
Therapeutic Efficacy of Pharmacological Ascorbate on Braf Inhibitor Resistant Melanoma Cells In Vitro and In Vivo

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Isolation and Culture of Human Cells
2.2. Signaling Pathway Inhibitors and Treatments
2.3. Viability Assay
2.4. Cell Cycle Analysis
2.5. ROS Reporter Assay
2.6. Western Blot
2.7. Colony Formation Assay
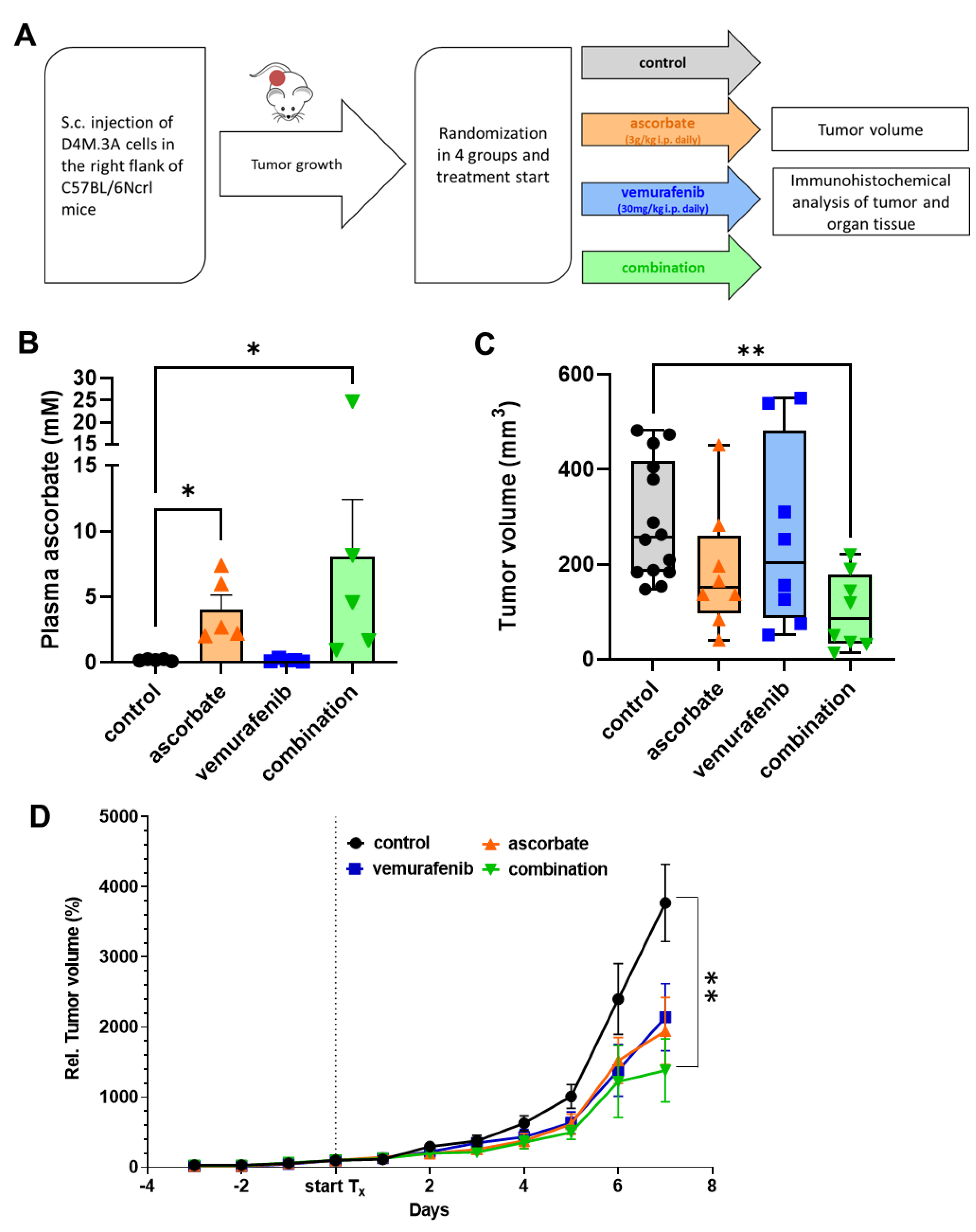
2.8. In Vivo Mouse Experiment
2.9. Plasma Ascorbate Assay
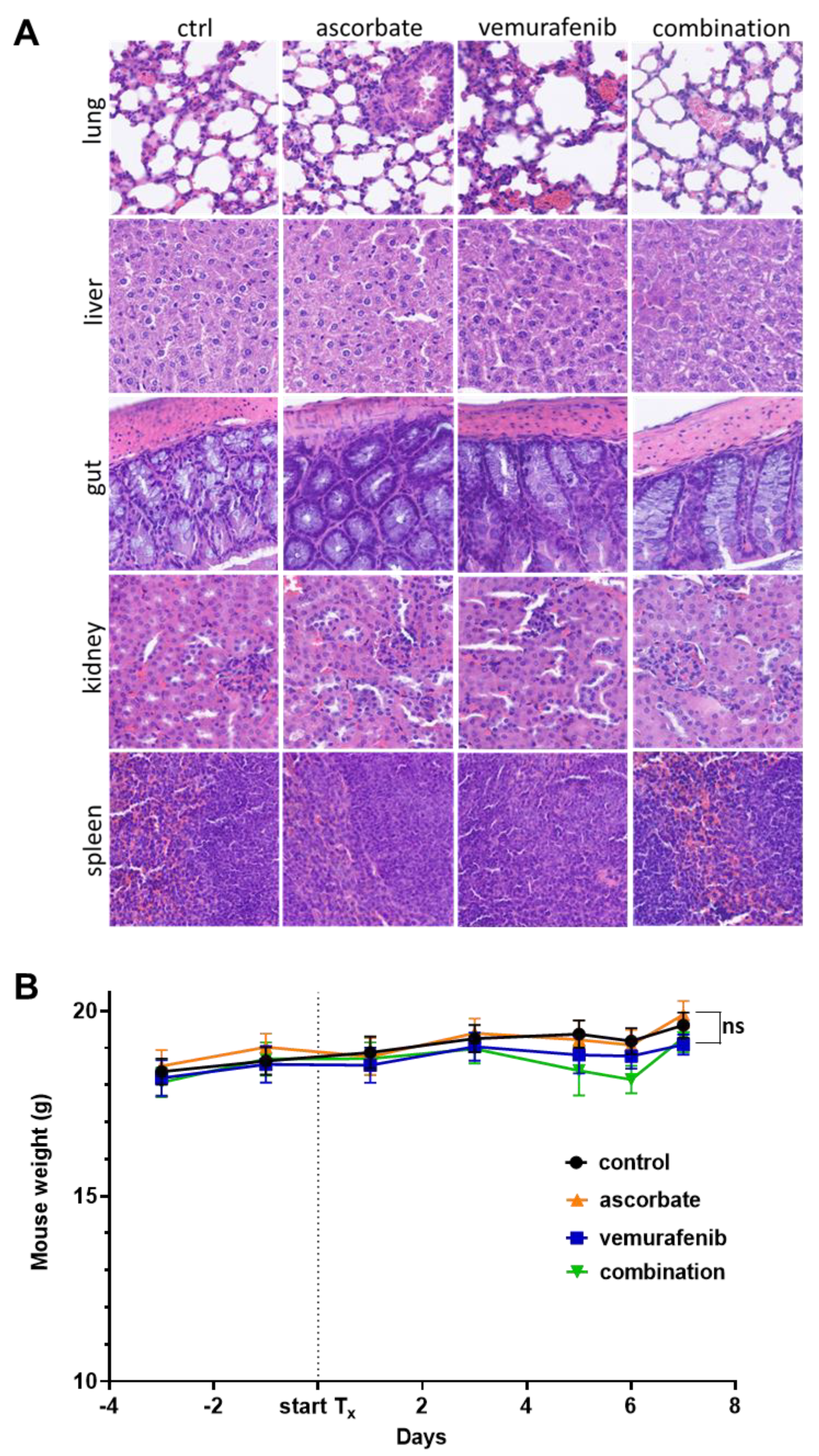
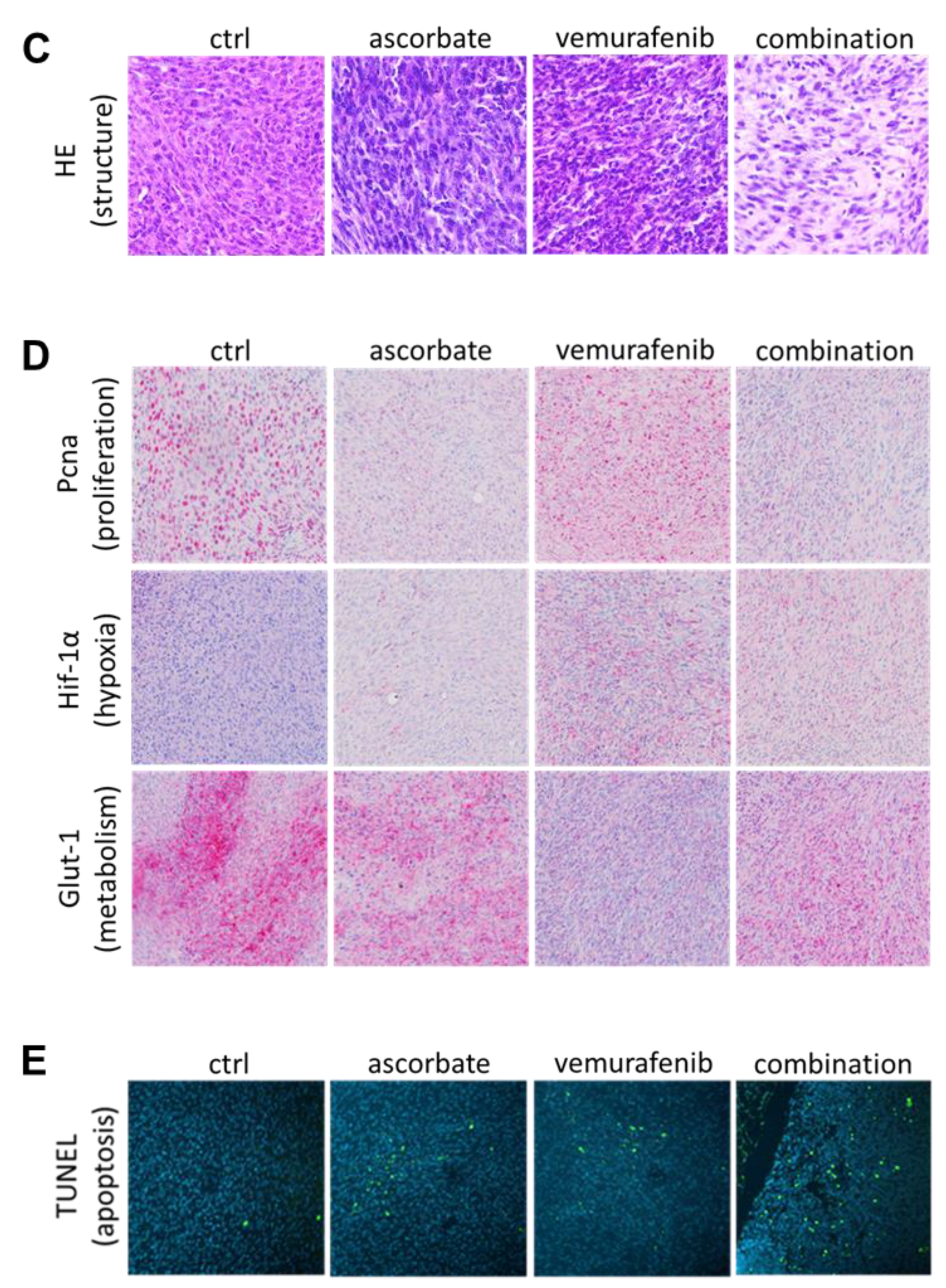
2.10. Immunohistochemistry of Mouse Tumors and Organs
2.11. TUNEL Staining
2.12. Statistics
3. Results
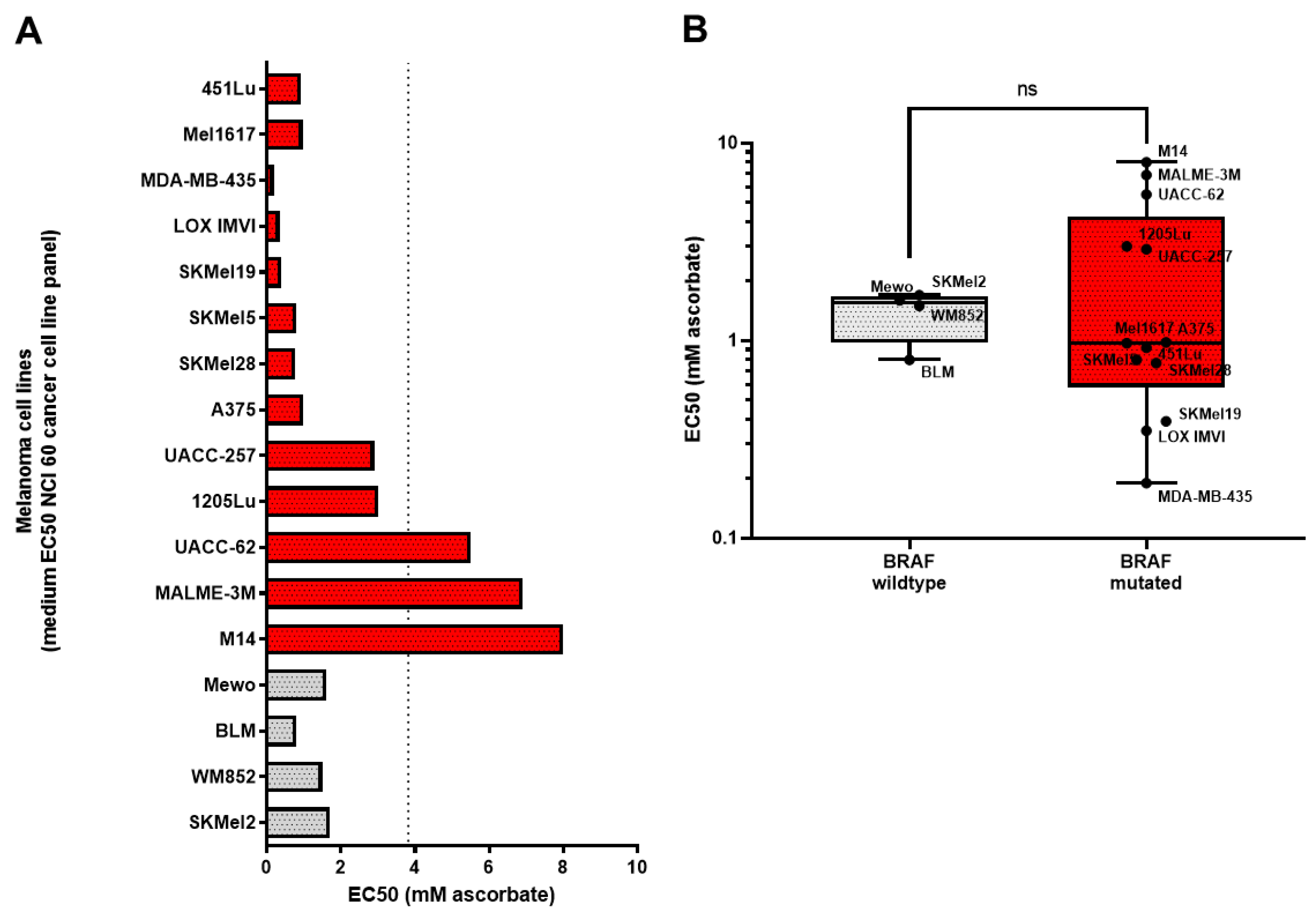
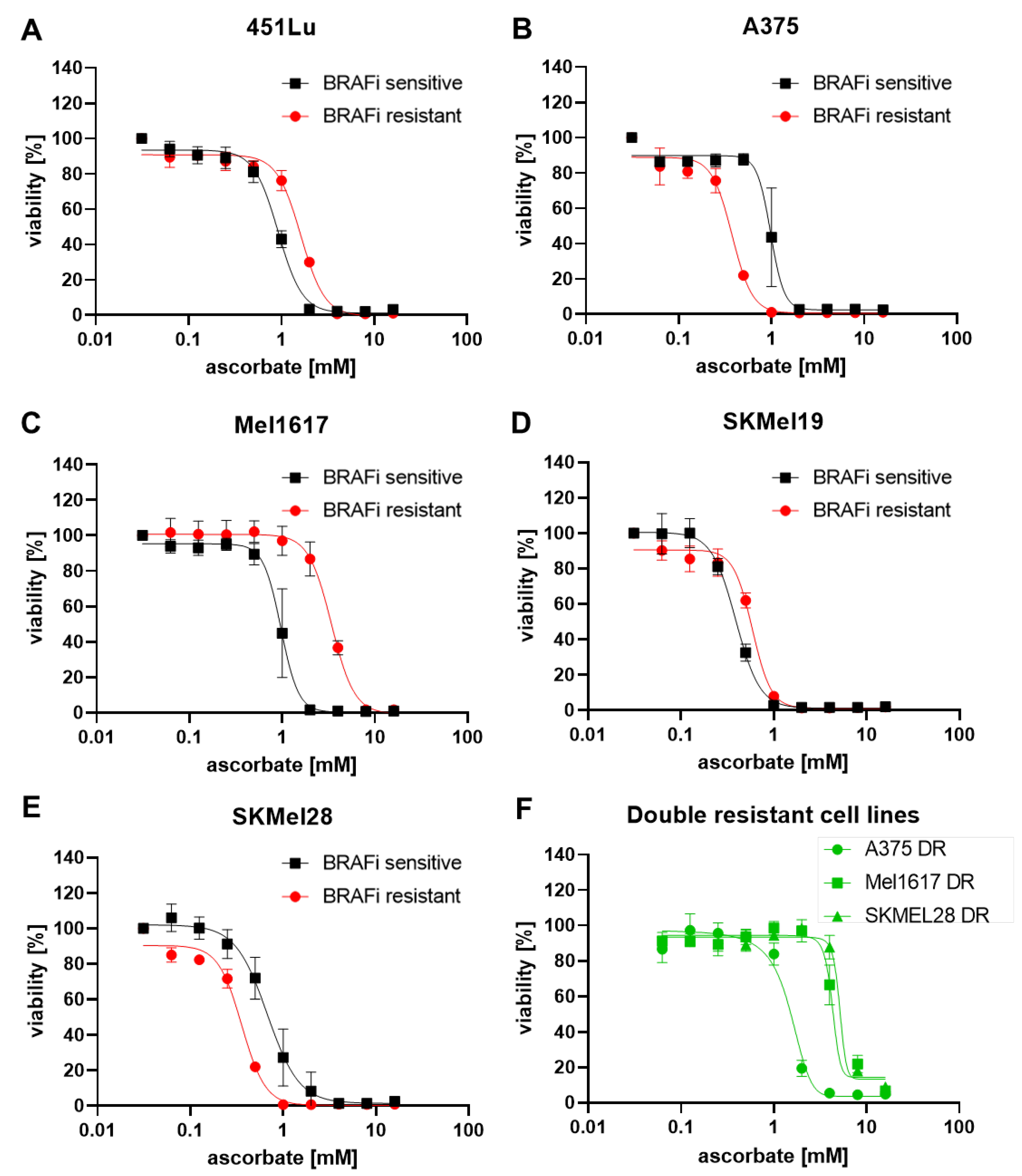
3.1. Growth Inhibitory Effects of Ascorbate on Melanoma Cells
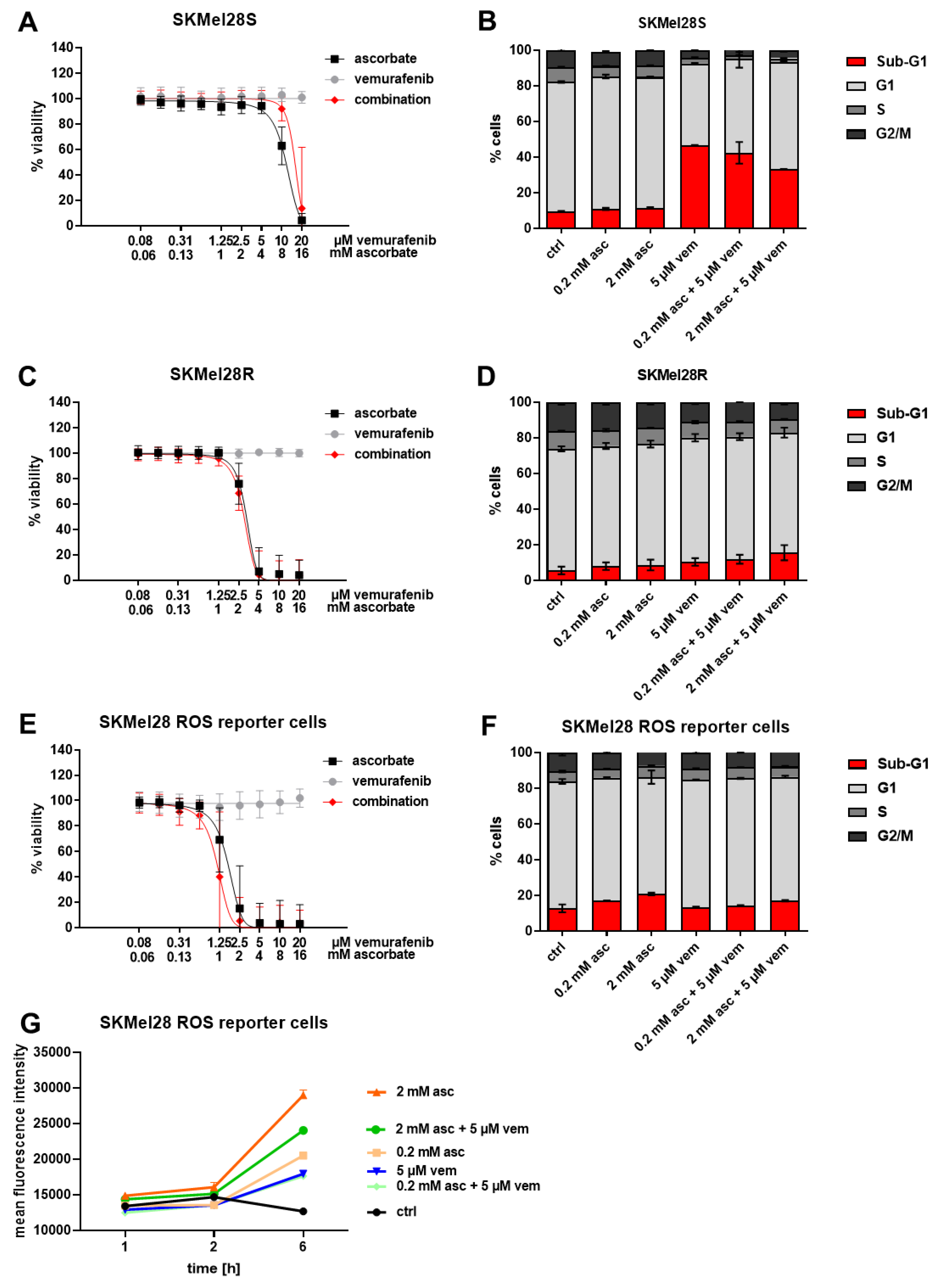
3.2. Effects of Ascorbate and Vemurafenib on Viability, Cell Cycle Distribution, and ROS Production of Melanoma Cells
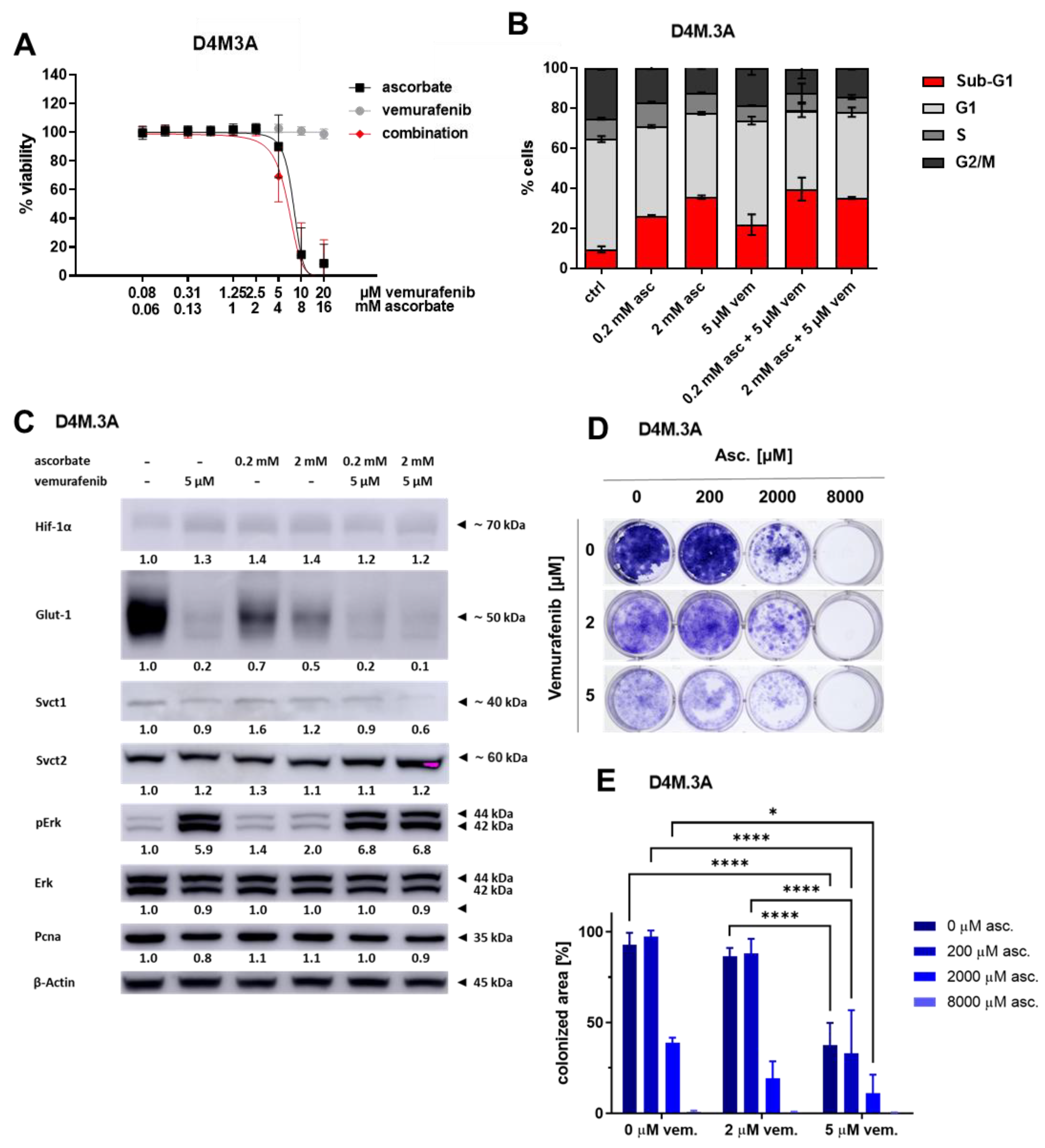
3.3. Effects of Ascorbate and Vemurafenib on Viability, Cell Cycle, and Protein Expression of the Mouse Melanoma Cell Line D4M.3A
3.4. Effects of Ascorbate and Vemurafenib In Vivo
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Paluncic, J.; Kovacevic, Z.; Jansson, P.J.; Kalinowski, D.; Merlot, A.M.; Huang, M.L.-H.; Lok, H.C.; Sahni, S.; Lane, D.J.R.; Des Richardson, R. Roads to melanoma: Key pathways and emerging players in melanoma progression and oncogenic signaling. Biochim. Biophys. Acta 2016, 1863, 770–784. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Apalla, Z.; Lallas, A.; Sotiriou, E.; Lazaridou, E.; Ioannides, D. Epidemiological trends in skin cancer. Dermatol. Pract. Concept. 2017, 7, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Luther, C.; Swami, U.; Zhang, J.; Milhem, M.; Zakharia, Y. Advanced stage melanoma therapies: Detailing the present and exploring the future. Crit. Rev. Oncol. Hematol. 2019, 133, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, K.; Hignett, E.; Khachemoune, A. Current and emerging treatment options for metastatic melanoma: A focused review. Dermatol. Online J. 2020, 26, 1. [Google Scholar] [CrossRef]
- Peyssonnaux, C.; Eychène, A. The Raf/MEK/ERK pathway: New concepts of activation. Biol. Cell 2001, 93, 53–62. [Google Scholar] [CrossRef]
- Avruch, J.; Khokhlatchev, A.; Kyriakis, J.M.; Luo, Z.; Tzivion, G.; Vavvas, D.; Zhang, X.F. Ras activation of the Raf kinase: Tyrosine kinase recruitment of the MAP kinase cascade. Recent Prog. Horm. Res. 2001, 56, 127–155. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, K.T.; Infante, J.R.; Daud, A.; Gonzalez, R.; Kefford, R.F.; Sosman, J.; Hamid, O.; Schuchter, L.; Cebon, J.; Ibrahim, N.; et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N. Engl. J. Med. 2012, 367, 1694–1703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Wan, P.T.; Garnett, M.J.; Roe, S.; Lee, S.; Niculescu-Duvaz, D.; Good, V.M.; Project, C.G.; Jones, C.; Marshall, C.J.; Springer, C.J.; et al. Mechanism of Activation of the RAF-ERK Signaling Pathway by Oncogenic Mutations of B-RAF. Cell 2004, 116, 855–867. [Google Scholar] [CrossRef] [Green Version]
- Niessner, H.; Sinnberg, T.; Kosnopfel, C.; Smalley, K.S.M.; Beck, D.; Praetorius, C.; Mai, M.; Beissert, S.; Kulms, D.; Schaller, M.; et al. BRAF Inhibitors Amplify the Proapoptotic Activity of MEK Inhibitors by Inducing ER Stress in NRAS-Mutant Melanoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 6203–6214. [Google Scholar] [CrossRef] [Green Version]
- Kim, G.; McKee, A.E.; Ning, Y.-M.; Hazarika, M.; Theoret, M.; Johnson, J.R.; Xu, Q.C.; Tang, S.; Sridhara, R.; Jiang, X.; et al. FDA approval summary: Vemurafenib for treatment of unresectable or metastatic melanoma with the BRAFV600E mutation. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 4994–5000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sosman, J.A.; Kim, K.B.; Schuchter, L.; Gonzalez, R.; Pavlick, A.C.; Weber, J.S.; McArthur, G.A.; Hutson, T.E.; Moschos, S.J.; Flaherty, K.T.; et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N. Engl. J. Med. 2012, 366, 707–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maio, M.; Lewis, K.; Demidov, L.; Mandalà, M.; Bondarenko, I.; Ascierto, P.A.; Herbert, C.; Mackiewicz, A.; Rutkowski, P.; Guminski, A.; et al. Adjuvant vemurafenib in resected, BRAF V600 mutation-positive melanoma (BRIM8): A randomised, double-blind, placebo-controlled, multicentre, phase 3 trial. Lancet Oncol. 2018, 19, 510–520. [Google Scholar] [CrossRef]
- Villani, A.; Scalvenzi, M.; Fabbrocini, G.; Ocampo-Candiani, J.; Ocampo-Garza, S.S. Looking into a Better Future: Novel Therapies for Metastatic Melanoma. Dermatol. Ther. 2021, 11, 751–767. [Google Scholar] [CrossRef]
- Cameron, E.; Pauling, L. Supplemental ascorbate in the supportive treatment of cancer: Prolongation of survival times in terminal human cancer. Proc. Natl. Acad. Sci. USA 1976, 73, 3685–3689. [Google Scholar] [CrossRef] [Green Version]
- Dattola, A.; Silvestri, M.; Bennardo, L.; Passante, M.; Scali, E.; Patruno, C.; Nisticò, S.P. Role of Vitamins in Skin Health: A Systematic Review. Curr. Nutr. Rep. 2020, 9, 226–235. [Google Scholar] [CrossRef]
- Chen, Q.; Espey, M.G.; Sun, A.Y.; Pooput, C.; Kirk, K.L.; Krishna, M.C.; Khosh, D.B.; Drisko, J.; Levine, M. Pharmacologic doses of ascorbate act as a prooxidant and decrease growth of aggressive tumor xenografts in mice. Proc. Natl. Acad. Sci. USA 2008, 105, 11105–11109. [Google Scholar] [CrossRef] [Green Version]
- Verrax, J.; Calderon, P.B. Pharmacologic concentrations of ascorbate are achieved by parenteral administration and exhibit antitumoral effects. Free Radic. Biol. Med. 2009, 47, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Espey, M.G.; Krishna, M.C.; Mitchell, J.B.; Corpe, C.P.; Buettner, G.R.; Shacter, E.; Levine, M. Pharmacologic ascorbic acid concentrations selectively kill cancer cells: Action as a pro-drug to deliver hydrogen peroxide to tissues. Proc. Natl. Acad. Sci. USA 2005, 102, 13604–13609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoenfeld, J.D.; Sibenaller, Z.A.; Mapuskar, K.A.; Wagner, B.A.; Cramer-Morales, K.L.; Furqan, M.; Sandhu, S.; Carlisle, T.L.; Smith, M.C.; Abu Hejleh, T.; et al. O2-and H2O2-Mediated Disruption of Fe Metabolism Causes the Differential Susceptibility of NSCLC and GBM Cancer Cells to Pharmacological Ascorbate. Cancer Cell 2017, 31, 487–500.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nauman, G.; Gray, J.C.; Parkinson, R.; Levine, M.; Paller, C.J. Systematic Review of Intravenous Ascorbate in Cancer Clinical Trials. Antioxidants 2018, 7, 89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.; Espey, M.G.; Sun, A.Y.; Lee, J.-H.; Krishna, M.C.; Shacter, E.; Choyke, P.L.; Pooput, C.; Kirk, K.L.; Buettner, G.R.; et al. Ascorbate in pharmacologic concentrations selectively generates ascorbate radical and hydrogen peroxide in extracellular fluid in vivo. Proc. Natl. Acad. Sci. USA 2007, 104, 8749–8754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.; Chapman, J.; Levine, M.; Polireddy, K.; Drisko, J.; Chen, Q. High-dose parenteral ascorbate enhanced chemosensitivity of ovarian cancer and reduced toxicity of chemotherapy. Sci. Transl. Med. 2014, 6, 222ra18. [Google Scholar] [CrossRef] [PubMed]
- Bram, S.; Froussard, P.; Guichard, M.; Jasmin, C.; Augery, Y.; Sinoussi-Barre, F.; Wray, W. Vitamin C preferential toxicity for malignant melanoma cells. Nature 1980, 284, 629–631. [Google Scholar] [CrossRef] [PubMed]
- Gardiner, N.S.; Duncan, J.R. Enhanced prostaglandin synthesis as a mechanism for inhibition of melanoma cell growth by ascorbic acid. Prostaglandins Leukot. Essent. Fat. Acids 1988, 34, 119–126. [Google Scholar] [CrossRef]
- Kang, J.S.; Cho, D.; Kim, Y.-I.; Hahm, E.; Yang, Y.; Kim, D.; Hur, D.; Park, H.; Bang, S.; Hwang, Y.I.; et al. L-ascorbic acid (vitamin C) induces the apoptosis of B16 murine melanoma cells via a caspase-8-independent pathway. Cancer Immunol. Immunother. 2003, 52, 693–698. [Google Scholar] [CrossRef]
- Jenkins, M.H.; Steinberg, S.M.; Alexander, M.P.; Fisher, J.L.; Ernstoff, M.S.; Turk, M.J.; Mullins, D.W.; Brinckerhoff, C.E. Multiple murine BRaf(V600E) melanoma cell lines with sensitivity to PLX4032. Pigment Cell Melanoma Res. 2014, 27, 495–501. [Google Scholar] [CrossRef] [Green Version]
- Sinnberg, T.; Makino, E.; Krueger, M.A.; Velic, A.; Macek, B.; Rothbauer, U.; Groll, N.; Pötz, O.; Czemmel, S.; Niessner, H.; et al. A Nexus Consisting of Beta-Catenin and Stat3 Attenuates BRAF Inhibitor Efficacy and Mediates Acquired Resistance to Vemurafenib. EBioMedicine 2016, 8, 132–149. [Google Scholar] [CrossRef] [Green Version]
- Gutscher, M.; Sobotta, M.C.; Wabnitz, G.H.; Ballikaya, S.; Meyer, A.J.; Samstag, Y.; Dick, T.P. Proximity-based protein thiol oxidation by H2O2-scavenging peroxidases. J. Biol. Chem. 2009, 284, 31532–31540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.; Sullivan, G.G.; Schrick, E.; Choi, I.-Y.; He, Z.; Lierman, J.; Lee, P.; Drisko, J.A.; Chen, Q. A convenient method for measuring blood ascorbate concentrations in patients receiving high-dose intravenous ascorbate. J. Am. Coll. Nutr. 2013, 32, 187–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, G.; Yan, Y.; Ma, Y.; Yang, Y. Vitamin C at high concentrations induces cytotoxicity in malignant melanoma but promotes tumor growth at low concentrations. Mol. Carcinog. 2017, 56, 1965–1976. [Google Scholar] [CrossRef]
- Padayatty, S.J.; Sun, H.; Wang, Y.; Riordan, H.D.; Hewitt, S.M.; Katz, A.; Wesley, R.A.; Levine, M. Vitamin C pharmacokinetics: Implications for oral and intravenous use. Ann. Intern. Med. 2004, 140, 533–537. [Google Scholar] [CrossRef] [PubMed]
- Sinnberg, T.; Noor, S.; Venturelli, S.; Berger, A.; Schuler, P.; Garbe, C.; Busch, C. The ROS-induced cytotoxicity of ascorbate is attenuated by hypoxia and HIF-1alpha in the NCI60 cancer cell lines. J. Cell. Mol. Med. 2014, 18, 530–541. [Google Scholar] [CrossRef] [PubMed]
- Serrano, O.K.; Parrow, N.L.; Violet, P.-C.; Yang, J.; Zornjak, J.; Basseville, A.; Levine, M. Antitumor effect of pharmacologic ascorbate in the B16 murine melanoma model. Free Radic. Biol. Med. 2015, 87, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Lv, H.; Wang, C.; Fang, T.; Li, T.; Lv, G.; Han, Q.; Yang, W.; Wang, H. Vitamin C preferentially kills cancer stem cells in hepatocellular carcinoma via SVCT-2. NPJ Precis. Oncol. 2018, 2, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santini, R.; Pietrobono, S.; Pandolfi, S.; Montagnani, V.; D’Amico, M.; Penachioni, J.Y.; Vinci, M.C.; Borgognoni, L.; Stecca, B. SOX2 regulates self-renewal and tumorigenicity of human melanoma-initiating cells. Oncogene 2014, 33, 4697–4708. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.M.; Liu, S.; Lu, H.; Zhang, H.; Zhang, P.J.; Gimotty, P.A.; Guerra, M.; Guo, W.; Xu, X. Acquired cancer stem cell phenotypes through Oct4-mediated dedifferentiation. Oncogene 2012, 31, 4898–4911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jamal, S.M.E.; Alamodi, A.; Wahl, R.U.; Grada, Z.; Shareef, M.A.; Hassan, S.-Y.; Murad, F.; Hassan, S.-L.; Santourlidis, S.; Gomez, C.R.; et al. Melanoma stem cell maintenance and chemo-resistance are mediated by CD133 signal to PI3K-dependent pathways. Oncogene 2020, 39, 5468–5478. [Google Scholar] [CrossRef] [PubMed]
- Marzagalli, M.; Moretti, R.M.; Messi, E.; Marelli, M.M.; Fontana, F.; Anastasia, A.; Bani, M.R.; Beretta, G.; Limonta, P. Targeting melanoma stem cells with the Vitamin E derivative δ-tocotrienol. Sci. Rep. 2018, 8, 587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Bao, S.; Wu, Q.; Wang, H.; Eyler, C.; Sathornsumetee, S.; Shi, Q.; Cao, Y.; Lathia, J.; McLendon, R.E.; et al. Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell 2009, 15, 501–513. [Google Scholar] [CrossRef] [Green Version]
- Hasmim, M.; Noman, M.Z.; Messai, Y.; Bordereaux, D.; Gros, G.; Baud, V.; Chouaib, S. Cutting edge: Hypoxia-induced Nanog favors the intratumoral infiltration of regulatory T cells and macrophages via direct regulation of TGF-β1. J. Immunol. 2013, 191, 5802–5806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yue, X.; Rao, A. TET family dioxygenases and the TET activator vitamin C in immune responses and cancer. Blood 2020, 136, 1394–1401. [Google Scholar] [CrossRef] [PubMed]
- Lian, C.G.; Xu, Y.; Ceol, C.; Wu, F.; Larson, A.; Dresser, K.; Xu, W.; Tan, L.; Hu, Y.; Zhan, Q.; et al. Loss of 5-hydroxymethylcytosine is an epigenetic hallmark of melanoma. Cell 2012, 150, 1135–1146. [Google Scholar] [CrossRef] [Green Version]
- Gustafson, C.B.; Yang, C.; Dickson, K.M.; Shao, H.; van Booven, D.; Harbour, J.W.; Liu, Z.-J.; Wang, G. Epigenetic reprogramming of melanoma cells by vitamin C treatment. Clin. Epigenet. 2015, 7, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johannessen, C.M.; Boehm, J.S.; Kim, S.Y.; Thomas, S.R.; Wardwell, L.; Johnson, L.A.; Emery, C.M.; Stransky, N.; Cogdill, A.P.; Barretina, J.; et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature 2010, 468, 968–972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nazarian, R.; Shi, H.; Wang, Q.; Kong, X.; Koya, R.C.; Lee, H.; Chen, Z.; Lee, M.-K.; Attar, N.; Sazegar, H.; et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 2010, 468, 973–977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amaral, T.; Sinnberg, T.; Meier, F.; Krepler, C.; Levesque, M.; Niessner, H.; Garbe, C. MAPK pathway in melanoma part II-secondary and adaptive resistance mechanisms to BRAF inhibition. Eur. J. Cancer 2017, 73, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Amaral, T.; Sinnberg, T.; Meier, F.; Krepler, C.; Levesque, M.; Niessner, H.; Garbe, C. The mitogen-activated protein kinase pathway in melanoma part I—Activation and primary resistance mechanisms to BRAF inhibition. Eur. J. Cancer 2017, 73, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Shen, Z.; Yang, Q.; Sui, F.; Pu, J.; Ma, J.; Ma, S.; Yao, D.; Ji, M.; Hou, P. Vitamin C kills thyroid cancer cells through ROS-dependent inhibition of MAPK/ERK and PI3K/AKT pathways via distinct mechanisms. Theranostics 2019, 9, 4461–4473. [Google Scholar] [CrossRef] [PubMed]
- Yun, J.; Mullarky, E.; Lu, C.; Bosch, K.N.; Kavalier, A.; Rivera, K.; Roper, J.; Chio, I.I.C.; Giannopoulou, E.G.; Rago, C.; et al. Vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells by targeting GAPDH. Science 2015, 350, 1391–1396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parmenter, T.J.; Kleinschmidt, M.; Kinross, K.M.; Bond, S.T.; Li, J.; Kaadige, M.R.; Rao, A.; Sheppard, K.E.; Hugo, W.; Pupo, G.M.; et al. Response of BRAF-mutant melanoma to BRAF inhibition is mediated by a network of transcriptional regulators of glycolysis. Cancer Discov. 2014, 4, 423–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trotta, A.P.; Gelles, J.D.; Serasinghe, M.N.; Loi, P.; Arbiser, J.L.; Chipuk, J.E. Disruption of mitochondrial electron transport chain function potentiates the pro-apoptotic effects of MAPK inhibition. J. Biol. Chem. 2017, 292, 11727–11739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haq, R.; Shoag, J.; Andreu-Perez, P.; Yokoyama, S.; Edelman, H.; Rowe, G.C.; Frederick, D.T.; Hurley, A.D.; Nellore, A.; Kung, A.L.; et al. Oncogenic BRAF regulates oxidative metabolism via PGC1α and MITF. Cancer Cell 2013, 23, 302–315. [Google Scholar] [CrossRef] [Green Version]
- Corazao-Rozas, P.; Guerreschi, P.; Jendoubi, M.; André, F.; Jonneaux, A.; Scalbert, C.; Garçon, G.; Malet-Martino, M.; Balayssac, S.; Rocchi, S.; et al. Mitochondrial oxidative stress is the Achille’s heel of melanoma cells resistant to Braf-mutant inhibitor. Oncotarget 2013, 4, 1986–1998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Leite de Oliveira, R.; Huijberts, S.; Bosdriesz, E.; Pencheva, N.; Brunen, D.; Bosma, A.; Song, J.-Y.; Zevenhoven, J.; Los-de Vries, G.T.; et al. An Acquired Vulnerability of Drug-Resistant Melanoma with Therapeutic Potential. Cell 2018, 173, 1413–1425.e14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cesi, G.; Walbrecq, G.; Zimmer, A.; Kreis, S.; Haan, C. ROS production induced by BRAF inhibitor treatment rewires metabolic processes affecting cell growth of melanoma cells. Mol. Cancer 2017, 16, 102. [Google Scholar] [CrossRef] [Green Version]
- Bauer, D.; Werth, F.; Nguyen, H.a.; Kiecker, F.; Eberle, J. Critical role of reactive oxygen species (ROS) for synergistic enhancement of apoptosis by vemurafenib and the potassium channel inhibitor TRAM-34 in melanoma cells. Cell Death Dis. 2017, 8, e2594. [Google Scholar] [CrossRef]
- Bael, T.E.; Peterson, B.L.; Gollob, J.A. Phase II trial of arsenic trioxide and ascorbic acid with temozolomide in patients with metastatic melanoma with or without central nervous system metastases. Melanoma Res. 2008, 18, 147–151. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niessner, H.; Burkard, M.; Leischner, C.; Renner, O.; Plöger, S.; Meraz-Torres, F.; Böcker, M.; Hirn, C.; Lauer, U.M.; Venturelli, S.; et al. Therapeutic Efficacy of Pharmacological Ascorbate on Braf Inhibitor Resistant Melanoma Cells In Vitro and In Vivo. Cells 2022, 11, 1229. https://doi.org/10.3390/cells11071229
Niessner H, Burkard M, Leischner C, Renner O, Plöger S, Meraz-Torres F, Böcker M, Hirn C, Lauer UM, Venturelli S, et al. Therapeutic Efficacy of Pharmacological Ascorbate on Braf Inhibitor Resistant Melanoma Cells In Vitro and In Vivo. Cells. 2022; 11(7):1229. https://doi.org/10.3390/cells11071229
Chicago/Turabian StyleNiessner, Heike, Markus Burkard, Christian Leischner, Olga Renner, Sarah Plöger, Francisco Meraz-Torres, Matti Böcker, Constanze Hirn, Ulrich M. Lauer, Sascha Venturelli, and et al. 2022. "Therapeutic Efficacy of Pharmacological Ascorbate on Braf Inhibitor Resistant Melanoma Cells In Vitro and In Vivo" Cells 11, no. 7: 1229. https://doi.org/10.3390/cells11071229