
D-Cysteine Activates Chaperone-Mediated Autophagy in Cerebellar Purkinje Cells via the Generation of Hydrogen Sulfide and Nrf2 Activation

 , , and
, , and 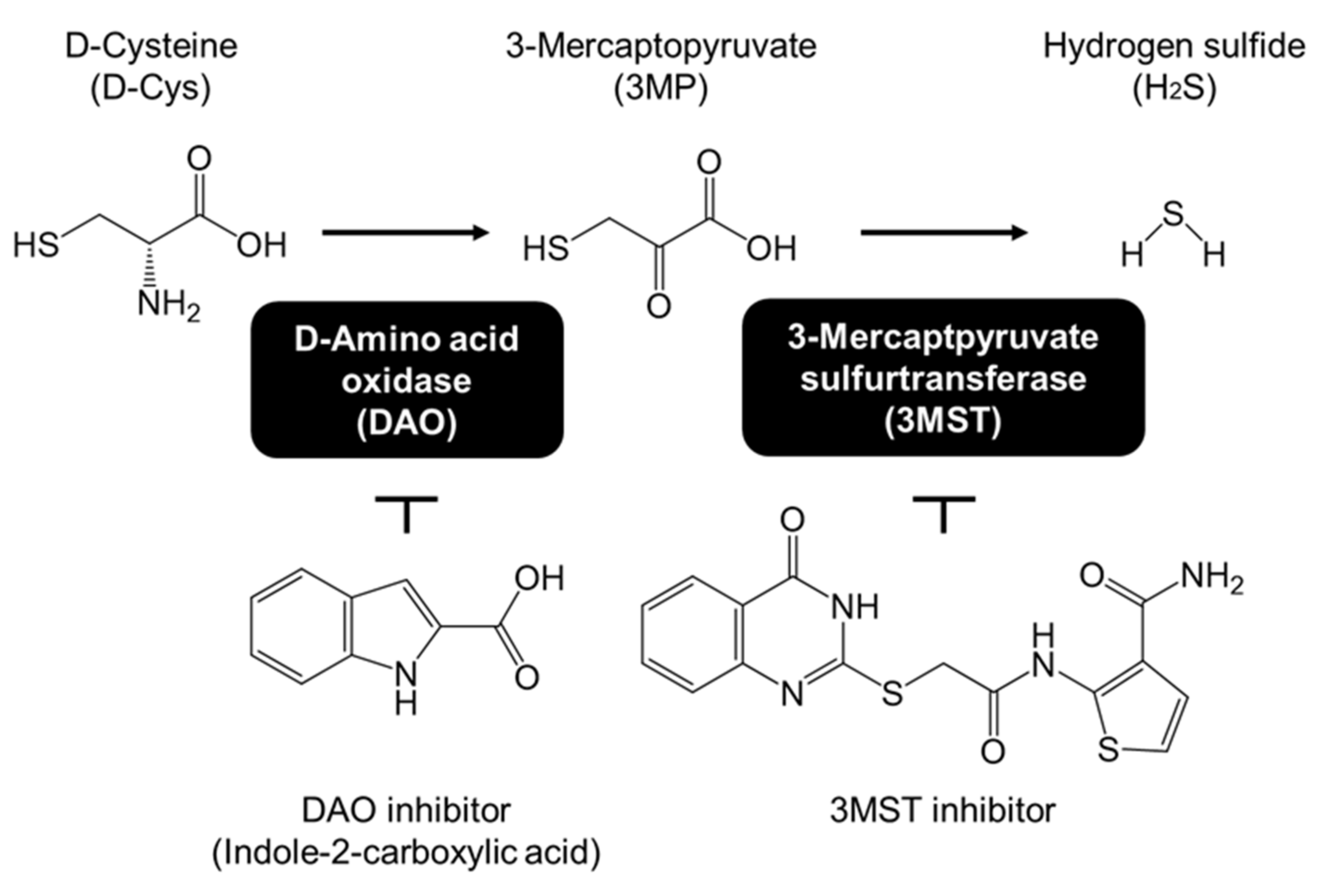{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture Experiments Using AD293 Cells
2.3. Experiments Using Primary Cerebellar Cultures
2.4. Long-Term Treatment with D-Cysteine in Mice
2.5. Immunoblot Analyses
2.6. Statistical Analyses
3. Results
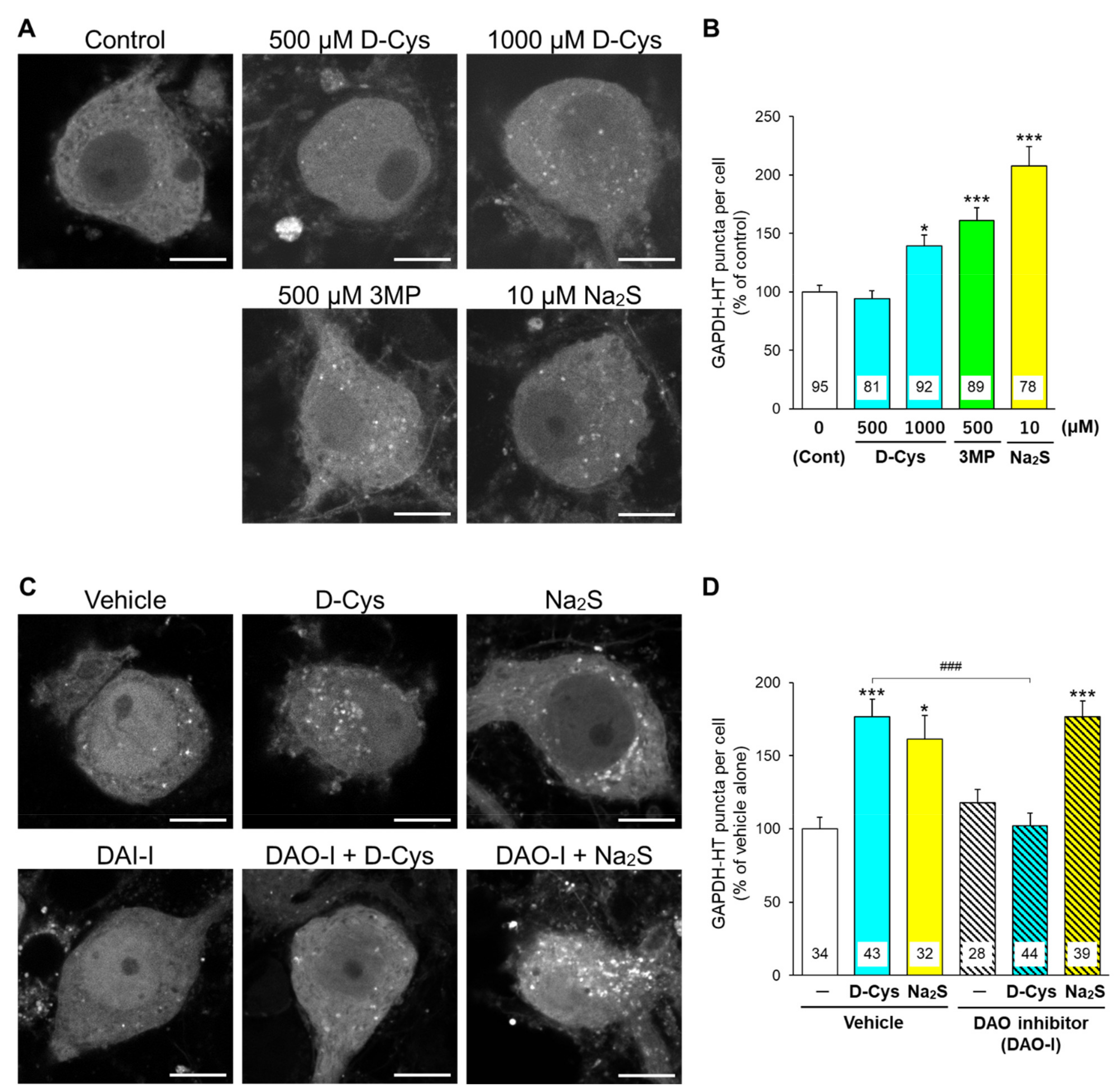
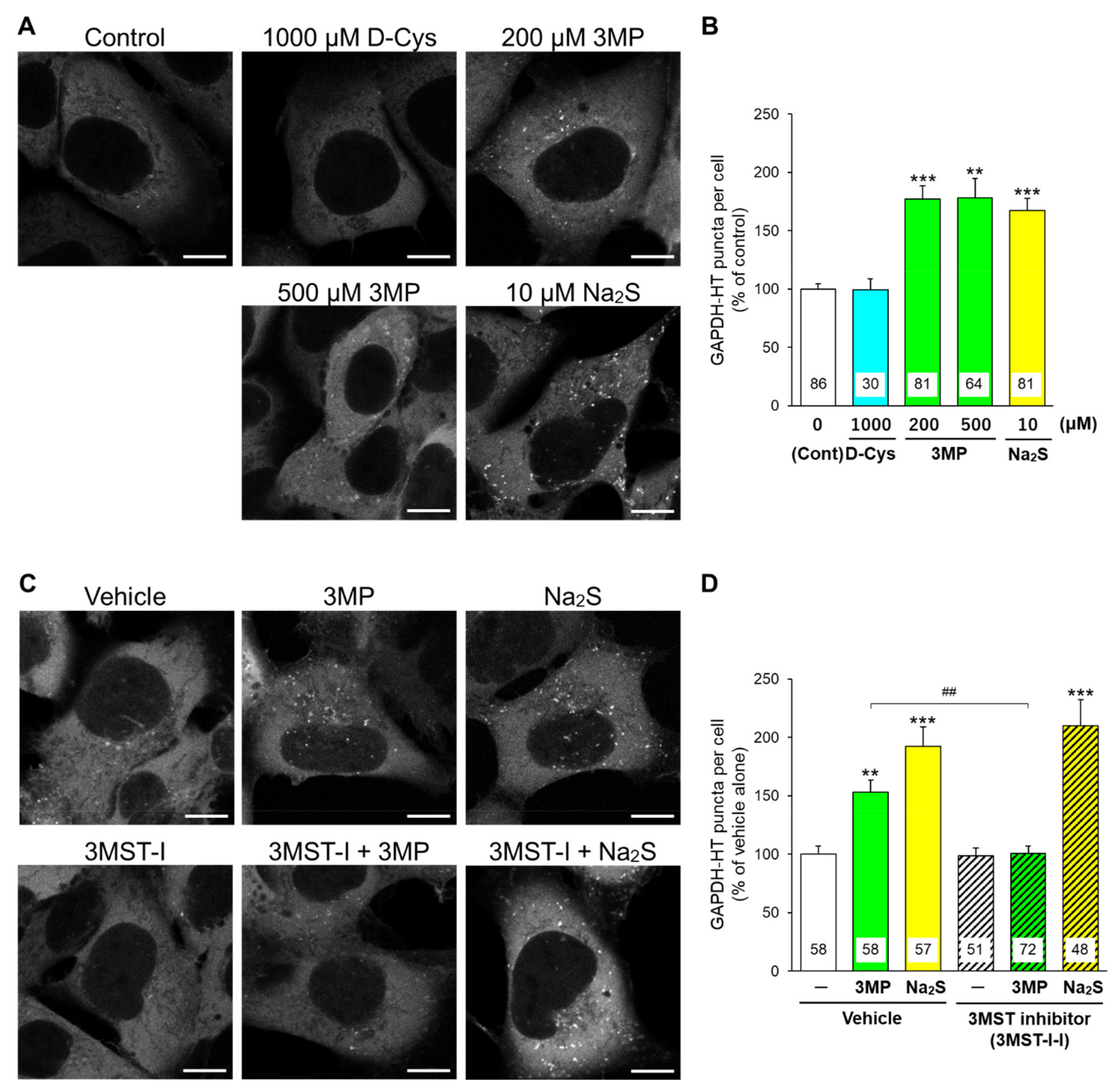
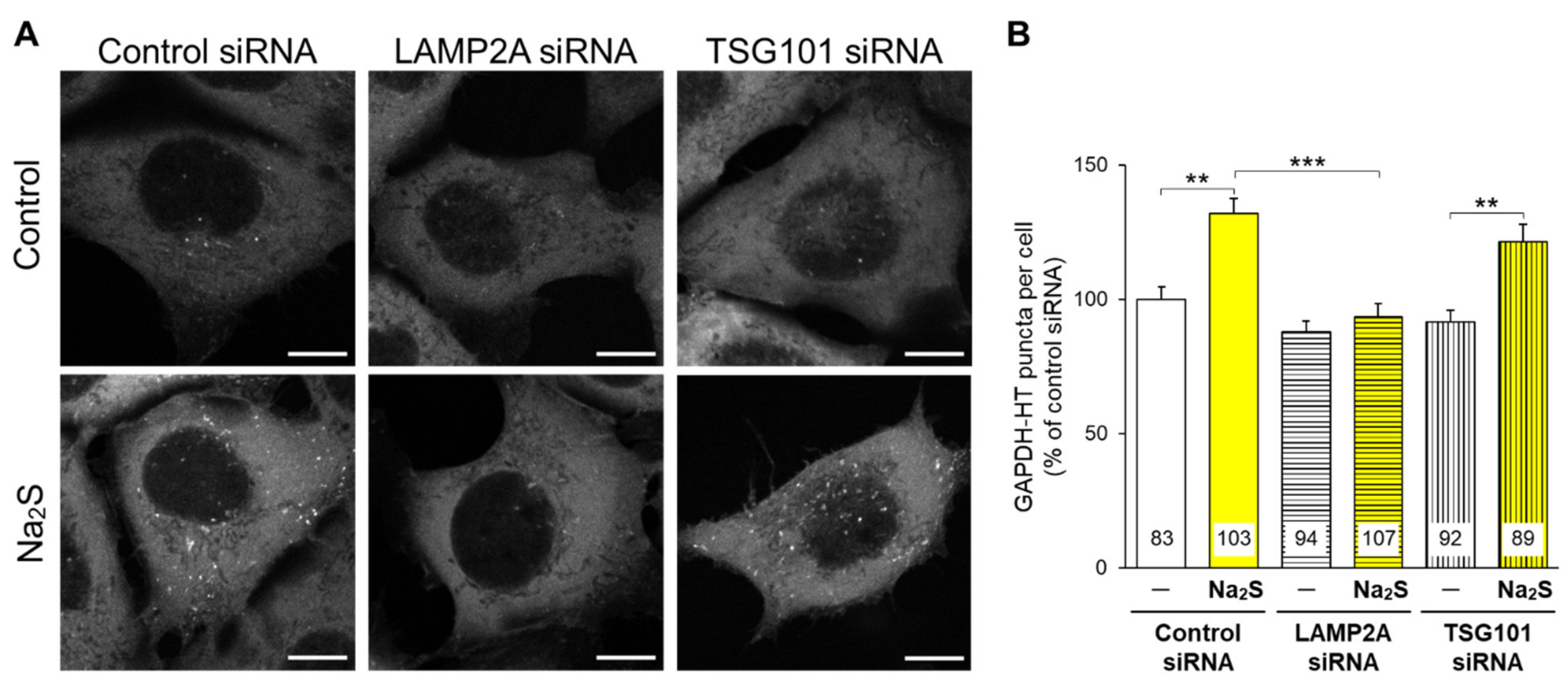
3.1. Effects of D-Cysteine, 3MP, and Na2S on CMA/Ma Activity in Primary Cultured Purkinje Cells and AD293 Cells
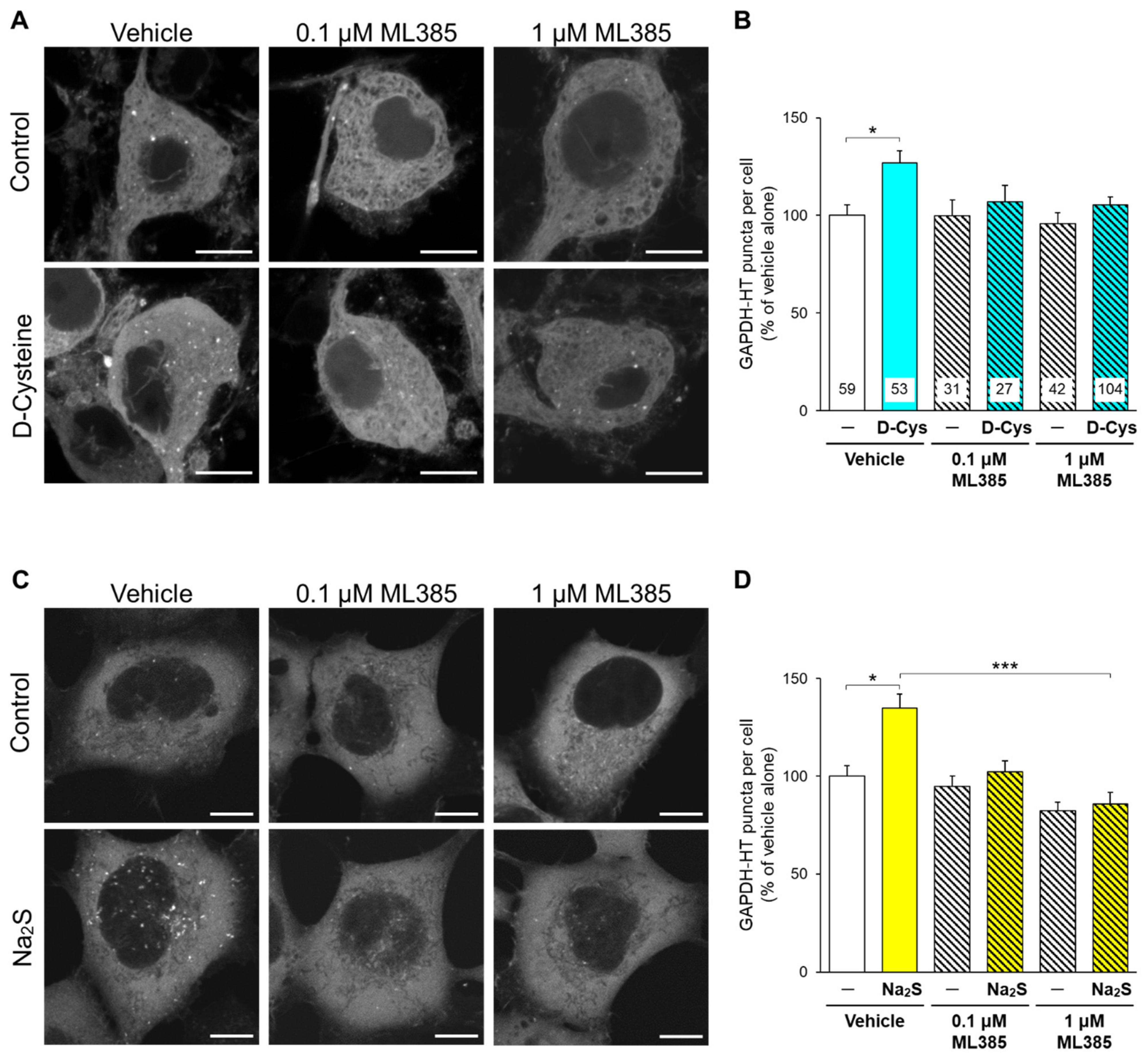
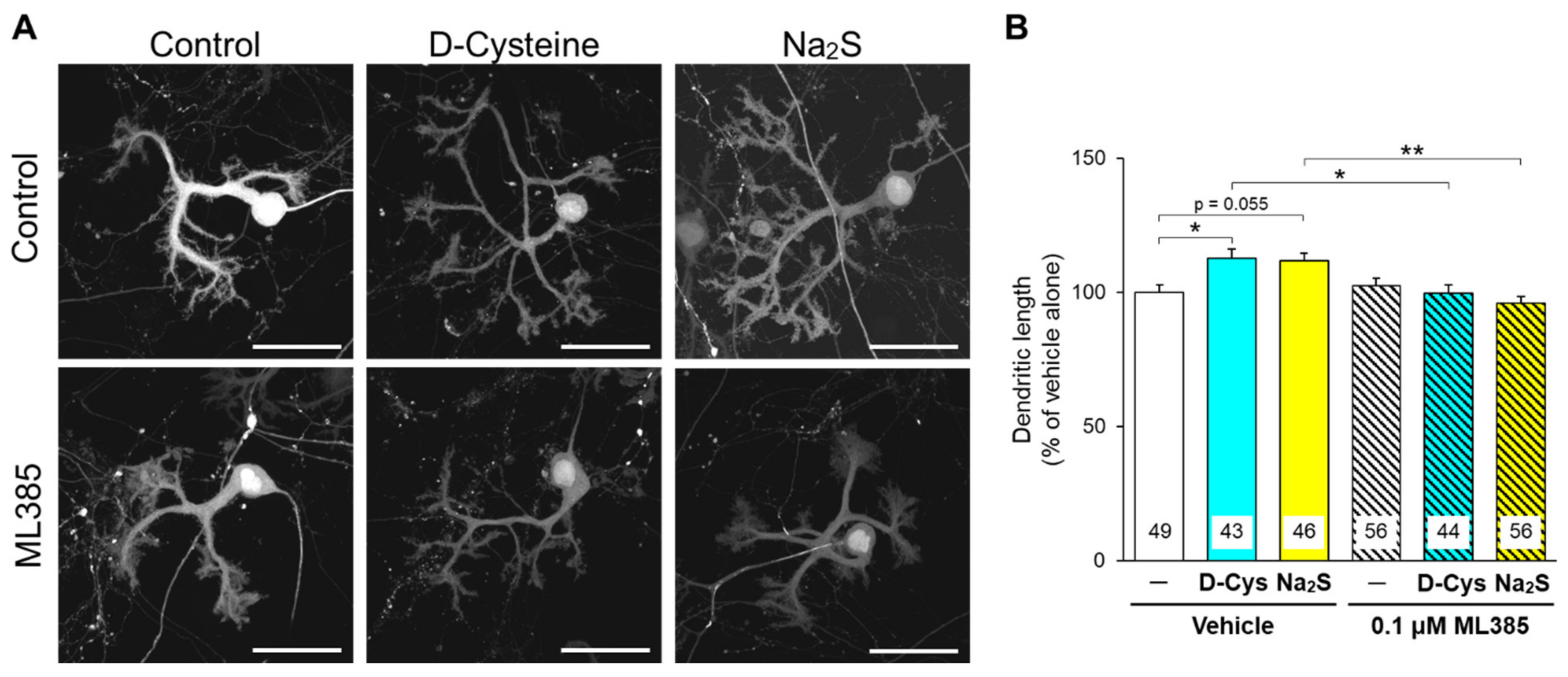
3.2. Involvement of Nrf2 in D-Cysteine—And Na2S-Induced Activation of CMA in Primary Cultured Purkinje Cells and AD293 Cells
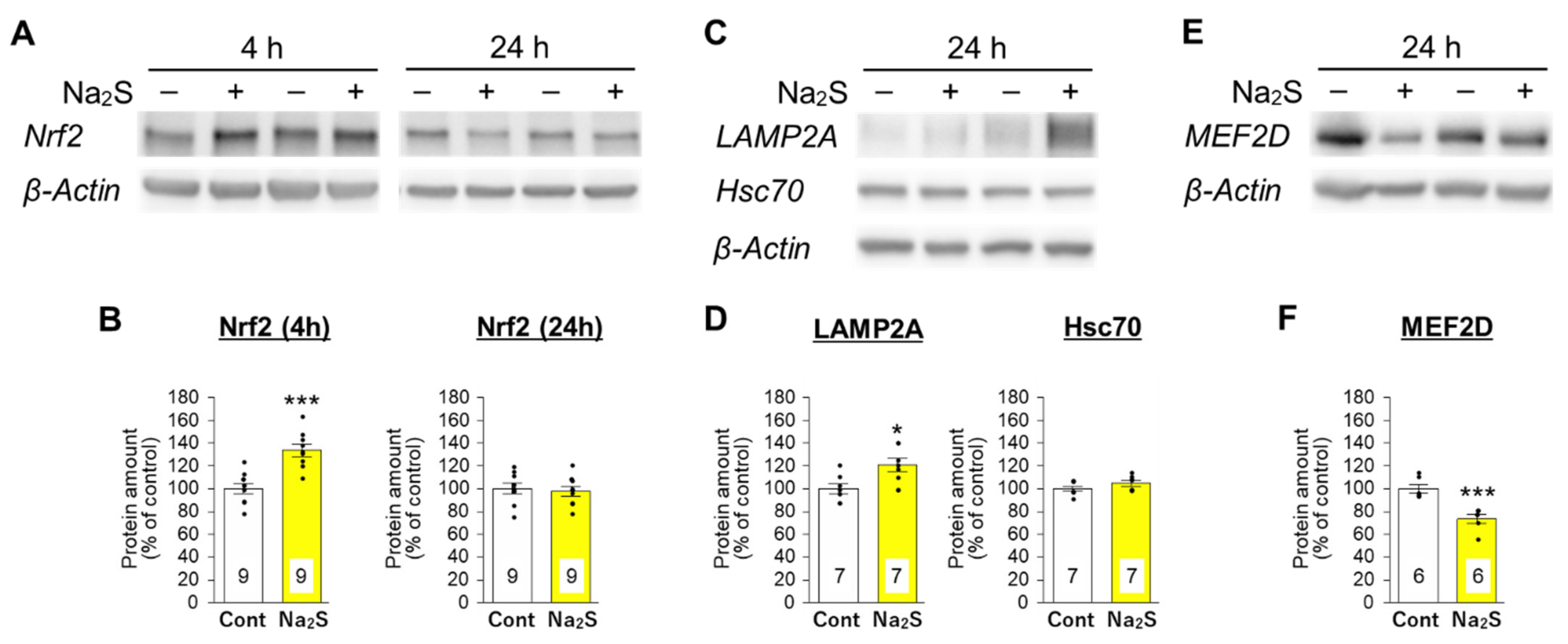
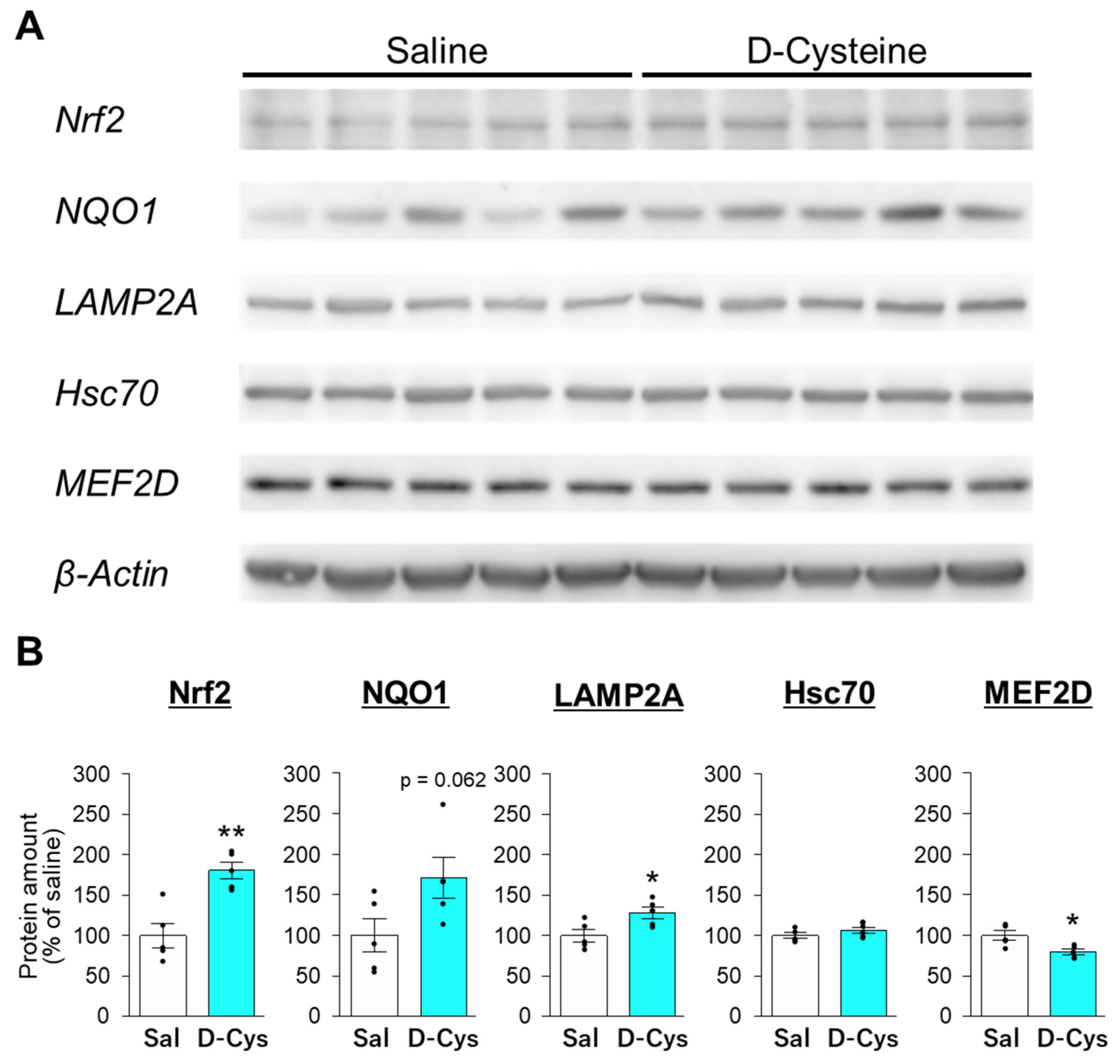
3.3. Effects of D-Cysteine and Na2S on the Expression of Nrf2 and CMA-Related Proteins in AD293 Cells and Mouse Cerebellum
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kimura, H. Hydrogen Sulfide (H2S) and Polysulfide (H2Sn) Signaling: The First 25 Years. Biomolecules 2021, 11, 896. [Google Scholar] [CrossRef] [PubMed]
- Shefa, U.; Kim, M.S.; Jeong, N.Y.; Jung, J. Antioxidant and Cell-Signaling Functions of Hydrogen Sulfide in the Central Nervous System. Oxid. Med. Cell. Longev. 2018, 2018, 1873962. [Google Scholar] [CrossRef] [PubMed]
- Klockgether, T.; Mariotti, C.; Paulson, H.L. Spinocerebellar ataxia. Nat. Rev. Dis. Prim. 2019, 5, 24. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.M.; Lu, C.; Wu, Z.Y. Spinocerebellar ataxia: Relationship between phenotype and genotype—A review. Clin. Genet. 2016, 90, 305–314. [Google Scholar] [CrossRef]
- Seki, T.; Shimahara, T.; Yamamoto, K.; Abe, N.; Amano, T.; Adachi, N.; Takahashi, H.; Kashiwagi, K.; Saito, N.; Sakai, N. Mutant γPKC found in spinocerebellar ataxia type 14 induces aggregate-independent maldevelopment of dendrites in primary cultured Purkinje cells. Neurobiol. Dis. 2009, 33, 260–273. [Google Scholar] [CrossRef]
- Seki, T.; Sato, M.; Kibe, Y.; Ohta, T.; Oshima, M.; Konno, A.; Hirai, H.; Kurauchi, Y.; Hisatsune, A.; Katsuki, H. Lysosomal dysfunction and early glial activation are involved in the pathogenesis of spinocerebellar ataxia type 21 caused by mutant transmembrane protein 240. Neurobiol. Dis. 2018, 120, 34–50. [Google Scholar] [CrossRef]
- Ohta, T.; Morikawa, Y.; Sato, M.; Konno, A.; Hirai, H.; Kurauchi, Y.; Hisatsune, A.; Katsuki, H.; Seki, T. Therapeutic potential of d-cysteine against in vitro and in vivo models of spinocerebellar ataxia. Exp. Neurol. 2021, 343, 113791. [Google Scholar] [CrossRef]
- Heck, D.H.; De Zeeuw, C.I.; Jaeger, D.; Khodakhah, K.; Person, A.L. The Neuronal Code(s) of the Cerebellum. J. Neurosci. 2013, 33, 17603–17609. [Google Scholar] [CrossRef] [Green Version]
- Cvetanovic, M.; Ingram, M.; Orr, H.; Opal, P. Early activation of microglia and astrocytes in mouse models of spinocerebellar ataxia type 1. Neuroscience 2015, 289, 289–299. [Google Scholar] [CrossRef] [Green Version]
- Shibuya, N.; Koike, S.; Tanaka, M.; Ishigami-Yuasa, M.; Kimura, Y.; Ogasawara, Y.; Fukui, K.; Nagahara, N.; Kimura, H. A novel pathway for the production of hydrogen sulfide from D-cysteine in mammalian cells. Nat. Commun. 2013, 4, 1366–1367. [Google Scholar] [CrossRef] [Green Version]
- Koga, R.; Miyoshi, Y.; Sakaue, H.; Hamase, K.; Konno, R. Mouse d-Amino-Acid Oxidase: Distribution and Physiological Substrates. Front. Mol. Biosci. 2017, 4, 82. [Google Scholar] [CrossRef] [Green Version]
- Pollegioni, L.; Sacchi, S.; Murtas, G. Human D-amino acid oxidase: Structure, function, and regulation. Front. Mol. Biosci. 2018, 5, 107. [Google Scholar] [CrossRef] [Green Version]
- Seki, T.; Sato, M.; Konno, A.; Hirai, H.; Kurauchi, Y.; Hisatsune, A.; Katsuki, H. d-Cysteine promotes dendritic development in primary cultured cerebellar Purkinje cells via hydrogen sulfide production. Mol. Cell. Neurosci. 2018, 93, 36–47. [Google Scholar] [CrossRef]
- Ciechanover, A.; Kwon, Y.T. Degradation of misfolded proteins in neurodegenerative diseases: Therapeutic targets and strategies. Exp. Mol. Med. 2015, 47, e147. [Google Scholar] [CrossRef] [Green Version]
- Seki, T.; Yoshino, K.I.; Tanaka, S.; Dohi, E.; Onji, T.; Yamamoto, K.; Hide, I.; Paulson, H.L.; Saito, N.; Sakai, N. Establishment of a novel fluorescence-based method to evaluate chaperone-mediated autophagy in a single neuron. PLoS ONE 2012, 7, e31232. [Google Scholar] [CrossRef] [Green Version]
- Kaushik, S.; Cuervo, A.M. The coming of age of chaperone-mediated autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 365–381. [Google Scholar] [CrossRef]
- Sato, M.; Ohta, T.; Morikawa, Y.; Konno, A.; Hirai, H.; Kurauchi, Y.; Hisatsune, A.; Katsuki, H.; Seki, T. Ataxic phenotype and neurodegeneration are triggered by the impairment of chaperone-mediated autophagy in cerebellar neurons. Neuropathol. Appl. Neurobiol. 2021, 47, 198–209. [Google Scholar] [CrossRef]
- Sahu, R.; Kaushik, S.; Clement, C.C.; Cannizzo, E.S.; Scharf, B.; Follenzi, A.; Potolicchio, I.; Nieves, E.; Cuervo, A.M.; Santambrogio, L. Microautophagy of Cytosolic Proteins by Late Endosomes. Dev. Cell 2011, 20, 131–139. [Google Scholar] [CrossRef] [Green Version]
- Sato, M.; Seki, T.; Konno, A.; Hirai, H.; Kurauchi, Y.; Hisatsune, A.; Katsuki, H. Fluorescent-based evaluation of chaperone-mediated autophagy and microautophagy activities in cultured cells. Genes Cells 2016, 21, 861–873. [Google Scholar] [CrossRef] [Green Version]
- Hanaoka, K.; Sasakura, K.; Suwanai, Y.; Toma-Fukai, S.; Shimamoto, K.; Takano, Y.; Shibuya, N.; Terai, T.; Komatsu, T.; Ueno, T.; et al. Discovery and Mechanistic Characterization of Selective Inhibitors of H2S-producing Enzyme: 3-Mercaptopyruvate Sulfurtransferase (3MST) Targeting Active-site Cysteine Persulfide. Sci. Rep. 2017, 7, 40227. [Google Scholar] [CrossRef]
- Seki, T.; Adachi, N.; Ono, Y.; Mochizuki, H.; Hiramoto, K.; Amano, T.; Matsubayashi, H.; Matsumoto, M.; Kawakami, H.; Saito, N.; et al. Mutant protein kinase Cγ found in spinocerebellar ataxia type 14 is susceptible to aggregation and causes cell death. J. Biol. Chem. 2005, 280, 29096–29106. [Google Scholar] [CrossRef] [Green Version]
- Konno, A.; Hirai, H. Efficient whole brain transduction by systemic infusion of minimally purified AAV-PHP.eB. J. Neurosci. Methods 2020, 346, 108914. [Google Scholar] [CrossRef]
- Pajares, M.; Rojo, A.I.; Arias, E.; Díaz-Carretero, A.; Cuervo, A.M.; Cuadrado, A. Transcription factor NFE2L2/NRF2 modulates chaperone-mediated autophagy through the regulation of LAMP2A. Autophagy 2018, 14, 1310–1322. [Google Scholar] [CrossRef]
- Xie, L.; Gu, Y.; Wen, M.; Zhao, S.; Wang, W.; Ma, Y.; Meng, G.; Han, Y.; Wang, Y.; Liu, G.; et al. Hydrogen Sulfide Induces Keap1 S-sulfhydration and Suppresses Diabetes-Accelerated Atherosclerosis via Nrf2 Activation. Diabetes 2016, 65, 3171–3184. [Google Scholar] [CrossRef] [Green Version]
- Vomund, S.; Schäfer, A.; Parnham, M.J.; Brüne, B.; Von Knethen, A. Nrf2, the Master Regulator of Anti-Oxidative Responses. Int. J. Mol. Sci. 2017, 18, 2772. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Venkannagari, S.; Oh, K.H.; Zhang, Y.Q.; Rohde, J.M.; Liu, L.; Nimmagadda, S.; Sudini, K.; Brimacombe, K.R.; Gajghate, S.; et al. Small molecule inhibitor of NRF2 selectively intervenes therapeutic resistance in KEAP1-deficient NSCLC tumors. ACS Chem. Biol. 2016, 11, 3214–3225. [Google Scholar] [CrossRef] [Green Version]
- Yang, Q.; She, H.; Gearing, M.; Colla, E.; Lee, M.; Shacka, J.J.; Mao, Z. Regulation of neuronal survival factor MEF2D by chaperone-mediated autophagy. Science 2009, 323, 124–127. [Google Scholar] [CrossRef] [Green Version]
- Dikic, I. Proteasomal and Autophagic Degradation Systems. Annu. Reivew Biochem. 2017, 86, 193–224. [Google Scholar] [CrossRef]
- Mizushima, N. Physiological Functions of Autophagy; Springer: Berlin/Heidelberg, Germany, 2009; pp. 71–84. [Google Scholar]
- Mizushima, N.; Levine, B. Autophagy in Human Diseases. N. Engl. J. Med. 2020, 383, 1564–1576. [Google Scholar] [CrossRef]
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 441, 885–889. [Google Scholar] [CrossRef]
- Komatsu, M.; Waguri, S.; Chiba, T.; Murata, S.; Iwata, J.I.; Tanida, I.; Ueno, T.; Koike, M.; Uchiyama, Y.; Kominami, E.; et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006, 441, 880–884. [Google Scholar] [CrossRef] [PubMed]
- Mo, Y.; Sun, Y.Y.; Liu, K.Y. Autophagy and inflammation in ischemic stroke. Neural Regen. Res. 2020, 15, 1388–1396. [Google Scholar] [CrossRef] [PubMed]
- Kimura, Y. Hydrogen sulfide protects neurons from oxidative stress. FASEB J. 2004, 16, 1165–1167. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wu, D.; Wang, H. Hydrogen sulfide plays an important protective role by influencing autophagy in diseases. Physiol. Res. 2019, 68, 335–345. [Google Scholar] [CrossRef]
- Wu, D.; Wang, H.; Teng, T.; Duan, S.; Ji, A.; Li, Y. Hydrogen sulfide and autophagy: A double edged sword. Pharmacol. Res. 2018, 131, 120–127. [Google Scholar] [CrossRef]
- Jiang, W.; Zou, W.; Hu, M.; Tian, Q.; Xiao, F.; Li, M.; Zhang, P.; Chen, Y.J.; Jiang, J.M. Hydrogen sulphide attenuates neuronal apoptosis of substantia nigra by re-establishing autophagic flux via promoting leptin signalling in a 6-hydroxydopamine rat model of Parkinson’s disease. Clin. Exp. Pharmacol. Physiol. 2022, 49, 122–133. [Google Scholar] [CrossRef]
- Zhu, Y.; Shui, M.; Liu, X.; Hu, W.; Wang, Y. Increased autophagic degradation contributes to the neuroprotection of hydrogen sulfide against cerebral ischemia/reperfusion injury. Metab. Brain Dis. 2017, 32, 1449–1458. [Google Scholar] [CrossRef]
- Zhang, M.; Shan, H.; Chang, P.; Wang, T.; Dong, W.; Chen, X.; Tao, L. Hydrogen Sulfide Offers Neuroprotection on Traumatic Brain Injury in Parallel with Reduced Apoptosis and Autophagy in Mice. PLoS ONE 2014, 9, e87241. [Google Scholar] [CrossRef]
- Shui, M.; Liu, X.; Zhu, Y.; Wang, Y. Exogenous hydrogen sulfide attenuates cerebral ischemia–reperfusion injury by inhibiting autophagy in mice. Can. J. Physiol. Pharmacol. 2016, 94, 1187–1192. [Google Scholar] [CrossRef] [Green Version]
- Kirchner, P.; Bourdenx, M.; Madrigal-Matute, J.; Tiano, S.; Diaz, A.; Bartholdy, B.A.; Will, B.; Cuervo, A.M. Proteome-wide analysis of chaperone-mediated autophagy targeting motifs. PLoS Biol. 2019, 17, e3000301. [Google Scholar] [CrossRef]
- Li, W.; Nie, T.; Xu, H.; Yang, J.; Yang, Q.; Mao, Z. Chaperone-mediated autophagy: Advances from bench to bedside. Neurobiol. Dis. 2019, 122, 41–48. [Google Scholar] [CrossRef]
- Sala, G.; Stefanoni, G.; Arosio, A.; Riva, C.; Melchionda, L.; Saracchi, E.; Fermi, S.; Brighina, L.; Ferrarese, C. Reduced expression of the chaperone-mediated autophagy carrier hsc70 protein in lymphomonocytes of patients with Parkinson’s disease. Brain Res. 2014, 1546, 46–52. [Google Scholar] [CrossRef]
- Bourdenx, M.; Martín-Segura, A.; Scrivo, A.; Rodriguez-Navarro, J.A.; Kaushik, S.; Tasset, I.; Diaz, A.; Storm, N.J.; Xin, Q.; Juste, Y.R.; et al. Chaperone-mediated autophagy prevents collapse of the neuronal metastable proteome. Cell 2021, 184, 2696–2714. [Google Scholar] [CrossRef]
- Xilouri, M.; Brekk, O.R.; Polissidis, A.; Chrysanthou-Piterou, M.; Kloukina, I.; Stefanis, L. Impairment of chaperone-mediated autophagy induces dopaminergic neurodegeneration in rats. Autophagy 2016, 12, 2230–2247. [Google Scholar] [CrossRef] [Green Version]
- Xilouri, M.; Brekk, O.R.; Landeck, N.; Pitychoutis, P.M.; Papasilekas, T.; Papadopoulou-Daifoti, Z.; Kirik, D.; Stefanis, L. Boosting chaperone-mediated autophagy in vivo mitigates α-synuclein-induced neurodegeneration. Brain 2013, 136, 2130–2146. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Sun, Y.; Cen, X.; Shan, B.; Zhao, Q.; Xie, T.; Wang, Z.; Hou, T.; Xue, Y.; Zhang, M.; et al. Metformin activates chaperone-mediated autophagy and improves disease pathologies in an Alzheimer disease mouse model. Protein Cell 2021, 12, 769–787. [Google Scholar] [CrossRef]
- Hori, Y.; Tsutsumi, R.; Nasu, K.; Boateng, A.; Ashikari, Y.; Sugiura, M.; Nakajima, M.; Kurauchi, Y.; Hisatsune, A.; Katsuki, H.; et al. Aromatic-Turmerone Analogs Protect Dopaminergic Neurons in Midbrain Slice Cultures through Their Neuroprotective Activities. Cells 2021, 10, 1090. [Google Scholar] [CrossRef]
- Lee, L.C.; Weng, Y.T.; Wu, Y.R.; Soong, B.W.; Tseng, Y.C.; Chen, C.M.; Lee-Chen, G.J. Downregulation of proteins involved in the endoplasmic reticulum stress response and Nrf2-ARE signaling in lymphoblastoid cells of spinocerebellar ataxia type 17. J. Neural Transm. 2014, 121, 601–610. [Google Scholar] [CrossRef]
- Anzovino, A.; Chiang, S.; Brown, B.E.; Hawkins, C.L.; Richardson, D.R.; Huang, M.L.H. Molecular Alterations in a Mouse Cardiac Model of Friedreich Ataxia: An Impaired Nrf2 Response Mediated via Upregulation of Keap1 and Activation of the Gsk3β Axis. Am. J. Pathol. 2017, 187, 2858–2875. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.F.; Wu, Z.H.; Gao, M.; Loor, J.J. Nuclear factor erythroid 2-related factor 2-antioxidant activation through the action of ataxia telangiectasia-mutated serine/threonine kinase is essential to counteract oxidative stress in bovine mammary epithelial cells. J. Dairy Sci. 2018, 101, 5317–5328. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.L.; Chang, J.C.; Lin, W.Y.; Li, C.C.; Hsieh, M.; Chen, H.W.; Wang, T.S.; Wu, W.T.; Liu, C.S.; Liu, K.L. Caffeic acid and resveratrol ameliorate cellular damage in cell and Drosophila models of spinocerebellar ataxia type 3 through upregulation of Nrf2 pathway. Free Radic. Biol. Med. 2018, 115, 309–317. [Google Scholar] [CrossRef]
- Genchi, G. An overview on d-amino acids. Amin. Acids 2017, 49, 1521–1533. [Google Scholar] [CrossRef]
- Wolosker, H. The Neurobiology of d-Serine Signaling. Adv. Pharmacol. 2018, 82, 325–348. [Google Scholar] [CrossRef]
- Hashimoto, K.; Fukushima, T.; Shimizu, E.; Komatsu, N.; Watanabe, H.; Shinoda, N.; Nakazato, M.; Kumakiri, C.; Okada, S.I.; Hasegawa, H.; et al. Decreased serum levels of D-serine in patients with schizophrenia: Evidence in support of the N-methyl-D-aspartate receptor hypofunction hypothesis of schizophrenia. Arch. Gen. Psychiatry 2003, 60, 572–576. [Google Scholar] [CrossRef] [Green Version]
- Kakegawa, W.; Miyoshi, Y.; Hamase, K.; Matsuda, S.; Matsuda, K.; Kohda, K.; Emi, K.; Motohashi, J.; Konno, R.; Zaitsu, K.; et al. D-Serine regulates cerebellar LTD and motor coordination through the δ2 glutamate receptor. Nat. Neurosci. 2011, 14, 603–611. [Google Scholar] [CrossRef]
- Semenza, E.R.; Harraz, M.M.; Abramson, E.; Malla, A.P.; Vasavda, C.; Gadalla, M.M.; Kornberg, M.D.; Snyder, S.H.; Roychaudhuri, R. D-cysteine is an endogenous regulator of neural progenitor cell dynamics in the mammalian brain. Proc. Natl. Acad. Sci. USA 2021, 118, e2110610118. [Google Scholar] [CrossRef]
- Chen, Z.; Zhang, W.; Selmi, C.; Ridgway, W.M.; Leung, P.S.C.; Zhang, F.; Gershwin, M.E. The myristoylated alanine-rich C-kinase substrates (MARCKS): A membrane-anchored mediator of the cell function. Autoimmun. Rev. 2021, 20, 102942. [Google Scholar] [CrossRef]
- McNamara, R.K.; Lenox, R.H. Comparative distribution of myristoylated alanine-rich C kinase substrate (MARCKS) and F1/GAP-43 gene expression in the adult rat brain. J. Comp. Neurol. 1998, 379, 48–71. [Google Scholar] [CrossRef]
- Cuadrado, A.; Manda, G.; Hassan, A.; Alcaraz, M.J.; Barbas, C.; Daiber, A.; Ghezzi, P.; León, R.; López, M.G.; Oliva, B.; et al. Transcription Factor NRF2 as a Therapeutic Target for Chronic Diseases: A Systems Medicine Approach. Pharmacol. Rev. 2018, 70, 348–383. [Google Scholar] [CrossRef] [Green Version]
- Ma, S.Y.; Sun, K.S.; Zhang, M.; Zhou, X.; Zheng, X.H.; Tian, S.Y.; Liu, Y.S.; Chen, L.; Gao, X.; Ye, J.; et al. Disruption of Plin5 degradation by CMA causes lipid homeostasis imbalance in NAFLD. Liver Int. 2020, 40, 2427–2438. [Google Scholar] [CrossRef]
- Yuan, Z.; Wang, S.; Tan, X.; Wang, D. New Insights into the Mechanisms of Chaperon-Mediated Autophagy and Implications for Kidney Diseases. Cells 2022, 11, 406. [Google Scholar] [CrossRef]
- Subedi, L.; Lee, J.H.; Yumnam, S.; Ji, E.; Kim, S.Y. Anti-Inflammatory Effect of Sulforaphane on LPS-Activated Microglia Potentially through JNK/AP-1/NF-κB Inhibition and Nrf2/HO-1 Activation. Cells 2019, 8, 194. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Kelly, M.G.; Yang, X.R.; Fernandez, T.G.; Wierzbicki, E.L.; Skrobach, A.; Doré, S. Nrf2 Deficiency Exacerbates Cognitive Impairment and Reactive Microgliosis in a Lipopolysaccharide-Induced Neuroinflammatory Mouse Model. Cell. Mol. Neurobiol. 2020, 40, 1185–1197. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ueda, E.; Ohta, T.; Konno, A.; Hirai, H.; Kurauchi, Y.; Katsuki, H.; Seki, T. D-Cysteine Activates Chaperone-Mediated Autophagy in Cerebellar Purkinje Cells via the Generation of Hydrogen Sulfide and Nrf2 Activation. Cells 2022, 11, 1230. https://doi.org/10.3390/cells11071230
Ueda E, Ohta T, Konno A, Hirai H, Kurauchi Y, Katsuki H, Seki T. D-Cysteine Activates Chaperone-Mediated Autophagy in Cerebellar Purkinje Cells via the Generation of Hydrogen Sulfide and Nrf2 Activation. Cells. 2022; 11(7):1230. https://doi.org/10.3390/cells11071230
Chicago/Turabian StyleUeda, Erika, Tomoko Ohta, Ayumu Konno, Hirokazu Hirai, Yuki Kurauchi, Hiroshi Katsuki, and Takahiro Seki. 2022. "D-Cysteine Activates Chaperone-Mediated Autophagy in Cerebellar Purkinje Cells via the Generation of Hydrogen Sulfide and Nrf2 Activation" Cells 11, no. 7: 1230. https://doi.org/10.3390/cells11071230