogene 2017, 36, 4929–4942. [Google Scholar] [CrossRef]
- King, H.W.; Michael, M.Z.; Gleadle, J.M. Hypoxic enhancement of exosome release by breast cancer cells. BMC Cancer 2012, 12, 421. [Google Scholar] [CrossRef] [Green Version]
- Xue, M.; Chen, W.; Xiang, A.; Wang, R.; Chen, H.; Pan, J.; Pang, H.; An, H.; Wang, X.; Hou, H.; et al. Hypoxic exosomes facilitate bladder tumor growth and development through transferring long non-coding RNA-UCA1. Mol. Cancer 2017, 16, 143. [Google Scholar] [CrossRef] [Green Version]
- Ramteke, A.; Ting, H.; Agarwal, C.; Mateen, S.; Somasagara, R.; Hussain, A.; Graner, M.; Frederick, B.; Agarwal, R.; Deep, G. Exosomes secreted under hypoxia enhance invasiveness and stemness of prostate cancer cells by targeting adherens junction molecules. Mol. Carcinog. 2015, 54, 554–565. [Google Scholar] [CrossRef] [Green Version]
- Moraes, L.A.; Kar, S.; Foo, S.L.; Gu, T.; Toh, Y.Q.; Ampomah, P.B.; Sachaphibulkij, K.; Yap, G.; Zharkova, O.; Lukman, H.M.; et al. Annexin-A1 enhances breast cancer growth and migration by promoting alternative macrophage polarization in the tumour microenvironment. Sci. Rep. 2017, 7, 17925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, W.; Zhou, Y.; Chen, X.; Ning, T.; Chen, H.; Guo, Q.; Zhang, Y.; Liu, P.; Li, C.; Chu, Y.; et al. Pancreatic cancer-targeting exosomes for enhancing immunotherapy and reprogramming tumor microenvironment. Biomaterials 2021, 268, 120546. [Google Scholar] [CrossRef] [PubMed]
- Logozzi, M.; Angelini, D.F.; Iessi, E.; Mizzoni, D.; Di Raimo, R.; Federici, C.; Lugini, L.; Borsellino, G.; Gentilucci, A.; Pierella, F.; et al. Increased PSA expression on prostate cancer exosomes in in vitro condition and in cancer patients. Cancer Lett. 2017, 403, 318–329. [Google Scholar] [CrossRef] [PubMed]
- Ceyhan, G.O.; Bergmann, F.; Kadihasanoglu, M.; Altintas, B.; Demir, I.E.; Hinz, U.; Müller, M.W.; Giese, T.; Büchler, M.W.; Giese, N.A.; et al. Pancreatic neuropathy and neuropathic pain—A comprehensive pathomorphological study of 546 cases. Gastroenterology 2009, 136, 177–186.e171. [Google Scholar] [CrossRef]
- Liebl, F.; Demir, I.E.; Mayer, K.; Schuster, T.; D’Haese, J.G.; Becker, K.; Langer, R.; Bergmann, F.; Wang, K.; Rosenberg, R.; et al. The impact of neural invasion severity in gastrointestinal malignancies: A clinicopathological study. Ann. Surg. 2014, 260, 900–907; discussion 907–908. [Google Scholar] [CrossRef] [Green Version]
- Bapat, A.A.; Munoz, R.M.; Von Hoff, D.D.; Han, H. Blocking Nerve Growth Factor Signaling Reduces the Neural Invasion Potential of Pancreatic Cancer Cells. PLoS ONE 2016, 11, e0165586. [Google Scholar] [CrossRef] [Green Version]
- Kim-Fuchs, C.; Le, C.P.; Pimentel, M.A.; Shackleford, D.; Ferrari, D.; Angst, E.; Hollande, F.; Sloan, E.K. Chronic stress accelerates pancreatic cancer growth and invasion: A critical role for beta-adrenergic signaling in the pancreatic microenvironment. Brain Behav. Immun. 2014, 40, 40–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renz, B.W.; Takahashi, R.; Tanaka, T.; Macchini, M.; Hayakawa, Y.; Dantes, Z.; Maurer, H.C.; Chen, X.; Jiang, Z.; Westphalen, C.B.; et al. β2 Adrenergic-Neurotrophin Feedforward Loop Promotes Pancreatic Cancer. Cancer Cell 2018, 33, 75–90.e77. [Google Scholar] [CrossRef] [Green Version]
- Orth, M.; Metzger, P.; Gerum, S.; Mayerle, J.; Schneider, G.; Belka, C.; Schnurr, M.; Lauber, K. Pancreatic ductal adenocarcinoma: Biological hallmarks, current status, and future perspectives of combined modality treatment approaches. Radiat. Oncol. 2019, 14, 141. [Google Scholar] [CrossRef] [Green Version]
- Sarantis, P.; Koustas, E.; Papadimitropoulou, A.; Papavassiliou, A.G.; Karamouzis, M.V. Pancreatic ductal adenocarcinoma: Treatment hurdles, tumor microenvironment and immunotherapy. World J. Gastrointest. Oncol. 2020, 12, 173–181. [Google Scholar] [CrossRef]
- Hessmann, E.; Buchholz, S.M.; Demir, I.E.; Singh, S.K.; Gress, T.M.; Ellenrieder, V.; Neesse, A. Microenvironmental Determinants of Pancreatic Cancer. Physiol. Rev. 2020, 100, 1707–1751. [Google Scholar] [CrossRef] [PubMed]
- Casado-Pelaez, M.; Bueno-Costa, A.; Esteller, M. Single cell cancer epigenetics. Trends Cancer 2022, 8, 820–838. [Google Scholar] [CrossRef] [PubMed]
- Jia, Q.; Chu, H.; Jin, Z.; Long, H.; Zhu, B. High-throughput single-cell sequencing in cancer research. Signal Transduct. Target. Ther. 2022, 7, 145. [Google Scholar] [CrossRef] [PubMed]
- Puleo, F.; Nicolle, R.; Blum, Y.; Cros, J.; Marisa, L.; Demetter, P.; Quertinmont, E.; Svrcek, M.; Elarouci, N.; Iovanna, J.; et al. Stratification of Pancreatic Ductal Adenocarcinomas Based on Tumor and Microenvironment Features. Gastroenterology 2018, 155, 1999–2013.e1993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moffitt, R.A.; Marayati, R.; Flate, E.L.; Volmar, K.E.; Loeza, S.G.; Hoadley, K.A.; Rashid, N.U.; Williams, L.A.; Eaton, S.C.; Chung, A.H.; et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet. 2015, 47, 1168–1178. [Google Scholar] [CrossRef]
- Steele, N.G.; Carpenter, E.S.; Kemp, S.B.; Sirihorachai, V.R.; The, S.; Delrosario, L.; Lazarus, J.; Amir, E.D.; Gunchick, V.; Espinoza, C.; et al. Multimodal Mapping of the Tumor and Peripheral Blood Immune Landscape in Human Pancreatic Cancer. Nat. Cancer 2020, 1, 1097–1112. [Google Scholar] [CrossRef]
- Elyada, E.; Bolisetty, M.; Laise, P.; Flynn, W.F.; Courtois, E.T.; Burkhart, R.A.; Teinor, J.A.; Belleau, P.; Biffi, G.; Lucito, M.S.; et al. Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov. 2019, 9, 1102–1123. [Google Scholar] [CrossRef] [Green Version]
- Ligorio, M.; Sil, S.; Malagon-Lopez, J.; Nieman, L.T.; Misale, S.; Di Pilato, M.; Ebright, R.Y.; Karabacak, M.N.; Kulkarni, A.S.; Liu, A.; et al. Stromal Microenvironment Shapes the Intratumoral Architecture of Pancreatic Cancer. Cell 2019, 178, 160–175.e127. [Google Scholar] [CrossRef]
- Dominguez, C.X.; Müller, S.; Keerthivasan, S.; Koeppen, H.; Hung, J.; Gierke, S.; Breart, B.; Foreman, O.; Bainbridge, T.W.; Castiglioni, A.; et al. Single-Cell RNA Sequencing Reveals Stromal Evolution into LRRC15. Cancer Discov. 2020, 10, 232–253. [Google Scholar] [CrossRef] [Green Version]
- Öhlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.J.; Bernard, V.; Semaan, A.; Monberg, M.E.; Huang, J.; Stephens, B.M.; Lin, D.; Rajapakshe, K.I.; Weston, B.R.; Bhutani, M.S.; et al. Elucidation of Tumor-Stromal Heterogeneity and the Ligand-Receptor Interactome by Single-Cell Transcriptomics in Real-world Pancreatic Cancer Biopsies. Clin. Cancer Res. 2021, 27, 5912–5921. [Google Scholar] [CrossRef] [PubMed]
- Freed-Pastor, W.A.; Lambert, L.J.; Ely, Z.A.; Pattada, N.B.; Bhutkar, A.; Eng, G.; Mercer, K.L.; Garcia, A.P.; Lin, L.; Rideout, W.M.; et al. The CD155/TIGIT axis promotes and maintains immune evasion in neoantigen-expressing pancreatic cancer. Cancer Cell 2021, 39, 1342–1360.e1314. [Google Scholar] [CrossRef] [PubMed]
- Werba, G.; Weissinger, D.; Kawaler, E.A.; Zhao, E.; Kalfakakou, D.; Dhara, S.; Wang, L.; Lim, H.B.; Oh, G.; Jing, X.; et al. Single-cell RNA sequencing reveals the effects of chemotherapy on human pancreatic adenocarcinoma and its tumor microenvironment. Nat. Commun. 2023, 14, 797. [Google Scholar] [CrossRef]
- Chu, X.; Yang, Y.; Tian, X. Crosstalk between Pancreatic Cancer Cells and Cancer-Associated Fibroblasts in the Tumor Microenvironment Mediated by Exosomal MicroRNAs. Int. J. Mol. Sci. 2022, 23, 9512. [Google Scholar] [CrossRef] [PubMed]
- Hsu, S.K.; Jadhao, M.; Liao, W.T.; Chang, W.T.; Lin, I.L.; Chiu, C.C. The Role of Exosomes in Pancreatic Ductal Adenocarcinoma Progression and Their Potential as Biomarkers. Cancers 2023, 15, 1776. [Google Scholar] [CrossRef]
- Xavier, C.P.R.; Castro, I.; Caires, H.R.; Ferreira, D.; Cavadas, B.; Pereira, L.; Santos, L.L.; Oliveira, M.J.; Vasconcelos, M.H. Chitinase 3-like-1 and fibronectin in the cargo of extracellular vesicles shed by human macrophages influence pancreatic cancer cellular response to gemcitabine. Cancer Lett. 2021, 501, 210–223. [Google Scholar] [CrossRef]
- Linton, S.S.; Abraham, T.; Liao, J.; Clawson, G.A.; Butler, P.J.; Fox, T.; Kester, M.; Matters, G.L. Tumor-promoting effects of pancreatic cancer cell exosomes on THP-1-derived macrophages. PLoS ONE 2018, 13, e0206759. [Google Scholar] [CrossRef] [Green Version]
- Yin, Z.; Ma, T.; Huang, B.; Lin, L.; Zhou, Y.; Yan, J.; Zou, Y.; Chen, S. Macrophage-derived exosomal microRNA-501-3p promotes progression of pancreatic ductal adenocarcinoma through the TGFBR3-mediated TGF-β signaling pathway. J. Exp. Clin. Cancer Res. 2019, 38, 310. [Google Scholar] [CrossRef] [Green Version]
- Costa-Silva, B.; Aiello, N.M.; Ocean, A.J.; Singh, S.; Zhang, H.; Thakur, B.K.; Becker, A.; Hoshino, A.; Mark, M.T.; Molina, H.; et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat. Cell Biol. 2015, 17, 816–826. [Google Scholar] [CrossRef]
- Ding, G.; Zhou, L.; Qian, Y.; Fu, M.; Chen, J.; Xiang, J.; Wu, Z.; Jiang, G.; Cao, L. Pancreatic cancer-derived exosomes transfer miRNAs to dendritic cells and inhibit RFXAP expression via miR-212-3p. Oncotarget 2015, 6, 29877–29888. [Google Scholar] [CrossRef] [Green Version]
- Zhou, M.; Chen, J.; Zhou, L.; Chen, W.; Ding, G.; Cao, L. Pancreatic cancer derived exosomes regulate the expression of TLR4 in dendritic cells via miR-203. Cell. Immunol. 2014, 292, 65–69. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wang, S.; Jia, S.; Ding, G.; Jiang, G.; Cao, L. Integrated Analysis of Long Non-Coding RNA and mRNA Expression Profile in Pancreatic Cancer Derived Exosomes Treated Dendritic Cells by Microarray Analysis. J. Cancer 2018, 9, 21–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takikawa, T.; Masamune, A.; Yoshida, N.; Hamada, S.; Kogure, T.; Shimosegawa, T. Exosomes Derived from Pancreatic Stellate Cells: MicroRNA Signature and Effects on Pancreatic Cancer Cells. Pancreas 2017, 46, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Wu, H.; Xiao, Y.; Liang, Z.; Liu, T. Upregulation of exosomal microRNA-21 in pancreatic stellate cells promotes pancreatic cancer cell migration and enhances Ras/ERK pathway activity. Int. J. Oncol. 2020, 56, 1025–1033. [Google Scholar] [CrossRef]
- Sung, J.S.; Kang, C.W.; Kang, S.; Jang, Y.; Chae, Y.C.; Kim, B.G.; Cho, N.H. ITGB4-mediated metabolic reprogramming of cancer-associated fibroblasts. Oncogene 2020, 39, 664–676. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhao, W.; Wang, Y.; Wang, H.; Liu, S. Extracellular vesicle-mediated crosstalk between pancreatic cancer and stromal cells in the tumor microenvironment. J. Nanobiotechnology 2022, 20, 208. [Google Scholar] [CrossRef]
- Le Large, T.Y.; Mato Prado, M.; Krell, J.; Bijlsma, M.F.; Meijer, L.L.; Kazemier, G.; Frampton, A.E.; Giovannetti, E. Bioinformatic analysis reveals pancreatic cancer molecular subtypes specific to the tumor and the microenvironment. Expert Rev. Mol. Diagn. 2016, 16, 733–736. [Google Scholar] [CrossRef] [Green Version]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.M.; Gingras, M.C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef]
- Collisson, E.A.; Sadanandam, A.; Olson, P.; Gibb, W.J.; Truitt, M.; Gu, S.; Cooc, J.; Weinkle, J.; Kim, G.E.; Jakkula, L.; et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat. Med. 2011, 17, 500–503. [Google Scholar] [CrossRef]
- Malinova, A.; Veghini, L.; Real, F.X.; Corbo, V. Cell Lineage Infidelity in PDAC Progression and Therapy Resistance. Front. Cell Dev. Biol. 2021, 9, 795251. [Google Scholar] [CrossRef]
- Gillen, S.; Schuster, T.; Büschenfelde, C.M.Z.; Friess, H.; Kleeff, J. Preoperative/neoadjuvant therapy in pancreatic cancer: A systematic review and meta-analysis of response and resection percentages. PLoS Med. 2010, 7, e1000267. [Google Scholar] [CrossRef] [Green Version]
- Werner, J.; Combs, S.E.; Springfeld, C.; Hartwig, W.; Hackert, T.; Büchler, M.W. Advanced-stage pancreatic cancer: Therapy options. Nat. Rev. Clin. Oncol. 2013, 10, 323–333. [Google Scholar] [CrossRef] [PubMed]
- Manji, G.A.; Olive, K.P.; Saenger, Y.M.; Oberstein, P. Current and Emerging Therapies in Metastatic Pancreatic Cancer. Clin. Cancer Res. 2017, 23, 1670–1678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teague, A.; Lim, K.H.; Wang-Gillam, A. Advanced pancreatic adenocarcinoma: A review of current treatment strategies and developing therapies. Ther. Adv. Med. Oncol. 2015, 7, 68–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouché, O.; Guimbaud, R.; Bécouarn, Y.; Adenis, A.; Raoul, J.L.; Gourgou-Bourgade, S.; de la Fouchardière, C.; et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef] [Green Version]
- Atashzar, M.R.; Baharlou, R.; Karami, J.; Abdollahi, H.; Rezaei, R.; Pourramezan, F.; Moghaddam, S.H.Z. Cancer stem cells: A review from origin to therapeutic implications. J. Cell. Physiol. 2020, 235, 790–803. [Google Scholar] [CrossRef]
- Pattabiraman, D.R.; Weinberg, R.A. Tackling the cancer stem cells-what challenges do they pose? Nat. Rev. Drug Discov. 2014, 13, 497–512. [Google Scholar] [CrossRef] [Green Version]
- Lytle, N.K.; Barber, A.G.; Reya, T. Stem cell fate in cancer growth, progression and therapy resistance. Nat. Rev. Cancer 2018, 18, 669–680. [Google Scholar] [CrossRef]
- Zhang, G.N.; Liang, Y.; Zhou, L.J.; Chen, S.P.; Chen, G.; Zhang, T.P.; Kang, T.; Zhao, Y.P. Combination of salinomycin and gemcitabine eliminates pancreatic cancer cells. Cancer Lett. 2011, 313, 137–144. [Google Scholar] [CrossRef]
- Venkatesha, V.A.; Parsels, L.A.; Parsels, J.D.; Zhao, L.; Zabludoff, S.D.; Simeone, D.M.; Maybaum, J.; Lawrence, T.S.; Morgan, M.A. Sensitization of pancreatic cancer stem cells to gemcitabine by Chk1 inhibition. Neoplasia 2012, 14, 519–525. [Google Scholar] [CrossRef] [Green Version]
- Brunner, T.B.; Scott-Brown, M. The role of radiotherapy in multimodal treatment of pancreatic carcinoma. Radiat. Oncol. 2010, 5, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roeder, F. Neoadjuvant radiotherapeutic strategies in pancreatic cancer. World J. Gastrointest. Oncol. 2016, 8, 186–197. [Google Scholar] [CrossRef]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Bao, S.; Wu, Q.; Sathornsumetee, S.; Hao, Y.; Li, Z.; Hjelmeland, A.B.; Shi, Q.; McLendon, R.E.; Bigner, D.D.; Rich, J.N. Stem cell-like glioma cells promote tumor angiogenesis through vascular endothelial growth factor. Cancer Res. 2006, 66, 7843–7848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diehn, M.; Cho, R.W.; Lobo, N.A.; Kalisky, T.; Dorie, M.J.; Kulp, A.N.; Qian, D.; Lam, J.S.; Ailles, L.E.; Wong, M.; et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 2009, 458, 780–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaupel, P. Tumor microenvironmental physiology and its implications for radiation oncology. Semin. Radiat. Oncol. 2004, 14, 198–206. [Google Scholar] [CrossRef]
- Nixon, N.A.; Blais, N.; Ernst, S.; Kollmannsberger, C.; Bebb, G.; Butler, M.; Smylie, M.; Verma, S. Current landscape of immunotherapy in the treatment of solid tumours, with future opportunities and challenges. Curr. Oncol. 2018, 25, e373–e384. [Google Scholar] [CrossRef] [Green Version]
- Luchini, C.; Brosens, L.A.A.; Wood, L.D.; Chatterjee, D.; Shin, J.I.; Sciammarella, C.; Fiadone, G.; Malleo, G.; Salvia, R.; Kryklyva, V.; et al. Comprehensive characterisation of pancreatic ductal adenocarcinoma with microsatellite instability: Histology, molecular pathology and clinical implications. Gut 2021, 70, 148–156. [Google Scholar] [CrossRef]
- Feig, C.; Gopinathan, A.; Neesse, A.; Chan, D.S.; Cook, N.; Tuveson, D.A. The pancreas cancer microenvironment. Clin. Cancer Res. 2012, 18, 4266–4276. [Google Scholar] [CrossRef] [Green Version]
- Liyanage, U.K.; Moore, T.T.; Joo, H.G.; Tanaka, Y.; Herrmann, V.; Doherty, G.; Drebin, J.A.; Strasberg, S.M.; Eberlein, T.J.; Goedegebuure, P.S.; et al. Prevalence of regulatory T cells is increased in peripheral blood and tumor microenvironment of patients with pancreas or breast adenocarcinoma. J. Immunol. 2002, 169, 2756–2761. [Google Scholar] [CrossRef] [Green Version]
- Tanida, T.; Tanemura, M.; Miyoshi, E.; Nagano, H.; Furukawa, K.; Nonaka, Y.; Akita, H.; Hama, N.; Wada, H.; Kawamoto, K.; et al. Pancreatic cancer immunotherapy using a tumor lysate vaccine, engineered to express α-gal epitopes, targets pancreatic cancer stem cells. Int. J. Oncol. 2015, 46, 78–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ercan, G.; Karlitepe, A.; Ozpolat, B. Pancreatic Cancer Stem Cells and Therapeutic Approaches. Anticancer Res. 2017, 37, 2761–2775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ying, H.; Dey, P.; Yao, W.; Kimmelman, A.C.; Draetta, G.F.; Maitra, A.; DePinho, R.A. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev. 2016, 30, 355–385. [Google Scholar] [CrossRef] [Green Version]
- Waters, A.M.; Der, C.J. KRAS: The Critical Driver and Therapeutic Target for Pancreatic Cancer. Cold Spring Harb. Perspect. Med. 2018, 8, a031435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raphael, B.J.; Hruban, R.H.; Aguirre, A.J.; Moffitt, R.A.; Yeh, J.J.; Stewart, C.; Robertson, A.G.; Cherniack, A.D.; Gupta, M.; Getz, G.; et al. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2017, 32, 185–203.e113. [Google Scholar] [CrossRef] [Green Version]
- Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Kamiyama, H.; Jimeno, A.; et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 2008, 321, 1801–1806. [Google Scholar] [CrossRef] [Green Version]
- Witkiewicz, A.K.; McMillan, E.A.; Balaji, U.; Baek, G.; Lin, W.C.; Mansour, J.; Mollaee, M.; Wagner, K.U.; Koduru, P.; Yopp, A.; et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat. Commun. 2015, 6, 6744. [Google Scholar] [CrossRef]
- Juiz, N.A.; Iovanna, J.; Dusetti, N. Pancreatic Cancer Heterogeneity Can Be Explained Beyond the Genome. Front. Oncol. 2019, 9, 246. [Google Scholar] [CrossRef] [Green Version]
- Falvo, D.J. A Lineage-Specific Epigenetic Memory of Inflammation Potentiates Kras-Driven Pancreatic Tumorigenesis; Grimont, A., Zumbo, P., Yang, J.L., Osterhoudt, A., Breves, S.L., Pan, G., Rendeiro, A.F., Wilkinson, J.E., Dündar, F., Elemento, O., et al., Eds.; CSH: New York, NY, USA, 2021. [Google Scholar] [CrossRef]
- Tang, B.; Raviv, A.; Esposito, D.; Flanders, K.C.; Daniel, C.; Nghiem, B.T.; Garfield, S.; Lim, L.; Mannan, P.; Robles, A.I.; et al. A flexible reporter system for direct observation and isolation of cancer stem cells. Stem Cell Rep. 2015, 4, 155–169. [Google Scholar] [CrossRef] [Green Version]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Galindo-Vega, A.; Maldonado-Lagunas, V.; Mitre-Aguilar, I.B.; Melendez-Zajgla, J. Tumor Microenvironment Role in Pancreatic Cancer Stem Cells. Cells 2023, 12, 1560. https://doi.org/10.3390/cells12121560
Galindo-Vega A, Maldonado-Lagunas V, Mitre-Aguilar IB, Melendez-Zajgla J. Tumor Microenvironment Role in Pancreatic Cancer Stem Cells. Cells. 2023; 12(12):1560. https://doi.org/10.3390/cells12121560
Chicago/Turabian StyleGalindo-Vega, Aaron, Vilma Maldonado-Lagunas, Irma B. Mitre-Aguilar, and Jorge Melendez-Zajgla. 2023. "Tumor Microenvironment Role in Pancreatic Cancer Stem Cells" Cells 12, no. 12: 1560. https://doi.org/10.3390/cells12121560