
Advanced Glycation End-Products (AGEs): Formation, Chemistry, Classification, Receptors, and Diseases Related to AGEs


Abstract
:1. Advanced Glycation End-Products (AGEs)—Introduction, Chemistry, Classification
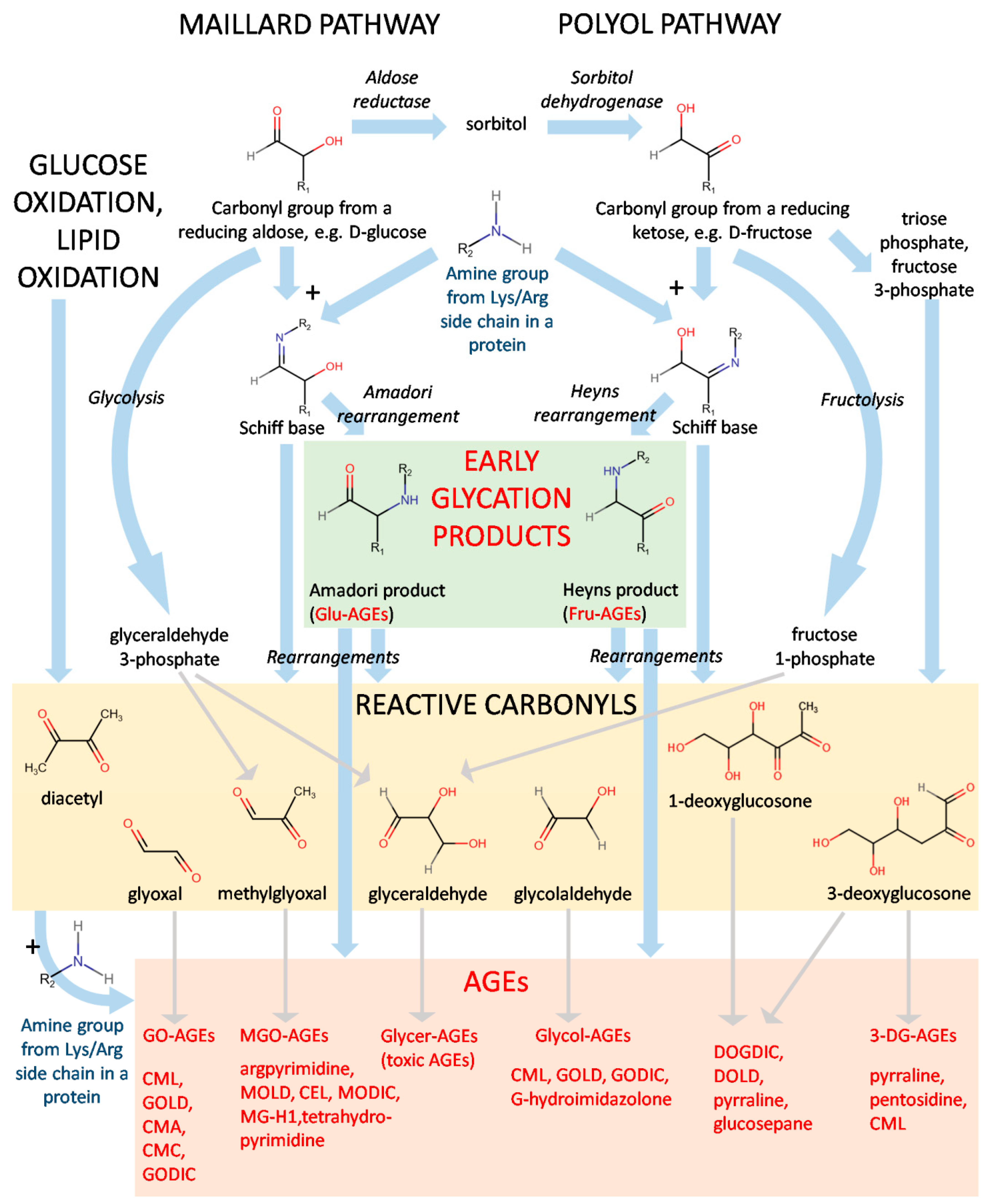
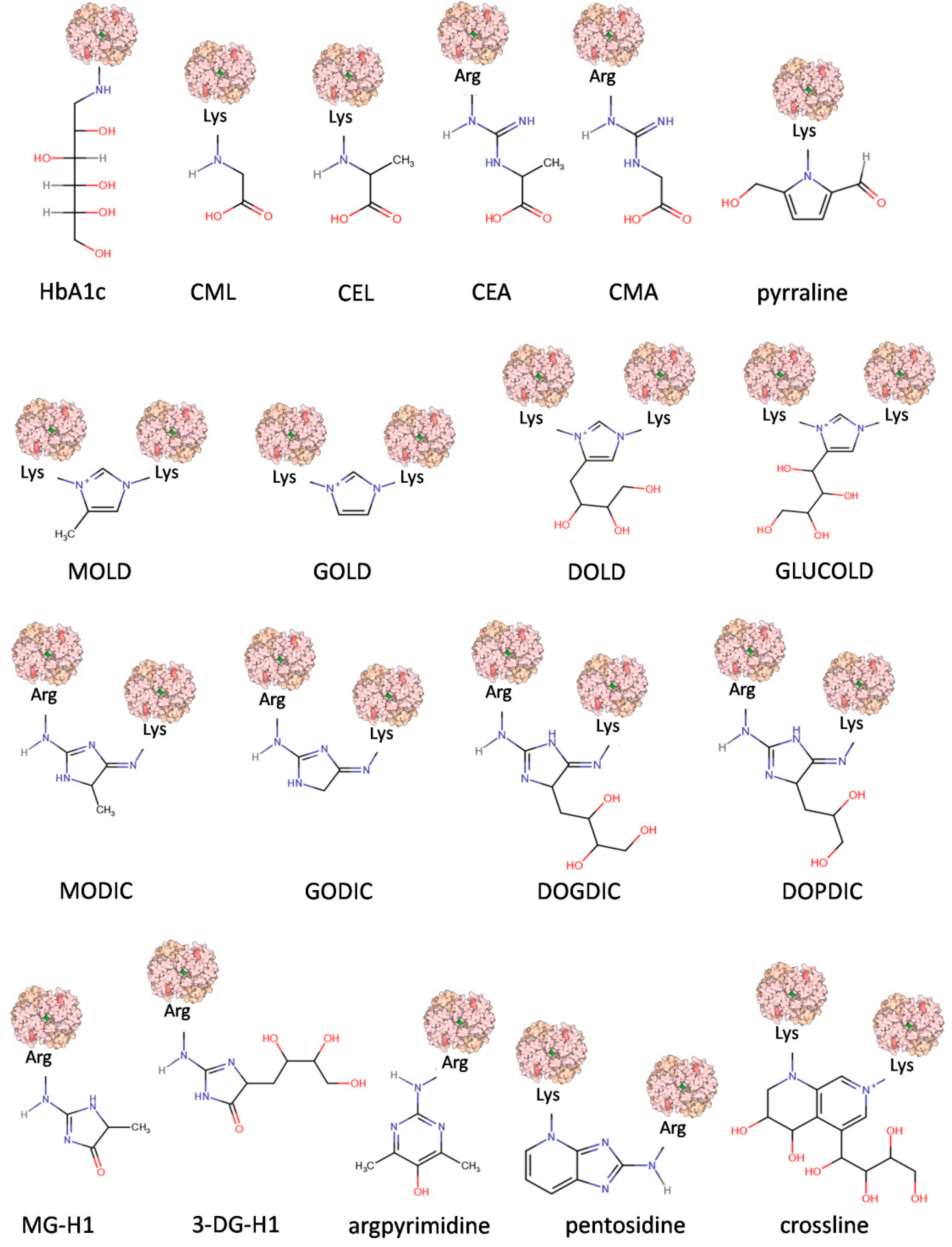
1.1. Formation and Chemistry of AGEs
1.2. Types of AGEs
1.2.1. Endogenous and Exogenous AGEs
1.2.2. Fluorescence and Crosslinking of AGEs
1.2.3. Low-Molecular-Weight AGEs (LMW) and High-Molecular-Weight (HMW) AGEs
1.2.4. Non-Toxic and Toxic AGEs (TAGEs)
2. AGE Receptors
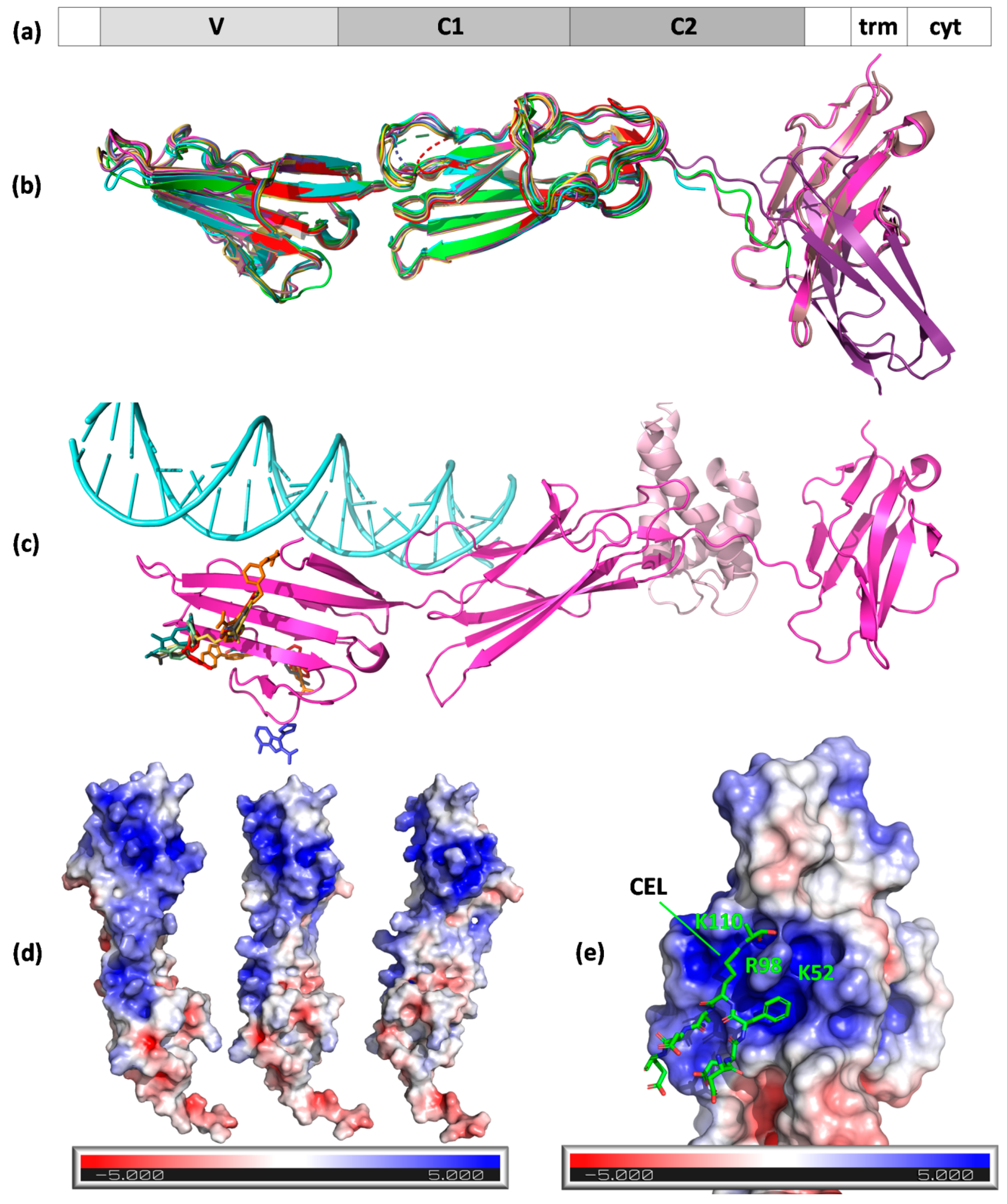
2.1. RAGE
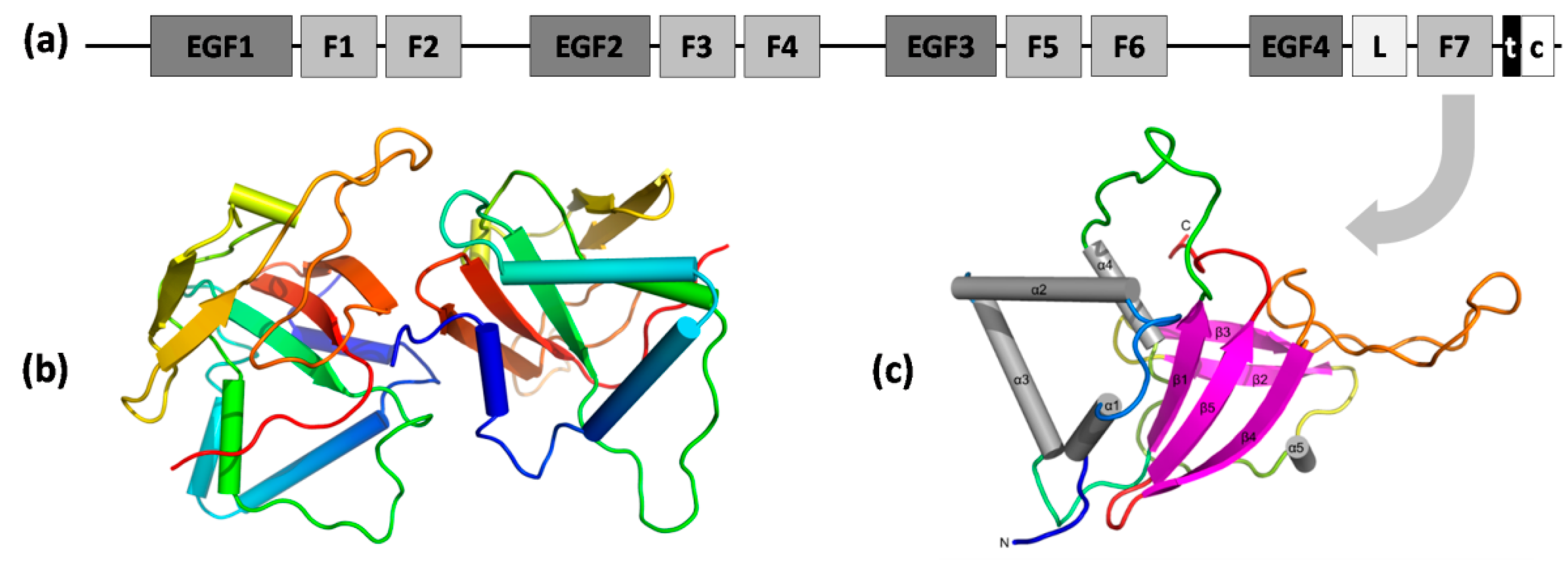
2.2. Stab1 and Stab2
2.3. Other Scavenger Receptors
2.4. AGE-R1, -R2, and -R3
3. Pathophysiological States and Diseases Induced by AGEs
3.1. Absorption
3.1.1. Absorption—Gut Microbiota
3.1.2. Excretion
3.1.3. Mechanisms of Action
3.2. Oxidative Stress and Inflammation States
3.2.1. Oxidative Stress
3.2.2. Inflammation States
3.3. Measurement of AGEs
3.4. Diseases
3.4.1. Diabetes Mellitus
3.4.2. CVD and CeVD
3.4.3. Neurodegenerative Diseases
3.4.4. Kidney Diseases
3.4.5. Collagen Tissues
3.4.6. Cancer
3.4.7. Liver Diseases
3.4.8. Infertility
3.4.9. Viral Infection—COVID-19
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| AGE | advanced glycation end-product |
| AGE-R1/R2/R3 | advanced glycation end-product receptor 1/2/3 |
| ALE | advanced lipoxidation end-product |
| CAD | coronary artery disease |
| CEL | Nε-(1-carboxyethyl)lysine |
| CeVD | cerebrovascular disease |
| CHD | coronary heart disease |
| CHF | chronic heart failure |
| CKD | chronic kidney disease |
| CMA | N7–(carboxymethyl)arginine |
| CML | Nε-(carboxymethyl)lysine |
| CVD | cardiovascular disease |
| dAGE | dietary advanced glycation end-product |
| FEEL-1/2 | fascilin, EGF-like, laminin-type EGF-like and link domain-containing scavenger receptor-1/-2 |
| GOLD | glyoxyl-derived lysine dimer |
| GSP | Glucosepane |
| HA | hyaluronic acid |
| HARE | hyaluronic acid receptor for endocytosis (Stab2) |
| HbA1c | glycated hemoglobin |
| LSRD | lifestyle-related disease |
| MG-H1 | methylglyoxal-hydroimidazolone |
| NAFLD | non-alcoholic fatty liver disease |
| NASH | non-alcoholic steatohepatitis |
| OST | N-oligosaccharyl transferase |
| PAD | peripheral artery disease |
| PD | Parkinson’s disease |
| RAGE | receptor for advanced glycation end-product |
| RNS | reactive nitrogen species |
| ROS | reactive oxygen species |
| SAF | skin autofluorescence |
| SEC | sinusoidal endothelial cell |
| SR | scavenger receptor |
| sRAGE | soluble receptor for advanced glycation end-product |
| Stab1/2 | stabilin-1/-2 |
| TAGE | toxic advanced glycation end-product |
| TIM-1 | TNO gastrointestinal in vitro model |
References
- Maillard, L.-C. Action Des Acides Aminés Sur Les Sucres: Formation Des Melanoidines Par Voie Méthodique. CR Acad. Sci. 1912, 154, 66–68. [Google Scholar]
- Rahbar, S.; Blumenfeld, O.; Ranney, H.M. Studies of an Unusual Hemoglobin in Patients with Diabetes Mellitus. Biochem. Biophys. Res. Commun. 1969, 36, 838–843. [Google Scholar] [CrossRef]
- Cerami, A.; Vlassara, H.; Brownlee, M. Role of Nonenzymatic Glycosylation in Atherogenesis. J. Cell. Biochem. 1986, 30, 111–120. [Google Scholar] [CrossRef]
- Shen, C.Y.; Lu, C.H.; Wu, C.H.; Li, K.J.; Kuo, Y.M.; Hsieh, S.C.; Yu, C.L. The Development of Maillard Reaction, and Advanced Glycation End Product (Age)-Receptor for Age (Rage) Signaling Inhibitors as Novel Therapeutic Strategies for Patients with Age-Related Diseases. Molecules 2020, 25, 5591. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, J.; Bains, Y.; Guha, S.; Kahn, A.; Hall, D.; Bose, N.; Gugliucci, A.; Kapahi, P. The Role of Advanced Glycation End Products in Aging and Metabolic Diseases: Bridging Association and Causality. Cell Metab. 2018, 28, 337–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dariya, B.; Nagaraju, G.P. Advanced Glycation End Products in Diabetes, Cancer and Phytochemical Therapy. Drug Discov. Today 2020, 25, 1614–1623. [Google Scholar] [CrossRef] [PubMed]
- Vistoli, G.; De Maddis, D.; Cipak, A.; Zarkovic, N.; Carini, M.; Aldini, G. Advanced Glycoxidation and Lipoxidation End Products (AGEs and ALEs): An Overview of Their Mechanisms of Formation. Free Radic. Res. 2013, 47, 3–27. [Google Scholar] [CrossRef] [Green Version]
- Heyns, K.; Beilfuss, W. Ketosylamine rearrangement of D-threo-pentulose (D-xylulose) with alpha-amino acids. Chem. Ber. 1970, 103, 2873–2876. [Google Scholar] [CrossRef]
- Ribeiro, R.T.; Macedo, M.P.; Raposo, J.F. HbA1c, Fructosamine, and Glycated Albumin in the Detection of Dysglycaemic Conditions. Curr. Diabetes Rev. 2016, 12, 14–19. [Google Scholar] [CrossRef]
- Anguizola, J.; Matsuda, R.; Barnaby, O.S.; Hoy, K.S.; Wa, C.; DeBolt, E.; Koke, M.; Hage, D.S. Review: Glycation of Human Serum Albumin. Clin. Chim. Acta 2013, 425, 64–76. [Google Scholar] [CrossRef] [Green Version]
- Heidari, F.; Rabizadeh, S.; Rajab, A.; Heidari, F.; Mouodi, M.; Mirmiranpour, H.; Esteghamati, A.; Nakhjavani, M. Advanced Glycation End-Products and Advanced Oxidation Protein Products Levels Are Correlates of Duration of Type 2 Diabetes. Life Sci. 2020, 260, 118422. [Google Scholar] [CrossRef] [PubMed]
- Nowotny, K.; Jung, T.; Höhn, A.; Weber, D.; Grune, T. Advanced Glycation End Products and Oxidative Stress in Type 2 Diabetes Mellitus. Biomolecules 2015, 5, 194–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, H.-Y.; Hou, N.-N.; Shi, J.-F.; Liu, Y.-P.; Kan, C.-X.; Han, F.; Sun, X.-D. Comprehensive Overview of Human Serum Albumin Glycation in Diabetes Mellitus. World J. Diabetes 2021, 12, 1057–1069. [Google Scholar] [CrossRef] [PubMed]
- Dorcely, B.; Katz, K.; Jagannathan, R.; Chiang, S.S.; Oluwadare, B.; Goldberg, I.J.; Bergman, M. Novel Biomarkers for Prediabetes, Diabetes, and Associated Complications. Diabetes Metab. Syndr. Obes. Targets Ther. 2017, 10, 345–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bunn, H.F.; Higgins, P.J. Reaction of Monosaccharides with Proteins: Possible Evolutionary Significance. Science 1981, 213, 222–224. [Google Scholar] [CrossRef] [PubMed]
- Bierhaus, A.; Hofmann, M.A.; Ziegler, R.; Nawroth, P.P. AGEs and Their Interaction with AGE-Receptors in Vascular Disease and Diabetes Mellitus. I. The AGE Concept. Cardiovasc. Res. 1998, 37, 586–600. [Google Scholar] [CrossRef] [Green Version]
- Laroque, D.; Inisan, C.; Berger, C.; Vouland, É.; Dufossé, L.; Guérard, F. Kinetic Study on the Maillard Reaction. Consideration of Sugar Reactivity. Food Chem. 2008, 111, 1032–1042. [Google Scholar] [CrossRef]
- Suárez, G.; Rajaram, R.; Oronsky, A.L.; Gawinowicz, M.A. Nonenzymatic Glycation of Bovine Serum Albumin by Fructose (Fructation). J. Biol. Chem. 1989, 264, 3674–3679. [Google Scholar] [CrossRef]
- Helsley, R.N.; Moreau, F.; Gupta, M.K.; Radulescu, A.; DeBosch, B.; Softic, S. Tissue-Specific Fructose Metabolism in Obesity and Diabetes. Curr. Diab. Rep. 2020, 20, 64. [Google Scholar] [CrossRef]
- Ott, C.; Jacobs, K.; Haucke, E.; Navarrete Santos, A.; Grune, T.; Simm, A. Role of Advanced Glycation End Products in Cellular Signaling. Redox Biol. 2014, 2, 411–429. [Google Scholar] [CrossRef] [Green Version]
- Thornalley, P.J.; Langborg, A.; Minhas, H.S. Formation of Glyoxal, Methylglyoxal and 8-Deoxyglucosone in the Glycation of Proteins by Glucose. Biochem. J. 1999, 344, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Perrone, A.; Giovino, A.; Benny, J.; Martinelli, F. Advanced Glycation End Products (AGEs): Biochemistry, Signaling, Analytical Methods, and Epigenetic Effects. Oxid. Med. Cell. Longev. 2020, 2020, 3818196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuzan, A. Toxicity of Advanced Glycation End Products (Review). Biomed. Rep. 2021, 14, 46. [Google Scholar] [CrossRef] [PubMed]
- Moheimani, F.; Morgan, P.E.; van Reyk, D.M.; Davies, M.J. Deleterious Effects of Reactive Aldehydes and Glycated Proteins on Macrophage Proteasomal Function: Possible Links between Diabetes and Atherosclerosis. Biochim. Biophys. Acta Mol. Basis Dis. 2010, 1802, 561–571. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, N.; Lüthen, R.; Häussinger, D.; Sebeková, K.; Schinzel, R.; Voelker, W.; Heidland, A.; Thornalley, P.J. Increased Protein Glycation in Cirrhosis and Therapeutic Strategies to Prevent It. Ann. N. Y. Acad. Sci. 2005, 1043, 718–724. [Google Scholar] [CrossRef]
- Kisugi, R.; Kouzuma, T.; Yamamoto, T.; Akizuki, S.; Miyamoto, H.; Someya, Y.; Yokoyama, J.; Abe, I.; Hirai, N.; Ohnishi, A. Structural and Glycation Site Changes of Albumin in Diabetic Patient with Very High Glycated Albumin. Clin. Chim. Acta. 2007, 382, 59–64. [Google Scholar] [CrossRef]
- Sharma, C.; Kaur, A.; Thind, S.S.; Singh, B.; Raina, S. Advanced Glycation End-Products (AGEs): An Emerging Concern for Processed Food Industries. J. Food Sci. Technol. 2015, 52, 7561–7576. [Google Scholar] [CrossRef] [Green Version]
- Verzijl, N.; Bank, R.A.; TeKoppele, J.M.; DeGroot, J. AGEing and Osteoarthritis: A Different Perspective. Curr. Opin. Rheumatol. 2003, 15, 616–622. [Google Scholar] [CrossRef]
- Vashishth, D.; Gibson, G.J.; Khoury, J.I.; Schaffler, M.B.; Kimura, J.; Fyhrie, D.P. Influence of Nonenzymatic Glycation on Biomechanical Properties of Cortical Bone. Bone 2001, 28, 195–201. [Google Scholar] [CrossRef]
- Cerami, C.; Founds, H.; Nicholl, I.; Mitsuhashi, T.; Giordano, D.; Vanpatten, S.; Lee, A.; Al-Abed, Y.; Vlassara, H.; Bucala, R.; et al. Tobacco Smoke Is a Source of Toxic Reactive Glycation Products. Proc. Natl. Acad. Sci. USA 1997, 94, 13915–13920. [Google Scholar] [CrossRef] [Green Version]
- Uribarri, J.; Woodruff, S.; Goodman, S.; Cai, W.; Chen, X.; Pyzik, R.; Yong, A.; Striker, G.E.; Vlassara, H. Advanced Glycation End Products in Foods and a Practical Guide to Their Reduction in the Diet. J. Am. Diet. Assoc. 2010, 110, 911–916.e12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koschinsky, T.; He, C.J.; Mitsuhashi, T.; Bucala, R.; Liu, C.; Buenting, C.; Heitmann, K.; Vlassara, H. Orally Absorbed Reactive Glycation Products (Glycotoxins): An Environmental Risk Factor in Diabetic Nephropathy. Proc. Natl. Acad. Sci. USA 1997, 94, 6474–6479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, C.; Sabol, J.; Mitsuhashi, T.; Vlassara, H. Dietary Glycotoxins: Inhibition of Reactive Products by Aminoguanidine Facilitates Renal Clearance and Reduces Tissue Sequestration. Diabetes 1999, 48, 1308–1315. [Google Scholar] [CrossRef]
- Cai, W.; He, J.C.; Zhu, L.; Chen, X.; Striker, G.E.; Vlassara, H. AGE-Receptor-1 Counteracts Cellular Oxidant Stress Induced by AGEs via Negative Regulation of P66shc-Dependent FKHRL1 Phosphorylation. Am. J. Physiol. Cell Physiol. 2008, 294, C145–C152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- AGE Database. Available online: https://lemchem.file3.wcms.tu-dresden.de// (accessed on 10 March 2022).
- Botros, N.; Sluik, D.; van Waateringe, R.P.; de Vries, J.H.M.; Geelen, A.; Feskens, E.J.M. Advanced Glycation End-Products (AGEs) and Associations with Cardio-Metabolic, Lifestyle, and Dietary Factors in a General Population: The NQplus Study. Diabetes. Metab. Res. Rev. 2017, 33, e2892. [Google Scholar] [CrossRef]
- Singh, R.; Barden, A.; Mori, T.; Beilin, L. Advanced Glycation End-Products: A Review. Diabetologia 2001, 44, 129–146. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, N.; Argirov, O.K.; Minhas, H.S.; Cordeiro, C.A.A.; Thornalley, P.J. Assay of Advanced Glycation Endproducts (AGEs): Surveying AGEs by Chromatographic Assay with Derivatization by 6-Aminoquinolyl-N-Hydroxysuccinimidyl-Carbamate and Application to Nε-Carboxymethyl-Lysine- and Nε-(1-Carboxyethyl)Lysine-Modified Albumin. Biochem. J. 2002, 364, 1–14. [Google Scholar] [CrossRef]
- Ashraf, J.M.; Ahmad, S.; Choi, I.; Ahmad, N.; Farhan, M.; Tatyana, G.; Shahab, U. Recent Advances in Detection of AGEs: Immunochemical, Bioanalytical and Biochemical Approaches. IUBMB Life 2015, 67, 897–913. [Google Scholar] [CrossRef]
- Thomas, C.J.; Cleland, T.P.; Sroga, G.E.; Vashishth, D. Accumulation of Carboxymethyl-Lysine (CML) in Human Cortical Bone. Bone 2018, 110, 128–133. [Google Scholar] [CrossRef]
- Deluyker, D.; Evens, L.; Bito, V. Advanced Glycation End Products (AGEs) and Cardiovascular Dysfunction: Focus on High Molecular Weight AGEs. Amino Acids 2017, 49, 1535–1541. [Google Scholar] [CrossRef]
- Băbţan, A.M.; Ilea, A.; Boşca, B.A.; Crişan, M.; Petrescu, N.B.; Collino, M.; Sainz, R.M.; Gerlach, J.Q.; Câmpian, R.S. Advanced Glycation End Products as Biomarkers in Systemic Diseases: Premises and Perspectives of Salivary Advanced Glycation End Products. Biomark. Med. 2019, 13, 479–495. [Google Scholar] [CrossRef] [PubMed]
- Salazar, J.; Navarro, C.; Ortega, Á.; Nava, M.; Morillo, D.; Torres, W.; Hernández, M.; Cabrera, M.; Angarita, L.; Ortiz, R.; et al. Advanced Glycation End Products: New Clinical and Molecular Perspectives. Int. J. Environ. Res. Public Health 2021, 18, 7236. [Google Scholar] [CrossRef] [PubMed]
- Gerdemann, A.; Lemke, H.D.; Nothdurft, A.; Heidland, A.; Münch, G.; Bahner, U.; Schinzel, R. Low-Molecular but Not High-Molecular Advanced Glycation End Products (AGEs) Are Removed by High-Flux Dialysis. Clin. Nephrol. 2000, 54, 276–283. [Google Scholar] [PubMed]
- Thomas, M.C.; Forbes, J.M.; MacIsaac, R.; Jerums, G.; Cooper, M.E. Low-Molecular Weight Advanced Glycation End Products: Markers of Tissue AGE Accumulation and More? Ann. N. Y. Acad. Sci. 2005, 1043, 644–654. [Google Scholar] [CrossRef]
- Grzebyk, E.; Knapik-Kordecka, M.; Piwowar, A. Advanced Glycation End-Products and Cathepsin Cysteine Protease in Type 2 Diabetic Patients. Pol. Arch. Med. Wewn. 2013, 123, 364–370. [Google Scholar] [CrossRef] [Green Version]
- Poulsen, M.W.; Andersen, J.M.; Hedegaard, R.V.; Madsen, A.N.; Krath, B.N.; Monošík, R.; Bak, M.J.; Nielsen, J.; Holst, B.; Skibsted, L.H.; et al. Short-Term Effects of Dietary Advanced Glycation End Products in Rats. Br. J. Nutr. 2016, 115, 629–636. [Google Scholar] [CrossRef]
- Hellwig, M.; Geissler, S.; Matthes, R.; Peto, A.; Silow, C.; Brandsch, M.; Henle, T. Transport of Free and Peptide-Bound Glycated Amino Acids: Synthesis, Transepithelial Flux at Caco-2 Cell Monolayers, and Interaction with Apical Membrane Transport Proteins. ChemBioChem 2011, 12, 1270–1279. [Google Scholar] [CrossRef]
- Takeuchi, M. Toxic AGEs (TAGE) Theory: A New Concept for Preventing the Development of Diseases Related to Lifestyle. Diabetol. Metab. Syndr. 2020, 12, 105. [Google Scholar] [CrossRef]
- Ahmed, M.U.; Dunn, J.A.; Walla, M.D.; Thorpe, S.R.; Baynes, J.W. Oxidative Degradation of Glucose Adducts to Protein. Formation of 3-(N(ε)-Lysino)-Lactic Acid from Model Compounds and Glycated Proteins. J. Biol. Chem. 1988, 263, 8816–8821. [Google Scholar] [CrossRef]
- Sato, T.; Shimogaito, N.; Wu, X.; Kikuchi, S.; Yamagishi, S.; Takeuchi, M. Toxic Advanced Glycation End Products (TAGE) Theory in Alzheimer’s Disease. Am. J. Alzheimers. Dis. Other Demen. 2006, 21, 197–208. [Google Scholar] [CrossRef]
- Takeuchi, M.; Yamagishi, S. TAGE (Toxic AGEs) Hypothesis in Various Chronic Diseases. Med. Hypotheses 2004, 63, 449–452. [Google Scholar] [CrossRef]
- Takeuchi, M.; Sakasai-Sakai, A.; Takata, T.; Takino, J.; Koriyama, Y.; Kikuchi, C.; Furukawa, A.; Nagamine, K.; Hori, T.; Matsunaga, T. Intracellular Toxic AGEs (TAGE) Triggers Numerous Types of Cell Damage. Biomolecules 2021, 11, 387. [Google Scholar] [CrossRef] [PubMed]
- WHO. Available online: https://www.who.int/news-room/detail/11-10-2016-who-urges-global-action-to-curtail-consumption-and-health-impacts-of-sugary-drinks (accessed on 10 March 2022).
- Tamura, Y.; Adachi, H.; Osuga, J.I.; Ohashi, K.; Yahagi, N.; Sekiya, M.; Okazaki, H.; Tomita, S.; Iizuka, Y.; Shimano, H.; et al. FEEL-1 and FEEL-2 Are Endocytic Receptors for Advanced Glycation End Products. J. Biol. Chem. 2003, 278, 12613–12617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neeper, M.; Schmidt, A.M.; Brett, J.; Yan, S.D.; Wang, F.; Pan, Y.C.; Elliston, K.; Stern, D.; Shaw, A. Cloning and Expression of a Cell Surface Receptor for Advanced Glycosylation End Products of Proteins. J. Biol. Chem. 1992, 267, 14998–15004. [Google Scholar] [CrossRef]
- Politz, O.; Gratchev, A.; McCourt, P.A.G.; Schledzewski, K.; Guillot, P.; Johansson, S.; Svineng, G.; Franke, P.; Kannicht, C.; Kzhyshkowska, J.; et al. Stabilin-1 and -2 Constitute a Novel Family of Fasciclin-like Hyaluronan Receptor Homologues. Biochem. J. 2002, 362, 155–164. [Google Scholar] [CrossRef]
- Matsumoto, A.; Naito, M.; Itakura, H.; Ikemoto, S.; Asaoka, H.; Hayakawa, I.; Kanamori, H.; Aburatani, H.; Takaku, F.; Suzuki, H.; et al. Human Macrophage Scavenger Receptors: Primary Structure, Expression, and Localization in Atherosclerotic Lesions. Proc. Natl. Acad. Sci. USA 1990, 87, 9133–9137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calvo, D.; Vega, M.A. Identification, Primary Structure, and Distribution of CLA-1, a Novel Member of the CD36/LIMPII Gene Family. J. Biol. Chem. 1993, 268, 18929–18935. [Google Scholar] [CrossRef]
- Sawamura, T.; Kume, N.; Aoyama, T.; Moriwaki, H.; Hoshikawa, H.; Aiba, Y.; Tanaka, T.; Miwa, S.; Katsura, Y.; Kita, T.; et al. An Endothelial Receptor for Oxidized Low-Density Lipoprotein. Nature 1997, 386, 73–77. [Google Scholar] [CrossRef]
- Yamagata, T.; Tsuru, T.; Momoi, M.Y.; Suwa, K.; Nozaki, Y.; Mukasa, T.; Ohashi, H.; Fukushima, Y.; Momoi, T. Genome Organization of Human 48-KDa Oligosaccharyltransferase (DDOST). Genomics 1997, 45, 535–540. [Google Scholar] [CrossRef]
- Sakai, K.; Hirai, M.; Minoshima, S.; Kudoh, J.; Fukuyama, R.; Shimizu, N. Isolation of CDNAs Encoding a Substrate for Protein Kinase C: Nucleotide Sequence and Chromosomal Mapping of the Gene for a Human 80K Protein. Genomics 1989, 5, 309–315. [Google Scholar] [CrossRef]
- Cherayil, B.J.; Chaitovitz, S.; Wong, C.; Pillai, S. Molecular Cloning of a Human Macrophage Lectin Specific for Galactose. Proc. Natl. Acad. Sci. USA 1990, 87, 7324–7328. [Google Scholar] [CrossRef] [Green Version]
- Jules, J.; Maiguel, D.; Hudson, B.I. Alternative Splicing of the RAGE Cytoplasmic Domain Regulates Cell Signaling and Function. PLoS ONE 2013, 8, e78267. [Google Scholar] [CrossRef] [Green Version]
- Kalea, A.Z.; Schmidt, A.M.; Hudson, B.I. Alternative Splicing of RAGE: Roles in Biology and Disease. Front. Biosci. (Landmark Ed.) 2011, 16, 2756–2770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kislinger, T.; Fu, C.; Huber, B.; Qu, W.; Taguchi, A.; Yan, S.D.; Hofmann, M.; Yan, S.F.; Pischetsrieder, M.; Stern, D.; et al. N(ε)-(Carboxymethyl)Lysine Adducts of Proteins Are Ligands for Receptor for Advanced Glycation End Products That Activate Cell Signaling Pathways and Modulate Gene Expression. J. Biol. Chem. 1999, 274, 31740–31749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horiuchi, S.; Sakamoto, Y.; Sakai, M. Scavenger Receptors for Oxidized and Glycated Proteins. Amino Acids 2003, 25, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Koch, M.; Chitayat, S.; Dattilo, B.M.; Schiefner, A.; Diez, J.; Chazin, W.J.; Fritz, G. Structural Basis for Ligand Recognition and Activation of RAGE. Structure 2010, 18, 1342–1352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, H.; Adsit, F.G.; Boyington, J.C. The 1.5 Å Crystal Structure of Human Receptor for Advanced Glycation Endproducts (RAGE) Ectodomains Reveals Unique Features Determining Ligand Binding. J. Biol. Chem. 2010, 285, 40762–40770. [Google Scholar] [CrossRef] [Green Version]
- Yatime, L.; Andersen, G.R. Structural Insights into the Oligomerization Mode of the Human Receptor for Advanced Glycation End-Products. FEBS J. 2013, 280, 6556–6568. [Google Scholar] [CrossRef] [Green Version]
- Sirois, C.M.; Jin, T.; Miller, A.L.; Bertheloot, D.; Nakamura, H.; Horvath, G.L.; Mian, A.; Jiang, J.; Schrum, J.; Bossaller, L.; et al. RAGE Is a Nucleic Acid Receptor That Promotes Inflammatory Responses to DNA. J. Exp. Med. 2013, 210, 2447–2463. [Google Scholar] [CrossRef] [Green Version]
- Yatime, L.; Betzer, C.; Jensen, R.K.; Mortensen, S.; Jensen, P.H.; Andersen, G.R. The Structure of the RAGE:S100A6 Complex Reveals a Unique Mode of Homodimerization for S100 Proteins. Structure 2016, 24, 2043–2052. [Google Scholar] [CrossRef] [Green Version]
- Kozlyuk, N.; Gilston, B.A.; Salay, L.E.; Gogliotti, R.D.; Christov, P.P.; Kim, K.; Ovee, M.; Waterson, A.G.; Chazin, W.J. A Fragment-Based Approach to Discovery of Receptor for Advanced Glycation End Products Inhibitors. Proteins Struct. Funct. Bioinforma. 2021, 89, 1399–1412. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Rai, V.; Singer, D.; Chabierski, S.; Xie, J.; Reverdatto, S.; Burz, D.S.; Schmidt, A.M.; Hoffmann, R.; Shekhtman, A. Advanced Glycation End Product Recognition by the Receptor for AGEs. Structure 2011, 19, 722–732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobori, T.; Ganesh, D.; Kumano-Kuramochi, M.; Torigoe, K.; Machida, S. Assay for Advanced Glycation End Products Generating Intracellular Oxidative Stress through Binding to Its Receptor. Anal. Biochem. 2020, 611, 114018. [Google Scholar] [CrossRef] [PubMed]
- Bucciarelli, L.G.; Wendt, T.; Rong, L.; Lalla, E.; Hofmann, M.A.; Goova, M.T.; Taguchi, A.; Yan, S.F.; Yan, S.D.; Stern, D.M.; et al. RAGE Is a Multiligand Receptor of the Immunoglobulin Superfamily: Implications for Homeostasis and Chronic Disease. Cell. Mol. Life Sci. 2002, 59, 1117–1128. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.M.; Yan, S.D.; Yan, S.F.; Stern, D.M. The Biology of the Receptor for Advanced Glycation End Products and Its Ligands. Biochim. Biophys. Acta Mol. Cell Res. 2000, 1498, 99–111. [Google Scholar] [CrossRef] [Green Version]
- Xie, J.; Méndez, J.D.; Méndez-Valenzuela, V.; Aguilar-Hernández, M.M. Cellular Signalling of the Receptor for Advanced Glycation End Products (RAGE). Cell. Signal. 2013, 25, 2185–2197. [Google Scholar] [CrossRef]
- Erusalimsky, J.D. The Use of the Soluble Receptor for Advanced Glycation-End Products (SRAGE) as a Potential Biomarker of Disease Risk and Adverse Outcomes. Redox Biol. 2021, 42, 101958. [Google Scholar] [CrossRef]
- Chiappalupi, S.; Salvadori, L.; Vukasinovic, A.; Donato, R.; Sorci, G.; Riuzzi, F. Targeting RAGE to Prevent SARS-CoV-2-Mediated Multiple Organ Failure: Hypotheses and Perspectives. Life Sci. 2021, 272, 119251. [Google Scholar] [CrossRef]
- Sellegounder, D.; Zafari, P.; Rajabinejad, M.; Taghadosi, M.; Kapahi, P. Advanced Glycation End Products (AGEs) and Its Receptor, RAGE, Modulate Age-Dependent COVID-19 Morbidity and Mortality. A Review and Hypothesis. Int. Immunopharmacol. 2021, 98, 107806. [Google Scholar] [CrossRef]
- Stilhano, R.S.; Costa, A.J.; Nishino, M.S.; Shams, S.; Bartolomeo, C.S.; Breithaupt-Faloppa, A.C.; Silva, E.A.; Ramirez, A.L.; Prado, C.M.; Ureshino, R.P. SARS-CoV-2 and the Possible Connection to ERs, ACE2, and RAGE: Focus on Susceptibility Factors. FASEB J. 2020, 34, 14103–14119. [Google Scholar] [CrossRef]
- Smedsrød, B.; Melkko, J.; Araki, N.; Sano, H.; Horiuchi, S. Advanced Glycation End Products Are Eliminated by Scavenger-Receptor-Mediated Endocytosis in Hepatic Sinusoidal Kupffer and Endothelial Cells. Biochem. J. 1997, 322, 567–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, B.; Svistounov, D.; Olsen, R.; Nagai, R.; Horiuchi, S.; Smedsrød, B. Advanced Glycation End Products Impair the Scavenger Function of Rat Hepatic Sinusoidal Endothelial Cells. Diabetologia 2002, 45, 1379–1388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shetty, S.; Lalor, P.F.; Adams, D.H. Liver Sinusoidal Endothelial Cells—Gatekeepers of Hepatic Immunity. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 555–567. [Google Scholar] [CrossRef] [PubMed]
- Hare, A.K.; Harris, E.N. Tissue-Specific Splice Variants of HARE/Stabilin-2 Are Expressed in Bone Marrow, Lymph Node, and Spleen. Biochem. Biophys. Res. Commun. 2015, 456, 257–261. [Google Scholar] [CrossRef] [Green Version]
- Adachi, H.; Tsujimoto, M. FEEL-1, a Novel Scavenger Receptor with in Vitro Bacteria-Binding and Angiogenesis-Modulating Activities. J. Biol. Chem. 2002, 277, 34264–34270. [Google Scholar] [CrossRef] [Green Version]
- Kzhyshkowska, J.; Mamidi, S.; Gratchev, A.; Kremmer, E.; Schmuttermaier, C.; Krusell, L.; Haus, G.; Utikal, J.; Schledzewski, K.; Scholtze, J.; et al. Novel Stabilin-1 Interacting Chitinase-like Protein (SI-CLP) Is up-Regulated in Alternatively Activated Macrophages and Secreted via Lysosomal Pathway. Blood 2006, 107, 3221–3228. [Google Scholar] [CrossRef] [Green Version]
- Harris, E.N.; Weigel, P.H. The Ligand-Binding Profile of HARE: Hyaluronan and Chondroitin Sulfates A, C, and D Bind to Overlapping Sites Distinct from the Sites for Heparin, Acetylated Low-Density Lipoprotein, Dermatan Sulfate, and CS-E. Glycobiology 2008, 18, 638–648. [Google Scholar] [CrossRef] [Green Version]
- Pandey, M.S.; Weigel, P.H. A Hyaluronan Receptor for Endocytosis (HARE) Link Domain N-Glycan Is Required for Extracellular Signal-Regulated Kinase (ERK) and Nuclear Factor-ΚB (NF-ΚB) Signaling in Response to the Uptake of Hyaluronan but Not Heparin, Dermatan Sulfate, or Acetylated. J. Biol. Chem. 2014, 289, 21807–21817. [Google Scholar] [CrossRef] [Green Version]
- Blundell, C.D.; Almond, A.; Mahoney, D.J.; DeAngelis, P.L.; Campbell, I.D.; Day, A.J. Towards a Structure for a TSG-6.Hyaluronan Complex by Modeling and NMR Spectroscopy: Insights into Other Members of the Link Module Superfamily. J. Biol. Chem. 2005, 280, 18189–18201. [Google Scholar] [CrossRef] [Green Version]
- Park, S.Y.; Jung, M.Y.; Kim, I.S. Stabilin-2 Mediates Homophilic Cell-Cell Interactions via Its FAS1 Domains. FEBS Lett. 2009, 583, 1375–1380. [Google Scholar] [CrossRef] [Green Version]
- Jung, M.-Y.; Park, S.-Y.; Kim, I.-S. Stabilin-2 Is Involved in Lymphocyte Adhesion to the Hepatic Sinusoidal Endothelium via the Interaction with AMβ2 Integrin. J. Leukoc. Biol. 2007, 82, 1156–1165. [Google Scholar] [CrossRef] [PubMed]
- Twarda-Clapa, A.; Labuzek, B.; Krzemien, D.; Musielak, B.; Grudnik, P.; Dubin, G.; Holak, T.A. Crystal Structure of the FAS1 Domain of the Hyaluronic Acid Receptor Stabilin-2. Acta Crystallogr. Sect. D Struct. Biol. 2018, 74, 695–701. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-Y.; Kim, S.-Y.; Jung, M.-Y.; Bae, D.-J.; Kim, I.-S. Epidermal Growth Factor-Like Domain Repeat of Stabilin-2 Recognizes Phosphatidylserine during Cell Corpse Clearance. Mol. Cell. Biol. 2008, 28, 5288–5298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandey, M.S.; Harris, E.N.; Weigel, J.A.; Weigel, P.H. The Cytoplasmic Domain of the Hyaluronan Receptor for Endocytosis (Hare) Contains Multiple Endocytic Motifs Targeting Coated Pit-Mediated Internalization. J. Biol. Chem. 2008, 283, 21453–21461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.Y.; Yun, Y.; Lim, J.S.; Kim, M.J.; Kim, S.Y.; Kim, J.E.; Kim, I.S. Stabilin-2 Modulates the Efficiency of Myoblast Fusion during Myogenic Differentiation and Muscle Regeneration. Nat. Commun. 2016, 7, 10871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirose, Y.; Saijou, E.; Sugano, Y.; Takeshita, F.; Nishimura, S.; Nonaka, H.; Chen, Y.R.; Sekine, K.; Kido, T.; Nakamura, T.; et al. Inhibition of Stabilin-2 Elevates Circulating Hyaluronic Acid Levels and Prevents Tumor Metastasis. Proc. Natl. Acad. Sci. USA 2012, 109, 4263–4268. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; McCourt, P.; Schledzewski, K.; Goerdt, S.; Moldenhauer, G.; Liu, X.; Smedsrød, B.; Sørensen, K.K. Endocytosis of Advanced Glycation End-Products in Bovine Choriocapillaris Endothelial Cells. Microcirculation 2009, 16, 640–655. [Google Scholar] [CrossRef]
- Campbell, F.; Bos, F.L.; Sieber, S.; Arias-Alpizar, G.; Koch, B.E.; Huwyler, J.; Kros, A.; Bussmann, J. Directing Nanoparticle Biodistribution through Evasion and Exploitation of Stab2-Dependent Nanoparticle Uptake. ACS Nano 2018, 12, 2138–2150. [Google Scholar] [CrossRef]
- Miller, C.M.; Donner, A.J.; Blank, E.E.; Egger, A.W.; Kellar, B.M.; Østergaard, M.E.; Seth, P.P.; Harris, E.N. Stabilin-1 and Stabilin-2 Are Specific Receptors for the Cellular Internalization of Phosphorothioate-Modified Antisense Oligonucleotides (ASOs) in the Liver. Nucleic Acids Res. 2016, 44, 2782–2794. [Google Scholar] [CrossRef]
- Banerji, S.; Wright, A.J.; Noble, M.; Mahoney, D.J.; Campbell, I.D.; Day, A.J.; Jackson, D.G. Structures of the Cd44-Hyaluronan Complex Provide Insight into a Fundamental Carbohydrate-Protein Interaction. Nat. Struct. Mol. Biol. 2007, 14, 234–239. [Google Scholar] [CrossRef]
- Kumar Pasupulati, A.; Chitra, P.S.; Reddy, G.B. Advanced Glycation End Products Mediated Cellular and Molecular Events in the Pathology of Diabetic Nephropathy. Biomol. Concepts 2016, 7, 293–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldin, A.; Beckman, J.A.; Schmidt, A.M.; Creager, M.A. Advanced Glycation End Products: Sparking the Development of Diabetic Vascular Injury. Circulation 2006, 114, 597–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stitt, A.W.; Bucala, R.; Vlassara, H. Atherogenesis and Advanced Glycation: Promotion, Progression, and Prevention. Ann. N. Y. Acad. Sci. 1997, 811, 115–129. [Google Scholar] [CrossRef] [PubMed]
- Acton, S.L.; Scherer, P.E.; Lodish, H.F.; Krieger, M. Expression Cloning of SR-BI, a CD36-Related Class B Scavenger Receptor. J. Biol. Chem. 1994, 269, 21003–21009. [Google Scholar] [CrossRef]
- Kawasaki, Y.; Nakagawa, A.; Nagaosa, K.; Shiratsuchi, A.; Nakanishi, Y. Phosphatidylserine Binding of Class B Scavenger Receptor Type I, a Phagocytosis Receptor of Testicular Sertoli Cells. J. Biol. Chem. 2002, 277, 27559–27566. [Google Scholar] [CrossRef] [Green Version]
- Philips, J.A.; Rubin, E.J.; Perrimon, N. Drosophila RNAi Screen Reveals CD36 Family Member Required for Mycobacterial Infection. Science 2005, 309, 1251–1253. [Google Scholar] [CrossRef] [Green Version]
- Scarselli, E.; Ansuini, H.; Cerino, R.; Roccasecca, R.M.; Acali, S.; Filocamo, G.; Traboni, C.; Nicosia, A.; Cortese, R.; Vitelli, A. The Human Scavenger Receptor Class B Type I Is a Novel Candidate Receptor for the Hepatitis C Virus. EMBO J. 2002, 21, 5017–5025. [Google Scholar] [CrossRef] [Green Version]
- Wei, C.; Wan, L.; Yan, Q.; Wang, X.; Zhang, J.; Yang, X.; Zhang, Y.; Fan, C.; Li, D.; Deng, Y.; et al. HDL-Scavenger Receptor B Type 1 Facilitates SARS-CoV-2 Entry. Nat. Metab. 2020, 2, 1391–1400. [Google Scholar] [CrossRef]
- Febbraio, M.; Hajjar, D.P.; Silverstein, R.L. CD36: A Class B Scavenger Receptor Involved in Angiogenesis, Atherosclerosis, Inflammation, and Lipid Metabolism. J. Clin. Investig. 2001, 108, 785–791. [Google Scholar] [CrossRef]
- Nakajou, K.; Horiuchi, S.; Sakai, M.; Hirata, K.; Tanaka, M.; Takeya, M.; Kai, T.; Otagiri, M. CD36 Is Not Involved in Scavenger Receptor-Mediated Endocytic Uptake of Glycolaldehyde- and Methylglyoxal-Modified Proteins by Liver Endothelial Cells. J. Biochem. 2005, 137, 607–616. [Google Scholar] [CrossRef]
- Horiuchi, S.; Unno, Y.; Usui, H.; Shikata, K.; Takaki, K.; Koito, W.; Sakamoto, Y.I.; Nagai, R.; Makino, K.; Sasao, A.; et al. Pathological Roles of Advanced Glycation End Product Receptors SR-A and CD36. Ann. N. Y. Acad. Sci. 2005, 1043, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Ohgami, N.; Nagai, R.; Miyazaki, A.; Ikemoto, M.; Arai, H.; Horiuchi, S.; Nakayama, H. Scavenger Receptor Class B Type I-Mediated Reverse Cholesterol Transport Is Inhibited by Advanced Glycation End Products. J. Biol. Chem. 2001, 276, 13348–13355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jono, T.; Miyazaki, A.; Nagai, R.; Sawamura, T.; Kitamura, T.; Horiuchi, S. Lectin-like Oxidized Low Density Lipoprotein Receptor-1 (LOX-1) Serves as an Endothelial Receptor for Advanced Glycation End Products (AGE). FEBS Lett. 2002, 511, 170–174. [Google Scholar] [CrossRef] [Green Version]
- Cominacini, L.; Rigoni, A.; Pasini, A.F.; Garbin, U.; Davoli, A.; Campagnola, M.; Pastorino, A.M.; Cascio, V.L.; Sawamura, T. The Binding of Oxidized Low Density Lipoprotein (Ox-LDL) to Ox-LDL Receptor-1 Reduces the Intracellular Concentration of Nitric Oxide in Endothelial Cells through an Increased Production of Superoxide. J. Biol. Chem. 2001, 276, 13750–13755. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Nagase, M.; Fujita, T.; Narumiya, S.; Masaki, T.; Sawamura, T. Diabetes Enhances Lectin-like Oxidized LDL Receptor-1 (LOX-1) Expression in the Vascular Endothelium: Possible Role of LOX-1 Ligand and AGE. Biochem. Biophys. Res. Commun. 2001, 287, 962–968. [Google Scholar] [CrossRef]
- Park, H.J.; Adsit, F.G.; Boyington, J.C. The 1.4 Å Crystal Structure of the Human Oxidized Low Density Lipoprotein Receptor Lox-1. J. Biol. Chem. 2005, 280, 13593–13599. [Google Scholar] [CrossRef] [Green Version]
- Nakano, S.; Sugihara, M.; Yamada, R.; Katayanagi, K.; Tate, S. Structural Implication for the Impaired Binding of W150A Mutant LOX-1 to Oxidized Low Density Lipoprotein, OxLDL. Biochim. Biophys. Acta 2012, 1824, 739–749. [Google Scholar] [CrossRef] [Green Version]
- Roboti, P.; High, S. The Oligosaccharyltransferase Subunits OST48, DAD1 and KCP2 Function as Ubiquitous and Selective Modulators of Mammalian N-Glycosylation. J. Cell Sci. 2012, 125, 3474–3484. [Google Scholar] [CrossRef] [Green Version]
- Pelletier, M.F.; Marcil, A.; Sevigny, G.; Jakob, C.A.; Tessier, D.C.; Chevet, E.; Menard, R.; Bergeron, J.J.M.; Thomas, D.Y. The Heterodimeric Structure of Glucosidase II Is Required for Its Activity, Solubility, and Localization in Vivo. Glycobiology 2000, 10, 815–827. [Google Scholar] [CrossRef] [Green Version]
- Fukushi, J.; Makagiansar, I.T.; Stallcup, W.B. NG2 Proteoglycan Promotes Endothelial Cell Motility and Angiogenesis via Engagement of Galectin-3 and Alpha3beta1 Integrin. Mol. Biol. Cell 2004, 15, 3580–3590. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, A.; Forbes, J.M. Diabetic Kidney Disease: A Role for Advanced Glycation End-Product Receptor 1 (AGE-R1)? Glycoconj. J. 2016, 33, 645–652. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.L.Y.; Forbes, J.M.; Cooper, M.E. AGE, RAGE, and ROS in Diabetic Nephropathy. Semin. Nephrol. 2007, 27, 130–143. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zhu, L.; Zheng, F.; Vlassara, H. Overexpression of AGE-Receptor-1 (AGE-R1) in Mice Prevent AGE Accumulation and Delays Diabetic Renal Injury. Diabetes 2005, 54, A21-B. [Google Scholar]
- Lu, C.; He, J.C.; Cai, W.; Liu, H.; Zhu, L.; Vlassara, H. Advanced Glycation Endproduct (AGE) Receptor 1 Is a Negative Regulator of the Inflammatory Response to AGE in Mesangial Cells. Proc. Natl. Acad. Sci. USA 2004, 101, 11767–11772. [Google Scholar] [CrossRef] [Green Version]
- Yap, F.Y.T.; Kantharidis, P.; Coughlan, M.T.; Slattery, R.; Forbes, J.M. Advanced Glycation End Products as Environmental Risk Factors for the Development of Type 1 Diabetes. Curr. Drug Targets 2012, 13, 526–540. [Google Scholar] [CrossRef]
- Li, Y.M.; Mitsuhashi, T.; Wojciechowicz, D.; Shimizu, N.; Li, J.; Stitt, A.; He, C.; Banerjee, D.; Vlassara, H. Molecular Identity and Cellular Distribution of Advanced Glycation Endproduct Receptors: Relationship of P60 to OST-48 and P90 to 80K-H Membrane Proteins. Proc. Natl. Acad. Sci. USA 1996, 93, 11047–11052. [Google Scholar] [CrossRef] [Green Version]
- Vlassara, H.; Li, Y.M.; Imani, F.; Wojciechowicz, D.; Yang, Z.; Liu, F.T.; Cerami, A. Identification of Galectin-3 as a High-Affinity Binding Protein for Advanced Glycation End Products (AGE): A New Member of the AGE-Receptor Complex. Mol. Med. 1995, 1, 634–646. [Google Scholar] [CrossRef] [Green Version]
- Casselmann, C.; Reimann, A.; Friedrich, I.; Schubert, A.; Silber, R.E.; Simm, A. Age-Dependent Expression of Advanced Glycation End Product Receptor Genes in the Human Heart. Gerontology 2004, 50, 127–134. [Google Scholar] [CrossRef]
- Gill, V.; Kumar, V.; Singh, K.; Kumar, A.; Kim, J.J. Advanced Glycation End Products (AGEs) May Be a Striking Link between Modern Diet and Health. Biomolecules 2019, 9, 888. [Google Scholar] [CrossRef] [Green Version]
- Nie, C.; Li, Y.; Qian, H.; Ying, H.; Wang, L. Advanced Glycation End Products in Food and Their Effects on Intestinal Tract. Crit. Rev. Food Sci. Nutr. 2022, 62, 3103–3115. [Google Scholar] [CrossRef]
- Zhao, D.; Li, L.; Le, T.T.; Larsen, L.B.; Su, G.; Liang, Y.; Li, B. Digestibility of Glyoxal-Glycated β-Casein and β-Lactoglobulin and Distribution of Peptide-Bound Advanced Glycation End Products in Gastrointestinal Digests. J. Agric. Food Chem. 2017, 65, 5778–5788. [Google Scholar] [CrossRef] [PubMed]
- Serin, Y.; Akbulut, G.; Uğur, H.; Yaman, M. Recent Developments in In-Vitro Assessment of Advanced Glycation End Products. Curr. Opin. Food Sci. 2021, 40, 136–143. [Google Scholar] [CrossRef]
- Clarke, R.E.; Dordevic, A.L.; Tan, S.M.; Ryan, L.; Coughlan, M.T. Dietary Advanced Glycation End Products and Risk Factors for Chronic Disease: A Systematic Review of Randomised Controlled Trials. Nutrients 2016, 8, 125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nowotny, K.; Schröter, D.; Schreiner, M.; Grune, T. Dietary Advanced Glycation End Products and Their Relevance for Human Health. Ageing Res. Rev. 2018, 47, 55–66. [Google Scholar] [CrossRef]
- Zhao, D.; Sheng, B.; Wu, Y.; Li, H.; Xu, D.; Nian, Y.; Mao, S.; Li, C.; Xu, X.; Zhou, G. Comparison of Free and Bound Advanced Glycation End Products in Food: A Review on the Possible Influence on Human Health. J. Agric. Food Chem. 2019, 67, 14007–14018. [Google Scholar] [CrossRef] [PubMed]
- Moscovici, A.M.; Joubran, Y.; Briard-Bion, V.; MacKie, A.; Dupont, D.; Lesmes, U. The Impact of the Maillard Reaction on the in Vitro Proteolytic Breakdown of Bovine Lactoferrin in Adults and Infants. Food Funct. 2014, 5, 1898–1908. [Google Scholar] [CrossRef] [PubMed]
- Hellwig, M.; Matthes, R.; Peto, A.; Lobner, J.; Henle, T. N-Epsilon-Fructosyllysine and N-Epsilon-Carboxymethyllysine, but Not Lysinoalanine, Are Available for Absorption after Simulated Gastrointestinal Digestion. Amino Acids 2014, 46, 289–299. [Google Scholar] [CrossRef]
- Geissler, S.; Michael, H.; Madlen, Z.; Fritz, M.; Thomas, H.; Matthias, B. Transport of the Advanced Glycation End Products Alanylpyrraline and Pyrralylalanine by the Human Proton-Coupled Peptide Transporter HPEPT1. J. Agric. Food Chem. 2010, 58, 2543–2547. [Google Scholar] [CrossRef]
- Chen, Y.; Guo, T.L. Dietary Advanced Glycation End-Products Elicit Toxicological Effects by Disrupting Gut Microbiome and Immune Homeostasis. J. Immunotoxicol. 2021, 18, 93–104. [Google Scholar] [CrossRef]
- Hellwig, M.; Auerbach, C.; Müller, N.; Samuel, P.; Kammann, S.; Beer, F.; Gunzer, F.; Henle, T. Metabolization of the Advanced Glycation End Product N-ϵ-Carboxymethyllysine (CML) by Different Probiotic E. Coli Strains. J. Agric. Food Chem. 2019, 67, 1963–1972. [Google Scholar] [CrossRef]
- Bui, T.P.N.; Troise, A.D.; Fogliano, V.; De Vos, W.M. Anaerobic Degradation of N-ϵ-Carboxymethyllysine, a Major Glycation End-Product, by Human Intestinal Bacteria. J. Agric. Food Chem. 2019, 67, 6594–6602. [Google Scholar] [CrossRef] [PubMed]
- Mastrocola, R.; Collotta, D.; Gaudioso, G.; Le Berre, M.; Cento, A.S.; Ferreira, G.A.; Chiazza, F.; Verta, R.; Bertocchi, I.; Manig, F.; et al. Effects of Exogenous Dietary Advanced Glycation End Products on the Cross-Talk Mechanisms Linking Microbiota to Metabolic Inflammation. Nutrients 2020, 12, 2497. [Google Scholar] [CrossRef] [PubMed]
- Yacoub, R.; Nugent, M.; Cai, W.; Nadkarni, G.N.; Chaves, L.D.; Abyad, S.; Honan, A.M.; Thomas, S.A.; Zheng, W.; Valiyaparambil, S.A.; et al. Advanced Glycation End Products Dietary Restriction Effects on Bacterial Gut Microbiota in Peritoneal Dialysis Patients; A Randomized Open Label Controlled Trial. PLoS ONE 2017, 12, e0184789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snelson, M.; Coughlan, M.T. Dietary Advanced Glycation End Products: Digestion, Metabolism and Modulation of Gut Microbial Ecology. Nutrients 2019, 11, 215. [Google Scholar] [CrossRef] [Green Version]
- Pugin, B.; Barcik, W.; Westermann, P.; Heider, A.; Wawrzyniak, M.; Hellings, P.; Akdis, C.A.; O’Mahony, L. A Wide Diversity of Bacteria from the Human Gut Produces and Degrades Biogenic Amines. Microb. Ecol. Health Dis. 2017, 28, 1353881. [Google Scholar] [CrossRef]
- Scheijen, J.L.J.M.; Hanssen, N.M.J.; van Greevenbroek, M.M.; Van der Kallen, C.J.; Feskens, E.J.M.; Stehouwer, C.D.A.; Schalkwijk, C.G. Dietary Intake of Advanced Glycation Endproducts Is Associated with Higher Levels of Advanced Glycation Endproducts in Plasma and Urine: The CODAM Study. Clin. Nutr. 2018, 37, 919–925. [Google Scholar] [CrossRef]
- Delgado-Andrade, C.; Tessier, F.J.; Niquet-Leridon, C.; Seiquer, I.; Navarro, M.P. Study of the Urinary and Faecal Excretion of N e -Carboxymethyllysine in Young Human Volunteers. Amino Acids 2012, 43, 595–602. [Google Scholar] [CrossRef]
- Palaseweenun, P.; Hagen-Plantinga, E.A.; Schonewille, J.T.; Koop, G.; Butre, C.; Jonathan, M.; Wierenga, P.A.; Hendriks, W.H. Urinary Excretion of Advanced Glycation End Products in Dogs and Cats. J. Anim. Physiol. Anim. Nutr. 2021, 105, 149–156. [Google Scholar] [CrossRef] [Green Version]
- Minekus, M.; Alminger, M.; Alvito, P.; Ballance, S.; Bohn, T.; Bourlieu, C.; Carrière, F.; Boutrou, R.; Corredig, M.; Dupont, D.; et al. A Standardised Static in Vitro Digestion Method Suitable for Food-an International Consensus. Food Funct. 2014, 5, 1113–1124. [Google Scholar] [CrossRef] [Green Version]
- Brodkorb, A.; Egger, L.; Alminger, M.; Alvito, P.; Assunção, R.; Ballance, S.; Bohn, T.; Bourlieu-Lacanal, C.; Boutrou, R.; Carrière, F.; et al. INFOGEST Static in Vitro Simulation of Gastrointestinal Food Digestion. Nat. Protoc. 2019, 14, 991–1014. [Google Scholar] [CrossRef]
- van der Lugt, T.; Opperhuizen, A.; Bast, A.; Vrolijk, M.F. Dietary Advanced Glycation Endproducts and the Gastrointestinal Tract. Nutrients 2020, 12, 2814. [Google Scholar] [CrossRef] [PubMed]
- Zeng, C.; Li, Y.; Ma, J.; Niu, L.; Tay, F.R. Clinical/Translational Aspects of Advanced Glycation End-Products. Trends Endocrinol. Metab. 2019, 30, 959–973. [Google Scholar] [CrossRef] [PubMed]
- Wautier, M.P.; Chappey, O.; Corda, S.; Stern, D.M.; Schmidt, A.M.; Wautier, J.L. Activation of NADPH Oxidase by AGE Links Oxidant Stress to Altered Gene Expression via RAGE. Am. J. Physiol. Endocrinol. Metab. 2001, 280, E685–E694. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.D.; Schmidt, A.M.; Anderson, G.M.; Zhang, J.; Brett, J.; Zou, Y.S.; Pinsky, D.; Stern, D. Enhanced Cellular Oxidant Stress by the Interaction of Advanced Glycation End Products with Their Receptors/Binding Proteins. J. Biol. Chem. 1994, 269, 9889–9897. [Google Scholar] [CrossRef]
- Lee, H.W.; Gu, M.J.; Kim, Y.; Lee, J.Y.; Lee, S.; Choi, I.W.; Ha, S.K. Glyoxal-Lysine Dimer, an Advanced Glycation End Product, Induces Oxidative Damage and Inflammatory Response by Interacting with RAGE. Antioxidants 2021, 10, 1486. [Google Scholar] [CrossRef]
- Moldogazieva, N.T.; Mokhosoev, I.M.; Mel’Nikova, T.I.; Porozov, Y.B.; Terentiev, A.A. Oxidative Stress and Advanced Lipoxidation and Glycation End Products (ALEs and AGEs) in Aging and Age-Related Diseases. Oxid. Med. Cell. Longev. 2019, 2019, 3085756. [Google Scholar] [CrossRef] [Green Version]
- Cepas, V.; Manig, F.; Mayo, J.C.; Hellwig, M.; Collota, D.; Sanmartino, V.; Carrocera-Pumarino, R.; Collino, M.; Henle, T.; Sainz, R.M. In Vitro Evaluation of the Toxicological Profile and Oxidative Stress of Relevant Diet-Related Advanced Glycation End Products and Related 1,2-Dicarbonyls. Oxid. Med. Cell. Longev. 2021, 2021, 9912240. [Google Scholar] [CrossRef]
- Snelson, M.; Tan, S.M.; Clarke, R.E.; De Pasquale, C.; Thallas-Bonke, V.; Nguyen, T.V.; Penfold, S.A.; Harcourt, B.E.; Sourris, K.C.; Lindblom, R.S.; et al. Processed Foods Drive Intestinal Barrier Permeability and Microvascular Diseases. Sci. Adv. 2021, 7, eabe4841. [Google Scholar] [CrossRef]
- Byun, K.; Yoo, Y.C.; Son, M.; Lee, J.; Jeong, G.B.; Park, Y.M.; Salekdeh, G.H.; Lee, B. Advanced Glycation End-Products Produced Systemically and by Macrophages: A Common Contributor to Inflammation and Degenerative Diseases. Pharmacol. Ther. 2017, 177, 44–55. [Google Scholar] [CrossRef]
- Garay-Sevilla, M.E.; Rojas, A.; Portero-Otin, M.; Uribarri, J. Dietary AGEs as Exogenous Boosters of Inflammation. Nutrients 2021, 13, 2802. [Google Scholar] [CrossRef]
- Song, Y.; Wang, Y.; Zhang, Y.; Geng, W.; Liu, W.; Gao, Y.; Li, S.; Wang, K.; Wu, X.; Kang, L.; et al. Advanced Glycation and Products Regulate Anabolic and Catabolic Activities via NLRP3-Inflammasome Activation in Human Nucleus Pulposus Cells. J. Cell. Mol. Med. 2017, 21, 1373–1387. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Chen, H.; Chai, Y. Advanced Glycation End Products (AGEs) Induce Apoptosis of Fibroblasts by Activation of NLRP3 Inflammasome via Reactive Oxygen Species (ROS) Signaling Pathway. Med. Sci. Monit. 2019, 25, 7499–7508. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.; Lu, A.-L.; Yao, X.-M.; Hua, Q.; Li, X.-Y.; Qin, L.; Zhang, H.-M.; Meng, G.-X.; Su, Q. Activation of NLRP3 Inflammasome by Advanced Glycation End Products Promotes Pancreatic Islet Damage. Oxid. Med. Cell. Longev. 2017, 2017, 9692546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Son, S.; Hwang, I.; Hyeokhan, S.; Shin, J.S.; Shin, O.S.; Yu, J.W. Advanced Glycation End Products Impair NLRP3 Inflammasome-Mediated Innate Immune Responses in Macrophages. J. Biol. Chem. 2017, 292, 20437–20448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Der Lugt, T.; Venema, K.; Van Leeuwen, S.; Vrolijk, M.F.; Opperhuizen, A.; Bast, A. Gastrointestinal Digestion of Dietary Advanced Glycation Endproducts Using an: In Vitro Model of the Gastrointestinal Tract (TIM-1). Food Funct. 2020, 11, 6297–6307. [Google Scholar] [CrossRef] [PubMed]
- de Vos, L.C.; Noordzij, M.J.; Mulder, D.J.; Smit, A.J.; Lutgers, H.L.; Dullaart, R.P.F.; Kamphuisen, P.W.; Zeebregts, C.J.; Lefrandt, J.D. Skin Autofluorescence as a Measure of Advanced Glycation End Products Deposition Is Elevated in Peripheral Artery Disease. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 131–138. [Google Scholar] [CrossRef] [Green Version]
- Varikasuvu, S.R.; Varshney, S.; Sulekar, H. Skin Autofluorescence as a Novel and Noninvasive Technology for Advanced Glycation End Products in Diabetic Foot Ulcers: A Systematic Review and Meta-Analysis. Adv. Skin Wound Care 2021, 34, 1–8. [Google Scholar] [CrossRef]
- Ying, L.; Shen, Y.; Zhang, Y.; Wang, Y.; Liu, Y.; Yin, J.; Wang, Y.; Yin, J.; Zhu, W.; Bao, Y.; et al. Advanced Glycation End Products via Skin Autofluorescence as Potential Marker of Carotid Atherosclerosis in Patients with Type 2 Diabetes. Nutr. Metab. Cardiovasc. Dis. 2021, 31, 3449–3456. [Google Scholar] [CrossRef]
- Stirban, A.; Heinemann, L. Skin Autofluorescence—A Non-Invasive Measurement for Assessing Cardiovascular Risk and Risk of Diabetes. Eur. Endocrinol. 2014, 10, 106–110. [Google Scholar] [CrossRef]
- Fokkens, B.T.; Smit, A.J. Skin Fluorescence as a Clinical Tool for Non-Invasive Assessment of Advanced Glycation and Long-Term Complications of Diabetes. Glycoconj. J. 2016, 33, 527–535. [Google Scholar] [CrossRef] [Green Version]
- Nentwich, M.M. Diabetic Retinopathy—Ocular Complications of Diabetes Mellitus. World J. Diabetes 2015, 6, 489. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Sugimoto, T. Advanced Glycation End Products, Diabetes, and Bone Strength. Curr. Osteoporos. Rep. 2016, 14, 320–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Lou, X.; Zhang, J.; Nie, R.; Liu, J.; Tu, P.; Duan, P. Association Between Early Markers of Renal Injury and Type 2 Diabetic Peripheral Neuropathy. Diabetes. Metab. Syndr. Obes. 2021, 14, 4391–4397. [Google Scholar] [CrossRef] [PubMed]
- Bjornstad, P.; Maahs, D.M.; Jensen, T.; Lanaspa, M.A.; Johnson, R.J.; Rewers, M.; Snell-Bergeon, J.K. Elevated Copeptin Is Associated with Atherosclerosis and Diabetic Kidney Disease in Adults with Type 1 Diabetes. J. Diabetes Complications 2016, 30, 1093–1096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Chen, L.-J.; Yu, J.; Wang, H.-J.; Zhang, F.; Liu, Q.; Wu, J. Involvement of Advanced Glycation End Products in the Pathogenesis of Diabetic Retinopathy. Cell. Physiol. Biochem. 2018, 48, 705–717. [Google Scholar] [CrossRef]
- Yaribeygi, H.; Sathyapalan, T.; Atkin, S.L.; Sahebkar, A. Molecular Mechanisms Linking Oxidative Stress and Diabetes Mellitus. Oxid. Med. Cell. Longev. 2020, 2020, 8609213. [Google Scholar] [CrossRef] [Green Version]
- He, C.J.; Koschinsky, T.; Buenting, C.; Vlassara, H. Presence of Diabetic Complications in Type 1 Diabetic Patients Correlates with Low Expression of Mononuclear Cell AGE-Receptor-1 and Elevated Serum AGE. Mol. Med. 2001, 7, 159–168. [Google Scholar] [CrossRef] [Green Version]
- Cai, W.; Ramdas, M.; Zhu, L.; Chen, X.; Striker, G.E.; Vlassara, H. Oral Advanced Glycation Endproducts (AGEs) Promote Insulin Resistance and Diabetes by Depletin the Antioxidant Defenses AGE Reseptor-1 and Sirtuin 1. Proc. Natl. Acad. Sci. USA 2012, 109, 15888–15893. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Yang, H. Relationship between Advanced Glycation End Products and Gestational Diabetes Mellitus. J. Matern. Neonatal Med. 2019, 32, 2783–2789. [Google Scholar] [CrossRef]
- Chiu, C.J.; Rabbani, N.; Rowan, S.; Chang, M.L.; Sawyer, S.; Hu, F.B.; Willett, W.; Thornalley, P.J.; Anwar, A.; Bar, L.; et al. Studies of Advanced Glycation End Products and Oxidation Biomarkers for Type 2 Diabetes. BioFactors 2018, 44, 281–288. [Google Scholar] [CrossRef]
- Zhao, J.; Randive, R.; Stewart, J. Molecular Mechanisms of AGE/RAGE-Mediated Fibrosis in the Diabetic Heart. World J. Diabetes 2014, 5, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Hegab, Z.; Gibbons, S.; Neyses, L.; Mamas, M. Role of Advanced Glycation End Products in Cardiovascular Disease. World J. Cardiol. 2012, 4, 90–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bucala, R.; Tracey, K.J.; Cerami, A. Advanced Glycosylation Products Quench Nitric Oxide and Mediate Defective Endothelium-Dependent Vasodilatation in Experimental Diabetes. J. Clin. Investig. 1991, 87, 432–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luévano-Contreras, C.; Gómez-Ojeda, A.; Macías-Cervantes, M.H.; Garay-Sevilla, M.E. Dietary Advanced Glycation End Products and Cardiometabolic Risk. Curr. Diab. Rep. 2017, 17, 63. [Google Scholar] [CrossRef]
- Wautier, J.L.; Zoukourian, C.; Chappey, O.; Wautier, M.P.; Guillausseau, P.J.; Cao, R.; Horl, O.; Stern, D.; Schmidt, A.M. Receptor-Mediated Endothelial Cell Dysfunction in Diabetic Vasculopathy. Soluble Receptor for Advanced Glycation End Products Blocks Hyperpermeability in Diabetic Rats. J. Clin. Investig. 1996, 97, 238–243. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, K.; Yamagishi, S.-I.; Adachi, H.; Matsui, T.; Kurita-Nakamura, Y.; Takeuchi, M.; Inoue, H.; Imaizumi, T. Serum Levels of Soluble Form of Receptor for Advanced Glycation End Products (SRAGE) Are Positively Associated with Circulating AGEs and Soluble Form of VCAM-1 in Patients with Type 2 Diabetes. Microvasc. Res. 2008, 76, 52–56. [Google Scholar] [CrossRef]
- Koska, J.; Saremi, A.; Howell, S.; Bahn, G.; De Courten, B.; Ginsberg, H.; Beisswenger, P.J.; Reaven, P.D. Advanced Glycation End Products, Oxidation Products, and Incident Cardiovascular Events in Patients with Type 2 Diabetes. Diabetes Care 2018, 41, 570–576. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, S.; Kapoor, D.; Vijayvergiya, R.; Sangwan, S.; Wangkheimayum, S.; Mehta, S.; Dhawan, V. Correlation between Soluble Receptor for Advanced Glycation End Products Levels and Coronary Artery Disease in Postmenopausal Nondiabetic Women. World J. Cardiol. 2021, 13, 130–143. [Google Scholar] [CrossRef]
- Safwat, N.A.; Kenny, M.A. Soluble Receptor for Advanced Glycation End Products as a Vasculopathy Biomarker in Sickle Cell Disease. Pediatr. Res. 2018, 84, 869–874. [Google Scholar] [CrossRef]
- Li, J.; Liu, D.; Sun, L.; Lu, Y.; Zhang, Z. Advanced Glycation End Products and Neurodegenerative Diseases: Mechanisms and Perspective. J. Neurol. Sci. 2012, 317, 1–5. [Google Scholar] [CrossRef]
- Mirlohi, M.S.; Yaghooti, H.; Shirali, S.; Aminasnafi, A.; Olapour, S. Increased Levels of Advanced Glycation End Products Positively Correlate with Iron Overload and Oxidative Stress Markers in Patients with β-Thalassemia Major. Ann. Hematol. 2018, 97, 679–684. [Google Scholar] [CrossRef] [PubMed]
- Madhu, P.; Mukhopadhyay, S. Distinct Types of Amyloid-β Oligomers Displaying Diverse Neurotoxicity Mechanisms in Alzheimer’s Disease. J. Cell. Biochem. 2021, 122, 1594–1608. [Google Scholar] [CrossRef] [PubMed]
- Akhter, F.; Chen, D.; Akhter, A.; Sosunov, A.A.; Chen, A.; McKhann, G.M.; Yan, S.F.; Yan, S.S. Du High Dietary Advanced Glycation End Products Impair Mitochondrial and Cognitive Function. J. Alzheimer’s Dis. 2020, 76, 165–178. [Google Scholar] [CrossRef]
- Jain, V.; Langham, M.C.; Wehrli, F.W. MRI Estimation of Global Brain Oxygen Consumption Rate. J. Cereb. Blood Flow Metab. 2010, 30, 1598–1607. [Google Scholar] [CrossRef]
- Burré, J.; Sharma, M.; Südhof, T.C. Cell Biology and Pathophysiology of α-Synuclein. Cold Spring Harb. Perspect. Med. 2018, 8, a024091. [Google Scholar] [CrossRef]
- Sharma, A.; Weber, D.; Raupbach, J.; Dakal, T.C.; Fließbach, K.; Ramirez, A.; Grune, T.; Wüllner, U. Advanced Glycation End Products and Protein Carbonyl Levels in Plasma Reveal Sex-Specific Differences in Parkinson’s and Alzheimer’s Disease. Redox Biol. 2020, 34, 101546. [Google Scholar] [CrossRef]
- Ko, S.Y.; Ko, H.A.; Chu, K.H.; Shieh, T.M.; Chi, T.C.; Chen, H.I.; Chang, W.C.; Chang, S.S. The Possible Mechanism of Advanced Glycation End Products (AGEs) for Alzheimer’s Disease. PLoS ONE 2015, 10, e0143345. [Google Scholar] [CrossRef]
- Liu, K.; Liu, Y.; Li, L.; Qin, P.; Iqbal, J.; Deng, Y.; Qing, H. Glycation Alter the Process of Tau Phosphorylation to Change Tau Isoforms Aggregation Property. Biochim. Biophys. Acta Mol. Basis Dis. 2016, 1862, 192–201. [Google Scholar] [CrossRef]
- Deane, R.; Yan, S.D.; Submamaryan, R.K.; LaRue, B.; Jovanovic, S.; Hogg, E.; Welch, D.; Manness, L.; Lin, C.; Yu, J.; et al. RAGE Mediates Amyloid-Beta Peptide Transport across the Blood-Brain Barrier and Accumulation in Brain. Nat. Med. 2003, 9, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Mariño, L.; Ramis, R.; Casasnovas, R.; Ortega-Castro, J.; Vilanova, B.; Frau, J.; Adrover, M. Unravelling the Effect of: N (ϵ)-(Carboxyethyl)Lysine on the Conformation, Dynamics and Aggregation Propensity of α-Synuclein. Chem. Sci. 2020, 11, 3332–3344. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.Y.; Deng, C.Q.; Wang, J.; Deng, X.J.; Xiao, Q.; Li, Y.; He, Q.; Fan, W.H.; Quan, F.Y.; Zhu, Y.P.; et al. Plasma Levels of Soluble Receptor for Advanced Glycation End Products in Alzheimer’s Disease. Int. J. Neurosci. 2017, 127, 454–458. [Google Scholar] [CrossRef] [PubMed]
- Stinghen, A.E.M.; Massy, Z.A.; Vlassara, H.; Striker, G.E.; Boullier, A. Uremic Toxicity of Advanced Glycation End Products in CKD. J. Am. Soc. Nephrol. 2016, 27, 354–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, M.E. Importance of Advanced Glycation End Products in Diabetes-Associated Cardiovascular and Renal Disease. Am. J. Hypertens. 2004, 17, S31–S38. [Google Scholar] [CrossRef] [PubMed]
- Yabuuchi, J.; Ueda, S.; Yamagishi, S.-I.; Nohara, N.; Nagasawa, H.; Wakabayashi, K.; Matsui, T.; Yuichiro, H.; Kadoguchi, T.; Otsuka, T.; et al. Association of Advanced Glycation End Products with Sarcopenia and Frailty in Chronic Kidney Disease. Sci. Rep. 2020, 10, 17647. [Google Scholar] [CrossRef]
- McNulty, M.; Mahmud, A.; Feely, J. Advanced Glycation End-Products and Arterial Stiffness in Hypertension. Am. J. Hypertens. 2007, 20, 242–247. [Google Scholar] [CrossRef] [Green Version]
- Taki, K.; Takayama, F.; Tsuruta, Y.; Niwa, T. Oxidative Stress, Advanced Glycation End Product, and Coronary Artery Calcification in Hemodialysis Patients. Kidney Int. 2006, 70, 218–224. [Google Scholar] [CrossRef] [Green Version]
- Belmokhtar, K.; Ortillon, J.; Jaisson, S.; Massy, Z.A.; Rombi, C.B.; Doue, M.; Maurice, P.; Fritz, G.; Gillery, P.; Schmidt, A.M.; et al. Receptor for Advanced Glycation End Products: A Key Molecule in the Genesis of Chronic Kidney Disease Vascular Calcification and a Potential Modulator of Sodium Phosphate Co-Transporter PIT-1 Expression. Nephrol. Dial. Transplant. 2019, 34, 2018–2030. [Google Scholar] [CrossRef]
- Brunt, V.E.; Casso, A.G.; Gioscia-Ryan, R.A.; Sapinsley, Z.J.; Ziemba, B.P.; Clayton, Z.S.; Bazzoni, A.E.; VanDongen, N.S.; Richey, J.J.; Hutton, D.A.; et al. Gut Microbiome-Derived Metabolite Trimethylamine N-Oxide Induces Aortic Stiffening and Increases Systolic Blood Pressure with Aging in Mice and Humans. Hypertension 2021, 78, 499–511. [Google Scholar] [CrossRef]
- Taguchi, K.; Fukami, K.; Elias, B.C.; Brooks, C.R. Dysbiosis-Related Advanced Glycation Endproducts and Trimethylamine N-Oxide in Chronic Kidney Disease. Toxins 2021, 13, 361. [Google Scholar] [CrossRef]
- Teissier, T.; Quersin, V.; Gnemmi, V.; Daroux, M.; Howsam, M.; Delguste, F.; Lemoine, C.; Fradin, C.; Schmidt, A.M.; Cauffiez, C.; et al. Knockout of Receptor for Advanced Glycation End-Products Attenuates Age-Related Renal Lesions. Aging Cell 2018, 18, e12850. [Google Scholar] [CrossRef]
- Nishad, R.; Tahaseen, V.; Kavvuri, R.; Motrapu, M.; Singh, A.K.; Peddi, K.; Pasupulati, A.K. Advanced-Glycation End-Products Induce Podocyte Injury and Contribute to Proteinuria. Front. Med. 2021, 8, 685447. [Google Scholar] [CrossRef] [PubMed]
- Saulnier, P.J.; Wheelock, K.M.; Howell, S.; Weil, E.J.; Tanamas, S.K.; Knowler, W.C.; Lemley, K.V.; Mauer, M.; Yee, B.; Nelson, R.G.; et al. Advanced Glycation End Products Predict Loss of Renal Function and Correlate With Lesions of Diabetic Kidney Disease in American Indians With Type 2 Diabetes. Diabetes 2016, 65, 3744–3753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, M.; Marumo, K. Collagen Cross-Links as a Determinant of Bone Quality: A Possible Explanation for Bone Fragility in Aging, Osteoporosis, and Diabetes Mellitus. Osteoporos. Int. 2010, 21, 195–214. [Google Scholar] [CrossRef] [PubMed]
- McKay, T.B.; Priyadarsini, S.; Karamichos, D. Mechanisms of Collagen Crosslinking in Diabetes and Keratoconus. Cells 2019, 8, 1239. [Google Scholar] [CrossRef] [Green Version]
- Andriotis, O.G.; Elsayad, K.; Smart, D.E.; Nalbach, M.; Davies, D.E.; Thurner, P.J. Hydration and Nanomechanical Changes in Collagen Fibrils Bearing Advanced Glycation End-Products. Biomed. Opt. Express 2019, 10, 1841–1855. [Google Scholar] [CrossRef]
- Hwang, K.R.; Murrell, G.A.C.; Millar, N.L.; Bonar, F.; Lam, P.; Walton, J.R. Advanced Glycation End Products in Idiopathic Frozen Shoulders. J. Shoulder Elb. Surg. 2016, 25, 981–988. [Google Scholar] [CrossRef] [Green Version]
- Khajuria, D.K.; Soliman, M.; Elfar, J.C.; Lewis, G.S.; Abraham, T.; Kamal, F.; Elbarbary, R.A. Aberrant Structure of Fibrillar Collagen and Elevated Levels of Advanced Glycation End Products Typify Delayed Fracture Healing in the Diet-Induced Obesity Mouse Model. Bone 2020, 137, 115436. [Google Scholar] [CrossRef]
- Papagrigoraki, A.; Maurelli, M.; Del Giglio, M.; Gisondi, P.; Girolomoni, G. Advanced Glycation End Products in the Pathogenesis of Psoriasis. Int. J. Mol. Sci. 2017, 18, 2471. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-S.; Wang, X.-J.; Feng, W.; Hua, K.-Q. Advanced Glycation End Products Decrease Collagen I Levels in Fibroblasts from the Vaginal Wall of Patients with POP via the RAGE, MAPK and NF-KappaB Pathways. Int. J. Mol. Med. 2017, 40, 987–998. [Google Scholar] [CrossRef] [Green Version]
- Van Putte, L.; De Schrijver, S.; Moortgat, P. The Effects of Advanced Glycation End Products (AGEs) on Dermal Wound Healing and Scar Formation: A Systematic Review. Scars Burn. Heal. 2016, 2, 1–14. [Google Scholar] [CrossRef]
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Baby, D.; Rajguru, J.; Patil, P.; Thakkannavar, S.; Pujari, V. Inflammation and Cancer. Ann. Afr. Med. 2019, 18, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Shahab, U.; Ahmad, M.K.; Mahdi, A.A.; Waseem, M.; Arif, B.; Moinuddin; Ahmad, S. The Receptor for Advanced Glycation End Products: A Fuel to Pancreatic Cancer. Semin. Cancer Biol. 2018, 49, 37–43. [Google Scholar] [CrossRef]
- Turner, D.P. The Role of Advanced Glycation End-Products in Cancer Disparity. Adv. Cancer Res. 2017, 133, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Malik, P.; Chaudhry, N.; Mittal, R.; Mukherjee, T.K. Role of Receptor for Advanced Glycation End Products in the Complication and Progression of Various Types of Cancers. Biochim. Biophys. Acta Gen. Subj. 2015, 1850, 1898–1904. [Google Scholar] [CrossRef]
- Omofuma, O.O.; Turner, D.P.; Peterson, L.L.; Merchant, A.T.; Zhang, J.; Steck, S.E. Dietary Advanced Glycation End-Products (AGEs) and Risk of Breast Cancer in the Prostate, Lung, Colorectal and Ovarian Cancer Screening Trial (PLCO). Cancer Prev. Res. 2020, 13, 601–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.; Pinney, S.M.; Mallick, P.; Ho, S.M.; Bracken, B.; Wu, T. Impact of Oxidative Stress Biomarkers and Carboxymethyllysine (an Advanced Glycation End Product) on Prostate Cancer: A Prospective Study. Clin. Genitourin. Cancer 2015, 13, e347–e351. [Google Scholar] [CrossRef] [Green Version]
- Abdel-Razik, A.; Shabana, W.; El Nakib, A.M.; Abdelsalam, M.; Abdelwahab, A.; Hasan, A.S.; Elzehery, R.; Elhelaly, R.; Fathy, A.A.; Mostafa, S.A.; et al. De Novo Hepatocellular Carcinoma in Hepatitis C-Related Cirrhosis: Are Advanced Glycation End Products a Key Driver? Front. Cell. Infect. Microbiol. 2021, 11, 662431. [Google Scholar] [CrossRef]
- Sasahira, T.; Kirita, T.; Bhawal, U.K.; Yamamoto, K.; Ohmori, H.; Fujii, K.; Kuniyasu, H. Receptor for Advanced Glycation End Products (RAGE) Is Important in the Prediction of Recurrence in Human Oral Squamous Cell Carcinoma. Histopathology 2007, 51, 166–172. [Google Scholar] [CrossRef]
- Khan, H.; Khan, M.S.; Ahmad, S. The in Vivo and in Vitro Approaches for Establishing a Link between Advanced Glycation End Products and Lung Cancer. J. Cell. Biochem. 2018, 119, 9099–9109. [Google Scholar] [CrossRef]
- Menini, S.; Iacobini, C.; de Latouliere, L.; Manni, I.; Ionta, V.; Blasetti Fantauzzi, C.; Pesce, C.; Cappello, P.; Novelli, F.; Piaggio, G.; et al. The Advanced Glycation End-Product Nϵ-Carboxymethyllysine Promotes Progression of Pancreatic Cancer: Implications for Diabetes-Associated Risk and Its Prevention. J. Pathol. 2018, 245, 197–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.J.; Yoo, J.W.; Kim, Y.K.; Choi, J.H.; Ha, T.Y.; Gil, M. Advanced Glycation End Products Promote Triple Negative Breast Cancer Cells via ERK and NF-ΚB Pathway. Biochem. Biophys. Res. Commun. 2018, 495, 2195–2201. [Google Scholar] [CrossRef] [PubMed]
- Deng, R.; Wu, H.; Ran, H.; Kong, X.; Hu, L.; Wang, X.; Su, Q. Glucose-Derived AGEs Promote Migration and Invasion of Colorectal Cancer by up-Regulating Sp1 Expression. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 1065–1074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rungratanawanich, W.; Qu, Y.; Wang, X.; Essa, M.M.; Song, B.J. Advanced Glycation End Products (AGEs) and Other Adducts in Aging-Related Diseases and Alcohol-Mediated Tissue Injury. Exp. Mol. Med. 2021, 53, 168–188. [Google Scholar] [CrossRef]
- Petriv, N.; Neubert, L.; Vatashchuk, M.; Timrott, K.; Suo, H.; Hochnadel, I.; Huber, R.; Petzold, C.; Hrushchenko, A.; Yatsenko, A.S.; et al. Increase of α-Dicarbonyls in Liver and Receptor for Advanced Glycation End Products on Immune Cells Are Linked to Nonalcoholic Fatty Liver Disease and Liver Cancer. Oncoimmunology 2021, 10, 1874159. [Google Scholar] [CrossRef]
- da Silva Pereira, E.N.G.; Paula, D.P.; de Araujo, B.P.; Da Fonseca, M.D.J.M.; Diniz, M.D.F.H.S.; Daliry, A.; Griep, R.H. Advanced Glycation End Product: A Potential Biomarker for Risk Stratification of Non-Alcoholic Fatty Liver Disease in ELSA-Brasil Study. World J. Gastroenterol. 2021, 27, 4913–4923. [Google Scholar] [CrossRef]
- Laudenslager, M.; Lazo, M.; Wang, D.; Selvin, E.; Chen, P.H.; Pankow, J.S.; Clark, J.M. Association between the Soluble Receptor for Advanced Glycation End Products (SRAGE) and NAFLD in Participants in the Atherosclerosis Risk in Communities Study. Dig. Liver Dis. 2021, 53, 873–878. [Google Scholar] [CrossRef]
- Zhu, J.L.; Cai, Y.Q.; Long, S.L.; Chen, Z.; Mo, Z.C. The Role of Advanced Glycation End Products in Human Infertility. Life Sci. 2020, 255, 117830. [Google Scholar] [CrossRef]
- Merhi, Z. Advanced Glycation End Products and Their Relevance in Female Reproduction. Hum. Reprod. 2014, 29, 135–145. [Google Scholar] [CrossRef] [Green Version]
- Thornton, K.; Merhi, Z.; Jindal, S.; Goldsammler, M.; Charron, M.J.; Buyuk, E. Dietary Advanced Glycation End Products (AGEs) Could Alter Ovarian Function in Mice. Mol. Cell. Endocrinol. 2020, 510, 110826. [Google Scholar] [CrossRef]
- Takahashi, N.; Harada, M.; Azhary, J.M.K.; Kunitomi, C.; Nose, E.; Terao, H.; Koike, H.; Wada-Hiraike, O.; Hirata, T.; Hirota, Y.; et al. Accumulation of Advanced Glycation End Products in Follicles Is Associated with Poor Oocyte Developmental Competence. Mol. Hum. Reprod. 2019, 25, 684–694. [Google Scholar] [CrossRef]
- Mallidis, C.; Agbaje, I.M.; Rogers, D.A.; Glenn, J.V.; Pringle, R.; Atkinson, A.B.; Steger, K.; Stitt, A.W.; McClure, N. Advanced Glycation End Products Accumulate in the Reproductive Tract of Men with Diabetes. Int. J. Androl. 2009, 32, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Nevin, C.; McNeil, L.; Ahmed, N.; Murgatroyd, C.; Brison, D.; Carroll, M. Investigating the Glycating Effects of Glucose, Glyoxal and Methylglyoxal on Human Sperm. Sci. Rep. 2018, 8, 9002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azhary, J.M.K.; Harada, M.; Kunitomi, C.; Kusamoto, A.; Takahashi, N.; Nose, E.; Oi, N.; Wada-Hiraike, O.; Urata, Y.; Hirata, T.; et al. Androgens Increase Accumulation of Advanced Glycation End Products in Granulosa Cells by Activating ER Stress in PCOS. Endocrinology 2020, 161, bqaa015. [Google Scholar] [CrossRef] [PubMed]
- Nejabati, H.R.; Mota, A.; Farzadi, L.; Ghojazadeh, M.; Fattahi, A.; Hamdi, K.; Nouri, M. Follicular Fluid PlGF/SFlt-1 Ratio and Soluble Receptor for Advanced Glycation End–Products Correlate with Ovarian Sensitivity Index in Women Undergoing A.R.T. J. Endocrinol. Investig. 2017, 40, 207–215. [Google Scholar] [CrossRef]
- Hao, L.; Noguchi, S.; Kamada, Y.; Sasaki, A.; Matsuda, M.; Shimizu, K.; Hiramatsu, Y.; Nakatsuka, M. Adverse Effects of Advanced Glycation End Products on Embryonal Development. Acta Med. Okayama 2008, 62, 93–99. [Google Scholar]
- Seven, B.; Tascı, Y.; Gülerman, C.; Candar, T.; Kokanalı, K.; Yüksel, Y.; Bardakcı, Y.; Engin-Ustün, Y. Do Follicular Fluid Advanced Glycation End Products Levels Affect the Ovarian Response in Unexplained Infertility? Gynecol. Endocrinol. 2021, 37, 802–806. [Google Scholar] [CrossRef]
- Rojas, A.; Schneider, I.; Lindner, C.; Gonzàlez, I.; Morales, M.A. Receptor for Advanced Glycation End-Products Axis and Coronavirus Disease 2019 in Inflammatory Bowel Diseases: A Dangerous Liaison? World J. Gastroenterol. 2021, 27, 2270–2280. [Google Scholar] [CrossRef]
- Oczypok, E.A.; Perkins, T.N.; Oury, T.D. All the “RAGE” in Lung Disease: The Receptor for Advanced Glycation Endproducts (RAGE) Is a Major Mediator of Pulmonary Inflammatory Responses. Paediatr. Respir. Rev. 2017, 23, 40–49. [Google Scholar] [CrossRef]
- Lim, A.; Radujkovic, A.; Weigand, M.A.; Merle, U. Soluble Receptor for Advanced Glycation End Products (SRAGE) as a Biomarker of COVID-19 Disease Severity and Indicator of the Need for Mechanical Ventilation, ARDS and Mortality. Ann. Intensive Care 2021, 11, 50. [Google Scholar] [CrossRef]
- Dozio, E.; Sitzia, C.; Pistelli, L.; Cardani, R.; Rigolini, R.; Ranucci, M.; Corsi Romanelli, M.M. Soluble Receptor for Advanced Glycation End Products and Its Forms in COVID-19 Patients with and without Diabetes Mellitus: A Pilot Study on Their Role as Disease Biomarkers. J. Clin. Med. 2020, 9, 3785. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Liu, H.; Wang, J.; Sun, B. Inhibitory Effect of Phenolic Compounds and Plant Extracts on the Formation of Advance Glycation End Products: A Comprehensive Review. Food Res. Int. 2020, 130, 108933. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, S.; Matsui, T.; Ishibashi, Y.; Isami, F.; Abe, Y.; Sakaguchi, T.; Higashimoto, Y. Phytochemicals Against Advanced Glycation End Products (AGEs) and the Receptor System. Curr. Pharm. Des. 2016, 23, 1135–1141. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Source | Precursor | Chemical Structure and Ability to Emit Fluorescence | Molecular Weight | Physiological Importance |
|---|---|---|---|---|
| -Endogenous | -Glucose-derived (Glu-AGEs) | -Fluorescent and crosslinked | -Low (LMW AGEs) | -Non-toxic |
| -Exogenous (dietary, dAGEs) | -Fructose-derived (Fru-AGEs) | -Fluorescent and non-crosslinked | -Toxic (TAGEs) | |
| -Glycolaldehyde-derived (Glycol-AGEs) | -Non-fluorescent and crosslinked | -High (HMW AGEs) | ||
| -Glyceraldehyde-derived (Glycer-AGEs) | -Non-fluorescent and non-crosslinked | |||
| -Methylglyoxal-derived (MGO-AGEs) | ||||
| -Glyoxal-derived (GO-AGEs) | ||||
| -3-Deoxyglucosone-derived (3-DG-AGEs) |
| Products | AGE 1 (kU/100 g) | Products | AGE (kU/100 g) |
|---|---|---|---|
| Meats | Carbohydrates | ||
| Beef, raw | 707 | Bread, pita | 53 |
| Beef, steak, pan fried with olive oil | 10,058 | Bread, Greek, hard, toasted | 607 |
| Chicken, breast, skinless, raw | 769 | Biscuit (McDonald’s) | 1470 |
| Chicken, boiled with lemon | 957 | Cookie, biscotti, vanilla almond | 3220 |
| Chicken, breast, breaded, deep-fried, 20 min | 9722 | Donut, chocolate iced, crème filled | 1803 |
| Chicken, skin, back or thigh, roasted, barbequed | 18,520 | Apple, baked | 43 |
| Chicken, skin, thigh, roasted | 11,149 | Apple, Macintosh | 13 |
| Turkey, ground, raw | 4957 | Fig, dried | 2663 |
| Turkey, ground, grilled, crust | 6351 | Carrots, canned | 10 |
| Bacon, fried, 5 min, no added oil | 91,577 | Tomato | 23 |
| Bacon, microwaved, 2 slices, 3 min | 9023 | Vegetables, grilled (broccoli, carrots, celery) | 226 |
| Lamb, leg, raw | 826 | Honey | 7 |
| Lamb, leg, broiled, 450°F, 1 min | 2431 | Fats | |
| Fish | Almonds, roasted | 6650 | |
| Salmon, fillet, microwaved | 912 | Butter, whipped | 26,480 |
| Salmon, fillet, broiled | 3347 | Margarine, tub | 17,520 |
| Trout, raw | 783 | Walnuts, roasted | 7887 |
| Trout, baked, 25 min | 2138 | Eggs | |
| Liquids | Egg white, large, 12 min | 63 | |
| Milk, whole (4% fat) | 5 | Egg yolk, large, 12 min | 1680 |
| Juice, apple | 2 | Egg, omelet, pan, low, butter, 13 min | 507 |
| Receptor Type | Receptor Name (Other Names, Uniprot ID) | Ligands (Other than AGEs) or Receptor Function | Ref. |
|---|---|---|---|
| RAGE | RAGE (AGER; Q15109) | S100A12, S100B, amyloid-β peptide precursor (ABPP), oligonucleotides | [56] |
| Scavenger, class H | Stabilin-1 (Stab1, FEEL1; Q9NY15) | acetylated low-density lipoprotein (AcLDL), Gram-positive and Gram-negative bacteria | [57] |
| Stabilin-2 (Stab2, FEEL2, HARE; Q8WWQ8) | hyaluronic acid (HA), heparin (Hep), chondroitin sulfate (CS), dermatan sulfate (DS), AcLDL, pro-collagen propeptides, oligonucleotides, phosphatidylserine (PS), bacteria | [57] | |
| Scavenger, class A | SR-AI (P21757) and transcript variant SR-AII | modified low-density lipoproteins (LDLs) | [58] |
| Scavenger, class B | SR-BI (Q8WTV0), CD36 | phospholipids, cholesterol (high-density lipoprotein; HDL), cholesterol ester, lipoproteins, PS | [59] |
| Scavenger, class E | OxLDL receptor 1 (LOX-1; P78380) | oxidatively modified low-density lipoprotein (oxLDL), HSP70 protein | [60] |
| AGE-Rs | AGE-R1 (OST-48/p60; P39656) | 48 kDa subunit of the oligosaccharyl transferase (OST) complex | [61] |
| AGE-R2 (80K-H/p90; P14314) | regulatory subunit β of glucosidase 2 | [62] | |
| AGE-R3 (galectin-3; P17931) | galactose-specific lectin that binds IgE | [63] |
| PDB ID | Title (Resolution, Space Group) | Protein Chain; Residues | Ligand | Ref. |
|---|---|---|---|---|
| 3CJJ | Crystal structure of human RAGE ligand-binding domain (1.85 Å, P21212) | A; 23–240 | None | [68] |
| 3O3U | Crystal Structure of Human RAGE (with MBP fusion) (1.50 Å, P21212) | N; 23–231 | None | [69] |
| 4LP4/4LP5 (4OF5, 4OFV) | Crystal structure of the human RAGE VC1 fragment in space group P62 (2.40 Å, P62)/Crystal structure of the full-length human RAGE extracellular domain (VC1C2 frag.) (3.80 Å, P65) | A, B; 23–231 /A, B; 23–323 | None | [70] |
| 4OI7 (4OI8 ) | RAGE recognizes nucleic acids and promotes inflammatory responses to DNA (3.10 Å, P 61) | A, B; 23–237 | DNA (chains: E, F) | [71] |
| 4P2Y (4YBH) | Crystal structure of the human RAGE ectodomain (fragment VC1C2) in complex with mouse (4P2Y)/human S100A6 (4YBH) (2.30/2.40 Å, I 2 2 2) | A; 23–323 | Murine S100-A6 (chain B, 91 aa)/Human S100-A6 (chain B, 92 aa) | [72] |
| 6XQ1–6XQ9, 7LMW | RAGE VC1 domain in complex with [inhibitor name] (1.51–2.50 Å, P 62) | A, B; 20–231 | Small molecule or fragment inhibitor 1 | [73] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Twarda-Clapa, A.; Olczak, A.; Białkowska, A.M.; Koziołkiewicz, M. Advanced Glycation End-Products (AGEs): Formation, Chemistry, Classification, Receptors, and Diseases Related to AGEs. Cells 2022, 11, 1312. https://doi.org/10.3390/cells11081312
Twarda-Clapa A, Olczak A, Białkowska AM, Koziołkiewicz M. Advanced Glycation End-Products (AGEs): Formation, Chemistry, Classification, Receptors, and Diseases Related to AGEs. Cells. 2022; 11(8):1312. https://doi.org/10.3390/cells11081312
Chicago/Turabian StyleTwarda-Clapa, Aleksandra, Aleksandra Olczak, Aneta M. Białkowska, and Maria Koziołkiewicz. 2022. "Advanced Glycation End-Products (AGEs): Formation, Chemistry, Classification, Receptors, and Diseases Related to AGEs" Cells 11, no. 8: 1312. https://doi.org/10.3390/cells11081312