
GBA Variants and Parkinson Disease: Mechanisms and Treatments


Abstract
:1. Introduction
2. Parkinson Disease
3. The GBA Gene
4. PD and the GBA Gene
4.1. The Link between GBA Mutations and PD
4.2. Presentation of GBA-PD
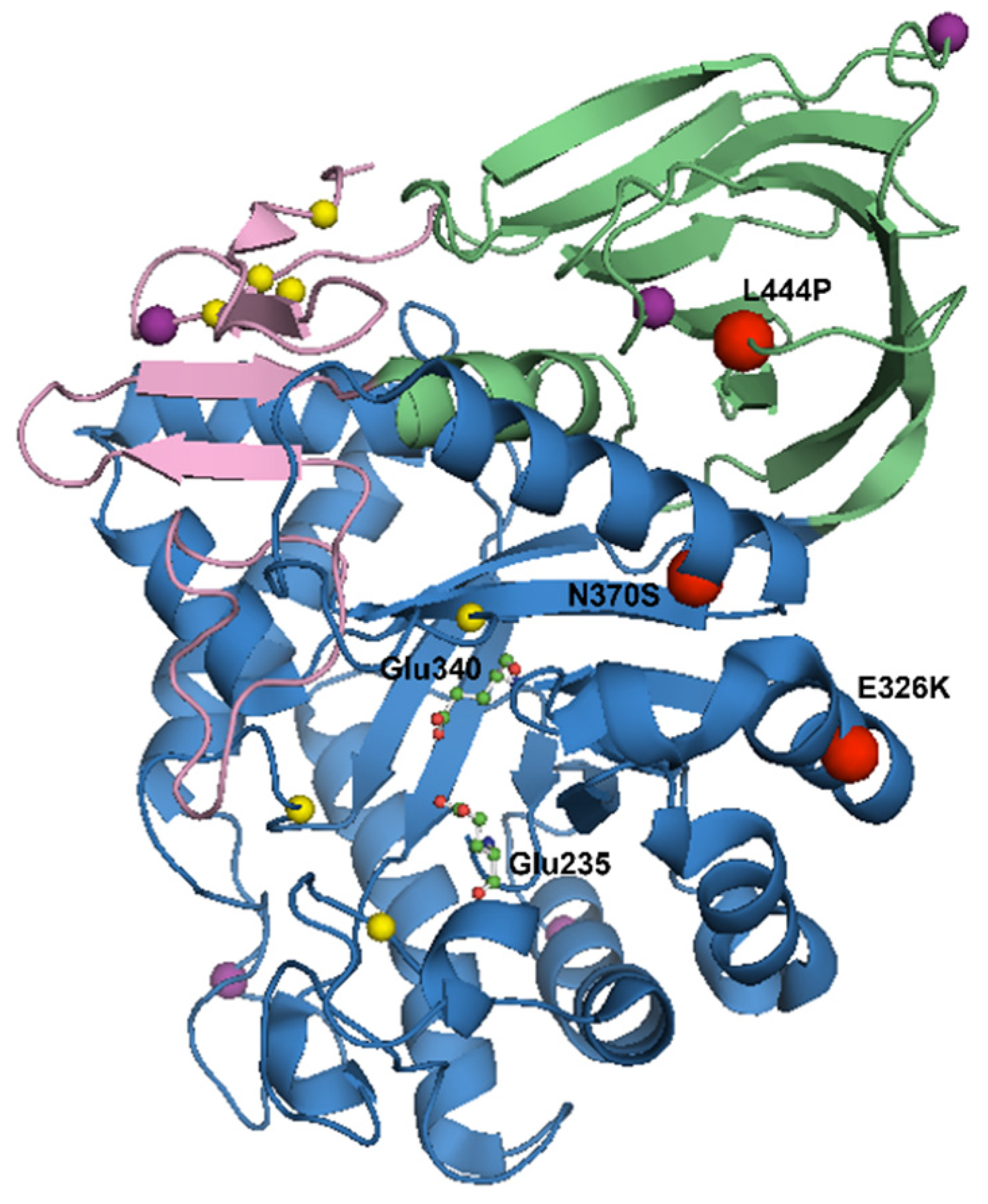
5. Mutations in the GBA Gene
6. GBA Activity and PD
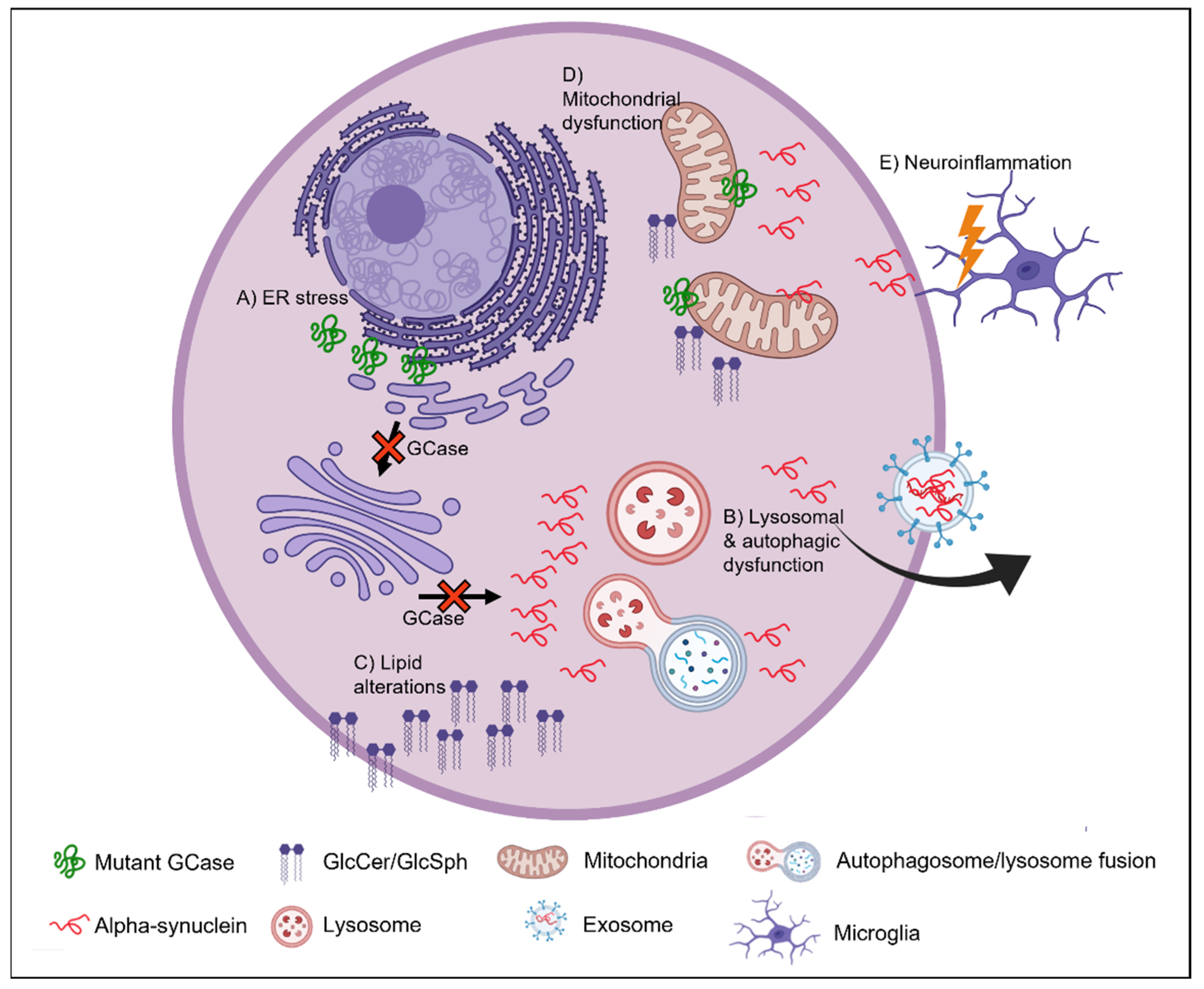
7. Mechanism Underlying GBA-PD
8. ER Stress
9. Autophagic-Lysosomal Pathway
10. Lipid Homeostasis
11. Mitochondrial Dysfunction
12. Neuroinflammation
13. GCase as a Therapeutic Target

{kind=link}
{kind=link}
| Treatment | Therapeutic Strategy | Drug Name | Phase in Drug Development | Reference |
|---|---|---|---|---|
| Substrate reduction | Reduce glycosphingolipid accumulation in the CNS | GZ667161 Venglustat Miglustat | Phase II completed for venglustat | [160,206,207] |
| Small molecule chaperones | Refold mutant GCase in the ER to improve trafficking to the lysosome and increase activity and stability while reducing ER stress | Ambroxol Isofagomine | Phase II completed for ambroxol | [39,90,138,211,212,213,214,215,216,217,218,219,220,221,222,223,224] |
| Gene therapy | Replace GCase activity and protein levels in the CNS | AAV-mediated delivery of recombinant GCase | Preclinical research ongoing Phase I/II ongoing for PR001 gene therapy (Prevail Therapeutics) | [111,120,205,209,210,225] |
| GCase activator | Increase GCase activity in the brain | BIA 28-6156/LTI-291 | Phase I completed | [205,226] |
| Transport vehicle modified recombinant GCase | Replace GCase activity and protein levels in the CNS | ETV:GBA | Preclinical research ongoing | [205] |
| Histone deacetylase inhibitors | Replace GCase activity and protein levels | LB-205 | Preclinical research ongoing | [227,228] |
14. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Braak, H. Neuropathological Staging of Brain Pathology in Sporadic Parkinson’s disease: Separating the Wheat from the Chaff. J. Park. Dis. 2017, 7, S71–S85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turpin, J.C.; Dubois, G.; Brice, A.; Masson, M.; Nadaud, M.C.; Boutry, J.M.; Schram, A.W.; Tager, J.M.; Baumann, N. Parkinsonian Symptomatology in a Patient with Type I (Adult) Gaucher’s Disease; Springer US: Boston, MA, USA, 1988; pp. 103–105. [Google Scholar]
- McKeran, R.O.; Bradbury, P.; Taylor, D.; Stern, G. Neurological involvement in type 1 (adult) Gaucher’s disease. J. Neurol. Neurosurg. Psychiatry 1985, 48, 172–175. [Google Scholar] [CrossRef] [PubMed]
- Sidransky, E.; Lopez, G. The link between the GBA gene and parkinsonism. Lancet Neurol. 2012, 11, 986–998. [Google Scholar] [CrossRef] [Green Version]
- Bultron, G.; Kacena, K.; Pearson, D.; Boxer, M.; Yang, R.; Sathe, S.; Pastores, G.; Mistry, P.K. The risk of Parkinson’s disease in type 1 Gaucher disease. J. Inherit. Metab. Dis. 2010, 33, 167–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gan-Or, Z.; Amshalom, I.; Kilarski, L.L.; Bar-Shira, A.; Gana-Weisz, M.; Mirelman, A.; Marder, K.; Bressman, S.; Giladi, N.; Orr-Urtreger, A. Differential effects of severe vs mild GBA mutations on Parkinson disease. Neurology 2015, 84, 880–887. [Google Scholar] [CrossRef] [Green Version]
- Sidransky, E.; Nalls, M.A.; Aasly, J.O.; Aharon-Peretz, J.; Annesi, G.; Barbosa, E.R.; Bar-Shira, A.; Berg, D.; Bras, J.; Brice, A.; et al. Multi-center analysis of glucocerebrosidase mutations in Parkinson disease. N. Engl. J. Med. 2009, 361, 1651–1661. [Google Scholar] [CrossRef] [Green Version]
- Tayebi, N.; Callahan, M.; Madike, V.; Stubblefield, B.K.; Orvisky, E.; Krasnewich, D.; Fillano, J.J.; Sidransky, E. Gaucher disease and parkinsonism: A phenotypic and genotypic characterization. Mol. Genet. Metab. 2001, 73, 313–321. [Google Scholar] [CrossRef]
- Hruska, K.S.; LaMarca, M.E.; Scott, C.R.; Sidransky, E. Gaucher disease: Mutation and polymorphism spectrum in the glucocerebrosidase gene (GBA). Hum. Mutat. 2008, 29, 567–583. [Google Scholar] [CrossRef]
- Beutler, E.; Beutler, L.; West, C. Mutations in the gene encoding cytosolic beta-glucosidase in Gaucher disease. J. Lab. Clin. Med. 2004, 144, 65–68. [Google Scholar] [CrossRef]
- Gegg, M.E.; Schapira, A.H.V. The role of glucocerebrosidase in Parkinson disease pathogenesis. FEBS J. 2018, 285, 3591–3603. [Google Scholar] [CrossRef] [Green Version]
- Balestrino, R.; Schapira, A.H.V. Parkinson disease. Eur. J. Neurol. 2020, 27, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Schapira, A.H.V.; Chaudhuri, K.R.; Jenner, P. Non-motor features of Parkinson disease. Nat. Rev. Neurosci. 2017, 18, 435–450. [Google Scholar] [CrossRef] [PubMed]
- Marsden, C.D. Parkinson’s disease. Lancet 1990, 335, 948–952. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef]
- Wakabayashi, K.; Tanji, K.; Odagiri, S.; Miki, Y.; Mori, F.; Takahashi, H. The Lewy body in Parkinson’s disease and related neurodegenerative disorders. Mol. Neurobiol. 2013, 47, 495–508. [Google Scholar] [CrossRef]
- Shahmoradian, S.H.; Lewis, A.J.; Genoud, C.; Hench, J.; Moors, T.E.; Navarro, P.P.; Castano-Diez, D.; Schweighauser, G.; Graff-Meyer, A.; Goldie, K.N.; et al. Lewy pathology in Parkinson’s disease consists of crowded organelles and lipid membranes. Nat. Neurosci. 2019, 22, 1099–1109. [Google Scholar] [CrossRef] [Green Version]
- Braak, H.; Tredici, K.D.; Rüb, U.; de Vos, R.A.I.; Jansen Steur, E.N.H.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Ono, K. The Oligomer Hypothesis in α-Synucleinopathy. Neurochem. Res. 2017, 42, 3362–3371. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Crowther, R.A.; Jakes, R.; Cairns, N.J.; Lantos, P.L.; Goedert, M. Filamentous alpha-synuclein inclusions link multiple system atrophy with Parkinson’s disease and dementia with Lewy bodies. Neurosci. Lett. 1998, 251, 205–208. [Google Scholar] [CrossRef]
- Alam, P.; Bousset, L.; Melki, R.; Otzen, D.E. α-Synuclein oligomers and fibrils: A spectrum of species, a spectrum of toxicities. J. Neurochem. 2019, 150, 522–534. [Google Scholar] [CrossRef] [Green Version]
- Cuervo, A.M.; Stefanis, L.; Fredenburg, R.; Lansbury, P.T.; Sulzer, D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science 2004, 305, 1292–1295. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Vicente, M.; Cuervo, A.M. Autophagy and neurodegeneration: When the cleaning crew goes on strike. Lancet Neurol. 2007, 6, 352–361. [Google Scholar] [CrossRef]
- Alvarez-Erviti, L.; Rodriguez-Oroz, M.C.; Cooper, J.M.; Caballero, C.; Ferrer, I.; Obeso, J.A.; Schapira, A.H. Chaperone-mediated autophagy markers in Parkinson disease brains. Arch. Neurol. 2010, 67, 1464–1472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dehay, B.; Bove, J.; Rodriguez-Muela, N.; Perier, C.; Recasens, A.; Boya, P.; Vila, M. Pathogenic lysosomal depletion in Parkinson’s disease. J. Neurosci. 2010, 30, 12535–12544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.Y.; Englund, E.; Holton, J.L.; Soulet, D.; Hagell, P.; Lees, A.J.; Lashley, T.; Quinn, N.P.; Rehncrona, S.; Bjorklund, A.; et al. Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat. Med. 2008, 14, 501–503. [Google Scholar] [CrossRef] [PubMed]
- Kordower, J.H.; Chu, Y.; Hauser, R.A.; Freeman, T.B.; Olanow, C.W. Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nat. Med. 2008, 14, 504–506. [Google Scholar] [CrossRef]
- Reczek, D.; Schwake, M.; Schröder, J.; Hughes, H.; Blanz, J.; Jin, X.; Brondyk, W.; Van Patten, S.; Edmunds, T.; Saftig, P. LIMP-2 Is a Receptor for Lysosomal Mannose-6-Phosphate-Independent Targeting of β-Glucocerebrosidase. Cell 2007, 131, 770–783. [Google Scholar] [CrossRef] [Green Version]
- Grabowski, G.A.; Gatt, S.; Horowitz, M. Acid beta-glucosidase: Enzymology and molecular biology of Gaucher disease. Crit Rev. Biochem. Mol. Biol. 1990, 25, 385–414. [Google Scholar] [CrossRef]
- Bergmann, J.E.; Grabowski, G.A. Posttranslational processing of human lysosomal acid beta-glucosidase: A continuum of defects in Gaucher disease type 1 and type 2 fibroblasts. Am. J. Hum. Genet. 1989, 44, 741–750. [Google Scholar]
- Lieberman, R.L. A Guided Tour of the Structural Biology of Gaucher Disease: Acid-beta-Glucosidase and Saposin C. Enzym. Res. 2011, 2011, 973231. [Google Scholar] [CrossRef] [Green Version]
- Dvir, H.; Harel, M.; McCarthy, A.A.; Toker, L.; Silman, I.; Futerman, A.H.; Sussman, J.L. X-ray structure of human acid-beta-glucosidase, the defective enzyme in Gaucher disease. EMBO Rep. 2003, 4, 704–709. [Google Scholar] [CrossRef] [PubMed]
- Horowitz, M.; Pasmanik-Chor, M.; Borochowitz, Z.; Falik-Zaccai, T.; Heldmann, K.; Carmi, R.; Parvari, R.; Beit-Or, H.; Goldman, B.; Peleg, L.; et al. Prevalence of glucocerebrosidase mutations in the Israeli Ashkenazi Jewish population. Hum. Mutat. 1998, 12, 240–244. [Google Scholar] [CrossRef]
- Charrow, J.; Andersson, H.C.; Kaplan, P.; Kolodny, E.H.; Mistry, P.; Pastores, G.; Rosenbloom, B.E.; Scott, C.R.; Wappner, R.S.; Weinreb, N.J.; et al. The gaucher registry: Demographics and disease characteristics of 1698 patients with gaucher disease. Arch. Intern. Med. 2000, 160, 2835–2843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grabowski, G.A. Phenotype, diagnosis, and treatment of Gaucher’s disease. Lancet 2008, 372, 1263–1271. [Google Scholar] [CrossRef]
- Beavan, M.; McNeill, A.; Proukakis, C.; Hughes, D.A.; Mehta, A.; Schapira, A.H. Evolution of prodromal clinical markers of Parkinson disease in a GBA mutation-positive cohort. JAMA Neurol. 2015, 72, 201–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, K.; Sidransky, E.; Verma, A.; Mixon, T.; Sandberg, G.D.; Wakefield, L.K.; Morrison, A.; Lwin, A.; Colegial, C.; Allman, J.M.; et al. Neuropathology provides clues to the pathophysiology of Gaucher disease. Mol. Genet. Metab. 2004, 82, 192–207. [Google Scholar] [CrossRef] [PubMed]
- Aflaki, E.; Stubblefield, B.K.; McGlinchey, R.P.; McMahon, B.; Ory, D.S.; Sidransky, E. A characterization of Gaucher iPS-derived astrocytes: Potential implications for Parkinson’s disease. Neurobiol. Dis. 2020, 134, 104647. [Google Scholar] [CrossRef]
- Maegawa, G.H.B.; Tropak, M.B.; Buttner, J.D.; Rigat, B.A.; Fuller, M.; Pandit, D.; Tang, L.I.; Kornhaber, G.J.; Hamuro, Y.; Clarke, J.T.R.; et al. Identification and Characterization of Ambroxol as an Enzyme Enhancement Agent for Gaucher Disease. J. Biol. Chem. 2009, 284, 23502–23516. [Google Scholar] [CrossRef] [Green Version]
- Mata, I.F.; Samii, A.; Schneer, S.H.; Roberts, J.W.; Griffith, A.; Leis, B.C.; Schellenberg, G.D.; Sidransky, E.; Bird, T.D.; Leverenz, J.B.; et al. Glucocerebrosidase gene mutations: A risk factor for Lewy body disorders. Arch. Neurol. 2008, 65, 379–382. [Google Scholar] [CrossRef] [Green Version]
- Alcalay, R.N.; Dinur, T.; Quinn, T.; Sakanaka, K.; Levy, O.; Waters, C.; Fahn, S.; Dorovski, T.; Chung, W.K.; Pauciulo, M.; et al. Comparison of Parkinson risk in Ashkenazi Jewish patients with Gaucher disease and GBA heterozygotes. JAMA Neurol. 2014, 71, 752–757. [Google Scholar] [CrossRef] [Green Version]
- Anheim, M.; Elbaz, A.; Lesage, S.; Durr, A.; Condroyer, C.; Viallet, F.; Pollak, P.; Bonaiti, B.; Bonaiti-Pellie, C.; Brice, A.; et al. Penetrance of Parkinson disease in glucocerebrosidase gene mutation carriers. Neurology 2012, 78, 417–420. [Google Scholar] [CrossRef] [PubMed]
- McNeill, A.; Duran, R.; Hughes, D.A.; Mehta, A.; Schapira, A.H. A clinical and family history study of Parkinson’s disease in heterozygous glucocerebrosidase mutation carriers. J. Neurol. Neurosurg. Psychiatry 2012, 83, 853–854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lwin, A.; Orvisky, E.; Goker-Alpan, O.; LaMarca, M.E.; Sidransky, E. Glucocerebrosidase mutations in subjects with parkinsonism. Mol. Genet. Metab. 2004, 81, 70–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Migdalska-Richards, A.; Schapira, A.H.V. The relationship between glucocerebrosidase mutations and Parkinson disease. J. Neurochem. 2016, 139, 77–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neumann, J.; Bras, J.; Deas, E.; O’Sullivan, S.S.; Parkkinen, L.; Lachmann, R.H.; Li, A.; Holton, J.; Guerreiro, R.; Paudel, R.; et al. Glucocerebrosidase mutations in clinical and pathologically proven Parkinson’s disease. Brain 2009, 132, 1783–1794. [Google Scholar] [CrossRef] [PubMed]
- Lesage, S.; Anheim, M.; Condroyer, C.; Pollak, P.; Durif, F.; Dupuits, C.; Viallet, F.; Lohmann, E.; Corvol, J.C.; Honore, A.; et al. Large-scale screening of the Gaucher’s disease-related glucocerebrosidase gene in Europeans with Parkinson’s disease. Hum. Mol. Genet. 2011, 20, 202–210. [Google Scholar] [CrossRef] [PubMed]
- Higgins, A.L.; Toffoli, M.; Mullin, S.; Lee, C.Y.; Koletsi, S.; Avenali, M.; Blandini, F.; Schapira, A.H. The remote assessment of parkinsonism supporting the ongoing development of interventions in Gaucher disease. Neurodegener. Dis. Manag. 2021, 11, 451–458. [Google Scholar] [CrossRef]
- Goker-Alpan, O.; Giasson, B.I.; Eblan, M.J.; Nguyen, J.; Hurtig, H.I.; Lee, V.M.Y.; Trojanowski, J.Q.; Sidransky, E. Glucocerebrosidase mutations are an important risk factor for Lewy body disorders. Neurology 2006, 67, 908–910. [Google Scholar] [CrossRef]
- Zhang, Y.; Sun, Q.Y.; Zhao, Y.W.; Shu, L.; Guo, J.F.; Xu, Q.; Yan, X.X.; Tang, B.S. Effect of GBA Mutations on Phenotype of Parkinson’s Disease: A Study on Chinese Population and a Meta-Analysis. Parkinson’s Dis. 2015, 2015, 916971. [Google Scholar] [CrossRef] [Green Version]
- Gan-Or, Z.; Bar-Shira, A.; Mirelman, A.; Gurevich, T.; Kedmi, M.; Giladi, N.; Orr-Urtreger, A. LRRK2 and GBA mutations differentially affect the initial presentation of Parkinson disease. Neurogenetics 2010, 11, 121–125. [Google Scholar] [CrossRef]
- Gan-Or, Z.; Giladi, N.; Rozovski, U.; Shifrin, C.; Rosner, S.; Gurevich, T.; Bar-Shira, A.; Orr-Urtreger, A. Genotype-phenotype correlations between GBA mutations and Parkinson disease risk and onset. Neurology 2008, 70, 2277–2283. [Google Scholar] [CrossRef] [PubMed]
- Malek, N.; Weil, R.S.; Bresner, C.; Lawton, M.A.; Grosset, K.A.; Tan, M.; Bajaj, N.; Barker, R.A.; Burn, D.J.; Foltynie, T.; et al. Features of GBA-associated Parkinson’s disease at presentation in the UK Tracking Parkinson’s study. J. Neurol. Neurosurg. Psychiatry 2018, 89, 702–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goker-Alpan, O.; Lopez, G.; Vithayathil, J.; Davis, J.; Hallett, M.; Sidransky, E. The spectrum of parkinsonian manifestations associated with glucocerebrosidase mutations. Arch. Neurol. 2008, 65, 1353–1357. [Google Scholar] [CrossRef] [PubMed]
- Brockmann, K.; Srulijes, K.; Pflederer, S.; Hauser, A.K.; Schulte, C.; Maetzler, W.; Gasser, T.; Berg, D. GBA-associated Parkinson’s disease: Reduced survival and more rapid progression in a prospective longitudinal study. Mov. Disord. Off. J. Mov. Disord. Soc. 2015, 30, 407–411. [Google Scholar] [CrossRef]
- Winder-Rhodes, S.E.; Evans, J.R.; Ban, M.; Mason, S.L.; Williams-Gray, C.H.; Foltynie, T.; Duran, R.; Mencacci, N.E.; Sawcer, S.J.; Barker, R.A. Glucocerebrosidase mutations influence the natural history of Parkinson’s disease in a community-based incident cohort. Brain 2013, 136, 392–399. [Google Scholar] [CrossRef]
- Petrucci, S.; Ginevrino, M.; Trezzi, I.; Monfrini, E.; Ricciardi, L.; Albanese, A.; Avenali, M.; Barone, P.; Bentivoglio, A.R.; Bonifati, V.; et al. GBA-Related Parkinson’s Disease: Dissection of Genotype-Phenotype Correlates in a Large Italian Cohort. Mov. Disord. 2020, 35, 2106–2111. [Google Scholar] [CrossRef]
- Alcalay, R.N.; Caccappolo, E.; Mejia-Santana, H.; Tang, M.; Rosado, L.; Orbe Reilly, M.; Ruiz, D.; Ross, B.; Verbitsky, M.; Kisselev, S.; et al. Cognitive performance of GBA mutation carriers with early-onset PD: The CORE-PD study. Neurology 2012, 78, 1434–1440. [Google Scholar] [CrossRef] [Green Version]
- Brockmann, K.; Srulijes, K.; Hauser, A.K.; Schulte, C.; Csoti, I.; Gasser, T.; Berg, D. GBA-associated PD presents with nonmotor characteristics. Neurology 2011, 77, 276–280. [Google Scholar] [CrossRef]
- Westbroek, W.; Gustafson, A.M.; Sidransky, E. Exploring the link between glucocerebrosidase mutations and parkinsonism. Trends Mol. Med. 2011, 17, 485–493. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.H.; Stubblefield, B.; Cookson, M.R.; Goldin, E.; Velayati, A.; Tayebi, N.; Sidransky, E. Aggregation of α-synuclein in brain samples from subjects with glucocerebrosidase mutations. Mol. Genet. Metab. 2011, 104, 185–188. [Google Scholar] [CrossRef] [Green Version]
- Tayebi, N.; Walker, J.; Stubblefield, B.; Orvisky, E.; LaMarca, M.E.; Wong, K.; Rosenbaum, H.; Schiffmann, R.; Bembi, B.; Sidransky, E. Gaucher disease with parkinsonian manifestations: Does glucocerebrosidase deficiency contribute to a vulnerability to parkinsonism? Mol. Genet. Metab. 2003, 79, 104–109. [Google Scholar] [CrossRef]
- Nishioka, K.; Ross, O.A.; Vilarino-Guell, C.; Cobb, S.A.; Kachergus, J.M.; Mann, D.M.; Snowden, J.; Richardson, A.M.; Neary, D.; Robinson, C.A.; et al. Glucocerebrosidase mutations in diffuse Lewy body disease. Parkinsonism Relat. Disord. 2011, 17, 55–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parkkinen, L.; Neumann, J.; O’Sullivan, S.S.; Holton, J.L.; Revesz, T.; Hardy, J.; Lees, A.J. Glucocerebrosidase mutations do not cause increased Lewy body pathology in Parkinson’s disease. Mol. Genet. Metab. 2011, 103, 410–412. [Google Scholar] [CrossRef] [PubMed]
- Blauwendraat, C.; Reed, X.; Krohn, L.; Heilbron, K.; Bandres-Ciga, S.; Tan, M.; Gibbs, J.R.; Hernandez, D.G.; Kumaran, R.; Langston, R.; et al. Genetic modifiers of risk and age at onset in GBA associated Parkinson’s disease and Lewy body dementia. Brain 2020, 143, 234–248. [Google Scholar] [CrossRef]
- Stoker, T.B.; Camacho, M.; Winder-Rhodes, S.; Liu, G.; Scherzer, C.R.; Foltynie, T.; Barker, R.A.; Williams-Gray, C.H. A common polymorphism in SNCA is associated with accelerated motor decline in GBA-Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 2020, 91, 673–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goker-Alpan, O.; Stubblefield, B.K.; Giasson, B.I.; Sidransky, E. Glucocerebrosidase is present in alpha-synuclein inclusions in Lewy body disorders. Acta Neuropathol. 2010, 120, 641–649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dandana, A.; Ben Khelifa, S.; Chahed, H.; Miled, A.; Ferchichi, S. Gaucher Disease: Clinical, Biological and Therapeutic Aspects. Pathobiology 2016, 83, 13–23. [Google Scholar] [CrossRef]
- Winfield, S.L.; Tayebi, N.; Martin, B.M.; Ginns, E.I.; Sidransky, E. Identification of three additional genes contiguous to the glucocerebrosidase locus on chromosome 1q21: Implications for Gaucher disease. Genome Res. 1997, 7, 1020–1026. [Google Scholar] [CrossRef] [Green Version]
- Huh, Y.E.; Chiang, M.S.R.; Locascio, J.J.; Liao, Z.; Liu, G.; Choudhury, K.; Kuras, Y.I.; Tuncali, I.; Videnovic, A.; Hunt, A.L.; et al. Beta-Glucocerebrosidase activity in GBA-linked Parkinson disease: The type of mutation matters. Neurology 2020, 95, e685–e696. [Google Scholar] [CrossRef]
- Cilia, R.; Tunesi, S.; Marotta, G.; Cereda, E.; Siri, C.; Tesei, S.; Zecchinelli, A.L.; Canesi, M.; Mariani, C.B.; Meucci, N.; et al. Survival and dementia in GBA-associated Parkinson’s disease: The mutation matters. Ann. Neurol. 2016, 80, 662–673. [Google Scholar] [CrossRef]
- Smith, L.; Mullin, S.; Schapira, A.H.V. Insights into the structural biology of Gaucher disease. Exp. Neurol. 2017, 298, 180–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duran, R.; Mencacci, N.E.; Angeli, A.V.; Shoai, M.; Deas, E.; Houlden, H.; Mehta, A.; Hughes, D.; Cox, T.M.; Deegan, P.; et al. The glucocerobrosidase E326K variant predisposes to Parkinson’s disease, but does not cause Gaucher’s disease. Mov. Disord. 2013, 28, 232–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chabas, A.; Gort, L.; Diaz-Font, A.; Montfort, M.; Santamaria, R.; Cidras, M.; Grinberg, D.; Vilageliu, L. Perinatal lethal phenotype with generalized ichthyosis in a type 2 Gaucher disease patient with the [L444P;E326K]/P182L genotype: Effect of the E326K change in neonatal and classic forms of the disease. Blood Cells Mol. Dis. 2005, 35, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Liou, B.; Grabowski, G.A. Is E326K glucocerebrosidase a polymorphic or pathological variant? Mol. Genet. Metab. 2012, 105, 528–529. [Google Scholar] [CrossRef]
- Berge-Seidl, V.; Pihlstrøm, L.; Maple-Grødem, J.; Forsgren, L.; Linder, J.; Larsen, J.P.; Tysnes, O.-B.; Toft, M. The GBA variant E326K is associated with Parkinson’s disease and explains a genome-wide association signal. Neurosci. Lett. 2017, 658, 48–52. [Google Scholar] [CrossRef]
- den Heijer, J.M.; Cullen, V.C.; Quadri, M.; Schmitz, A.; Hilt, D.C.; Lansbury, P.; Berendse, H.W.; van de Berg, W.D.J.; de Bie, R.M.A.; Boertien, J.M.; et al. A Large-Scale Full GBA1 Gene Screening in Parkinson’s Disease in the Netherlands. Mov. Disord. 2020, 35, 1667–1674. [Google Scholar] [CrossRef]
- Ruskey, J.A.; Greenbaum, L.; Roncière, L.; Alam, A.; Spiegelman, D.; Liong, C.; Levy, O.A.; Waters, C.; Fahn, S.; Marder, K.S.; et al. Increased yield of full GBA sequencing in Ashkenazi Jews with Parkinson’s disease. Eur. J. Med. Genet. 2019, 62, 65–69. [Google Scholar] [CrossRef]
- Davis, M.Y.; Johnson, C.O.; Leverenz, J.B.; Weintraub, D.; Trojanowski, J.Q.; Chen-Plotkin, A.; Van Deerlin, V.M.; Quinn, J.F.; Chung, K.A.; Peterson-Hiller, A.L.; et al. Association of GBA Mutations and the E326K Polymorphism With Motor and Cognitive Progression in Parkinson Disease. JAMA Neurol. 2016, 73, 1217–1224. [Google Scholar] [CrossRef] [Green Version]
- Stoker, T.B.; Camacho, M.; Winder-Rhodes, S.; Liu, G.; Scherzer, C.R.; Foltynie, T.; Evans, J.; Breen, D.P.; Barker, R.A.; Williams-Gray, C.H. Impact of GBA1 variants on long-term clinical progression and mortality in incident Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 2020, 91, 695–702. [Google Scholar] [CrossRef] [Green Version]
- Greuel, A.; Trezzi, J.-P.; Glaab, E.; Ruppert, M.C.; Maier, F.; Jäger, C.; Hodak, Z.; Lohmann, K.; Ma, Y.; Eidelberg, D.; et al. GBA Variants in Parkinson’s Disease: Clinical, Metabolomic, and Multimodal Neuroimaging Phenotypes. Mov. Disord. 2020, 35, 2201–2210. [Google Scholar] [CrossRef]
- Mallett, V.; Ross, J.P.; Alcalay, R.N.; Ambalavanan, A.; Sidransky, E.; Dion, P.A.; Rouleau, G.A.; Gan-Or, Z. GBA p.T369M substitution in Parkinson disease: Polymorphism or association? A meta-analysis. Neurology. Genet. 2016, 2, e104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenbloom, B.; Balwani, M.; Bronstein, J.M.; Kolodny, E.; Sathe, S.; Gwosdow, A.R.; Taylor, J.S.; Cole, J.A.; Zimran, A.; Weinreb, N.J. The incidence of Parkinsonism in patients with type 1 Gaucher disease: Data from the ICGG Gaucher Registry. Blood Cells Mol. Dis. 2011, 46, 95–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aharon-Peretz, J.; Rosenbaum, H.; Gershoni-Baruch, R. Mutations in the glucocerebrosidase gene and Parkinson’s disease in Ashkenazi Jews. N. Engl. J. Med. 2004, 351, 1972–1977. [Google Scholar] [CrossRef] [PubMed]
- Chetrit, E.B.; Alcalay, R.N.; Steiner-Birmanns, B.; Altarescu, G.; Phillips, M.; Elstein, D.; Zimran, A. Phenotype in patients with Gaucher disease and Parkinson disease. Blood Cells Mol. Dis. 2013, 50, 218–221. [Google Scholar] [CrossRef]
- Fernandes, H.J.R.; Hartfield, E.M.; Christian, H.C.; Emmanoulidou, E.; Zheng, Y.; Booth, H.; Bogetofte, H.; Lang, C.; Ryan, B.J.; Sardi, S.P.; et al. ER Stress and Autophagic Perturbations Lead to Elevated Extracellular α-Synuclein in GBA-N370S Parkinson’s iPSC-Derived Dopamine Neurons. Stem Cell Rep. 2016, 6, 342–356. [Google Scholar] [CrossRef] [Green Version]
- Alfonso, P.; Rodriguez-Rey, J.C.; Ganan, A.; Perez-Calvo, J.I.; Giralt, M.; Giraldo, P.; Pocovi, M. Expression and functional characterization of mutated glucocerebrosidase alleles causing Gaucher disease in Spanish patients. Blood Cells Mol. Dis. 2004, 32, 218–225. [Google Scholar] [CrossRef]
- Grace, M.E.; Newman, K.M.; Scheinker, V.; Bergfussman, A.; Grabowski, G.A. Analysis of Human Acid Beta-Glucosidase by Site-Directed Mutagenesis and Heterologous Expression. J. Biol. Chem. 1994, 269, 2283–2291. [Google Scholar] [CrossRef]
- Ohashi, T.; Hong, C.M.; Weiler, S.; Tomich, J.M.; Aerts, J.M.; Tager, J.M.; Barranger, J.A. Characterization of human glucocerebrosidase from different mutant alleles. J. Biol. Chem. 1991, 266, 3661–3667. [Google Scholar] [CrossRef]
- Sanchez-Martinez, A.; Beavan, M.; Gegg, M.E.; Chau, K.Y.; Whitworth, A.J.; Schapira, A.H. Parkinson disease-linked GBA mutation effects reversed by molecular chaperones in human cell and fly models. Sci. Rep. 2016, 6, 31380. [Google Scholar] [CrossRef] [Green Version]
- Maor, G.; Rencus-Lazar, S.; Filocamo, M.; Steller, H.; Segal, D.; Horowitz, M. Unfolded protein response in Gaucher disease: From human to Drosophila. Orphanet J. Rare Dis. 2013, 8, 140. [Google Scholar] [CrossRef] [Green Version]
- Schöndorf, D.C.; Aureli, M.; McAllister, F.E.; Hindley, C.J.; Mayer, F.; Schmid, B.; Sardi, S.P.; Valsecchi, M.; Hoffmann, S.; Schwarz, L.K.; et al. iPSC-derived neurons from GBA1-associated Parkinson’s disease patients show autophagic defects and impaired calcium homeostasis. Nat. Commun. 2014, 5, 4028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ron, I.; Horowitz, M. ER retention and degradation as the molecular basis underlying Gaucher disease heterogeneity. Hum. Mol. Genet. 2005, 14, 2387–2398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bendikov-Bar, I.; Ron, I.; Filocamo, M.; Horowitz, M. Characterization of the ERAD process of the L444P mutant glucocerebrosidase variant. Blood Cells Mol. Dis. 2011, 46, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Migdalska-Richards, A.; Wegrzynowicz, M.; Rusconi, R.; Deangeli, G.; Di Monte, D.A.; Spillantini, M.G.; Schapira, A.H.V. The L444P Gba1 mutation enhances alpha-synuclein induced loss of nigral dopaminergic neurons in mice. Brain A J. Neurol. 2017, 140, 2706–2721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yun, S.P.; Kim, D.; Kim, S.; Kim, S.; Karuppagounder, S.S.; Kwon, S.-H.; Lee, S.; Kam, T.-I.; Lee, S.; Ham, S.; et al. α-Synuclein accumulation and GBA deficiency due to L444P GBA mutation contributes to MPTP-induced parkinsonism. Mol. Neurodegener. 2018, 13, 1. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Deng, L.; Zhong, Y.; Yi, M. The Association between E326K of GBA and the Risk of Parkinson’s Disease. Parkinson’s Dis. 2018, 2018, 1048084. [Google Scholar] [CrossRef] [Green Version]
- Horowitz, M.; Pasmanik-Chor, M.; Ron, I.; Kolodny, E.H. The enigma of the E326K mutation in acid beta-glucocerebrosidase. Mol. Genet. Metab. 2011, 104, 35–38. [Google Scholar] [CrossRef]
- Montfort, M.; Chabás, A.; Vilageliu, L.; Grinberg, D. Functional analysis of 13 GBA mutant alleles identified in Gaucher disease patients: Pathogenic changes and “modifier” polymorphisms. Hum. Mutat. 2004, 23, 567–575. [Google Scholar] [CrossRef]
- Alcalay, R.N.; Levy, O.A.; Waters, C.C.; Fahn, S.; Ford, B.; Kuo, S.H.; Mazzoni, P.; Pauciulo, M.W.; Nichols, W.C.; Gan-Or, Z.; et al. Glucocerebrosidase activity in Parkinson’s disease with and without GBA mutations. Brain A J. Neurol. 2015, 138, 2648–2658. [Google Scholar] [CrossRef] [Green Version]
- Grace, M.E.; Ashton-Prolla, P.; Pastores, G.M.; Soni, A.; Desnick, R.J. Non-pseudogene-derived complex acid beta-glucosidase mutations causing mild type 1 and severe type 2 gaucher disease. J. Clin. Investig. 1999, 103, 817–823. [Google Scholar] [CrossRef] [Green Version]
- Malini, E.; Grossi, S.; Deganuto, M.; Rosano, C.; Parini, R.; Dominisini, S.; Cariati, R.; Zampieri, S.; Bembi, B.; Filocamo, M.; et al. Functional analysis of 11 novel GBA alleles. Eur. J. Hum. Genet. EJHG 2014, 22, 511–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alcalay, R.N.; Wolf, P.; Chiang, M.S.R.; Helesicova, K.; Zhang, X.K.; Merchant, K.; Hutten, S.J.; Scherzer, C.; Caspell-Garcia, C.; Blauwendraat, C.; et al. Longitudinal Measurements of Glucocerebrosidase activity in Parkinson’s patients. Ann. Clin. Transl. Neurol. 2020, 7, 1816–1830. [Google Scholar] [CrossRef] [PubMed]
- Torralba, M.A.; Pérez-Calvo, J.I.; Pastores, G.M.; Cenarro, A.; Giraldo, P.; Pocoví, M. Identification and Characterization of a Novel Mutation c.1090G>T (G325W) and Nine Common Mutant Alleles Leading to Gaucher Disease in Spanish Patients. Blood Cells Mol. Dis. 2001, 27, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Liou, B.; Kazimierczuk, A.; Zhang, M.; Scott, C.R.; Hegde, R.S.; Grabowski, G.A. Analyses of variant acid beta-glucosidases: Effects of Gaucher disease mutations. J. Biol. Chem. 2006, 281, 4242–4253. [Google Scholar] [CrossRef] [Green Version]
- Gegg, M.E.; Burke, D.; Heales, S.J.; Cooper, J.M.; Hardy, J.; Wood, N.W.; Schapira, A.H. Glucocerebrosidase deficiency in substantia nigra of parkinson disease brains. Ann. Neurol. 2012, 72, 455–463. [Google Scholar] [CrossRef] [Green Version]
- Gundner, A.L.; Duran-Pacheco, G.; Zimmermann, S.; Ruf, I.; Moors, T.; Baumann, K.; Jagasia, R.; van de Berg, W.D.J.; Kremer, T. Path mediation analysis reveals GBA impacts Lewy body disease status by increasing alpha-synuclein levels. Neurobiol. Dis. 2019, 121, 205–213. [Google Scholar] [CrossRef]
- Murphy, K.E.; Gysbers, A.M.; Abbott, S.K.; Tayebi, N.; Kim, W.S.; Sidransky, E.; Cooper, A.; Garner, B.; Halliday, G.M. Reduced glucocerebrosidase is associated with increased α-synuclein in sporadic Parkinson’s disease. Brain A J. Neurol. 2014, 137, 834–848. [Google Scholar] [CrossRef] [Green Version]
- Cullen, V.; Sardi, P.; Ng, J.; Xu, Y.H.; Sun, Y.; Tomlinson, J.J.; Kolodziej, P.; Kahn, I.; Saftig, P.; Woulfe, J.; et al. Acid beta-Glucosidase Mutants Linked to Gaucher Disease, Parkinson Disease, and Lewy Body Dementia Alter alpha-Synuclein Processing. Ann. Neurol. 2011, 69, 940–953. [Google Scholar] [CrossRef]
- Fishbein, I.; Kuo, Y.M.; Giasson, B.I.; Nussbaum, R.L. Augmentation of phenotype in a transgenic Parkinson mouse heterozygous for a Gaucher mutation. Brain A J. Neurol. 2014, 137, 3235–3247. [Google Scholar] [CrossRef] [Green Version]
- Sardi, S.P.; Clarke, J.; Kinnecom, C.; Tamsett, T.J.; Li, L.; Stanek, L.M.; Passini, M.A.; Grabowski, G.A.; Schlossmacher, M.G.; Sidman, R.L.; et al. CNS expression of glucocerebrosidase corrects alpha-synuclein pathology and memory in a mouse model of Gaucher-related synucleinopathy. Proc. Natl. Acad. Sci. USA 2011, 108, 12101–12106. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.H.; Xu, K.; Sun, Y.; Liou, B.; Quinn, B.; Li, R.H.; Xue, L.; Zhang, W.; Setchell, K.D.; Witte, D.; et al. Multiple pathogenic proteins implicated in neuronopathic Gaucher disease mice. Hum. Mol. Genet. 2014, 23, 3943–3957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.H.; Sun, Y.; Ran, H.; Quinn, B.; Witte, D.; Grabowski, G.A. Accumulation and distribution of alpha-synuclein and ubiquitin in the CNS of Gaucher disease mouse models. Mol. Genet. Metab. 2011, 102, 436–447. [Google Scholar] [CrossRef] [Green Version]
- Ginns, E.I.; Mak, S.K.; Ko, N.; Karlgren, J.; Akbarian, S.; Chou, V.P.; Guo, Y.; Lim, A.; Samuelsson, S.; LaMarca, M.L.; et al. Neuroinflammation and alpha-synuclein accumulation in response to glucocerebrosidase deficiency are accompanied by synaptic dysfunction. Mol. Genet. Metab. 2014, 111, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Manning-Boğ, A.B.; Schüle, B.; Langston, J.W. Alpha-synuclein-glucocerebrosidase interactions in pharmacological Gaucher models: A biological link between Gaucher disease and parkinsonism. NeuroToxicology 2009, 30, 1127–1132. [Google Scholar] [CrossRef] [PubMed]
- Cleeter, M.W.; Chau, K.Y.; Gluck, C.; Mehta, A.; Hughes, D.A.; Duchen, M.; Wood, N.W.; Hardy, J.; Mark Cooper, J.; Schapira, A.H. Glucocerebrosidase inhibition causes mitochondrial dysfunction and free radical damage. Neurochem. Int. 2013, 62, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abul Khair, S.B.; Dhanushkodi, N.R.; Ardah, M.T.; Chen, W.; Yang, Y.; Haque, M.E. Silencing of Glucocerebrosidase Gene in Drosophila Enhances the Aggregation of Parkinson’s Disease Associated alpha-Synuclein Mutant A53T and Affects Locomotor Activity. Front. Neurosci. 2018, 12, 81. [Google Scholar] [CrossRef] [Green Version]
- Bae, E.-J.; Yang, N.Y.; Lee, C.; Lee, H.-J.; Kim, S.; Sardi, S.P.; Lee, S.-J. Loss of glucocerebrosidase 1 activity causes lysosomal dysfunction and α-synuclein aggregation. Exp. Mol. Med. 2015, 47, e153. [Google Scholar] [CrossRef] [Green Version]
- Jo, J.; Yang, L.; Tran, H.D.; Yu, W.; Sun, A.X.; Chang, Y.Y.; Jung, B.C.; Lee, S.J.; Saw, T.Y.; Xiao, B.; et al. Lewy Body-like Inclusions in Human Midbrain Organoids Carrying Glucocerebrosidase and alpha-Synuclein Mutations. Ann. Neurol. 2021, 90, 490–505. [Google Scholar] [CrossRef]
- Sardi, P.S.; Shihabuddin, L.S.; Sidman, R.L.; Cheng, S.H. Augmenting CNS Glucocerebrosidase Activity as a Therapeutic Strategy for Parkinsonism and Other Gaucher-Related Synucleinopathies. Mol. Ther. 2013, 21, S14–S15. [Google Scholar] [CrossRef] [Green Version]
- Woodard, C.M.; Campos, B.A.; Kuo, S.H.; Nirenberg, M.J.; Nestor, M.W.; Zimmer, M.; Mosharov, E.V.; Sulzer, D.; Zhou, H.Y.; Paull, D.; et al. iPSC-Derived Dopamine Neurons Reveal Differences between Monozygotic Twins Discordant for Parkinson’s Disease. Cell Rep. 2014, 9, 1173–1182. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Hertz, E.; Zhang, X.; Leinartaite, L.; Lundius, E.G.; Li, J.; Svenningsson, P. Overexpression of alpha-synuclein simultaneously increases glutamate NMDA receptor phosphorylation and reduces glucocerebrosidase activity. Neurosci. Lett. 2016, 611, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Mazzulli, J.R.; Xu, Y.H.; Sun, Y.; Knight, A.L.; McLean, P.J.; Caldwell, G.A.; Sidransky, E.; Grabowski, G.A.; Krainc, D. Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 2011, 146, 37–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henderson, M.X.; Sedor, S.; McGeary, I.; Cornblath, E.J.; Peng, C.; Riddle, D.M.; Li, H.L.; Zhang, B.; Brown, H.J.; Olufemi, M.F.; et al. Glucocerebrosidase Activity Modulates Neuronal Susceptibility to Pathological alpha-Synuclein Insult. Neuron 2020, 105, 822–836.e7. [Google Scholar] [CrossRef]
- Dermentzaki, G.; Dimitriou, E.; Xilouri, M.; Michelakakis, H.; Stefanis, L. Loss of β-Glucocerebrosidase Activity Does Not Affect Alpha-Synuclein Levels or Lysosomal Function in Neuronal Cells. PLoS ONE 2013, 8, e60674. [Google Scholar] [CrossRef] [Green Version]
- Huebecker, M.; Moloney, E.B.; van der Spoel, A.C.; Priestman, D.A.; Isacson, O.; Hallett, P.J.; Platt, F.M. Reduced sphingolipid hydrolase activities, substrate accumulation and ganglioside decline in Parkinson’s disease. Mol. Neurodegener. 2019, 14, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rocha, E.M.; Smith, G.A.; Park, E.; Cao, H.; Brown, E.; Hallett, P.; Isacson, O. Progressive decline of glucocerebrosidase in aging and Parkinson’s disease. Ann. Clin. Transl. Neurol. 2015, 2, 433–438. [Google Scholar] [CrossRef]
- Moors, T.E.; Paciotti, S.; Ingrassia, A.; Quadri, M.; Breedveld, G.; Tasegian, A.; Chiasserini, D.; Eusebi, P.; Duran-Pacheco, G.; Kremer, T.; et al. Characterization of Brain Lysosomal Activities in GBA-Related and Sporadic Parkinson’s Disease and Dementia with Lewy Bodies. Mol. Neurobiol. 2019, 56, 1344–1355. [Google Scholar] [CrossRef] [Green Version]
- Chiasserini, D.; Paciotti, S.; Eusebi, P.; Persichetti, E.; Tasegian, A.; Kurzawa-Akanbi, M.; Chinnery, P.F.; Morris, C.M.; Calabresi, P.; Parnetti, L.; et al. Selective loss of glucocerebrosidase activity in sporadic Parkinson’s disease and dementia with Lewy bodies. Mol. Neurodegener. 2015, 10, 15. [Google Scholar] [CrossRef] [Green Version]
- Parnetti, L.; Chiasserini, D.; Persichetti, E.; Eusebi, P.; Varghese, S.; Qureshi, M.M.; Dardis, A.; Deganuto, M.; De Carlo, C.; Castrioto, A.; et al. Cerebrospinal fluid lysosomal enzymes and alpha-synuclein in Parkinson’s disease. Mov. Disord. Off. J. Mov. Disord. Soc. 2014, 29, 1019–1027. [Google Scholar] [CrossRef] [Green Version]
- Atashrazm, F.; Hammond, D.; Perera, G.; Dobson-Stone, C.; Mueller, N.; Pickford, R.; Kim, W.S.; Kwok, J.B.; Lewis, S.J.G.; Halliday, G.M.; et al. Reduced glucocerebrosidase activity in monocytes from patients with Parkinson’s disease. Sci. Rep. 2018, 8, 15446. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Song, W.; Brancati, G.; Segatori, L. Inhibition of endoplasmic reticulum-associated degradation rescues native folding in loss of function protein misfolding diseases. J. Biol. Chem. 2011, 286, 43454–43464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braunstein, H.; Maor, G.; Chicco, G.; Filocamo, M.; Zimran, A.; Horowitz, M. UPR activation and CHOP mediated induction of GBA1 transcription in Gaucher disease. Blood Cells Mol. Dis. 2018, 68, 21–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurzawa-Akanbi, M.; Hanson, P.S.; Blain, P.G.; Lett, D.J.; McKeith, I.G.; Chinnery, P.F.; Morris, C.M. Glucocerebrosidase Mutations alter the endoplasmic reticulum and lysosomes in Lewy body disease. J. Neurochem. 2012, 123, 298–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korkotian, E.; Schwarz, A.; Pelled, D.; Schwarzmann, G.; Segal, M.; Futerman, A.H. Elevation of intracellular glucosylceramide levels results in an increase in endoplasmic reticulum density and in functional calcium stores in cultured neurons. J. Biol. Chem. 1999, 274, 21673–21678. [Google Scholar] [CrossRef] [Green Version]
- Stojkovska, I.; Wani, W.Y.; Zunke, F.; Belur, N.R.; Pavlenko, E.A.; Mwenda, N.; Sharma, K.; Francelle, L.; Mazzulli, J.R. Rescue of α-synuclein aggregation in Parkinson’s patient neurons by synergistic enhancement of ER proteostasis and protein trafficking. Neuron 2022, 110, 436–451.e411. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Emr, S.D. Autophagy as a regulated pathway of cellular degradation. Science 2000, 290, 1717–1721. [Google Scholar] [CrossRef]
- Yang, S.Y.; Beavan, M.; Chau, K.Y.; Taanman, J.W.; Schapira, A.H. A Human Neural Crest Stem Cell-Derived Dopaminergic Neuronal Model Recapitulates Biochemical Abnormalities in GBA1 Mutation Carriers. Stem Cell Rep. 2017, 8, 728–742. [Google Scholar] [CrossRef]
- Rocha, E.M.; Smith, G.A.; Park, E.; Cao, H.; Graham, A.-R.; Brown, E.; McLean, J.R.; Hayes, M.A.; Beagan, J.; Izen, S.C.; et al. Sustained Systemic Glucocerebrosidase Inhibition Induces Brain α-Synuclein Aggregation, Microglia and Complement C1q Activation in Mice. Antioxid. Redox Signal. 2015, 23, 550–564. [Google Scholar] [CrossRef] [Green Version]
- Osellame, L.D.; Rahim, A.A.; Hargreaves, I.P.; Gegg, M.E.; Richard-Londt, A.; Brandner, S.; Waddington, S.N.; Schapira, A.H.; Duchen, M.R. Mitochondria and quality control defects in a mouse model of Gaucher disease—Links to Parkinson’s disease. Cell Metab. 2013, 17, 941–953. [Google Scholar] [CrossRef] [Green Version]
- Du, T.T.; Wang, L.; Duan, C.L.; Lu, L.L.; Zhang, J.L.; Gao, G.; Qiu, X.B.; Wang, X.M.; Yang, H. GBA deficiency promotes SNCA/alpha-synuclein accumulation through autophagic inhibition by inactivated PPP2A. Autophagy 2015, 11, 1803–1820. [Google Scholar] [CrossRef] [Green Version]
- Magalhaes, J.; Gegg, M.E.; Migdalska-Richards, A.; Doherty, M.K.; Whitfield, P.D.; Schapira, A.H.V. Autophagic lysosome reformation dysfunction in glucocerebrosidase deficient cells: Relevance to Parkinson disease. Hum. Mol. Genet. 2016, 25, 3432–3445. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.Y.; Gegg, M.; Chau, D.; Schapira, A. Glucocerebrosidase activity, cathepsin D and monomeric alpha-synuclein interactions in a stem cell derived neuronal model of a PD associated GBA1 mutation. Neurobiol. Dis. 2020, 134, 104620. [Google Scholar] [CrossRef] [PubMed]
- Klucken, J.; Poehler, A.-M.; Ebrahimi-Fakhari, D.; Schneider, J.; Nuber, S.; Rockenstein, E.; Schlötzer-Schrehardt, U.; Hyman, B.T.; McLean, P.J.; Masliah, E.; et al. Alpha-synuclein aggregation involves a bafilomycin A(1)-sensitive autophagy pathway. Autophagy 2012, 8, 754–766. [Google Scholar] [CrossRef] [Green Version]
- Kuo, S.H.; Tasset, I.; Cheng, M.M.; Diaz, A.; Pan, M.K.; Lieberman, O.J.; Hutten, S.J.; Alcalay, R.N.; Kim, S.; Ximénez-Embún, P.; et al. Mutant glucocerebrosidase impairs α-synuclein degradation by blockade of chaperone-mediated autophagy. Sci. Adv. 2022, 8, eabm6393. [Google Scholar] [CrossRef] [PubMed]
- Emmanouilidou, E.; Melachroinou, K.; Roumeliotis, T.; Garbis, S.D.; Ntzouni, M.; Margaritis, L.H.; Stefanis, L.; Vekrellis, K. Cell-produced alpha-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J. Neurosci. 2010, 30, 6838–6851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bae, E.J.; Yang, N.Y.; Song, M.; Lee, C.S.; Lee, J.S.; Jung, B.C.; Lee, H.J.; Kim, S.; Masliah, E.; Sardi, S.P.; et al. Glucocerebrosidase depletion enhances cell-to-cell transmission of alpha-synuclein. Nat. Commun. 2014, 5, 4755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gegg, M.E.; Verona, G.; Schapira, A.H.V. Glucocerebrosidase deficiency promotes release of alpha-synuclein fibrils from cultured neurons. Hum. Mol. Genet. 2020, 29, 1716–1728. [Google Scholar] [CrossRef]
- Alvarez-Erviti, L.; Seow, Y.; Schapira, A.H.; Gardiner, C.; Sargent, I.L.; Wood, M.J.A.; Cooper, J.M. Lysosomal dysfunction increases exosome-mediated alpha-synuclein release and transmission. Neurobiol. Dis. 2011, 42, 360–367. [Google Scholar] [CrossRef] [Green Version]
- Cerri, S.; Ghezzi, C.; Ongari, G.; Croce, S.; Avenali, M.; Zangaglia, R.; Di Monte, D.A.; Valente, E.M.; Blandini, F. GBA Mutations Influence the Release and Pathological Effects of Small Extracellular Vesicles from Fibroblasts of Patients with Parkinson’s Disease. Int. J. Mol. Sci. 2021, 22, 2215. [Google Scholar] [CrossRef]
- Jewett, K.A.; Thomas, R.E.; Phan, C.Q.; Lin, B.; Milstein, G.; Yu, S.; Bettcher, L.F.; Neto, F.C.; Djukovic, D.; Raftery, D.; et al. Glucocerebrosidase reduces the spread of protein aggregation in a Drosophila melanogaster model of neurodegeneration by regulating proteins trafficked by extracellular vesicles. PLoS Genet. 2021, 17, e1008859. [Google Scholar] [CrossRef]
- Thomas, R.E.; Vincow, E.S.; Merrihew, G.E.; MacCoss, M.J.; Davis, M.Y.; Pallanck, L.J. Glucocerebrosidase deficiency promotes protein aggregation through dysregulation of extracellular vesicles. PLoS Genet. 2018, 14, e1007694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Migdalska-Richards, A.; Wegrzynowicz, M.; Harrison, I.F.; Verona, G.; Bellotti, V.; Spillantini, M.G.; Schapira, A.H.V. L444P Gba1 mutation increases formation and spread of α-synuclein deposits in mice injected with mouse α-synuclein pre-formed fibrils. PLoS ONE 2020, 15, e0238075. [Google Scholar] [CrossRef] [PubMed]
- Farmer, B.C.; Walsh, A.E.; Kluemper, J.C.; Johnson, L.A. Lipid Droplets in Neurodegenerative Disorders. Front. Neurosci. 2020, 14, 742. [Google Scholar] [CrossRef] [PubMed]
- Barber, M.; Baldwin, A.; Gilbert, R.; Muller, H. Glucocerebrosidase rescues alpha-synuclein from amyloid formation. bioRxiv 2018. [Google Scholar] [CrossRef]
- Galvagnion, C.; Brown, J.W.P.; Ouberai, M.M.; Flagmeier, P.; Vendruscolo, M.; Buell, A.K.; Sparr, E.; Dobson, C.M. Chemical properties of lipids strongly affect the kinetics of the membrane-induced aggregation of α-synuclein. Proc. Natl. Acad. Sci. USA 2016, 113, 7065–7070. [Google Scholar] [CrossRef] [Green Version]
- Farfel-Becker, T.; Vitner, E.B.; Kelly, S.L.; Bame, J.R.; Duan, J.; Shinder, V.; Merrill, A.H., Jr.; Dobrenis, K.; Futerman, A.H. Neuronal accumulation of glucosylceramide in a mouse model of neuronopathic Gaucher disease leads to neurodegeneration. Hum. Mol. Genet. 2014, 23, 843–854. [Google Scholar] [CrossRef] [Green Version]
- Melo, F.; Caballero, L.; Zamorano, E.; Ventura, N.; Navarro, C.; Doll, I.; Zamorano, P.; Cornejo, A. The Cytotoxic Effect of alpha-Synuclein Aggregates. Chemphyschem 2021, 22, 526–532. [Google Scholar] [CrossRef]
- Fabelo, N.; Martin, V.; Santpere, G.; Marin, R.; Torrent, L.; Ferrer, I.; Diaz, M. Severe alterations in lipid composition of frontal cortex lipid rafts from Parkinson’s disease and incidental Parkinson’s disease. Mol. Med. 2011, 17, 1107–1118. [Google Scholar] [CrossRef]
- Sardi, S.P.; Viel, C.; Clarke, J.; Treleaven, C.M.; Richards, A.M.; Park, H.; Olszewski, M.A.; Dodge, J.C.; Marshall, J.; Makino, E.; et al. Glucosylceramide synthase inhibition alleviates aberrations in synucleinopathy models. Proc. Natl. Acad. Sci. USA 2017, 114, 2699–2704. [Google Scholar] [CrossRef] [Green Version]
- Nagata, M.; Izumi, Y.; Ishikawa, E.; Kiyotake, R.; Doi, R.; Iwai, S.; Omahdi, Z.; Yamaji, T.; Miyamoto, T.; Bamba, T.; et al. Intracellular metabolite β-glucosylceramide is an endogenous Mincle ligand possessing immunostimulatory activity. Proc. Natl. Acad. Sci. USA 2017, 114, E3285–E3294. [Google Scholar] [CrossRef] [Green Version]
- Orvisky, E.; Park, J.K.; LaMarca, M.E.; Ginns, E.I.; Martin, B.M.; Tayebi, N.; Sidransky, E. Glucosylsphingosine accumulation in tissues from patients with Gaucher disease: Correlation with phenotype and genotype. Mol. Genet. Metab. 2002, 76, 262–270. [Google Scholar] [CrossRef]
- Hallett, P.J.; Huebecker, M.; Brekk, O.R.; Moloney, E.B.; Rocha, E.M.; Priestman, D.A.; Platt, F.M.; Isacson, O. Glycosphingolipid levels and glucocerebrosidase activity are altered in normal aging of the mouse brain. Neurobiol. Aging 2018, 67, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Galvagnion, C.; Cerri, S.; Schapira, A.H.V.; Blandini, F.; Di Monte, D.A. Sphingolipid changes in Parkinson L444P GBA mutation fibroblasts promote α-synuclein aggregation. bioRxiv 2020. [Google Scholar] [CrossRef] [PubMed]
- Hein, L.K.; Duplock, S.; Hopwood, J.J.; Fuller, M. Lipid composition of microdomains is altered in a cell model of Gaucher disease. J. Lipid Res. 2008, 49, 1725–1734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varela, A.R.; Ventura, A.E.; Carreira, A.C.; Fedorov, A.; Futerman, A.H.; Prieto, M.; Silva, L.C. Pathological levels of glucosylceramide change the biophysical properties of artificial and cell membranes. Phys. Chem. Chem. Phys. 2016, 19, 340–346. [Google Scholar] [CrossRef]
- Gegg, M.E.; Sweet, L.; Wang, B.H.; Shihabuddin, L.S.; Sardi, S.P.; Schapira, A.H. No evidence for substrate accumulation in Parkinson brains with GBA mutations. Mov. Disord. 2015, 30, 1085–1089. [Google Scholar] [CrossRef]
- Clark, L.N.; Chan, R.; Cheng, R.; Liu, X.; Park, N.; Parmalee, N.; Kisselev, S.; Cortes, E.; Torres, P.A.; Pastores, G.M.; et al. Gene-wise association of variants in four lysosomal storage disorder genes in neuropathologically confirmed Lewy body disease. PLoS ONE 2015, 10, e0125204. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, O.; Svennerholm, L. Accumulation of glucosylceramide and glucosylsphingosine (psychosine) in cerebrum and cerebellum in infantile and juvenile Gaucher disease. J. Neurochem. 1982, 39, 709–718. [Google Scholar] [CrossRef]
- Taguchi, Y.V.; Liu, J.; Ruan, J.; Pacheco, J.; Zhang, X.; Abbasi, J.; Keutzer, J.; Mistry, P.K.; Chandra, S.S. Glucosylsphingosine Promotes alpha-Synuclein Pathology in Mutant GBA-Associated Parkinson’s Disease. J. Neurosci. 2017, 37, 9617–9631. [Google Scholar] [CrossRef] [Green Version]
- Paul, A.; Jacoby, G.; Laor Bar-Yosef, D.; Beck, R.; Gazit, E.; Segal, D. Glucosylceramide Associated with Gaucher Disease Forms Amyloid-like Twisted Ribbon Fibrils That Induce alpha-Synuclein Aggregation. ACS Nano 2021, 15, 11854–11868. [Google Scholar] [CrossRef]
- Zunke, F.; Moise, A.C.; Belur, N.R.; Gelyana, E.; Stojkovska, I.; Dzaferbegovic, H.; Toker, N.J.; Jeon, S.; Fredriksen, K.; Mazzulli, J.R. Reversible Conformational Conversion of α-Synuclein into Toxic Assemblies by Glucosylceramide. Neuron 2018, 97, 92–107.e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glajch, K.E.; Moors, T.E.; Chen, Y.; Bechade, P.A.; Nam, A.Y.; Rajsombath, M.M.; McCaffery, T.D.; Dettmer, U.; Weihofen, A.; Hirst, W.D.; et al. Wild-type GBA1 increases the alpha-synuclein tetramer-monomer ratio, reduces lipid-rich aggregates, and attenuates motor and cognitive deficits in mice. Proc. Natl. Acad. Sci. USA 2021, 118, e2103425118. [Google Scholar] [CrossRef] [PubMed]
- Guedes, L.C.; Chan, R.B.; Gomes, M.A.; Conceicao, V.A.; Machado, R.B.; Soares, T.; Xu, Y.; Gaspar, P.; Carrico, J.A.; Alcalay, R.N.; et al. Serum lipid alterations in GBA-associated Parkinson’s disease. Parkinsonism Relat. Disord. 2017, 44, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Moloney, E.B.; Moskites, A.; Ferrari, E.J.; Isacson, O.; Hallett, P.J. The glycoprotein GPNMB is selectively elevated in the substantia nigra of Parkinson’s disease patients and increases after lysosomal stress. Neurobiol. Dis. 2018, 120, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Murugesan, V.; Liu, J.; Yang, R.; Lin, H.; Lischuk, A.; Pastores, G.; Zhang, X.; Chuang, W.L.; Mistry, P.K. Validating glycoprotein non-metastatic melanoma B (gpNMB, osteoactivin), a new biomarker of Gaucher disease. Blood Cells Mol. Dis. 2018, 68, 47–53. [Google Scholar] [CrossRef]
- Schapira, A.H.; Gegg, M. Mitochondrial contribution to Parkinson’s disease pathogenesis. Parkinsons Dis. 2011, 2011, 159160. [Google Scholar] [CrossRef] [Green Version]
- Schapira, A.H.; Cooper, J.M.; Dexter, D.; Clark, J.B.; Jenner, P.; Marsden, C.D. Mitochondrial complex I deficiency in Parkinson’s disease. J. Neurochem. 1990, 54, 823–827. [Google Scholar]
- Schapira, A.H.; Cooper, J.M.; Dexter, D.; Jenner, P.; Clark, J.B.; Marsden, C.D. Mitochondrial complex I deficiency in Parkinson’s disease. Lancet 1989, 1, 1269. [Google Scholar] [CrossRef]
- Schapira, A.H. Mitochondria in the aetiology and pathogenesis of Parkinson’s disease. Lancet Neurol. 2008, 7, 97–109. [Google Scholar] [CrossRef]
- Kinghorn, K.J.; Grönke, S.; Castillo-Quan, J.I.; Woodling, N.S.; Li, L.; Sirka, E.; Gegg, M.; Mills, K.; Hardy, J.; Bjedov, I.; et al. A Drosophila Model of Neuronopathic Gaucher Disease Demonstrates Lysosomal-Autophagic Defects and Altered mTOR Signalling and Is Functionally Rescued by Rapamycin. J. Neurosci. 2016, 36, 11654–11670. [Google Scholar] [CrossRef] [Green Version]
- Schöndorf, D.C.; Ivanyuk, D.; Baden, P.; Sanchez-Martinez, A.; De Cicco, S.; Yu, C.; Giunta, I.; Schwarz, L.K.; Di Napoli, G.; Panagiotakopoulou, V.; et al. The NAD+ Precursor Nicotinamide Riboside Rescues Mitochondrial Defects and Neuronal Loss in iPSC and Fly Models of Parkinson’s Disease. Cell Rep. 2018, 23, 2976–2988. [Google Scholar] [CrossRef] [PubMed]
- Tansey, M.G.; Goldberg, M.S. Neuroinflammation in Parkinson’s disease: Its role in neuronal death and implications for therapeutic intervention. Neurobiol. Dis. 2010, 37, 510–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, M.J.; Myer, B.J.; Khokher, A.M.; Rushton, N.; Cox, T.M. Pro-inflammatory cytokines and the pathogenesis of Gaucher’s disease: Increased release of interleukin-6 and interleukin-10. QJM Mon. J. Assoc. Physicians 1997, 90, 19–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mistry, P.K.; Liu, J.; Yang, M.; Nottoli, T.; McGrath, J.; Jain, D.; Zhang, K.; Keutzer, J.; Chuang, W.L.; Mehal, W.Z.; et al. Glucocerebrosidase gene-deficient mouse recapitulates Gaucher disease displaying cellular and molecular dysregulation beyond the macrophage. Proc. Natl. Acad. Sci. USA 2010, 107, 19473–19478. [Google Scholar] [CrossRef] [Green Version]
- Vitner, E.B.; Salomon, R.; Farfel-Becker, T.; Meshcheriakova, A.; Ali, M.; Klein, A.D.; Platt, F.M.; Cox, T.M.; Futerman, A.H. RIPK3 as a potential therapeutic target for Gaucher’s disease. Nat. Med. 2014, 20, 204–208. [Google Scholar] [CrossRef]
- Keatinge, M.; Bui, H.; Menke, A.; Chen, Y.C.; Sokol, A.M.; Bai, Q.; Ellett, F.; Da Costa, M.; Burke, D.; Gegg, M.; et al. Glucocerebrosidase 1 deficient Danio rerio mirror key pathological aspects of human Gaucher disease and provide evidence of early microglial activation preceding alpha-synuclein-independent neuronal cell death. Hum. Mol. Genet. 2015, 24, 6640–6652. [Google Scholar] [CrossRef] [Green Version]
- Vitner, E.B.; Farfel-Becker, T.; Eilam, R.; Biton, I.; Futerman, A.H. Contribution of brain inflammation to neuronal cell death in neuronopathic forms of Gaucher’s disease. Brain A J. Neurol. 2012, 135, 1724–1735. [Google Scholar] [CrossRef]
- Enquist, I.B.; Lo Bianco, C.; Ooka, A.; Nilsson, E.; Mansson, J.E.; Ehinger, M.; Richter, J.; Brady, R.O.; Kirik, D.; Karlsson, S. Murine models of acute neuronopathic Gaucher disease. Proc. Natl. Acad. Sci. USA 2007, 104, 17483–17488. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Erviti, L.; Couch, Y.; Richardson, J.; Cooper, J.M.; Wood, M.J. Alpha-synuclein release by neurons activates the inflammatory response in a microglial cell line. Neurosci. Res. 2011, 69, 337–342. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, T.; Pei, Z.; Miller, D.S.; Wu, X.; Block, M.L.; Wilson, B.; Zhang, W.; Zhou, Y.; Hong, J.S.; et al. Aggregated alpha-synuclein activates microglia: A process leading to disease progression in Parkinson’s disease. FASEB J. 2005, 19, 533–542. [Google Scholar] [CrossRef]
- Lee, E.J.; Woo, M.S.; Moon, P.G.; Baek, M.C.; Choi, I.Y.; Kim, W.K.; Junn, E.; Kim, H.S. Alpha-synuclein activates microglia by inducing the expressions of matrix metalloproteinases and the subsequent activation of protease-activated receptor-1. J. Immunol. 2010, 185, 615–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, C.; Ho, D.H.; Suk, J.E.; You, S.; Michael, S.; Kang, J.; Joong Lee, S.; Masliah, E.; Hwang, D.; Lee, H.J.; et al. Neuron-released oligomeric alpha-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat. Commun. 2013, 4, 1562. [Google Scholar] [CrossRef] [Green Version]
- Thome, A.D.; Harms, A.S.; Volpicelli-Daley, L.A.; Standaert, D.G. microRNA-155 Regulates Alpha-Synuclein-Induced Inflammatory Responses in Models of Parkinson Disease. J. Neurosci. 2016, 36, 2383–2390. [Google Scholar] [CrossRef] [PubMed]
- Cavaliere, F.; Cerf, L.; Dehay, B.; Ramos-Gonzalez, P.; De Giorgi, F.; Bourdenx, M.; Bessede, A.; Obeso, J.A.; Matute, C.; Ichas, F.; et al. In vitro alpha-synuclein neurotoxicity and spreading among neurons and astrocytes using Lewy body extracts from Parkinson disease brains. Neurobiol. Dis. 2017, 103, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Loria, F.; Vargas, J.Y.; Bousset, L.; Syan, S.; Salles, A.; Melki, R.; Zurzolo, C. Alpha-Synuclein transfer between neurons and astrocytes indicates that astrocytes play a role in degradation rather than in spreading. Acta Neuropathol. 2017, 134, 789–808. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, S.G.; Eblan, M.J.; Gutti, U.; Hruska, K.S.; Stubblefield, B.K.; Goker-Alpan, O.; LaMarca, M.E.; Sidransky, E. Glucocerebrosidase mutations in Chinese subjects from Taiwan with sporadic Parkinson disease. Mol. Genet. Metab. 2007, 91, 195–200. [Google Scholar] [CrossRef] [Green Version]
- Angeli, A.; Mencacci, N.E.; Duran, R.; Aviles-Olmos, I.; Kefalopoulou, Z.; Candelario, J.; Rusbridge, S.; Foley, J.; Pradhan, P.; Jahanshahi, M.; et al. Genotype and phenotype in Parkinson’s disease: Lessons in heterogeneity from deep brain stimulation. Mov. Disord. 2013, 28, 1370–1375. [Google Scholar] [CrossRef] [PubMed]
- Shemesh, E.; Deroma, L.; Bembi, B.; Deegan, P.; Hollak, C.; Weinreb, N.J.; Cox, T.M. Enzyme replacement and substrate reduction therapy for Gaucher disease. Cochrane Database Syst. Rev. 2015, 2015, CD010324. [Google Scholar] [CrossRef]
- Chauhan, A.; Tikoo, A.; Kapur, A.K.; Singh, M. The taming of the cell penetrating domain of the HIV Tat: Myths and realities. J. Control. Release Off. J. Control. Release Soc. 2007, 117, 148–162. [Google Scholar] [CrossRef] [Green Version]
- Gillmeister, M.P.; Betenbaugh, M.J.; Fishman, P.S. Cellular trafficking and photochemical internalization of cell penetrating peptide linked cargo proteins: A dual fluorescent labeling study. Bioconjug. Chem. 2011, 22, 556–566. [Google Scholar] [CrossRef]
- Fu, A.; Wang, Y.; Zhan, L.; Zhou, R. Targeted delivery of proteins into the central nervous system mediated by rabies virus glycoprotein-derived peptide. Pharm. Res. 2012, 29, 1562–1569. [Google Scholar] [CrossRef] [PubMed]
- Fu, A.; Zhang, M.; Gao, F.; Xu, X.; Chen, Z. A novel peptide delivers plasmids across blood-brain barrier into neuronal cells as a single-component transfer vector. PLoS ONE 2013, 8, e59642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gramlich, P.A.; Westbroek, W.; Feldman, R.A.; Awad, O.; Mello, N.; Remington, M.P.; Sun, Y.; Zhang, W.; Sidransky, E.; Betenbaugh, M.J.; et al. A peptide-linked recombinant glucocerebrosidase for targeted neuronal delivery: Design, production, and assessment. J. Biotechnol. 2016, 221, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ysselstein, D.; Young, T.J.; Nguyen, M.; Padmanabhan, S.; Hirst, W.D.; Dzamko, N.; Krainc, D. Evaluation of Strategies for Measuring Lysosomal Glucocerebrosidase Activity. Mov. Disord. 2021, 36, 2719–2730. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Yun, S.P.; Lee, S.; Umanah, G.E.; Bandaru, V.V.R.; Yin, X.; Rhee, P.; Karuppagounder, S.S.; Kwon, S.-H.; Lee, H.; et al. GBA1 deficiency negatively affects physiological α-synuclein tetramers and related multimers. Proc. Natl. Acad. Sci. USA 2018, 115, 798–803. [Google Scholar] [CrossRef] [Green Version]
- Peterschmitt, M.J.; Saiki, H.; Hatano, T.; Gasser, T.; Isaacson, S.H.; Gaemers, S.J.M.; Minini, P.; Saubadu, S.; Sharma, J.; Walbillic, S.; et al. Safety, Pharmacokinetics, and Pharmacodynamics of Oral Venglustat in Patients with Parkinson’s Disease and a GBA Mutation: Results from Part 1 of the Randomized, Double-Blinded, Placebo-Controlled MOVES-PD Trial. J. Parkinson’s Dis. 2022, 12, 557–570. [Google Scholar] [CrossRef]
- Hudry, E.; Vandenberghe, L.H. Therapeutic AAV Gene Transfer to the Nervous System: A Clinical Reality. Neuron 2019, 101, 839–862. [Google Scholar] [CrossRef] [Green Version]
- Rocha, E.M.; Smith, G.A.; Park, E.; Cao, H.; Brown, E.; Hayes, M.A.; Beagan, J.; McLean, J.R.; Izen, S.C.; Perez-Torres, E.; et al. Glucocerebrosidase gene therapy prevents alpha-synucleinopathy of midbrain dopamine neurons. Neurobiol. Dis. 2015, 82, 495–503. [Google Scholar] [CrossRef] [Green Version]
- Sucunza, D.; Rico, A.J.; Roda, E.; Collantes, M.; González-Aseguinolaza, G.; Rodríguez-Pérez, A.I.; Peñuelas, I.; Vázquez, A.; Labandeira-García, J.L.; Broccoli, V.; et al. Glucocerebrosidase Gene Therapy Induces Alpha-Synuclein Clearance and Neuroprotection of Midbrain Dopaminergic Neurons in Mice and Macaques. Int. J. Mol. Sci. 2021, 22, 4825. [Google Scholar] [CrossRef]
- Mullin, S.; Smith, L.; Lee, K.; D’Souza, G.; Woodgate, P.; Elflein, J.; Hallqvist, J.; Toffoli, M.; Streeter, A.; Hosking, J.; et al. Ambroxol for the Treatment of Patients with Parkinson Disease with and without Glucocerebrosidase Gene Mutations: A Nonrandomized, Noncontrolled Trial. JAMA Neurol. 2020, 77, 427–434. [Google Scholar] [CrossRef] [Green Version]
- Zimran, A.; Altarescu, G.; Elstein, D. Pilot study using ambroxol as a pharmacological chaperone in type 1 Gaucher disease. Blood Cells Mol. Dis. 2013, 50, 134–137. [Google Scholar] [CrossRef] [PubMed]
- McNeill, A.; Magalhaes, J.; Shen, C.; Chau, K.Y.; Hughes, D.; Mehta, A.; Foltynie, T.; Cooper, J.M.; Abramov, A.Y.; Gegg, M.; et al. Ambroxol improves lysosomal biochemistry in glucocerebrosidase mutation-linked Parkinson disease cells. Brain 2014, 137, 1481–1495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magalhaes, J.; Gegg, M.E.; Migdalska-Richards, A.; Schapira, A.H. Effects of ambroxol on the autophagy-lysosome pathway and mitochondria in primary cortical neurons. Sci. Rep. 2018, 8, 1385. [Google Scholar] [CrossRef] [PubMed]
- Migdalska-Richards, A.; Ko, W.K.; Li, Q.; Bezard, E.; Schapira, A.H. Oral ambroxol increases brain glucocerebrosidase activity in a nonhuman primate. Synapse 2017, 71, e21967. [Google Scholar] [CrossRef] [PubMed]
- Migdalska-Richards, A.; Daly, L.; Bezard, E.; Schapira, A.H. Ambroxol effects in glucocerebrosidase and alpha-synuclein transgenic mice. Ann. Neurol. 2016, 80, 766–775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maor, G.; Cabasso, O.; Krivoruk, O.; Rodriguez, J.; Steller, H.; Segal, D.; Horowitz, M. The contribution of mutant GBA to the development of Parkinson disease in Drosophila. Hum. Mol. Genet. 2016, 25, 2712–2727. [Google Scholar] [CrossRef] [Green Version]
- Aflaki, E.; Borger, D.K.; Moaven, N.; Stubblefield, B.K.; Rogers, S.A.; Patnaik, S.; Schoenen, F.J.; Westbroek, W.; Zheng, W.; Sullivan, P.; et al. A New Glucocerebrosidase Chaperone Reduces α-Synuclein and Glycolipid Levels in iPSC-Derived Dopaminergic Neurons from Patients with Gaucher Disease and Parkinsonism. J. Neurosci. 2016, 36, 7441–7452. [Google Scholar] [CrossRef]
- Bendikov-Bar, I.; Maor, G.; Filocamo, M.; Horowitz, M. Ambroxol as a pharmacological chaperone for mutant glucocerebrosidase. Blood Cells Mol. Dis. 2013, 50, 141–145. [Google Scholar] [CrossRef] [Green Version]
- Mazzulli, J.R.; Zunke, F.; Tsunemi, T.; Toker, N.J.; Jeon, S.; Burbulla, L.F.; Patnaik, S.; Sidransky, E.; Marugan, J.J.; Sue, C.M.; et al. Activation of β-Glucocerebrosidase Reduces Pathological α-Synuclein and Restores Lysosomal Function in Parkinson’s Patient Midbrain Neurons. J. Neurosci. 2016, 36, 7693–7706. [Google Scholar] [CrossRef]
- Shanmuganathan, M.; Britz-McKibbin, P. Inhibitor screening of pharmacological chaperones for lysosomal beta-glucocerebrosidase by capillary electrophoresis. Anal. Bioanal. Chem. 2011, 399, 2843–2853. [Google Scholar] [CrossRef]
- Patnaik, S.; Zheng, W.; Choi, J.H.; Motabar, O.; Southall, N.; Westbroek, W.; Lea, W.A.; Velayati, A.; Goldin, E.; Sidransky, E.; et al. Discovery, structure-activity relationship, and biological evaluation of noninhibitory small molecule chaperones of glucocerebrosidase. J. Med. Chem. 2012, 55, 5734–5748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, T.; Shimoda, M.; Ito, K.; Hanai, S.; Aizawa, H.; Kato, T.; Kawasaki, K.; Yamaguchi, T.; Ryoo, H.D.; Goto-Inoue, N.; et al. Expression of Human Gaucher Disease Gene GBA Generates Neurodevelopmental Defects and ER Stress in Drosophila Eye. PLoS ONE 2013, 8, e69147, Erratum in PLoS ONE 2015, 10, e0135619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richter, F.; Fleming, S.M.; Watson, M.; Lemesre, V.; Pellegrino, L.; Ranes, B.; Zhu, C.; Mortazavi, F.; Mulligan, C.K.; Sioshansi, P.C.; et al. A GCase chaperone improves motor function in a mouse model of synucleinopathy. Neurotherapeutics 2014, 11, 840–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morabito, G.; Giannelli, S.G.; Ordazzo, G.; Bido, S.; Castoldi, V.; Indrigo, M.; Cabassi, T.; Cattaneo, S.; Luoni, M.; Cancellieri, C.; et al. AAV-PHP.B-Mediated Global-Scale Expression in the Mouse Nervous System Enables GBA1 Gene Therapy for Wide Protection from Synucleinopathy. Mol. Ther. 2017, 25, 2727–2742. [Google Scholar] [CrossRef] [Green Version]
- den Heijer, J.M.; Kruithof, A.C.; van Amerongen, G.; de Kam, M.L.; Thijssen, E.; Grievink, H.W.; Moerland, M.; Walker, M.; Been, K.; Skerlj, R.; et al. A randomized single and multiple ascending dose study in healthy volunteers of LTI-291, a centrally penetrant glucocerebrosidase activator. Br. J. Clin. Pharmacol. 2021, 87, 3561–3573. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Yang, C.; Chen, M.; Ye, D.Y.; Lonser, R.R.; Brady, R.O.; Zhuang, Z. Histone deacetylase inhibitors prevent the degradation and restore the activity of glucocerebrosidase in Gaucher disease. Proc. Natl. Acad. Sci. USA 2011, 108, 21200–21205. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Rahimpour, S.; Lu, J.; Pacak, K.; Ikejiri, B.; Brady, R.O.; Zhuang, Z. Histone deacetylase inhibitors increase glucocerebrosidase activity in Gaucher disease by modulation of molecular chaperones. Proc. Natl. Acad. Sci. USA 2013, 110, 966–971. [Google Scholar] [CrossRef] [Green Version]
- Jung, O.; Patnaik, S.; Marugan, J.; Sidransky, E.; Westbroek, W. Progress and potential of non-inhibitory small molecule chaperones for the treatment of Gaucher disease and its implications for Parkinson disease. Expert Rev. Proteom. 2016, 13, 471–479. [Google Scholar] [CrossRef] [Green Version]
- Bendikov-Bar, I.; Maor, G.; Horowitz, M. Processing and maturation of human glucocerebrosidase. In Advances in Gaucher Disease: Basic and Clinical Perspectives; Future Medicine Ltd.: London, UK, 2013; pp. 140–157. [Google Scholar] [CrossRef]
- Khanna, R.; Benjamin, E.R.; Pellegrino, L.; Schilling, A.; Rigat, B.A.; Soska, R.; Nafar, H.; Ranes, B.E.; Feng, J.; Lun, Y.; et al. The pharmacological chaperone isofagomine increases the activity of the Gaucher disease L444P mutant form of beta-glucosidase. FEBS J. 2010, 277, 1618–1638. [Google Scholar] [CrossRef]
- Spilman, P.; Podlutskaya, N.; Hart, M.J.; Debnath, J.; Gorostiza, O.; Bredesen, D.; Richardson, A.; Strong, R.; Galvan, V. Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-beta levels in a mouse model of Alzheimer’s disease. PLoS ONE 2010, 5, e9979. [Google Scholar] [CrossRef] [Green Version]
- Decressac, M.; Björklund, A. mTOR inhibition alleviates L-DOPA-induced dyskinesia in parkinsonian rats. J. Parkinson’s Dis. 2013, 3, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Fog, C.K.; Zago, P.; Malini, E.; Solanko, L.M.; Peruzzo, P.; Bornaes, C.; Magnoni, R.; Mehmedbasic, A.; Petersen, N.H.T.; Bembi, B.; et al. The heat shock protein amplifier arimoclomol improves refolding, maturation and lysosomal activity of glucocerebrosidase. EBioMedicine 2018, 38, 142–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]


| Mutation | Penetrance of Mutation | Location of Mutation | Effect on GCase | GD | GBA-PD | References |
|---|---|---|---|---|---|---|
| N370S | 0.08–71.8% | Interface of domains II and III | Loss of GCase activity Activation of the UPR Alpha-synuclein pathology | Generally mild, non-neuronopathic GD | Lower disease penetrance and a milder clinical phenotype | [5,6,7,40,47,52,73,80,83,84,85,86,87,88,89,90,91,92,93] |
| L444P | 0.06–18.8% | Domain II | Loss of GCase activity Activation of the UPR Alpha-synuclein pathology | Generally severe, neuronopathic GD | Higher disease penetrance and a worse clinical phenotype | [5,6,7,40,47,52,73,80,83,87,88,89,90,91,92,94,95,96] |
| E326K | 2.8–3.88% | Surface of domain III | Reduces GCase activity to a lesser extent than GD-causing mutations | No clinical manifestation | Worse clinical phenotype | [44,73,74,75,79,80,93,97,98,99,100,101,102,103,104] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smith, L.; Schapira, A.H.V. GBA Variants and Parkinson Disease: Mechanisms and Treatments. Cells 2022, 11, 1261. https://doi.org/10.3390/cells11081261
Smith L, Schapira AHV. GBA Variants and Parkinson Disease: Mechanisms and Treatments. Cells. 2022; 11(8):1261. https://doi.org/10.3390/cells11081261
Chicago/Turabian StyleSmith, Laura, and Anthony H. V. Schapira. 2022. "GBA Variants and Parkinson Disease: Mechanisms and Treatments" Cells 11, no. 8: 1261. https://doi.org/10.3390/cells11081261