
Live-Cell Imaging of the Contractile Velocity and Transient Intracellular Ca2+ Fluctuations in Human Stem Cell-Derived Cardiomyocytes

 ,
,
Abstract
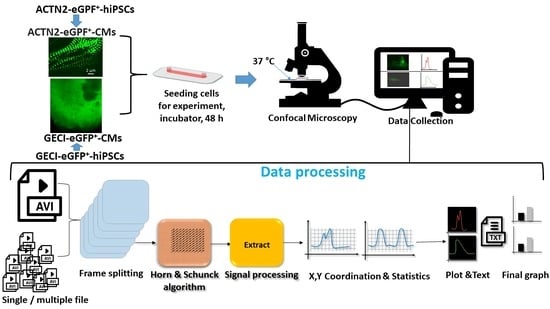
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Differentiation of hiPSCs to Cardiomyocytes
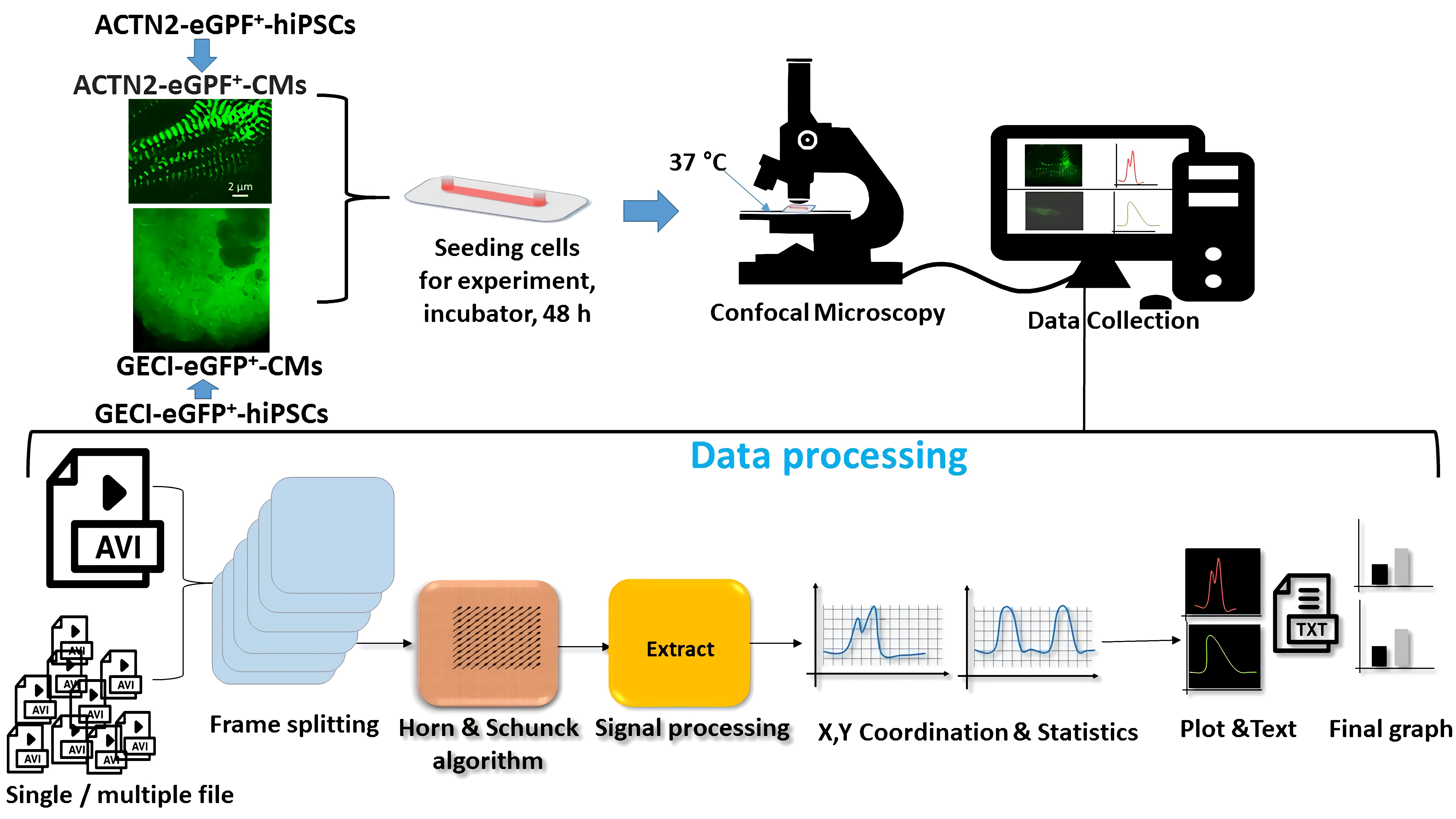
2.2. Generation of the GECI-eGFP+-hiPSCs
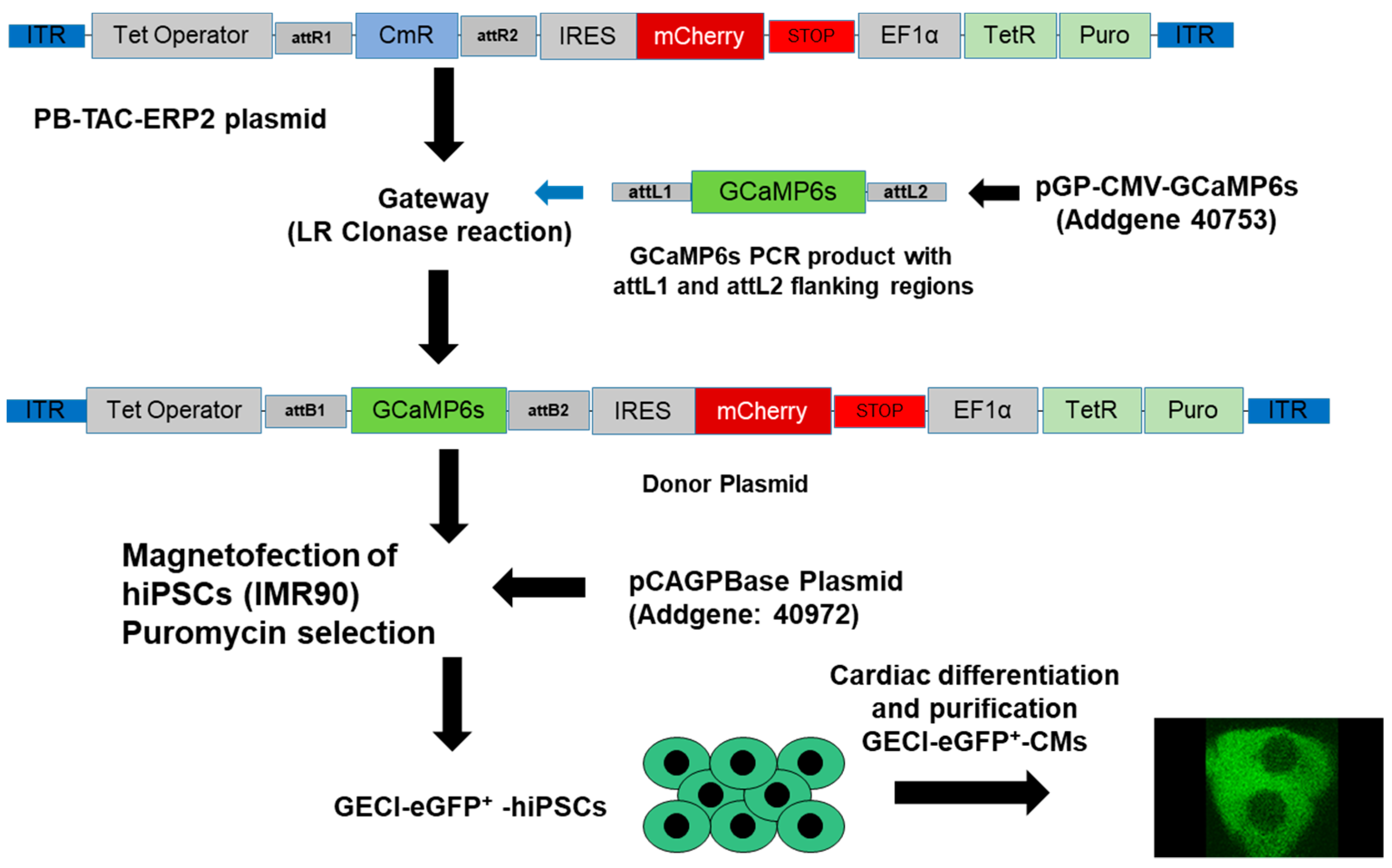
2.3. Generation of ACTN2-copGFP+-hiPSC Line
2.4. Video Analyzer 1.9
2.5. Statistical Analysis
3. Results
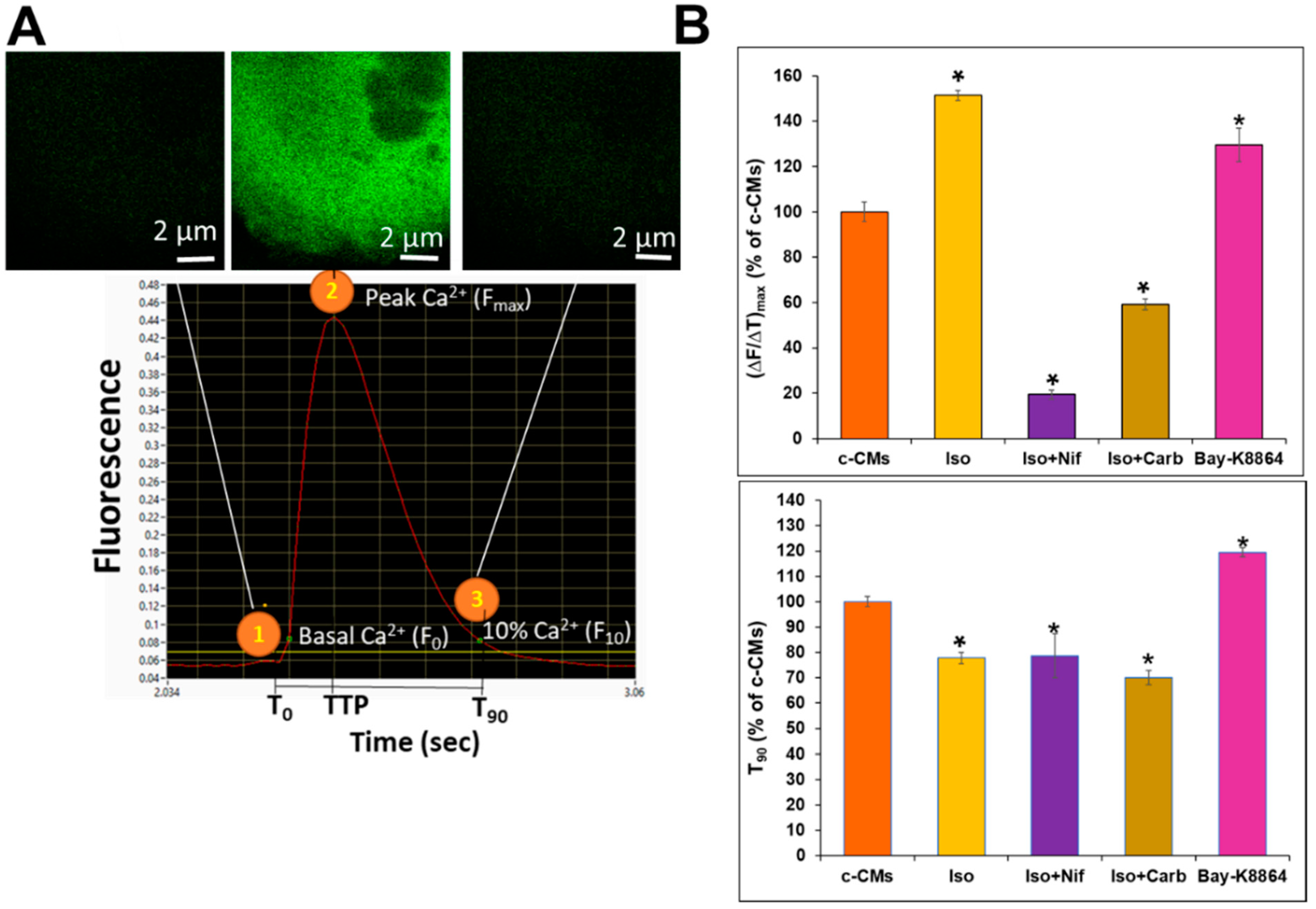
3.1. Live-Cell Imaging of Intracellular Ca2+ Alterations during Contraction of GECI-eGFP+-CMs
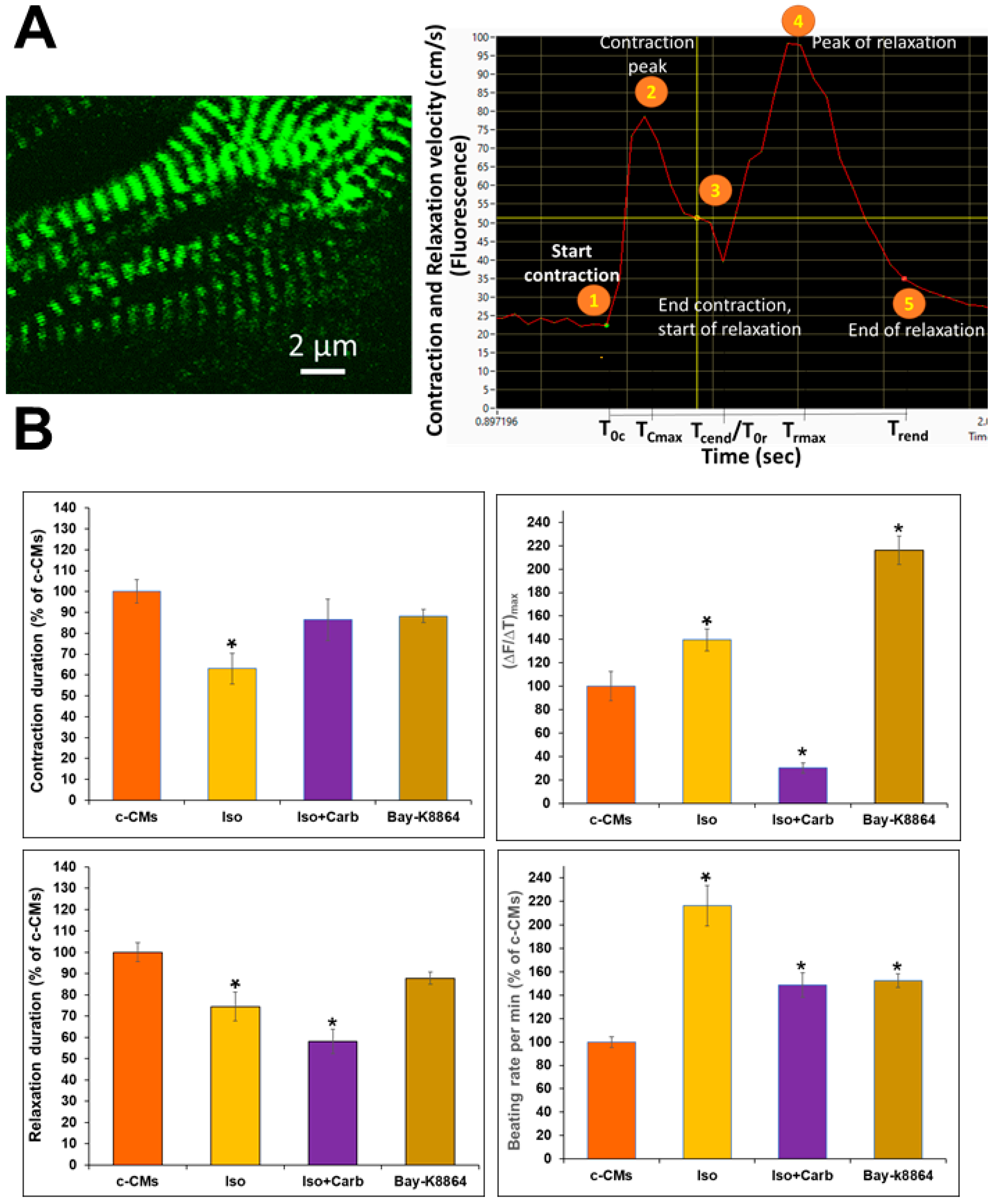
3.2. Live-Time Imaging of the Contractile Velocity of ACTN2-copGFP+-CMs in the Presence or Absence of Different Agonists and Antagonists
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lazzarini, V.; Mentz, R.J.; Fiuzat, M.; Metra, M.; O’Connor, C.M. Heart failure in elderly patients: Distinctive features and unresolved issues. Eur. J. Heart Fail. 2013, 15, 717–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lein, E.S.; Hawrylycz, M.J.; Ao, N.; Ayres, M.; Bensinger, A.; Bernard, A.; Boe, A.F.; Boguski, M.S.; Brockway, K.S.; Byrnes, E.J.; et al. Genome-Wide atlas of gene expression in the adult mouse brain. Nature 2007, 445, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Harvey, P.A.; Leinwand, L.A. The cell biology of disease: Cellular mechanisms of cardiomyopathy. J. Cell Biol. 2011, 194, 355–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyon, R.C.; Zanella, F.; Omens, J.H.; Sheikh, F. Mechanotransduction in cardiac hypertrophy and failure. Circ. Res. 2015, 116, 1462–1476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, P.M.; Sykes, B.D. Targeting the sarcomere to correct muscle function. Nat. Rev. Drug Discov. 2015, 14, 313–328. [Google Scholar] [CrossRef] [PubMed]
- Janssen, P.M.L. Myocardial relaxation in human heart failure: Why sarcomere kinetics should be center-stage. Arch. Biochem. Biophys. 2019, 661, 145–148. [Google Scholar] [CrossRef] [PubMed]
- Bai, S.W.; Herrera-Abreu, M.T.; Rohn, J.L.; Racine, V.; Tajadura, V.; Suryavanshi, N.; Bechtel, S.; Wiemann, S.; Baum, B.; Ridley, A.J. Identification and characterization of a set of conserved and new regulators of cytoskeletal organization, cell morphology and migration. BMC Biol. 2011, 9, 54. [Google Scholar] [CrossRef] [Green Version]
- Hwang, J.; Pallas, D.C. STRIPAK complexes: Structure, biological function, and involvement in human diseases. Int. J. Biochem. Cell Biol. 2013, 47, 118–148. [Google Scholar] [CrossRef] [Green Version]
- Doss, M.X.; Sachinidis, A. Current Challenges of iPSC-Based Disease Modeling and Therapeutic Implications. Cells 2019, 8, 403. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro, A.J.S.; Guth, B.D.; Engwall, M.; Eldridge, S.; Foley, C.M.; Guo, L.; Gintant, G.; Koerner, J.; Parish, S.T.; Pierson, J.B.; et al. Considerations for an In Vitro, Cell-Based Testing Platform for Detection of Drug-Induced Inotropic Effects in Early Drug Development. Part 2: Designing and Fabricating Microsystems for Assaying Cardiac Contractility With Physiological Relevance Using Human iPSC-Cardiomyocytes. Front. Pharmacol. 2019, 10, 934. [Google Scholar] [CrossRef] [Green Version]
- Sachinidis, A.; Albrecht, W.; Nell, P.; Cherianidou, A.; Hewitt, N.J.; Edlund, K.; Hengstler, J.G. Road Map for Development of Stem Cell-Based Alternative Test Methods. Trends Mol. Med. 2019, 25, 470–481. [Google Scholar] [CrossRef] [PubMed]
- Sachinidis, A. Cardiotoxicity and Heart Failure: Lessons from Human-Induced Pluripotent Stem Cell-Derived Cardiomyocytes and Anticancer Drugs. Cells 2020, 9, 1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, A.; McKeithan, W.L.; Serrano, R.; Kitani, T.; Burridge, P.W.; Del Álamo, J.C.; Mercola, M.; Wu, J.C. Use of human induced pluripotent stem cell-derived cardiomyocytes to assess drug cardiotoxicity. Nat. Protoc. 2018, 13, 3018–3041. [Google Scholar] [CrossRef] [PubMed]
- Chaudhari, U.; Ellis, J.K.; Wagh, V.; Nemade, H.; Hescheler, J.; Keun, H.C.; Sachinidis, A. Metabolite signatures of doxorubicin induced toxicity in human induced pluripotent stem cell-derived cardiomyocytes. Amino Acids 2017, 49, 1955–1963. [Google Scholar] [CrossRef]
- Chaudhari, U.; Nemade, H.; Gaspar, J.A.; Hescheler, J.; Hengstler, J.G.; Sachinidis, A. MicroRNAs as early toxicity signatures of doxorubicin in human-induced pluripotent stem cell-derived cardiomyocytes. Arch. Toxicol. 2016, 90, 3087–3098. [Google Scholar] [CrossRef] [Green Version]
- Chaudhari, U.; Nemade, H.; Sureshkumar, P.; Vinken, M.; Ates, G.; Rogiers, V.; Hescheler, J.; Hengstler, J.G.; Sachinidis, A. Functional cardiotoxicity assessment of cosmetic compounds using human-induced pluripotent stem cell-derived cardiomyocytes. Arch. Toxicol. 2018, 92, 371–381. [Google Scholar] [CrossRef]
- Chaudhari, U.; Nemade, H.; Wagh, V.; Gaspar, J.A.; Ellis, J.K.; Srinivasan, S.P.; Spitkovski, D.; Nguemo, F.; Louisse, J.; Bremer, S.; et al. Identification of genomic biomarkers for anthracycline-induced cardiotoxicity in human iPSC-derived cardiomyocytes: An in vitro repeated exposure toxicity approach for safety assessment. Arch. Toxicol. 2016, 90, 2763–2777. [Google Scholar] [CrossRef]
- Nemade, H.; Chaudhari, U.; Acharya, A.; Hescheler, J.; Hengstler, J.G.; Papadopoulos, S.; Sachinidis, A. Cell death mechanisms of the anti-cancer drug etoposide on human cardiomyocytes isolated from pluripotent stem cells. Arch. Toxicol. 2018, 92, 1507–1524. [Google Scholar] [CrossRef] [Green Version]
- Hortigon-Vinagre, M.P.; Zamora, V.; Burton, F.L.; Smith, G.L. The Use of Voltage Sensitive Dye di-4-ANEPPS and Video-Based Contractility Measurements to Assess Drug Effects on Excitation–Contraction Coupling in Human-Induced Pluripotent Stem Cell–Derived Cardiomyocytes. J. Cardiovasc. Pharmacol. 2021, 77, 280–290. [Google Scholar] [CrossRef]
- Bedut, S.; Seminatore-Nole, C.; Lamamy, V.; Caignard, S.; Boutin, J.A.; Nosjean, O.; Stephan, J.-P.; Coge, F. High-throughput drug profiling with voltage- and calcium-sensitive fluorescent probes in human iPSC-derived cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 2016, 311, H44–H53. [Google Scholar] [CrossRef] [Green Version]
- El-Battrawy, I.; Zhao, Z.; Lan, H.; Cyganek, L.; Tombers, C.; Li, X.; Buljubasic, F.; Lang, S.; Tiburcy, M.; Zimmermann, W.-H.; et al. Electrical dysfunctions in human-induced pluripotent stem cell-derived cardiomyocytes from a patient with an arrhythmogenic right ventricular cardiomyopathy. EP Europace 2018, 20, f46–f56. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, M.K.B.; Vos, M.A.; Mirams, G.R.; Duker, G.; Sartipy, P.; de Boer, T.P.; van Veen, T.A.B. Application of human stem cell-derived cardiomyocytes in safety pharmacology requires caution beyond hERG. J. Mol. Cell 2012, 52, 998–1008. [Google Scholar] [CrossRef] [PubMed]
- Christidi, E.; Huang, H.M.; Brunham, L.R. CRISPR/Cas9-mediated genome editing in human stem cell-derived cardiomyocytes: Applications for cardiovascular disease modelling and cardiotoxicity screening. Drug Discov. Today Technol. 2018, 28, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Lian, X.; Hsiao, C.; Wilson, G.; Zhu, K.; Hazeltine, L.B.; Azarin, S.M.; Raval, K.K.; Zhang, J.; Kamp, T.J.; Palecek, S.P. Robust cardiomyocyte differentiation from human pluripotent stem cells via temporal modulation of canonical Wnt signaling. Proc. Natl. Acad. Sci. USA 2012, 109, E1848–E1857. [Google Scholar] [CrossRef] [Green Version]
- Tohyama, S.; Hattori, F.; Sano, M.; Hishiki, T.; Nagahata, Y.; Matsuura, T.; Hashimoto, H.; Suzuki, T.; Yamashita, H.; Satoh, Y.; et al. Distinct Metabolic Flow Enables Large-Scale Purification of Mouse and Human Pluripotent Stem Cell-Derived Cardiomyocytes. Cell Stem Cell 2013, 12, 127–137. [Google Scholar] [CrossRef] [Green Version]
- Nemade, H.; Acharya, A.; Chaudhari, U.; Nembo, E.; Nguemo, F.; Riet, N.; Abken, H.; Hescheler, J.; Papadopoulos, S.; Sachinidis, A. Cyclooxygenases Inhibitors Efficiently Induce Cardiomyogenesis in Human Pluripotent Stem Cells. Cells 2020, 9, 554. [Google Scholar] [CrossRef] [Green Version]
- Correia, C.; Koshkin, A.; Duarte, P.; Hu, D.; Teixeira, A.; Domian, I.; Serra, M.; Alves, P.M. Distinct carbon sources affect structural and functional maturation of cardiomyocytes derived from human pluripotent stem cells. Sci. Rep. 2017, 7, 8590. [Google Scholar] [CrossRef]
- Horn, B.K.P.; Schunck, B.G. Determining optical flow. Artif. Intell. 1981, 17, 185–203. [Google Scholar] [CrossRef] [Green Version]
- Jensen, E.C. Overview of Live-Cell Imaging: Requirements and Methods Used. Anat. Rec. 2013, 296, 1–8. [Google Scholar] [CrossRef]
- Iannetti, E.F.; Prigione, A.; Smeitink, J.A.M.; Koopman, W.J.H.; Beyrath, J.; Renkema, H. Live-Imaging Readouts and Cell Models for Phenotypic Profiling of Mitochondrial Function. Front. Genet. 2019, 10, 131. [Google Scholar] [CrossRef] [Green Version]
- Nasu, Y.; Shen, Y.; Kramer, L.; Campbell, R.E. Structure- and mechanism-guided design of single fluorescent protein-based biosensors. Nat. Chem. Biol. 2021, 17, 509–518. [Google Scholar] [CrossRef] [PubMed]
- Huebsch, N.; Loskill, P.; Mandegar, M.A.; Marks, N.C.; Sheehan, A.S.; Ma, Z.; Mathur, A.; Nguyen, T.N.; Yoo, J.C.; Judge, L.M.; et al. Automated Video-Based Analysis of Contractility and Calcium Flux in Human-Induced Pluripotent Stem Cell-Derived Cardiomyocytes Cultured over Different Spatial Scales. Tissue Eng. Part C Methods 2015, 21, 467–479. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Zhou, Y.; Bao, X.; Chen, C.; Randolph, L.N.; Du, J.; Lian, X.L. An Ultrasensitive Calcium Reporter System via CRISPR-Cas9-Mediated Genome Editing in Human Pluripotent Stem Cells. iScience 2018, 9, 27–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, W.Z.; Filice, D.; Palpant, N.J.; Laflamme, M.A. Methods for assessing the electromechanical integration of human pluripotent stem cell-derived cardiomyocyte grafts. Methods Mol. Biol. 2014, 1181, 229–247. [Google Scholar] [CrossRef] [Green Version]
- Li, E.S.; Saha, M.S. Optimizing Calcium Detection Methods in Animal Systems: A Sandbox for Synthetic Biology. Biomolecules 2021, 11, 343. [Google Scholar] [CrossRef]
- Germond, A.; Fujita, H.; Ichimura, T.; Watanabe, T.M. Design and development of genetically encoded fluorescent sensors to monitor intracellular chemical and physical parameters. Biophys. Rev. 2016, 8, 121–138. [Google Scholar] [CrossRef] [Green Version]
- De Michele, R.; Carimi, F.; Frommer, W.B. Mitochondrial biosensors. Int. J. Biochem. Cell Biol. 2014, 48, 39–44. [Google Scholar] [CrossRef]
- Gökerküçük, E.B.; Tramier, M.; Bertolin, G. Imaging Mitochondrial Functions: From Fluorescent Dyes to Genetically-Encoded Sensors. Genes 2020, 11, 125. [Google Scholar] [CrossRef] [Green Version]
- Bassett, J.J.; Monteith, G.R. Genetically Encoded Calcium Indicators as Probes to Assess the Role of Calcium Channels in Disease and for High-Throughput Drug Discovery. Adv. Pharmacol. 2017, 79, 141–171. [Google Scholar] [CrossRef] [Green Version]
- Bootman, M.D.; Allman, S.; Rietdorf, K.; Bultynck, G. Deleterious effects of calcium indicators within cells; an inconvenient truth. Cell Calcium 2018, 73, 82–87. [Google Scholar] [CrossRef] [Green Version]
- Roberts, B.; Hendershott, M.C.; Arakaki, J.; Gerbin, K.A.; Malik, H.; Nelson, A.; Gehring, J.; Hookway, C.; Ludmann, S.A.; Yang, R.; et al. Fluorescent Gene Tagging of Transcriptionally Silent Genes in hiPSCs. Stem Cell Rep. 2019, 12, 1145–1158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribeiro, M.C.; Slaats, R.H.; Schwach, V.; Rivera-Arbelaez, J.M.; Tertoolen, L.G.J.; van Meer, B.J.; Molenaar, R.; Mummery, C.L.; Claessens, M.; Passier, R. A cardiomyocyte show of force: A fluorescent alpha-actinin reporter line sheds light on human cardiomyocyte contractility versus substrate stiffness. J. Mol. Cell. Cardiol. 2020, 141, 54–64. [Google Scholar] [CrossRef] [PubMed]
- Pasqualin, C.; Gannier, F.; Yu, A.; Malécot, C.O.; Bredeloux, P.; Maupoil, V. SarcOptiM for ImageJ: High-frequency online sarcomere length computing on stimulated cardiomyocytes. Am. J. Physiol. Cell Physiol. 2016, 311, C277–C283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Psaras, Y.; Margara, F.; Cicconet, M.; Sparrow, A.J.; Repetti, G.G.; Schmid, M.; Steeples, V.; Wilcox, J.A.L.; Bueno-Orovio, A.; Redwood, C.S.; et al. CalTrack: High-Throughput Automated Calcium Transient Analysis in Cardiomyocytes. Circ. Res. 2021, 129, 326–341. [Google Scholar] [CrossRef] [PubMed]
- Sala, L.; van Meer, B.J.; Tertoolen, L.G.J.; Bakkers, J.; Bellin, M.; Davis, R.P.; Denning, C.; Dieben, M.A.E.; Eschenhagen, T.; Giacomelli, E.; et al. MUSCLEMOTION: A Versatile Open Software Tool to Quantify Cardiomyocyte and Cardiac Muscle Contraction In Vitro and In Vivo. Circ. Res. 2018, 122, e5–e16. [Google Scholar] [CrossRef] [PubMed]
- Toepfer, C.N.; Sharma, A.; Cicconet, M.; Garfinkel, A.C.; Mücke, M.; Neyazi, M.; Willcox, J.A.L.; Agarwal, R.; Schmid, M.; Rao, J.; et al. SarcTrack. Circ. Res. 2019, 124, 1172–1183. [Google Scholar] [CrossRef]
- Thomas, G.; Chung, M.; Cohen, C.J. A dihydropyridine (Bay k 8644) that enhances calcium currents in guinea pig and calf myocardial cells. A new type of positive inotropic agent. Circ. Res. 1985, 56, 87–96. [Google Scholar] [CrossRef] [Green Version]
- Greenstein, J.L.; Hinch, R.; Winslow, R.L. Mechanisms of excitation-contraction coupling in an integrative model of the cardiac ventricular myocyte. Biophys. J. 2006, 90, 77–91. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.M.; Hartzell, C.; Narlow, M.; Dudley, S.C. Stem Cell-Derived Cardiomyocytes Demonstrate Arrhythmic Potential. Circulation 2002, 106, 1294–1299. [Google Scholar] [CrossRef] [Green Version]
- Roof, S.R.; Shannon, T.R.; Janssen, P.M.; Ziolo, M.T. Effects of increased systolic Ca2+ and phospholamban phosphorylation during β-adrenergic stimulation on Ca2+ transient kinetics in cardiac myocytes. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H1570–H1578. [Google Scholar] [CrossRef] [Green Version]
- Stoehr, A.; Neuber, C.; Baldauf, C.; Vollert, I.; Friedrich, F.W.; Flenner, F.; Carrier, L.; Eder, A.; Schaaf, S.; Hirt, M.N.; et al. Automated analysis of contractile force and Ca2+ transients in engineered heart tissue. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H1353–H1363. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Acharya, A.; Nemade, H.; Rajendra Prasad, K.; Khan, K.; Hescheler, J.; Blackburn, N.; Hemmersbach, R.; Papadopoulos, S.; Sachinidis, A. Live-Cell Imaging of the Contractile Velocity and Transient Intracellular Ca2+ Fluctuations in Human Stem Cell-Derived Cardiomyocytes. Cells 2022, 11, 1280. https://doi.org/10.3390/cells11081280
Acharya A, Nemade H, Rajendra Prasad K, Khan K, Hescheler J, Blackburn N, Hemmersbach R, Papadopoulos S, Sachinidis A. Live-Cell Imaging of the Contractile Velocity and Transient Intracellular Ca2+ Fluctuations in Human Stem Cell-Derived Cardiomyocytes. Cells. 2022; 11(8):1280. https://doi.org/10.3390/cells11081280
Chicago/Turabian StyleAcharya, Aviseka, Harshal Nemade, Krishna Rajendra Prasad, Khadija Khan, Jürgen Hescheler, Nick Blackburn, Ruth Hemmersbach, Symeon Papadopoulos, and Agapios Sachinidis. 2022. "Live-Cell Imaging of the Contractile Velocity and Transient Intracellular Ca2+ Fluctuations in Human Stem Cell-Derived Cardiomyocytes" Cells 11, no. 8: 1280. https://doi.org/10.3390/cells11081280