
Selectin-Mediated Signaling—Shedding Light on the Regulation of Integrin Activity in Neutrophils


{kind=link}
{kind=link}
{kind=link}
Abstract
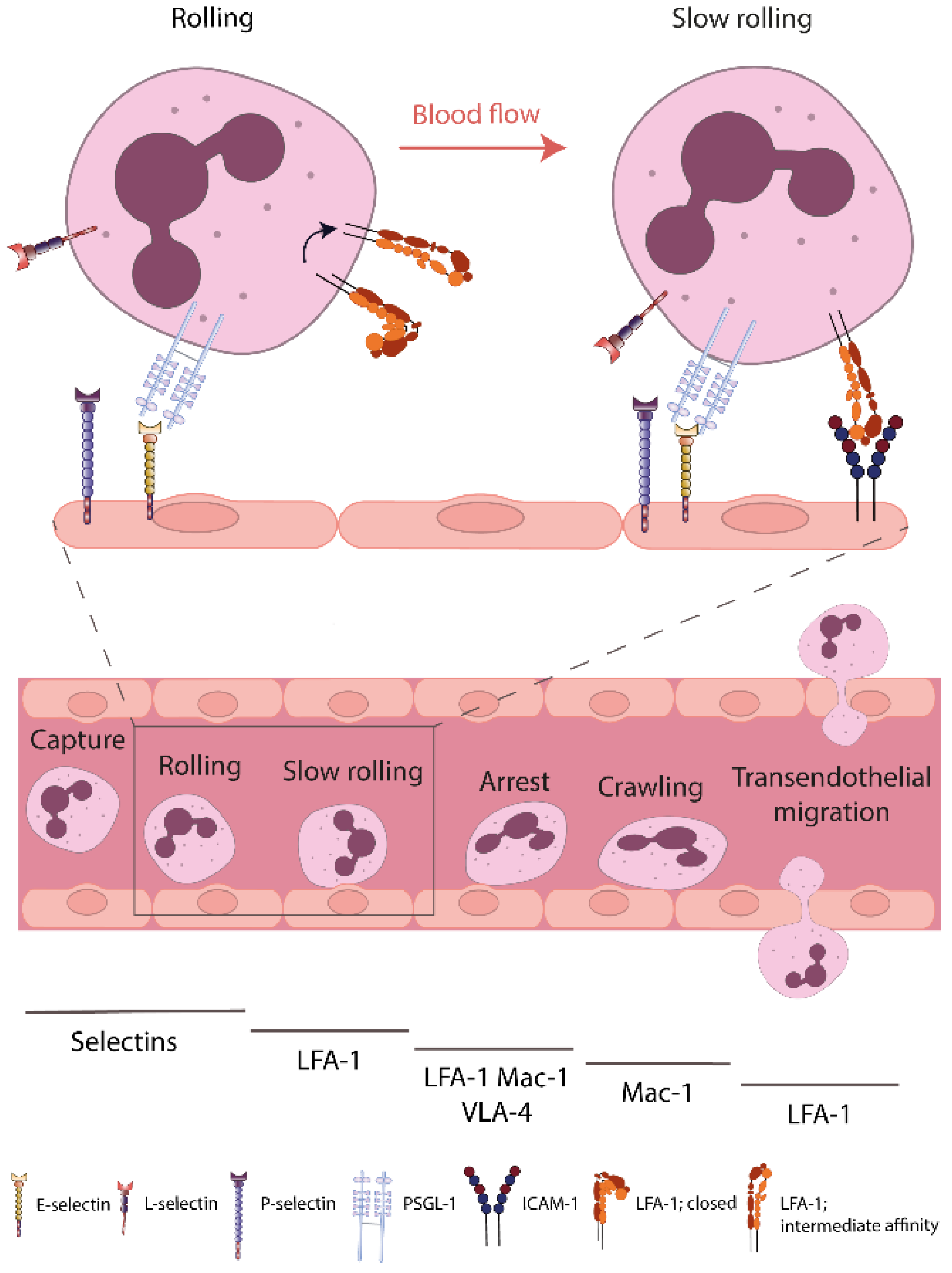
:1. Introduction
2. Integrins—Big Players during Each Step of Neutrophil Recruitment
2.1. Integrins and Their Role within the Leukocyte Recruitment Cascade
2.2. Integrin Characteristic Trait: Conformational Changes and Clustering
3. Selectins and PSGL-1: The Underestimated Basis for Neutrophil–Endothelial Interactions
3.1. Selectin-Ligand Engagement Induces Inside-Out Signaling in Neutrophils
3.2. L-Selectin and Its Special Role during Integrin Activation and Leukocyte Recruitment
3.3. Integrin-Deactivation: Important Mechanisms to Keep the Balance
4. The Crucial Role of Selectins and Integrins in the Development of Human Diseases
Author Contributions
Funding
Conflicts of Interest
References
- Ley, K.; Laudanna, C.; Cybulsky, M.I.; Nourshargh, S. Getting to the site of inflammation: The leukocyte adhesion cascade updated. Nat. Rev. Immunol. 2007, 7, 678–689. [Google Scholar] [CrossRef] [PubMed]
- Liew, P.X.; Kubes, P. The Neutrophil’s Role during Health and Disease. Physiol. Rev. 2019, 99, 1223–1248. [Google Scholar] [CrossRef] [PubMed]
- Margraf, A.; Lowell, C.A.; Zarbock, A. Neutrophils in acute inflammation—Current concepts and translational implications. Blood 2021, 139, 2130–2144. [Google Scholar] [CrossRef] [PubMed]
- Das, J.; Sharma, A.; Jindal, A.; Aggarwal, V.; Rawat, A. Leukocyte adhesion defect: Where do we stand circa 2019? Genes Dis. 2020, 7, 107–114. [Google Scholar] [CrossRef]
- Schmidt, S.; Moser, M.; Sperandio, M. The molecular basis of leukocyte recruitment and its deficiencies. Mol. Immunol. 2013, 55, 49–58. [Google Scholar] [CrossRef]
- McEver, R.P. Selectins: Initiators of leucocyte adhesion and signalling at the vascular wall. Cardiovasc. Res. 2015, 107, 331–339. [Google Scholar] [CrossRef] [Green Version]
- Luo, B.H.; Carman, C.V.; Springer, T.A. Structural basis of integrin regulation and signaling. Annu. Rev. Immunol. 2007, 25, 619–647. [Google Scholar] [CrossRef] [Green Version]
- Morse, E.M.; Brahme, N.N.; Calderwood, D.A. Integrin cytoplasmic tail interactions. Biochemistry 2014, 53, 810–820. [Google Scholar] [CrossRef]
- Legate, K.R.; Fassler, R. Mechanisms that regulate adaptor binding to beta-integrin cytoplasmic tails. J. Cell Sci. 2009, 122, 187–198. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Hu, L.; Fan, Z. beta2 integrin activation and signal transduction in leukocyte recruitment. Am. J. Physiol. Cell Physiol. 2021, 321, C308–C316. [Google Scholar] [CrossRef]
- Abram, C.L.; Lowell, C.A. The ins and outs of leukocyte integrin signaling. Annu. Rev. Immunol. 2009, 27, 339–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Podolnikova, N.P.; Podolnikov, A.V.; Haas, T.A.; Lishko, V.K.; Ugarova, T.P. Ligand recognition specificity of leukocyte integrin alphaMbeta2 (Mac-1, CD11b/CD18) and its functional consequences. Biochemistry 2015, 54, 1408–1420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parkos, C.A. Cell adhesion and migration. I. Neutrophil adhesive interactions with intestinal epithelium. Am. J. Physiol. 1997, 273, G763–G768. [Google Scholar] [CrossRef]
- Futosi, K.; Fodor, S.; Mocsai, A. Reprint of Neutrophil cell surface receptors and their intracellular signal transduction pathways. Int. Immunopharmacol. 2013, 17, 1185–1197. [Google Scholar] [CrossRef]
- Eriksson, E.E.; Xie, X.; Werr, J.; Thoren, P.; Lindbom, L. Importance of primary capture and L-selectin-dependent secondary capture in leukocyte accumulation in inflammation and atherosclerosis in vivo. J. Exp. Med. 2001, 194, 205–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sperandio, M.; Smith, M.L.; Forlow, S.B.; Olson, T.S.; Xia, L.; McEver, R.P.; Ley, K. P-selectin glycoprotein ligand-1 mediates L-selectin-dependent leukocyte rolling in venules. J. Exp. Med. 2003, 197, 1355–1363. [Google Scholar] [CrossRef] [PubMed]
- McEver, R.P.; Cummings, R.D. Role of PSGL-1 binding to selectins in leukocyte recruitment. J. Clin. Invest. 1997, 100, S97–S103. [Google Scholar] [CrossRef]
- Hidalgo, A.; Peired, A.J.; Wild, M.; Vestweber, D.; Frenette, P.S. Complete identification of E-selectin ligands on neutrophils reveals distinct functions of PSGL-1, ESL-1, and CD44. Immunity 2007, 26, 477–489. [Google Scholar] [CrossRef] [Green Version]
- Zarbock, A.; Lowell, C.A.; Ley, K. Spleen tyrosine kinase Syk is necessary for E-selectin-induced alpha(L)beta(2) integrin-mediated rolling on intercellular adhesion molecule-1. Immunity 2007, 26, 773–783. [Google Scholar] [CrossRef] [Green Version]
- Kuwano, Y.; Spelten, O.; Zhang, H.; Ley, K.; Zarbock, A. Rolling on E- or P-selectin induces the extended but not high-affinity conformation of LFA-1 in neutrophils. Blood 2010, 116, 617–624. [Google Scholar] [CrossRef] [Green Version]
- Lefort, C.T.; Rossaint, J.; Moser, M.; Petrich, B.G.; Zarbock, A.; Monkley, S.J.; Critchley, D.R.; Ginsberg, M.H.; Fassler, R.; Ley, K. Distinct roles for talin-1 and kindlin-3 in LFA-1 extension and affinity regulation. Blood 2012, 119, 4275–4282. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.J.; Hedrick, J.; Zlotnik, A.; Siani, M.A.; Thompson, D.A.; Butcher, E.C. Chemokines and the arrest of lymphocytes rolling under flow conditions. Science 1998, 279, 381–384. [Google Scholar] [CrossRef] [PubMed]
- Herter, J.M.; Rossaint, J.; Block, H.; Welch, H.; Zarbock, A. Integrin activation by P-Rex1 is required for selectin-mediated slow leukocyte rolling and intravascular crawling. Blood 2013, 121, 2301–2310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillipson, M.; Heit, B.; Colarusso, P.; Liu, L.; Ballantyne, C.M.; Kubes, P. Intraluminal crawling of neutrophils to emigration sites: A molecularly distinct process from adhesion in the recruitment cascade. J. Exp. Med. 2006, 203, 2569–2575. [Google Scholar] [CrossRef] [PubMed]
- Vestweber, D. Adhesion and signaling molecules controlling the transmigration of leukocytes through endothelium. Immunol. Rev. 2007, 218, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Vestweber, D. How leukocytes cross the vascular endothelium. Nat. Rev. Immunol. 2015, 15, 692–704. [Google Scholar] [CrossRef]
- de Oliveira, S.; Rosowski, E.E.; Huttenlocher, A. Neutrophil migration in infection and wound repair: Going forward in reverse. Nat. Rev. Immunol. 2016, 16, 378–391. [Google Scholar] [CrossRef] [Green Version]
- Buckley, C.D.; Ross, E.A.; McGettrick, H.M.; Osborne, C.E.; Haworth, O.; Schmutz, C.; Stone, P.C.; Salmon, M.; Matharu, N.M.; Vohra, R.K.; et al. Identification of a phenotypically and functionally distinct population of long-lived neutrophils in a model of reverse endothelial migration. J. Leukoc. Biol. 2006, 79, 303–311. [Google Scholar] [CrossRef]
- Wang, J.; Hossain, M.; Thanabalasuriar, A.; Gunzer, M.; Meininger, C.; Kubes, P. Visualizing the function and fate of neutrophils in sterile injury and repair. Science 2017, 358, 111–116. [Google Scholar] [CrossRef] [Green Version]
- He, W.; Holtkamp, S.; Hergenhan, S.M.; Kraus, K.; de Juan, A.; Weber, J.; Bradfield, P.; Grenier, J.M.P.; Pelletier, J.; Druzd, D.; et al. Circadian Expression of Migratory Factors Establishes Lineage-Specific Signatures that Guide the Homing of Leukocyte Subsets to Tissues. Immunity 2018, 49, 1175–1190.e1177. [Google Scholar] [CrossRef] [Green Version]
- Pick, R.; He, W.; Chen, C.S.; Scheiermann, C. Time-of-Day-Dependent Trafficking and Function of Leukocyte Subsets. Trends Immunol. 2019, 40, 524–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimaoka, M.; Xiao, T.; Liu, J.H.; Yang, Y.; Dong, Y.; Jun, C.D.; McCormack, A.; Zhang, R.; Joachimiak, A.; Takagi, J.; et al. Structures of the alpha L I domain and its complex with ICAM-1 reveal a shape-shifting pathway for integrin regulation. Cell 2003, 112, 99–111. [Google Scholar] [CrossRef] [Green Version]
- Arnaout, M.A.; Mahalingam, B.; Xiong, J.P. Integrin structure, allostery, and bidirectional signaling. Annu. Rev. Cell Dev. Biol. 2005, 21, 381–410. [Google Scholar] [CrossRef] [Green Version]
- Takagi, J.; Petre, B.M.; Walz, T.; Springer, T.A. Global conformational rearrangements in integrin extracellular domains in outside-in and inside-out signaling. Cell 2002, 110, 599–611. [Google Scholar] [CrossRef] [Green Version]
- Fan, Z.; Ley, K. Leukocyte arrest: Biomechanics and molecular mechanisms of beta2 integrin activation. Biorheology 2015, 52, 353–377. [Google Scholar] [CrossRef] [Green Version]
- Schurpf, T.; Springer, T.A. Regulation of integrin affinity on cell surfaces. EMBO J. 2011, 30, 4712–4727. [Google Scholar] [CrossRef] [Green Version]
- Salas, A.; Shimaoka, M.; Kogan, A.N.; Harwood, C.; von Andrian, U.H.; Springer, T.A. Rolling adhesion through an extended conformation of integrin alphaLbeta2 and relation to alpha I and beta I-like domain interaction. Immunity 2004, 20, 393–406. [Google Scholar] [CrossRef] [Green Version]
- Sen, M.; Yuki, K.; Springer, T.A. An internal ligand-bound, metastable state of a leukocyte integrin, alphaXbeta2. J. Cell Biol. 2013, 203, 629–642. [Google Scholar] [CrossRef] [PubMed]
- Adair, B.D.; Xiong, J.P.; Maddock, C.; Goodman, S.L.; Arnaout, M.A.; Yeager, M. Three-dimensional EM structure of the ectodomain of integrin {alpha}V{beta}3 in a complex with fibronectin. J. Cell Biol. 2005, 168, 1109–1118. [Google Scholar] [CrossRef]
- Honore, S.; Pichard, V.; Penel, C.; Rigot, V.; Prevt, C.; Marvaldi, J.; Briand, C.; Rognoni, J.B. Outside-in regulation of integrin clustering processes by ECM components per se and their involvement in actin cytoskeleton organization in a colon adenocarcinoma cell line. Histochem. Cell Biol. 2000, 114, 323–335. [Google Scholar] [CrossRef]
- Vestweber, D.; Blanks, J.E. Mechanisms that regulate the function of the selectins and their ligands. Physiol. Rev. 1999, 79, 181–213. [Google Scholar] [CrossRef] [PubMed]
- Watson, M.L.; Kingsmore, S.F.; Johnston, G.I.; Siegelman, M.H.; Le Beau, M.M.; Lemons, R.S.; Bora, N.S.; Howard, T.A.; Weissman, I.L.; McEver, R.P.; et al. Genomic organization of the selectin family of leukocyte adhesion molecules on human and mouse chromosome 1. J. Exp. Med. 1990, 172, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Kansas, G.S. Selectins and their ligands: Current concepts and controversies. Blood 1996, 88, 3259–3287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahn, J.; Walcheck, B.; Migaki, G.I.; Jutila, M.A.; Kishimoto, T.K. Calmodulin regulates L-selectin adhesion molecule expression and function through a protease-dependent mechanism. Cell 1998, 92, 809–818. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Miner, J.J.; Yago, T.; Yao, L.; Lupu, F.; Xia, L.; McEver, R.P. Differential regulation of human and murine P-selectin expression and function in vivo. J. Exp. Med. 2010, 207, 2975–2987. [Google Scholar] [CrossRef] [Green Version]
- McEver, R.P.; Moore, K.L.; Cummings, R.D. Leukocyte trafficking mediated by selectin-carbohydrate interactions. J. Biol. Chem. 1995, 270, 11025–11028. [Google Scholar] [CrossRef] [Green Version]
- Homeister, J.W.; Thall, A.D.; Petryniak, B.; Maly, P.; Rogers, C.E.; Smith, P.L.; Kelly, R.J.; Gersten, K.M.; Askari, S.W.; Cheng, G.; et al. The alpha(1,3)fucosyltransferases FucT-IV and FucT-VII exert collaborative control over selectin-dependent leukocyte recruitment and lymphocyte homing. Immunity 2001, 15, 115–126. [Google Scholar] [CrossRef] [Green Version]
- Lowe, J.B. Selectin ligands, leukocyte trafficking, and fucosyltransferase genes. Kidney Int. 1997, 51, 1418–1426. [Google Scholar] [CrossRef] [Green Version]
- Crutchfield, K.L.; Shinde Patil, V.R.; Campbell, C.J.; Parkos, C.A.; Allport, J.R.; Goetz, D.J. CD11b/CD18-coated microspheres attach to E-selectin under flow. J. Leukoc. Biol. 2000, 67, 196–205. [Google Scholar] [CrossRef]
- Katayama, Y.; Hidalgo, A.; Chang, J.; Peired, A.; Frenette, P.S. CD44 is a physiological E-selectin ligand on neutrophils. J. Exp. Med. 2005, 201, 1183–1189. [Google Scholar] [CrossRef] [Green Version]
- Xia, L.J.; Sperandio, M.; Yago, S.; McDaniel, J.M.; Cummings, R.D.; Pearson-White, S.; Ley, K.; McEver, R. P-selectin glycoprotein ligand-1-deficient mice have impaired leukocyte tethering to E-selectin under flow. J. Clin. Investig. 2002, 109, 939–950. [Google Scholar] [CrossRef] [PubMed]
- Norman, K.E.; Moore, K.L.; Mcever, R.P.; Ley, K. Leukocyte Rolling in-Vivo Is Mediated by P-Selectin Glycoprotein Ligand-1. Blood 1995, 86, 4417–4421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- del Conde, I.; Shrimpton, C.N.; Thiagarajan, P.; Lopez, J.A. Tissue-factor-bearing microvesicles arise from lipid rafts and fuse with activated platelets to initiate coagulation. Blood 2005, 106, 1604–1611. [Google Scholar] [CrossRef] [PubMed]
- Borges, E.; Eytner, R.; Moll, T.; Steegmaier, M.; Campbell, M.A.; Ley, K.; Mossmann, H.; Vestweber, D. The P-selectin glycoprotein ligand-1 is important for recruitment of neutrophils into inflamed mouse peritoneum. Blood 1997, 90, 1934–1942. [Google Scholar] [CrossRef]
- Ramachandran, V.; Nollert, M.U.; Qiu, H.Y.; Liu, W.J.; Cummings, R.D.; Zhu, C.; McEver, R.P. Tyrosine replacement in P-selectin glycoprotein ligand-1 affects distinct kinetic and mechanical properties of bonds with P- and L-selectin. Proc. Natl. Acad. Sci. USA 1999, 96, 13771–13776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westmuckett, A.D.; Thacker, K.M.; Moore, K.L. Tyrosine Sulfation of Native Mouse Psgl-1 Is Required for Optimal Leukocyte Rolling on P-Selectin In Vivo. PLoS ONE 2011, 6, e20406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tenno, M.; Ohtsubo, K.; Hagen, F.K.; Ditto, D.; Zarbock, A.; Schaerli, P.; von Andrian, U.H.; Ley, K.; Le, D.; Tabak, L.A.; et al. Initiation of protein O glycosylation by the polypeptide GalNAcT-1 in vascular biology and humoral immunity. Mol. Cell Biol. 2007, 27, 8783–8796. [Google Scholar] [CrossRef] [Green Version]
- Block, H.; Ley, K.; Zarbock, A. Severe Impairment of Leukocyte Recruitment in ppGalNAcT-1-Deficient Mice. J. Immunol. 2012, 188, 5674–5681. [Google Scholar] [CrossRef] [Green Version]
- Sundd, P.; Pospieszalska, M.K.; Ley, K. Neutrophil rolling at high shear: Flattening, catch bond behavior, tethers and slings. Mol. Immunol. 2013, 55, 59–69. [Google Scholar] [CrossRef] [Green Version]
- Sundd, P.; Gutierrez, E.; Pospieszalska, M.K.; Zhang, H.; Groisman, A.; Ley, K. Quantitative dynamic footprinting microscopy reveals mechanisms of neutrophil rolling. Nat. Methods 2010, 7, 821–824. [Google Scholar] [CrossRef] [Green Version]
- Marshall, B.T.; Long, M.; Piper, J.W.; Yago, T.; McEver, R.P.; Zhu, C. Direct observation of catch bonds involving cell-adhesion molecules. Nature 2003, 423, 190–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dembo, M.; Torney, D.C.; Saxman, K.; Hammer, D. The reaction-limited kinetics of membrane-to-surface adhesion and detachment. Proc. R Soc. Lond B Biol. Sci. 1988, 234, 55–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarangapani, K.K.; Yago, T.; Klopocki, A.G.; Lawrence, M.B.; Fieger, C.B.; Rosen, S.D.; McEver, R.P.; Zhu, C. Low force decelerates L-selectin dissociation from P-selectin glycoprotein ligand-1 and endoglycan. J. Biol. Chem. 2004, 279, 2291–2298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morikis, V.A.; Chase, S.; Wun, T.; Chaikof, E.L.; Magnani, J.L.; Simon, S.I. Selectin catch-bonds mechanotransduce integrin activation and neutrophil arrest on inflamed endothelium under shear flow. Blood 2017, 130, 2101–2110. [Google Scholar] [CrossRef] [PubMed]
- Morikis, V.A.; Simon, S.I. Neutrophil Mechanosignaling Promotes Integrin Engagement With Endothelial Cells and Motility Within Inflamed Vessels. Front. Immunol. 2018, 9, 2774. [Google Scholar] [CrossRef]
- Morikis, V.A.; Hernandez, A.A.; Magnani, J.L.; Sperandio, M.; Simon, S.I. Targeting Neutrophil Adhesive Events to Address Vaso-Occlusive Crisis in Sickle Cell Patients. Front. Immunol. 2021, 12, 663886. [Google Scholar] [CrossRef]
- Shao, J.Y.; Hochmuth, R.M. Micropipette suction for measuring piconewton forces of adhesion and tether formation from neutrophil membranes. Biophy. J. 1996, 71, 2892–2901. [Google Scholar] [CrossRef]
- Cugno, A.; Marki, A.; Ley, K. Biomechanics of Neutrophil Tethers. Life 2021, 11, 515. [Google Scholar] [CrossRef]
- Marki, A.; Buscher, K.; Mikulski, Z.; Pries, A.; Ley, K. Rolling neutrophils form tethers and slings under physiologic conditions in vivo. J. Leukoc. Biol. 2018, 103, 67–70. [Google Scholar] [CrossRef] [Green Version]
- Lo, S.K.; Lee, S.; Ramos, R.A.; Lobb, R.; Rosa, M.; Chi-Rosso, G.; Wright, S.D. Endothelial-leukocyte adhesion molecule 1 stimulates the adhesive activity of leukocyte integrin CR3 (CD11b/CD18, Mac-1, alpha m beta 2) on human neutrophils. J. Exp. Med. 1991, 173, 1493–1500. [Google Scholar] [CrossRef] [Green Version]
- Yago, T.; Shao, B.; Miner, J.J.; Yao, L.; Klopocki, A.G.; Maeda, K.; Coggeshall, K.M.; McEver, R.P. E-selectin engages PSGL-1 and CD44 through a common signaling pathway to induce integrin alphaLbeta2-mediated slow leukocyte rolling. Blood 2010, 116, 485–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stadtmann, A.; Germena, G.; Block, H.; Boras, M.; Rossaint, J.; Sundd, P.; Lefort, C.; Fisher, C.I.; Buscher, K.; Gelschefarth, B.; et al. The PSGL-1-L-selectin signaling complex regulates neutrophil adhesion under flow. J. Exp. Med. 2013, 210, 2171–2180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zarbock, A.; Abram, C.L.; Hundt, M.; Altman, A.; Lowell, C.A.; Ley, K. PSGL-1 engagement by E-selectin signals through Src kinase Fgr and ITAM adapters DAP12 and FcR gamma to induce slow leukocyte rolling. J. Exp. Med. 2008, 205, 2339–2347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urzainqui, A.; Serrador, J.M.; Viedma, F.; Yanez-Mo, M.; Rodriguez, A.; Corbi, A.L.; Alonso-Lebrero, J.L.; Luque, A.; Deckert, M.; Vazquez, J.; et al. ITAM-based interaction of ERM proteins with Syk mediates signaling by the leukocyte adhesion receptor PSGL-1. Immunity 2002, 17, 401–412. [Google Scholar] [CrossRef] [Green Version]
- Alonso-Lebrero, J.L.; Serrador, J.M.; Dominguez-Jimenez, C.; Barreiro, O.; Luque, A.; del Pozo, M.A.; Snapp, K.; Kansas, G.; Schwartz-Albiez, R.; Furthmayr, H.; et al. Polarization and interaction of adhesion molecules P-selectin glycoprotein ligand 1 and intercellular adhesion molecule 3 with moesin and ezrin in myeloid cells. Blood 2000, 95, 2413–2419. [Google Scholar] [CrossRef]
- Mueller, H.; Stadtmann, A.; Van Aken, H.; Hirsch, E.; Wang, D.; Ley, K.; Zarbock, A. Tyrosine kinase Btk regulates E-selectin-mediated integrin activation and neutrophil recruitment by controlling phospholipase C (PLC) gamma2 and PI3Kgamma pathways. Blood 2010, 115, 3118–3127. [Google Scholar] [CrossRef] [Green Version]
- Block, H.; Herter, J.M.; Rossaint, J.; Stadtmann, A.; Kliche, S.; Lowell, C.A.; Zarbock, A. Crucial role of SLP-76 and ADAP for neutrophil recruitment in mouse kidney ischemia-reperfusion injury. J. Exp. Med. 2012, 209, 407–421. [Google Scholar] [CrossRef] [Green Version]
- Yago, T.; Liu, Z.; Ahamed, J.; McEver, R.P. Cooperative PSGL-1 and CXCR2 signaling in neutrophils promotes deep vein thrombosis in mice. Blood 2018, 132, 1426–1437. [Google Scholar] [CrossRef] [Green Version]
- DiVietro, J.A.; Smith, M.J.; Smith, B.R.; Petruzzelli, L.; Larson, R.S.; Lawrence, M.B. Immobilized IL-8 triggers progressive activation of neutrophils rolling in vitro on P-selectin and intercellular adhesion molecule-1. J. Immunol. 2001, 167, 4017–4025. [Google Scholar] [CrossRef] [Green Version]
- Stadtmann, A.; Brinkhaus, L.; Mueller, H.; Rossaint, J.; Bolomini-Vittori, M.; Bergmeier, W.; Van Aken, H.; Wagner, D.D.; Laudanna, C.; Ley, K.; et al. Rap1a activation by CalDAG-GEFI and p38 MAPK is involved in E-selectin-dependent slow leukocyte rolling. Eur. J. Immunol. 2011, 41, 2074–2085. [Google Scholar] [CrossRef] [Green Version]
- Rossaint, J.; Oehmichen, J.; Van Aken, H.; Reuter, S.; Pavenstadt, H.J.; Meersch, M.; Unruh, M.; Zarbock, A. FGF23 signaling impairs neutrophil recruitment and host defense during CKD. J. Clin. Investig. 2016, 126, 962–974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kempf, T.; Zarbock, A.; Widera, C.; Butz, S.; Stadtmann, A.; Rossaint, J.; Bolomini-Vittori, M.; Korf-Klingebiel, M.; Napp, L.C.; Hansen, B.; et al. GDF-15 is an inhibitor of leukocyte integrin activation required for survival after myocardial infarction in mice. Nat. Med. 2011, 17, 581–588. [Google Scholar] [CrossRef] [PubMed]
- Hill, K.; Krugmann, S.; Andrews, S.R.; Coadwell, W.J.; Finan, P.; Welch, H.C.; Hawkins, P.T.; Stephens, L.R. Regulation of P-Rex1 by phosphatidylinositol (3,4,5)-trisphosphate and Gbetagamma subunits. J. Biol. Chem. 2005, 280, 4166–4173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Svensson, L.; Howarth, K.; McDowall, A.; Patzak, I.; Evans, R.; Ussar, S.; Moser, M.; Metin, A.; Fried, M.; Tomlinson, I.; et al. Leukocyte adhesion deficiency-III is caused by mutations in KINDLIN3 affecting integrin activation. Nat. Med. 2009, 15, 306–312. [Google Scholar] [CrossRef] [Green Version]
- Moser, M.; Legate, K.R.; Zent, R.; Fassler, R. The tail of integrins, talin, and kindlins. Science 2009, 324, 895–899. [Google Scholar] [CrossRef]
- Lee, H.S.; Lim, C.J.; Puzon-McLaughlin, W.; Shattil, S.J.; Ginsberg, M.H. RIAM activates integrins by linking talin to ras GTPase membrane-targeting sequences. J. Biol. Chem. 2009, 284, 5119–5127. [Google Scholar] [CrossRef] [Green Version]
- Bromberger, T.; Klapproth, S.; Rohwedder, I.; Weber, J.; Pick, R.; Mittmann, L.; Min-Weissenhorn, S.J.; Reichel, C.A.; Scheiermann, C.; Sperandio, M.; et al. Binding of Rap1 and Riam to Talin1 Fine-Tune beta2 Integrin Activity During Leukocyte Trafficking. Front. Immunol. 2021, 12, 702345. [Google Scholar] [CrossRef]
- Swanson, K.D.; Tang, Y.; Ceccarelli, D.F.; Poy, F.; Sliwa, J.P.; Neel, B.G.; Eck, M.J. The Skap-hom dimerization and PH domains comprise a 3’-phosphoinositide-gated molecular switch. Mol. Cell. 2008, 32, 564–575. [Google Scholar] [CrossRef] [Green Version]
- Menasche, G.; Kliche, S.; Chen, E.J.; Stradal, T.E.; Schraven, B.; Koretzky, G. RIAM links the ADAP/SKAP-55 signaling module to Rap1, facilitating T-cell-receptor-mediated integrin activation. Mol. Cell. Biol. 2007, 27, 4070–4081. [Google Scholar] [CrossRef] [Green Version]
- Boras, M.; Volmering, S.; Bokemeyer, A.; Rossaint, J.; Block, H.; Bardel, B.; Van Marck, V.; Heitplatz, B.; Kliche, S.; Reinhold, A.; et al. Skap2 is required for beta2 integrin-mediated neutrophil recruitment and functions. J. Exp. Med. 2017, 214, 851–874. [Google Scholar] [CrossRef]
- Vadillo, E.; Chanez-Paredes, S.; Vargas-Robles, H.; Guerrero-Fonseca, I.M.; Castellanos-Martinez, R.; Garcia-Ponce, A.; Nava, P.; Giron-Perez, D.A.; Santos-Argumedo, L.; Schnoor, M. Intermittent rolling is a defect of the extravasation cascade caused by Myosin1e-deficiency in neutrophils. Proc. Natl. Acad. Sci. USA 2019, 116, 26752–26758. [Google Scholar] [CrossRef] [PubMed]
- Conrad, C.; Yildiz, D.; Cleary, S.J.; Margraf, A.; Cook, L.; Schlomann, U.; Panaretou, B.; Bowser, J.L.; Karmouty-Quintana, H.; Li, J.; et al. ADAM8 signaling drives neutrophil migration and ARDS severity. JCI Insight 2022, 7, e149870. [Google Scholar] [CrossRef] [PubMed]
- Miner, J.J.; Xia, L.; Yago, T.; Kappelmayer, J.; Liu, Z.; Klopocki, A.G.; Shao, B.; McDaniel, J.M.; Setiadi, H.; Schmidtke, D.W.; et al. Separable requirements for cytoplasmic domain of PSGL-1 in leukocyte rolling and signaling under flow. Blood 2008, 112, 2035–2045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stadtmann, A.; Block, H.; Volmering, S.; Abram, C.; Sohlbach, C.; Boras, M.; Lowell, C.A.; Zarbock, A. Cross-Talk between Shp1 and PIPKIgamma Controls Leukocyte Recruitment. J. Immunol. 2015, 195, 1152–1161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.; Carman, C.V.; Yang, W.; Salas, A.; Springer, T.A. The primacy of affinity over clustering in regulation of adhesiveness of the integrin {alpha}L{beta}2. J. Cell Biol. 2004, 167, 1241–1253. [Google Scholar] [CrossRef]
- Ivetic, A.; Deka, J.; Ridley, A.; Ager, A. The cytoplasmic tail of L-selectin interacts with members of the Ezrin-Radixin-Moesin (ERM) family of proteins: Cell activation-dependent binding of Moesin but not Ezrin. J. Biol. Chem. 2002, 277, 2321–2329. [Google Scholar] [CrossRef] [Green Version]
- Killock, D.J.; Parsons, M.; Zarrouk, M.; Ameer-Beg, S.M.; Ridley, A.J.; Haskard, D.O.; Zvelebil, M.; Ivetic, A. In Vitro and in Vivo Characterization of Molecular Interactions between Calmodulin, Ezrin/Radixin/Moesin, and L-selectin. J. Biol. Chem. 2009, 284, 8833–8845. [Google Scholar] [CrossRef] [Green Version]
- Ivetic, A. Signals regulating L-selectin-dependent leucocyte adhesion and transmigration. Int. J. Biochem. Cell Biol. 2013, 45, 550–555. [Google Scholar] [CrossRef]
- Stein, J.V.; Cheng, G.; Stockton, B.M.; Fors, B.P.; Butcher, E.C.; von Andrian, U.H. L-selectin-mediated leukocyte adhesion in vivo: Microvillous distribution determines tethering efficiency, but not rolling velocity. J. Exp. Med. 1999, 189, 37–50. [Google Scholar] [CrossRef] [Green Version]
- Zollner, O.; Lenter, M.C.; Blanks, J.E.; Borges, E.; Steegmaier, M.; Zerwes, H.G.; Vestweber, D. L-selectin from human, but not from mouse neutrophils binds directly to E-selectin. J. Cell. Biol. 1997, 136, 707–716. [Google Scholar] [CrossRef]
- Smalley, D.M.; Ley, K. L-selectin: Mechanisms and physiological significance of ectodomain cleavage. J. Cell Mol. Med. 2005, 9, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Kishimoto, T.K.; Jutila, M.A.; Berg, E.L.; Butcher, E.C. Neutrophil Mac-1 and MEL-14 adhesion proteins inversely regulated by chemotactic factors. Science 1989, 245, 1238–1241. [Google Scholar] [CrossRef] [PubMed]
- Schleiffenbaum, B.; Spertini, O.; Tedder, T.F. Soluble L-selectin is present in human plasma at high levels and retains functional activity. J. Cell Biol. 1992, 119, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Allport, J.R.; Ding, H.T.; Ager, A.; Steeber, D.A.; Tedder, T.F.; Luscinskas, F.W. L-selectin shedding does not regulate human neutrophil attachment, rolling, or transmigration across human vascular endothelium in vitro. J. Immunol. 1997, 158, 4365–4372. [Google Scholar]
- Rzeniewicz, K.; Newe, A.; Rey Gallardo, A.; Davies, J.; Holt, M.R.; Patel, A.; Charras, G.T.; Stramer, B.; Molenaar, C.; Tedder, T.F.; et al. L-selectin shedding is activated specifically within transmigrating pseudopods of monocytes to regulate cell polarity in vitro. Proc. Natl. Acad. Sci. USA 2015, 112, E1461–E1470. [Google Scholar] [CrossRef] [Green Version]
- Cappenberg, A.; Margraf, A.; Thomas, K.; Bardel, B.; McCreedy, D.A.; Van Marck, V.; Mellmann, A.; Lowell, C.A.; Zarbock, A. L-selectin shedding affects bacterial clearance in the lung: A new regulatory pathway for integrin outside-in signaling. Blood 2019, 134, 1445–1457. [Google Scholar] [CrossRef]
- Li, Y.; Brazzell, J.; Herrera, A.; Walcheck, B. ADAM17 deficiency by mature neutrophils has differential effects on L-selectin shedding. Blood 2006, 108, 2275–2279. [Google Scholar] [CrossRef]
- Singhal, A.; Dhankani, P.; Gupta, K.L.; Mazumder, J.; Adithya, R.; Dikshit, M.; Kumar, S. Rho signaling inhibition mitigates lung injury via targeting neutrophil recruitment and selectin-AKT signaling. Biochim. Biophys Acta Mol. Cell Res. 2021, 1868, 119122. [Google Scholar] [CrossRef]
- Rahman, I.; Collado Sanchez, A.; Davies, J.; Rzeniewicz, K.; Abukscem, S.; Joachim, J.; Hoskins Green, H.L.; Killock, D.; Sanz, M.J.; Charras, G.; et al. L-selectin regulates human neutrophil transendothelial migration. J. Cell Sci. 2021, 134, jcs250340. [Google Scholar] [CrossRef]
- Walcheck, B.; Kahn, J.; Fisher, J.M.; Wang, B.B.; Fisk, R.S.; Payan, D.G.; Feehan, C.; Betageri, R.; Darlak, K.; Spatola, A.F.; et al. Neutrophil rolling altered by inhibition of L-selectin shedding in vitro. Nature 1996, 380, 720–723. [Google Scholar] [CrossRef]
- Pavalko, F.M.; Walker, D.M.; Graham, L.; Goheen, M.; Doerschuk, C.M.; Kansas, G.S. The cytoplasmic domain of L-selectin interacts with cytoskeletal proteins via alpha-actinin: Receptor positioning in microvilli does not require interaction with alpha-actinin. J. Cell Biol. 1995, 129, 1155–1164. [Google Scholar] [CrossRef] [PubMed]
- Waddell, T.K.; Fialkow, L.; Chan, C.K.; Kishimoto, T.K.; Downey, G.P. Signaling functions of L-selectin. Enhancement of tyrosine phosphorylation and activation of MAP kinase. J. Biol. Chem. 1995, 270, 15403–15411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, C.E.; Pearson, D.N.; Camphausen, R.T.; Staunton, D.E.; Simon, S.I. Shear-dependent capping of L-selectin and P-selectin glycoprotein ligand 1 by E-selectin signals activation of high-avidity beta2-integrin on neutrophils. J. Immunol. 2004, 172, 7780–7790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giblin, P.A.; Hwang, S.T.; Katsumoto, T.R.; Rosen, S.D. Ligation of L-selectin on T lymphocytes activates beta1 integrins and promotes adhesion to fibronectin. J. Immunol. 1997, 159, 3498–3507. [Google Scholar] [PubMed]
- Opal, S.M.; DePalo, V.A. Anti-inflammatory cytokines. Chest 2000, 117, 1162–1172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Margraf, A.; Cappenberg, A.; Vadillo, E.; Ludwig, N.; Thomas, K.; Korner, K.; Zondler, L.; Rossaint, J.; Germena, G.; Hirsch, E.; et al. ArhGAP15, a RacGAP, Acts as a Temporal Signaling Regulator of Mac-1 Affinity in Sterile Inflammation. J. Immunol. 2020, 205, 1365–1375. [Google Scholar] [CrossRef]
- Plutzky, J.; Neel, B.G.; Rosenberg, R.D. Isolation of a src homology 2-containing tyrosine phosphatase. Proc. Natl. Acad Sci. USA 1992, 89, 1123–1127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, M.C.; Shultz, L.D. Motheaten, an immunodeficient mutant of the mouse. I. Genetics and pathology. J. Hered. 1975, 66, 250–258. [Google Scholar] [CrossRef]
- Koo, G.C.; Rosen, H.; Sirotina, A.; Ma, X.D.; Shultz, L.D. Anti-CD11b antibody prevents immunopathologic changes in viable moth-eaten bone marrow chimeric mice. J. Immunol. 1993, 151, 6733–6741. [Google Scholar]
- Rossaint, J.; Kuhne, K.; Skupski, J.; Van Aken, H.; Looney, M.R.; Hidalgo, A.; Zarbock, A. Directed transport of neutrophil-derived extracellular vesicles enables platelet-mediated innate immune response. Nat. Commun. 2016, 7, 13464. [Google Scholar] [CrossRef] [PubMed]
- Rittirsch, D.; Huber-Lang, M.S.; Flierl, M.A.; Ward, P.A. Immunodesign of experimental sepsis by cecal ligation and puncture. Nat. Protoc. 2009, 4, 31–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bittner, S.; Afzali, A.M.; Wiendl, H.; Meuth, S.G. Myelin oligodendrocyte glycoprotein (MOG35-55) induced experimental autoimmune encephalomyelitis (EAE) in C57BL/6 mice. J. Vis. Exp. 2014, 86, e51275. [Google Scholar] [CrossRef] [Green Version]
- Anderson, D.C.; Springer, T.A. Leukocyte adhesion deficiency: An inherited defect in the Mac-1, LFA-1, and p150,95 glycoproteins. Annu. Rev. Med. 1987, 38, 175–194. [Google Scholar] [CrossRef] [PubMed]
- Kuijpers, T.W.; Van Lier, R.A.; Hamann, D.; de Boer, M.; Thung, L.Y.; Weening, R.S.; Verhoeven, A.J.; Roos, D. Leukocyte adhesion deficiency type 1 (LAD-1)/variant. A novel immunodeficiency syndrome characterized by dysfunctional beta2 integrins. J. Clin. Investig. 1997, 100, 1725–1733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanna, S.; Etzioni, A. Leukocyte adhesion deficiencies. Ann. N. Y. Acad. Sci. 2012, 1250, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Almarza Novoa, E.; Kasbekar, S.; Thrasher, A.J.; Kohn, D.B.; Sevilla, J.; Nguyen, T.; Schwartz, J.D.; Bueren, J.A. Leukocyte adhesion deficiency-I: A comprehensive review of all published cases. J. Allergy Clin. Immunol. Pract. 2018, 6, 1418–1420.e1410. [Google Scholar] [CrossRef]
- Phillips, M.L.; Schwartz, B.R.; Etzioni, A.; Bayer, R.; Ochs, H.D.; Paulson, J.C.; Harlan, J.M. Neutrophil adhesion in leukocyte adhesion deficiency syndrome type 2. J. Clin. Investig. 1995, 96, 2898–2906. [Google Scholar] [CrossRef]
- Peters, T.; Bloch, W.; Pabst, O.; Wickenhauser, C.; Uthoff-Hachenberg, C.; Schmidt, S.V.; Varga, G.; Grabbe, S.; Kess, D.; Oreshkova, T.; et al. Adaptive immune response to model antigens is impaired in murine leukocyte-adhesion deficiency-1 revealing elevated activation thresholds in vivo. Clin. Dev. Immunol. 2012, 2012, 450738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Etzioni, A. Adhesion molecule deficiencies and their clinical significance. Cell Adhes Commun. 1994, 2, 257–260. [Google Scholar] [CrossRef]
- Etzioni, A.; Gershoni-Baruch, R.; Pollack, S.; Shehadeh, N. Leukocyte adhesion deficiency type II: Long-term follow-up. J. Allergy Clin. Immunol. 1998, 102, 323–324. [Google Scholar] [CrossRef]
- Luhn, K.; Wild, M.K.; Eckhardt, M.; Gerardy-Schahn, R.; Vestweber, D. The gene defective in leukocyte adhesion deficiency II encodes a putative GDP-fucose transporter. Nat. Genet. 2001, 28, 69–72. [Google Scholar] [CrossRef] [PubMed]
- Etzioni, A.; Frydman, M.; Pollack, S.; Avidor, I.; Phillips, M.L.; Paulson, J.C.; Gershoni-Baruch, R. Brief report: Recurrent severe infections caused by a novel leukocyte adhesion deficiency. N. Engl. J. Med. 1992, 327, 1789–1792. [Google Scholar] [CrossRef] [PubMed]
- von Andrian, U.H.; Berger, E.M.; Ramezani, L.; Chambers, J.D.; Ochs, H.D.; Harlan, J.M.; Paulson, J.C.; Etzioni, A.; Arfors, K.E. In vivo behavior of neutrophils from two patients with distinct inherited leukocyte adhesion deficiency syndromes. J. Clin Investig. 1993, 91, 2893–2897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marquardt, T.; Brune, T.; Luhn, K.; Zimmer, K.P.; Korner, C.; Fabritz, L.; van der Werft, N.; Vormoor, J.; Freeze, H.H.; Louwen, F.; et al. Leukocyte adhesion deficiency II syndrome, a generalized defect in fucose metabolism. J. Pediatr. 1999, 134, 681–688. [Google Scholar] [CrossRef]
- Yakubenia, S.; Frommhold, D.; Scholch, D.; Hellbusch, C.C.; Korner, C.; Petri, B.; Jones, C.; Ipe, U.; Bixel, M.G.; Krempien, R.; et al. Leukocyte trafficking in a mouse model for leukocyte adhesion deficiency II/congenital disorder of glycosylation IIc. Blood 2008, 112, 1472–1481. [Google Scholar] [CrossRef] [PubMed]
- Mory, A.; Feigelson, S.W.; Yarali, N.; Kilic, S.S.; Bayhan, G.I.; Gershoni-Baruch, R.; Etzioni, A.; Alon, R. Kindlin-3: A new gene involved in the pathogenesis of LAD-III. Blood 2008, 112, 2591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergmeier, W.; Goerge, T.; Wang, H.W.; Crittenden, J.R.; Baldwin, A.C.; Cifuni, S.M.; Housman, D.E.; Graybiel, A.M.; Wagner, D.D. Mice lacking the signaling molecule CalDAG-GEFI represent a model for leukocyte adhesion deficiency type III. J. Clin. Investig. 2007, 117, 1699–1707. [Google Scholar] [CrossRef] [Green Version]
- Etzioni, A. Defects in the leukocyte adhesion cascade. Clin. Rev. Allergy Immunol. 2010, 38, 54–60. [Google Scholar] [CrossRef]
- Wild, M.K.; Luhn, K.; Marquardt, T.; Vestweber, D. Leukocyte adhesion deficiency II: Therapy and genetic defect. Cells Tissues Organs 2002, 172, 161–173. [Google Scholar] [CrossRef]
- Krstic, D.; Knuesel, I. Deciphering the mechanism underlying late-onset Alzheimer disease. Nat. Rev. Neurol. 2013, 9, 25–34. [Google Scholar] [CrossRef] [Green Version]
- Czirr, E.; Wyss-Coray, T. The immunology of neurodegeneration. J. Clin Investig. 2012, 122, 1156–1163. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, M.; Kipnis, J.; Rivest, S.; Prat, A. How do immune cells support and shape the brain in health, disease, and aging? J. Neurosci. 2013, 33, 17587–17596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zenaro, E.; Pietronigro, E.; Della Bianca, V.; Piacentino, G.; Marongiu, L.; Budui, S.; Turano, E.; Rossi, B.; Angiari, S.; Dusi, S.; et al. Neutrophils promote Alzheimer’s disease-like pathology and cognitive decline via LFA-1 integrin. Nat. Med. 2015, 21, 880–886. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Nesbitt, W.S.; Westein, E. Dynamics of platelet thrombus formation. J. Thromb. Haemost. 2009, 7, 17–20. [Google Scholar] [CrossRef]
- Blann, A.D.; Nadar, S.K.; Lip, G.Y. The adhesion molecule P-selectin and cardiovascular disease. Eur. Heart J. 2003, 24, 2166–2179. [Google Scholar] [CrossRef] [Green Version]
- Totani, L.; Evangelista, V. Platelet-leukocyte interactions in cardiovascular disease and beyond. Arter. Thromb. Vasc. Biol. 2010, 30, 2357–2361. [Google Scholar] [CrossRef]
- Falati, S.; Liu, Q.; Gross, P.; Merrill-Skoloff, G.; Chou, J.; Vandendries, E.; Celi, A.; Croce, K.; Furie, B.C.; Furie, B. Accumulation of tissue factor into developing thrombi in vivo is dependent upon microparticle P-selectin glycoprotein ligand 1 and platelet P-selectin. J. Exp. Med. 2003, 197, 1585–1598. [Google Scholar] [CrossRef]
- Matsui, N.M.; Borsig, L.; Rosen, S.D.; Yaghmai, M.; Varki, A.; Embury, S.H. P-selectin mediates the adhesion of sickle erythrocytes to the endothelium. Blood 2001, 98, 1955–1962. [Google Scholar] [CrossRef] [Green Version]
- Dominical, V.M.; Samsel, L.; Nichols, J.S.; Costa, F.F.; McCoy, J.P., Jr.; Conran, N.; Kato, G.J. Prominent role of platelets in the formation of circulating neutrophil-red cell heterocellular aggregates in sickle cell anemia. Haematologica 2014, 99, e214–e217. [Google Scholar] [CrossRef]
- Bennewitz, M.F.; Tutuncuoglu, E.; Gudapati, S.; Brzoska, T.; Watkins, S.C.; Monga, S.P.; Pradhan-Sundd, T.; Sundd, P. P-selectin-deficient mice to study pathophysiology of sickle cell disease. Blood Adv. 2020, 4, 266–273. [Google Scholar] [CrossRef]
- Vats, R.; Kaminski, T.W.; Ju, E.M.; Brozska, T.; Tutuncuoglu, E.; Tejero, J.; Novelli, E.M.; Sundd, P.; Pradhan-Sundd, T. P-selectin deficiency promotes liver senescence in sickle cell disease mice. Blood 2021, 137, 2676–2680. [Google Scholar] [CrossRef] [PubMed]
- Manne, B.K.; Denorme, F.; Middleton, E.A.; Portier, I.; Rowley, J.W.; Stubben, C.; Petrey, A.C.; Tolley, N.D.; Guo, L.; Cody, M.; et al. Platelet gene expression and function in patients with COVID-19. Blood 2020, 136, 1317–1329. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cappenberg, A.; Kardell, M.; Zarbock, A. Selectin-Mediated Signaling—Shedding Light on the Regulation of Integrin Activity in Neutrophils. Cells 2022, 11, 1310. https://doi.org/10.3390/cells11081310
Cappenberg A, Kardell M, Zarbock A. Selectin-Mediated Signaling—Shedding Light on the Regulation of Integrin Activity in Neutrophils. Cells. 2022; 11(8):1310. https://doi.org/10.3390/cells11081310
Chicago/Turabian StyleCappenberg, Anika, Marina Kardell, and Alexander Zarbock. 2022. "Selectin-Mediated Signaling—Shedding Light on the Regulation of Integrin Activity in Neutrophils" Cells 11, no. 8: 1310. https://doi.org/10.3390/cells11081310