
Conditions That Simulate the Environment of Atopic Dermatitis Enhance Susceptibility of Human Keratinocytes to Vaccinia Virus

 , , ,
, , ,  , and
, and {kind=link}
{kind=link}
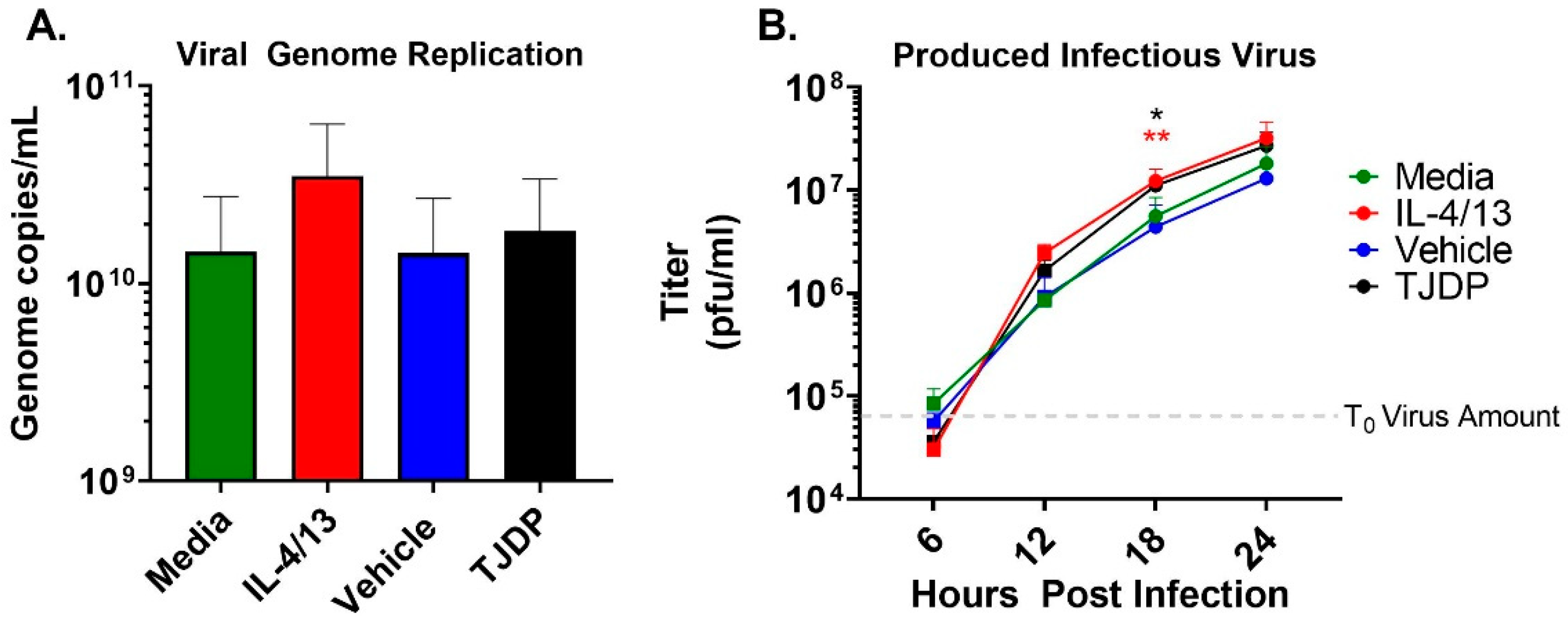{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Virus
2.2. Cytokines and Other Treatments
2.3. Transepithelial Electrical Resistance (TEER) Measurements
2.4. Plaque Assays and Quantification of Viral Replication (Titering/qPCR)
2.5. Viral Binding Assay
2.6. Plaque Reduction Assay
3. Results
4. Discussion
5. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nutten, S. Atopic Dermatitis: Global Epidemiology and Risk Factors. Ann. Nutr. Metab. 2015, 66 (Suppl. S1), 8–16. [Google Scholar] [CrossRef] [PubMed]
- Beck, L.A.; Boguniewicz, M.; Hata, T.; Schneider, L.C.; Hanifin, J.; Gallo, R.; Paller, A.; Lieff, S.; Reese, J.; Zaccaro, D.; et al. Phenotype of atopic dermatitis subjects with a history of eczema herpeticum. J. Allergy Clin. Immunol. 2009, 124, 260–269.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoeger, P.; Lenz, W.; Boutonnier, A.; Fournier, J.-M. Staphylococcal Skin Colonization in Children with Atopic Dermatitis: Prevalence, Persistence, and Transmission of Toxigenic and Nontoxigenic Strains. J. Infect. Dis. 1992, 165, 1064–1068. [Google Scholar] [CrossRef] [PubMed]
- Reed, J.L.; Scott, D.E.; Bray, M. Eczema vaccinatum. Clin. Infect. Dis. 2012, 54, 832–840. [Google Scholar] [CrossRef] [Green Version]
- Olsen, J.R.; Piguet, V.; Gallacher, J.; Francis, N.A. Molluscum contagiosum and associations with atopic eczema in children: A retrospective longitudinal study in primary care. Br. J. Gen. Pract. 2016, 66, e53–e58. [Google Scholar] [CrossRef] [Green Version]
- Ong, P.Y.; Leung, D.Y.M. Bacterial and Viral Infections in Atopic Dermatitis: A Comprehensive Review. Clin. Rev. Allergy Immunol. 2016, 51, 329–337. [Google Scholar] [CrossRef]
- Walsh, S.R.; Dolin, R. Vaccinia viruses: Vaccines against smallpox and vectors against infectious diseases and tumors. Expert Rev. Vaccines 2011, 10, 1221–1240. [Google Scholar] [CrossRef]
- Vora, S.; Damon, I.; Fulginiti, V.; Weber, S.G.; Kahana, M.; Stein, S.L.; Gerber, S.I.; Garcia-Houchins, S.; Lederman, E.; Hruby, D.; et al. Severe Eczema Vaccinatum in a Household Contact of a Smallpox Vaccinee. Clin. Infect. Dis. 2008, 46, 1555–1561. [Google Scholar] [CrossRef] [Green Version]
- Oyoshi, M.K.; Elkhal, A.; Kumar, L.; Scott, J.E.; Koduru, S.; He, R.; Leung, D.Y.M.; Howell, M.D.; Oettgen, H.C.; Murphy, G.F.; et al. Vaccinia virus inoculation in sites of allergic skin inflam-mation elicits a vigorous cutaneous IL-17 response. Proc. Natl. Acad. Sci. USA 2009, 106, 14954–14959. [Google Scholar] [CrossRef] [Green Version]
- Howell, M.D.; Jones, J.F.; Kisich, K.O.; Streib, J.E.; Gallo, R.L.; Leung, D.Y.M. Selective Killing of Vaccinia Virus by LL-37: Implications for Eczema Vaccinatum. J. Immunol. 2004, 172, 1763–1767. [Google Scholar] [CrossRef]
- Liu, L.; Xu, Z.; Fuhlbrigge, R.C.; Pena-Cruz, V.; Lieberman, J.; Kupper, T.S. Vaccinia virus induces strong immunoregulatory cyto-kine production in healthy human epidermal keratinocytes: A novel strategy for immune evasion. J. Virol. 2005, 79, 7363–7370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.; Sultana, I.; Takeda, K.; Reed, J.L. Cutaneous Deficiency of Filaggrin and STAT3 Exacerbates Vaccinia Disease In Vivo. PLoS ONE 2017, 12, e0170070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leung, D.Y.M. Human atopic dermatitis complicated by eczema herpeticum is associated with abnormalities in IFN-gamma response (vol 127, pg 965, 2011). J. Allergy Clin. Immunol. 2012, 130, 452. [Google Scholar]
- Howell, M.D.; Gallo, R.L.; Boguniewicz, M.; Jones, J.F.; Wong, C.; Streib, J.E.; Leung, D.Y.M. Cytokine milieu of atopic dermatitis skin sub-verts the innate immune response to vaccinia virus. Immunity 2006, 24, 341–348. [Google Scholar] [CrossRef] [Green Version]
- Duangkhae, P.; Erdos, G.; Ryman, K.D.; Watkins, S.; Falo, L.D.; Marques, E.T.; Barratt-Boyes, S.M. Interplay between Keratinocytes and Myeloid Cells Drives Dengue Virus Spread in Human Skin. J. Investig. Dermatol. 2018, 138, 618–626. [Google Scholar] [CrossRef] [Green Version]
- De Benedetto, A.; Slifka, M.K.; Rafaels, N.M.; Kuo, I.H.; Georas, S.N.; Boguniewicz, M.; Hata, T.; Schneider, L.C.; Hanifin, J.M.; Gallo, R.L.; et al. Reductions in claudin-1 may enhance susceptibility to herpes simplex virus 1 infections in atopic dermatitis. J. Allergy Clin. Immunol. 2011, 128, 242–246.e5. [Google Scholar] [CrossRef] [Green Version]
- De Benedetto, A.; Rafaels, N.M.; McGirt, L.Y.; Ivanov, A.I.; Georas, S.N.; Cheadle, C.; Berger, A.E.; Zhang, K.; Vidyasagar, S.; Yoshida, T.; et al. Tight junction defects in patients with atopic dermatitis. J. Allergy Clin. Immunol. 2011, 127, 773–786.e7. [Google Scholar] [CrossRef] [Green Version]
- Sandilands, A.; Terron-Kwiatkowski, A.; Hull, P.; O’Regan, G.M.; Clayton, T.H.; Watson, R.M.; Carrick, T.; Evans, A.T.; Liao, H.; Zhao, Y.; et al. Comprehensive analysis of the gene encoding filaggrin uncovers prevalent and rare mutations in ichthyosis vulgaris and atopic eczema. Nat. Genet. 2007, 39, 650–654. [Google Scholar] [CrossRef]
- Aleksza, M.; Irinyi, B.; Lukacs, A.; Antal-Szalmas, P.; Hunyadi, J.; Szegedi, A. Increased frequency of intracellular interleukin (IL)-13 and IL-10, but not IL-4, expressing CD4+ and CD8+ peripheral T cells of patients with atopic dermatitis. Br. J. Dermatol. 2002, 147, 1135–1141. [Google Scholar] [CrossRef]
- Jeong, C.-W.; Ahn, K.-S.; Rho, N.-K.; Park, Y.-D.; Lee, D.-Y.; Lee, J.-H.; Lee, E.-S.; Yang, J.-M. Differential in vivo cytokine mRNA expression in lesional skin of intrinsic vs. extrinsic atopic dermatitis patients using semiquantitative RT-PCR. Clin. Exp. Allergy 2003, 33, 1717–1724. [Google Scholar] [CrossRef]
- Aguado, B.; Selmes, I.P.; Smith, G.L. Nucleotide sequence of 21.8 kbp of variola major virus strain Harvey and comparison with vaccinia virus. J. Gen. Virol. 1992, 73 Pt 11, 2887–2902. [Google Scholar] [CrossRef] [PubMed]
- Moss, B. Poxviridae: The viruses and their replication. In Fields Virology; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2007; pp. 2905–2946. [Google Scholar]
- Moran, M.C.; Pandya, R.P.; Leffler, K.A.; Yoshida, T.; Beck, L.A.; Brewer, M.G. Characterization of Human Keratinocyte Cell Lines for Barrier Studies. JID Innov. 2021, 1, 100018. [Google Scholar] [CrossRef] [PubMed]
- Smits, J.P.H.; Niehues, H.; Rikken, G.; Van Vlijmen-Willems, I.M.J.J.; Van De Zande, G.W.H.J.F.; Zeeuwen, P.; Schalkwijk, J.; Bogaard, E.H.V.D. Immortalized N/TERT keratinocytes as an alternative cell source in 3D human epidermal models. Sci. Rep. 2017, 7, 11838. [Google Scholar] [CrossRef] [PubMed]
- Poumay, Y.; Roland, I.H.; Leclercq-Smekens, M.; Leloup, R. Basal detachment of the epidermis using dispase: Tissue spatial or-ganization and fate of integrin alpha 6 beta 4 and hemidesmosomes. J. Investig. Dermatol. 1994, 102, 111–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poumay, Y.; Pittelkow, M.R. Cell-Density and Culture Factors Regulate Keratinocyte Commitment to Differentiation and Expression of Suprabasal K1/K10 Keratins. J. Investig. Dermatol. 1995, 104, 271–276. [Google Scholar] [CrossRef] [Green Version]
- Brewer, M.G.; Yoshida, T.; Kuo, F.I.; Fridy, S.; Beck, L.A.; De Benedetto, A. Antagonistic Effects of IL-4 on IL-17A-Mediated En-hancement of Epidermal Tight Junction Function. Int. J. Mol. Sci. 2019, 20, 4070. [Google Scholar] [CrossRef] [Green Version]
- Dower, K.; Rubins, K.H.; Hensley, L.E.; Connor, J.H. Development of Vaccinia reporter viruses for rapid, high content analysis of viral function at all stages of gene expression. Antivir. Res. 2011, 91, 72–80. [Google Scholar] [CrossRef] [Green Version]
- Brewer, M.G.; Anderson, E.A.; Pandya, R.P.; De Benedetto, A.; Yoshida, T.; Hilimire, T.A.; Martinez-Sobrido, L.; Beck, L.A.; Miller, B.L. Peptides Derived from the Tight Junction Protein CLDN1 Disrupt the Skin Barrier and Promote Responsiveness to an Epicutaneous Vaccine. J. Investig. Dermatol. 2019, 140, 361–369.e3. [Google Scholar] [CrossRef]
- Baker, J.L.; Ward, B.M. Development and comparison of a quantitative TaqMan-MGB real-time PCR assay to three other methods of quantifying vaccinia virions. J. Virol. Methods 2014, 196, 126–132. [Google Scholar] [CrossRef] [Green Version]
- Tsoi, L.C.; Rodriguez, E.; Degenhardt, F.; Baurecht, H.; Wehkamp, U.; Volks, N.; Szymczak, S.; Swindell, W.R.; Sarkar, M.; Raja, K.; et al. Atopic Dermatitis Is an IL-13–Dominant Disease with Greater Molecular Heterogeneity Compared to Psoriasis. J. Investig. Dermatol. 2019, 139, 1480–1489. [Google Scholar] [CrossRef] [Green Version]
- De Benedetto, A.; Rafaels, N.M.; Dubois, M.; Bolognino, M.; Leung, D.Y.; Barnes, K.C.; Slifka, M.K.; Beck, L.A. Reductions in an Epidermal Tight Junction Protein Enhance Atopic Dermatitis Subjects’ Susceptibility to HSV Infections. J. Allergy Clin. Immun. 2011, 127, AB137. [Google Scholar] [CrossRef]
- David, M.; Ford, D.; Bertoglio, J.; Maizel, A.L.; Pierre, J. Induction of the IL-13 receptor alpha2-chain by IL-4 and IL-13 in human keratinocytes: Involvement of STAT6, ERK and p38 MAPK pathways. Oncogene 2001, 20, 6660–6668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hershey, G.K. IL-13 receptors and signaling pathways: An evolving web. J. Allergy Clin. Immunol. 2003, 111, 677–690. [Google Scholar] [CrossRef] [PubMed]
- Nelms, K.; Keegan, A.D.; Zamorano, J.; Ryan, J.J.; Paul, W.E. THE IL-4 RECEPTOR: Signaling Mechanisms and Biologic Functions. Annu. Rev. Immunol. 1999, 17, 701–738. [Google Scholar] [CrossRef] [Green Version]
- Rubins, K.H.; Hensley, L.E.; Relman, D.A.; Brown, P.O. Stunned Silence: Gene Expression Programs in Human Cells Infected with Monkeypox or Vaccinia Virus. PLoS ONE 2011, 6, e15615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chisholm, S.E.; Reyburn, H.T. Recognition of Vaccinia Virus-Infected Cells by Human Natural Killer Cells Depends on Natural Cytotoxicity Receptors. J. Virol. 2006, 80, 2225–2233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.; Guo, Z.S.; O’Malley, M.E.; Yin, X.; Zeh, H.J.; Bartlett, D.L. A new recombinant vaccinia with targeted deletion of three viral genes: Its safety and efficacy as an oncolytic virus. Gene Ther. 2007, 14, 638–647. [Google Scholar] [CrossRef] [PubMed]
- Jackson, R.J.; Ramsay, A.J.; Christensen, C.D.; Beaton, S.; Hall, D.F.; Ramshaw, I.A. Expression of mouse interleukin-4 by a recom-binant ectromelia virus suppresses cytolytic lymphocyte responses and overcomes genetic resistance to mousepox. J. Virol. 2001, 75, 1205–1210. [Google Scholar] [CrossRef] [Green Version]
- Kawakami, Y.; Tomimori, Y.; Yumoto, K.; Hasegawa, S.; Ando, T.; Tagaya, Y.; Crotty, S.; Kawakami, T. Inhibition of NK cell activity by IL-17 allows vaccinia virus to induce severe skin lesions in a mouse model of eczema vaccinatum. J. Exp. Med. 2009, 206, 1219–1225. [Google Scholar] [CrossRef] [Green Version]
- Suárez-Fariñas, M.; Tintle, S.J.; Shemer, A.; Chiricozzi, A.; Nograles, K.; Cardinale, I.; Duan, S.; Bowcock, A.M.; Krueger, J.G.; Guttman-Yassky, E. Nonlesional atopic dermatitis skin is characterized by broad terminal differentiation defects and variable immune abnormalities. J. Allergy Clin. Immunol. 2011, 127, 954–964.e4. [Google Scholar] [CrossRef] [Green Version]
- Silverberg, J.I. Public Health Burden and Epidemiology of Atopic Dermatitis. Dermatol. Clin. 2017, 35, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Toufighi, K.; Yang, J.-S.; Luis, N.M.; Benitah, S.A.; Lehner, B.; Serrano, L.; Kiel, C. Dissecting the Calcium-Induced Differentiation of Human Primary Keratinocytes Stem Cells by Integrative and Structural Network Analyses. PLoS Comput. Biol. 2015, 11, e1004256. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, E.; Green, H. Changes in keratin gene expression during terminal differentiation of the keratinocyte. Cell 1980, 19, 1033–1042. [Google Scholar] [CrossRef]
- Junttila, I.S. Tuning the Cytokine Responses: An Update on Interleukin (IL)-4 and IL-13 Receptor Complexes. Front. Immunol. 2018, 9, 888. [Google Scholar] [CrossRef]
- Omori-Miyake, M.; Yamashita, M.; Tsunemi, Y.; Kawashima, M.; Yagi, J. In Vitro Assessment of IL-4- or IL-13-Mediated Changes in the Structural Components of Keratinocytes in Mice and Humans. J. Investig. Dermatol. 2014, 134, 1342–1350. [Google Scholar] [CrossRef] [Green Version]
- Goleva, E.; Berdyshev, E.; Leung, D.Y. Epithelial barrier repair and prevention of allergy. J. Clin. Investig. 2019, 129, 1463–1474. [Google Scholar] [CrossRef] [Green Version]
- Szabo, I.; Wetzel, M.A.; Rogers, T.J. Cell-Density-Regulated Chemotactic Responsiveness of Keratinocytes In Vitro. J. Investig. Dermatol. 2001, 117, 1083–1090. [Google Scholar] [CrossRef] [Green Version]
- Bourke, C.D.; Prendergast, C.; Sanin, D.E.; Oulton, T.; Hall, R.; Mountford, A.P. Epidermal keratinocytes initiate wound healing and pro-inflammatory immune responses following percutaneous schistosome infection. Int. J. Parasitol. 2015, 45, 215–224. [Google Scholar] [CrossRef] [Green Version]
- Lowell, S.; Jones, P.; Le Roux, I.; Dunne, J.; Watt, F.M. Stimulation of human epidermal differentiation by Delta–Notch signalling at the boundaries of stem-cell clusters. Curr. Biol. 2000, 10, 491–500. [Google Scholar] [CrossRef] [Green Version]
- Sharma, D.P.; Ramsay, A.J.; Maguire, D.J.; Rolph, M.S.; Ramshaw, I.A. Interleukin-4 mediates down regulation of antiviral cytokine expression and cytotoxic T-lymphocyte responses and exacerbates vaccinia virus infection in vivo. J. Virol. 1996, 70, 7103–7107. [Google Scholar] [CrossRef] [Green Version]
- Kohonencorish, M.R.J.; King, N.J.C.; Woodhams, C.E.; Ramshaw, I.A. Immunodeficient Mice Recover from Infection with Vaccinia Virus Expressing Interferon-Gamma. Eur. J. Immunol. 1990, 20, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Chovatiya, R.; Paller, A.S. JAK inhibitors in the treatment of atopic dermatitis. J. Allergy Clin. Immunol. 2021, 148, 927–940. [Google Scholar] [CrossRef] [PubMed]
- Curtis, J.R.; Xie, F.; Yun, H.; Bernatsky, S.; Winthrop, K.L. Real-world comparative risks of herpes virus infections in tofacitinib and biologic-treated patients with rheumatoid arthritis. Ann. Rheum. Dis. 2016, 75, 1843–1847. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brewer, M.G.; Monticelli, S.R.; Moran, M.C.; Miller, B.L.; Beck, L.A.; Ward, B.M. Conditions That Simulate the Environment of Atopic Dermatitis Enhance Susceptibility of Human Keratinocytes to Vaccinia Virus. Cells 2022, 11, 1337. https://doi.org/10.3390/cells11081337
Brewer MG, Monticelli SR, Moran MC, Miller BL, Beck LA, Ward BM. Conditions That Simulate the Environment of Atopic Dermatitis Enhance Susceptibility of Human Keratinocytes to Vaccinia Virus. Cells. 2022; 11(8):1337. https://doi.org/10.3390/cells11081337
Chicago/Turabian StyleBrewer, Matthew G., Stephanie R. Monticelli, Mary C. Moran, Benjamin L. Miller, Lisa A. Beck, and Brian M. Ward. 2022. "Conditions That Simulate the Environment of Atopic Dermatitis Enhance Susceptibility of Human Keratinocytes to Vaccinia Virus" Cells 11, no. 8: 1337. https://doi.org/10.3390/cells11081337