
Necroptosis and Prostate Cancer: Molecular Mechanisms and Therapeutic Potential


Abstract
:1. Introduction
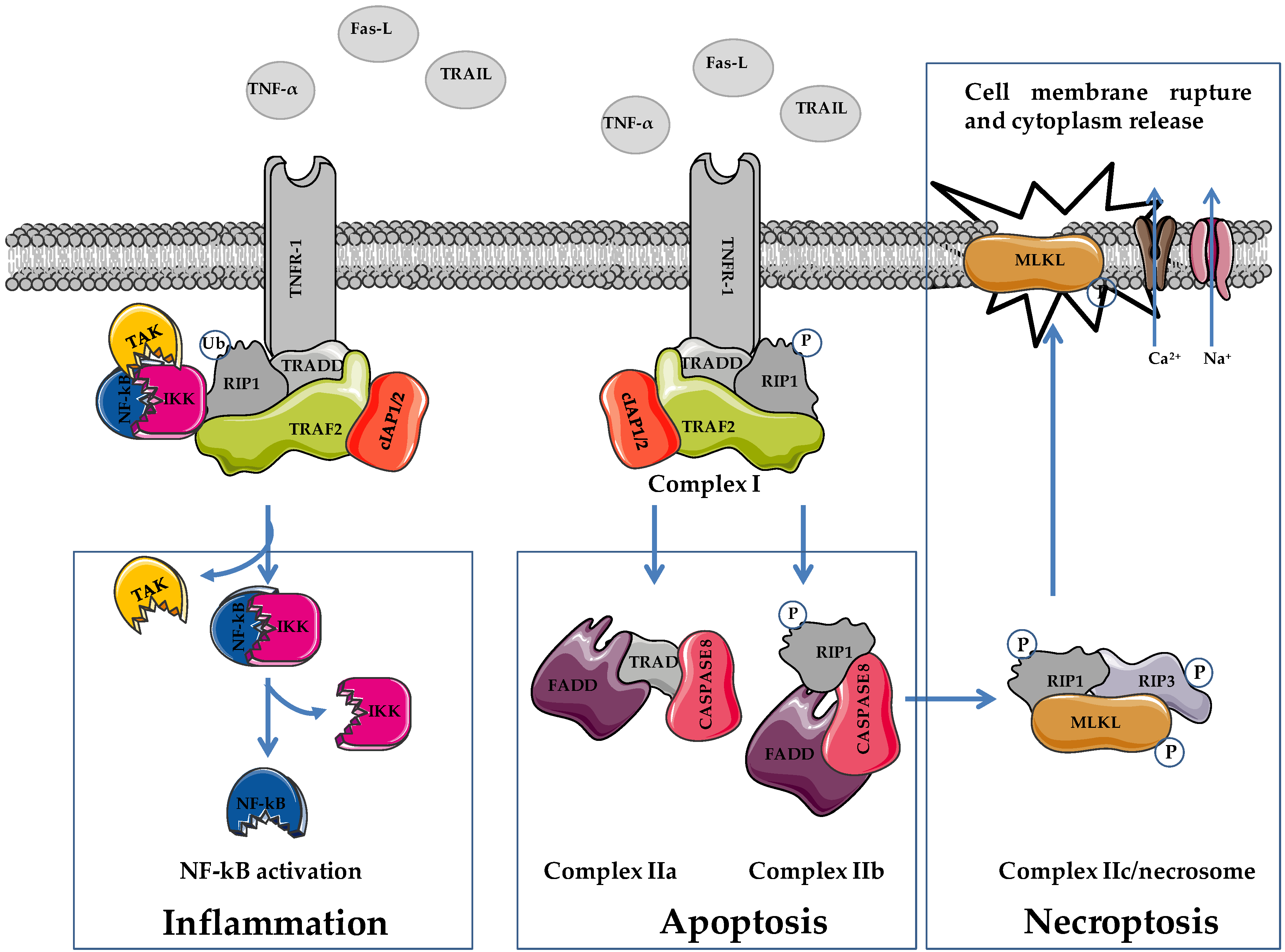
2. Apoptosis and Necroptosis: Overview on Molecular Mechanisms
3. Necroptosis and Necroptosis Inducers in Prostate Cancer
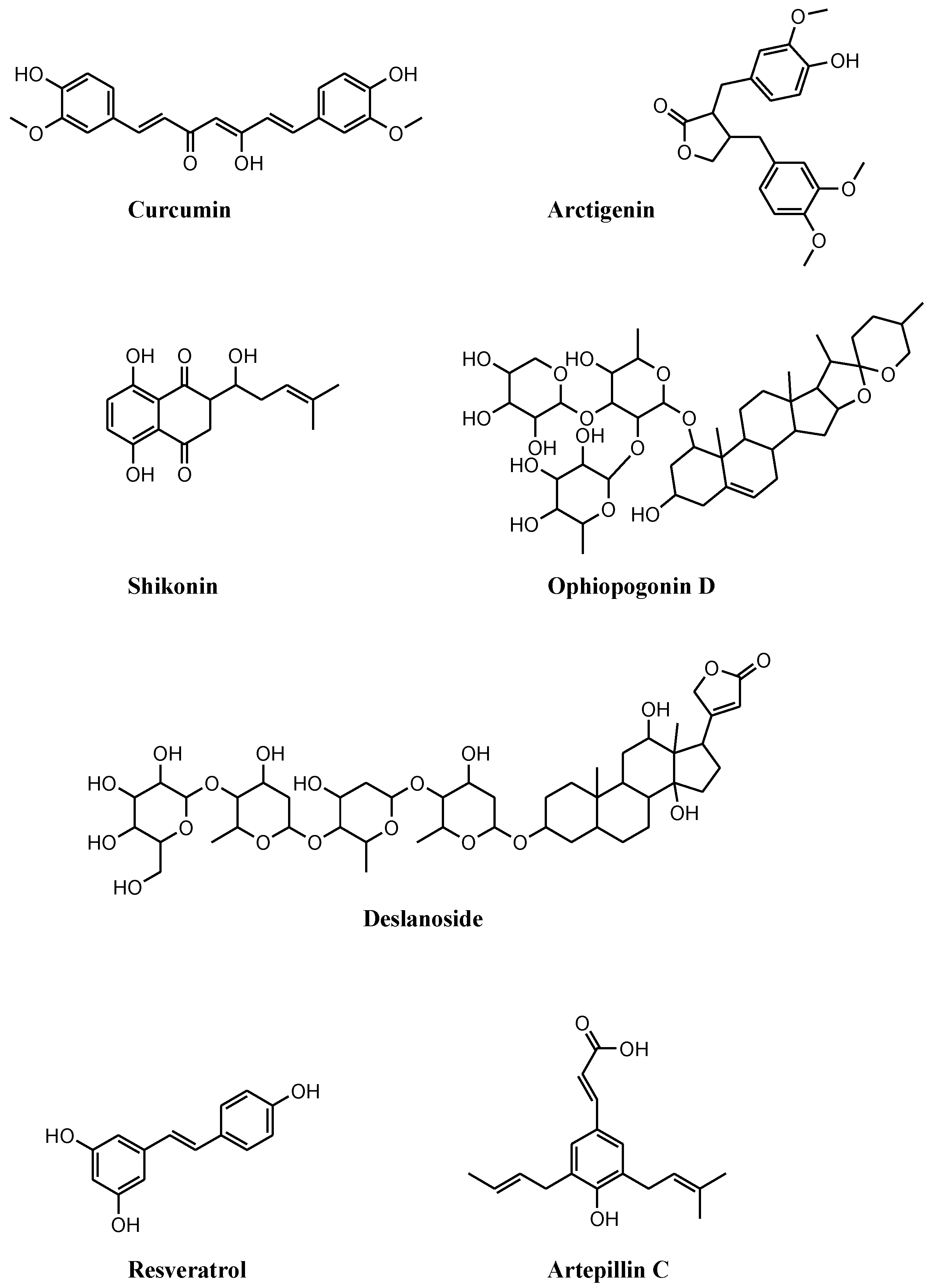
3.1. Compounds of Natural Origin
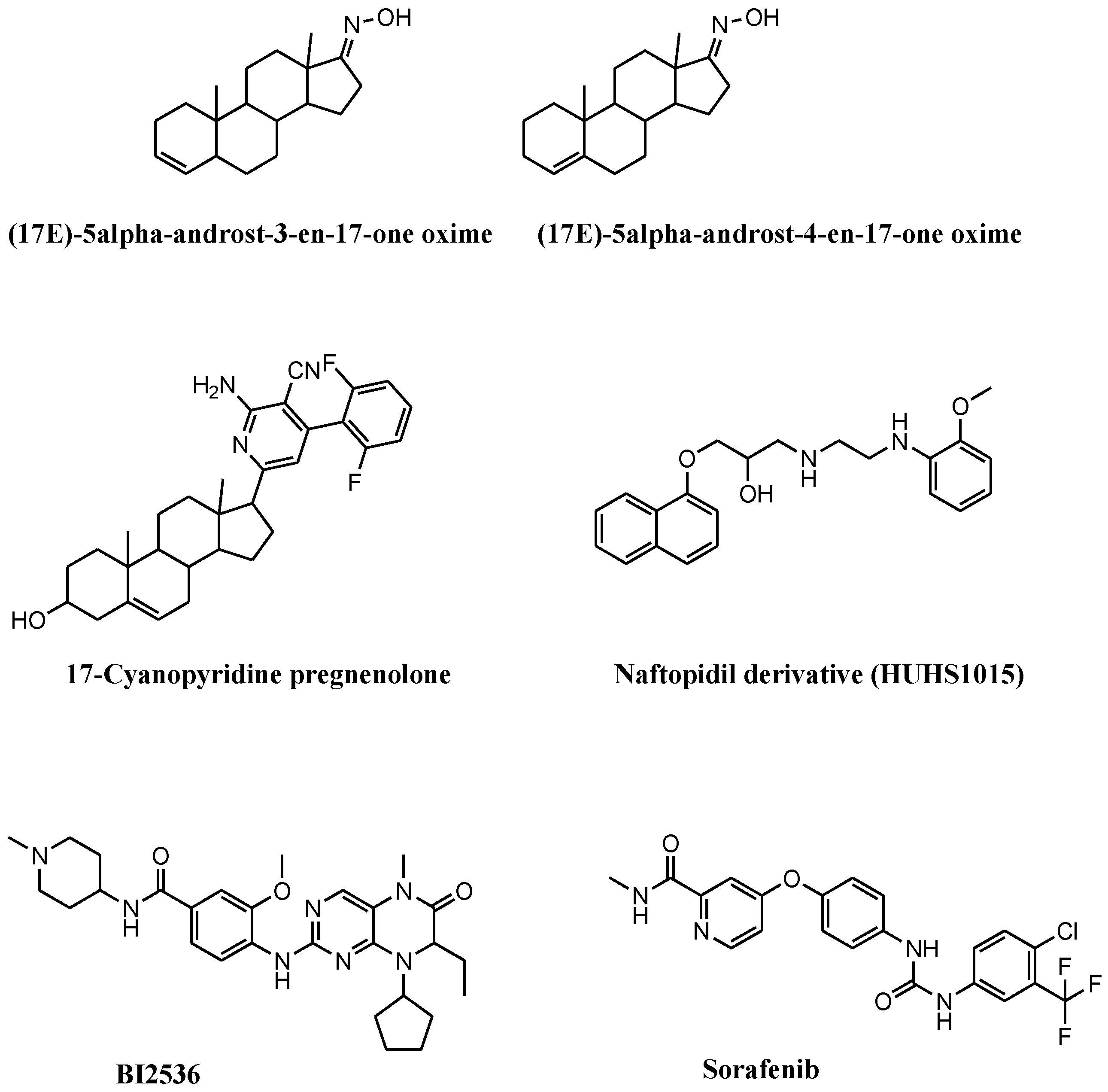
3.2. Synthetic and Semisynthetic Small Molecules
3.3. Selenium and Selenium-Based Nanoparticles
4. Ongoing Clinical Trials Containing Necroptosis Inducers in Prostate Cancer
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tang, D.; Kang, R.; Berghe, T.V.; Vandenabeele, P.; Kroemer, G. The molecular machinery of regulated cell death. Cell Res. 2019, 29, 347–364. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.J.; Reich, C.F.; Pisetsky, D.S. Release of DNA from dead and dying lymphocyte and monocyte cell lines in vitro. Scand. J. Immunol. 2004, 60, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Kang, R.; Zeng, L.; Zhu, S.; Xie, Y.; Liu, J.; Wen, Q.; Cao, L.; Xie, M.; Ran, Q.; Kroemer, G.; et al. Lipid peroxidation drives gasdermin D-mediated pyroptosis in lethal polymicrobial sepsis. Cell Host. Microbe. 2018, 24, 97–108.e104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canli, Ö.; Alankuş, Y.B.; Grootjans, S.; Vegi, N.; Hültner, L.; Hoppe, P.S.; Schroeder, T.; Vandenabeele, P.; Bornkamm, G.W.; Greten, F.R. Glutathione peroxidase 4 prevents necroptosis in mouse erythroid precursors. Blood J. Am. Soc. Hematol. 2016, 127, 139–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haberzettl, P.; Hill, B.G. Oxidized lipids activate autophagy in a JNK-dependent manner by stimulating the endoplasmic reticulum stress response. Redox Biol. 2013, 1, 56–64. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Wu, N.; Jiang, N. Autophagy provides a conceptual therapeutic framework for bone metastasis from prostate cancer. Cell Death Dis. 2021, 12, 909. [Google Scholar] [CrossRef] [PubMed]
- Zaffaroni, N.; Beretta, G.L. Nanoparticles for Ferroptosis Therapy in Cancer. Pharmaceutics 2021, 13, 1785. [Google Scholar] [CrossRef]
- Zaffaroni, N.; Beretta, G.L. Ferroptosis inducers for prostate cancer therapy. Curr. Med. Chem. 2022, in press. [Google Scholar] [CrossRef]
- Goodall, M.L.; Cramer, S.D.; Thorburn, A. Autophagy RIPs into cell death. Cell Cycle 2016, 15, 3014–3015. [Google Scholar] [CrossRef] [Green Version]
- Karki, R.; Sharma, B.R.; Lee, E.; Banoth, B.; Malireddi, R.K.S.; Samir, P.; Tuladhar, S.; Mummareddy, H.; Burton, A.R.; Vogel, P.; et al. Interferon regulatory factor 1 regulates PANoptosis to prevent colorectal cancer. JCI Insight 2020, 5, e136720. [Google Scholar] [CrossRef]
- Place, D.E.; Lee, S.; Kanneganti, T.D. PANoptosis in microbial infection. Curr. Opin. Microbiol. 2021, 59, 42–49. [Google Scholar] [CrossRef]
- Heidaryan, F.; Bamehr, H.; Babaabasi, B.; Emamvirdizadeh, A.; Mohammadzadeh, N.; Khalili, A. The Trend of ripk1/ripk3 and mlkl Mediated Necroptosis Pathway in Patients with Different Stages of Prostate Cancer as Promising Progression Biomarkers. Clin. Lab. 2020, 66. [Google Scholar] [CrossRef]
- Liu, W.; Jin, W.; Zhu, S.; Chen, Y.; Liu, B. Targeting regulated cell death (RCD) with small-molecule compounds in cancer therapy: A revisited review of apoptosis, autophagy-dependent cell death and necroptosis. Drug Discov. Today 2022, 27, 612–625. [Google Scholar] [CrossRef]
- Carneiro, B.A.; El-Deiry, W.S. Targeting apoptosis in cancer therapy. Nat. Rev. Clin. Oncol. 2020, 17, 395–417. [Google Scholar] [CrossRef]
- Fulda, S.; Vucic, D. Targeting IAP proteins for therapeutic intervention in cancer. Nat. Rev. Drug Discov. 2012, 11, 109–124. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Wan, P.; Choksi, S.; Liu, Z.G. Necroptosis and tumor progression. Trends Cancer 2022, 8, 21–27. [Google Scholar] [CrossRef]
- Nie, Z.; Chen, M.; Gao, Y.; Huang, D.; Cao, H.; Peng, Y.; Guo, N.; Zhang, S. Regulated Cell Death in Urinary Malignancies. Front. Cell Dev. Biol. 2021, 9, 789004. [Google Scholar] [CrossRef]
- Mohammad, R.M.; Muqbil, I.; Lowe, L.; Yedjou, C.; Hsu, H.Y.; Lin, L.T.; Siegelin, M.D.; Fimognari, C.; Kumar, N.B.; Dou, Q.P.; et al. Broad targeting of resistance to apoptosis in cancer. Semin. Cancer Biol. 2015, 35, S78–S103. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Nuhn, P.; De Bono, J.S.; Fizazi, K.; Freedland, S.J.; Grilli, M.; Kantoff, P.W.; Sonpavde, G.; Sternberg, C.N.; Yegnasubramanian, S.; Antonarakis, E.S. Update on systemic prostate cancer therapies: Management of metastatic castration resistant prostate cancer in the era of precision oncology. Eur. Urol. 2019, 75, 88–99. [Google Scholar] [CrossRef]
- Martin-Sanchez, D.; Fontecha-Barriuso, M.; Sanchez-Niño, M.D.; Ramos, A.M.; Cabello, R.; Gonzalez-Enguita, C.; Linkermann, A.; Sanz, A.B.; Ortiz, A. Cell death-based approaches in treatment of the urinary tract-associated diseases: A fight for survival in the killing fields. Cell Death Dis. 2018, 9, 118. [Google Scholar] [CrossRef]
- Degterev, A.; Huang, Z.; Boyce, M.; Li, Y.; Jagtap, P.; Mizushima, N.; Cuny, G.D.; Mitchison, T.J.; Moskowitz, M.A.; Yuan, J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 2005, 1, 112–119. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Liu, X.; Xie, X.; Ren, Y.; Shao, Z.; Zhang, N.; Li, L.; Ding, X.; Zhang, L. The role of necroptosis in disease and treatment. MedComm 2021, 2, 730–755. [Google Scholar] [CrossRef]
- Davidovich, P.; Kearney, C.J.; Martin, S.J. Inflammatory outcomes of apoptosis, necrosis and necroptosis. Biol. Chem. 2014, 395, 1163–1171. [Google Scholar] [CrossRef]
- Vanden Berghe, T.; Linkermann, A.; Jouan-Lanhouet, S.; Walczak, H.; Vandenabeele, P. Regulated necrosis: The expanding network of non-apoptotic cell death pathways. Nat. Rev. Mol. Cell Biol. 2014, 15, 135–147. [Google Scholar] [CrossRef]
- Laster, S.M.; Wood, J.G.; Gooding, L.R. Tumor necrosis factor can induce both apoptic and necrotic forms of cell lysis. J. Immunol. 1988, 141, 2629–2634. [Google Scholar]
- Vercammen, D.; Brouckaert, G.; Denecker, G.; Van de Craen, M.; Declercq, W.; Fiers, W.; Vandenabeele, P. Dual signaling of the Fas receptor: Initiation of both apoptotic and necrotic cell death pathways. J. Exp. Med. 1998, 188, 919–930. [Google Scholar] [CrossRef] [Green Version]
- Mishra, A.P.; Salehi, B.; Sharifi-Rad, M.; Pezzani, R.; Kobarfard, F.; Sharifi-Rad, J.; Nigam, M. Programmed Cell Death, from a Cancer Perspective: An Overview. Mol. Diagn. Ther. 2018, 22, 281–295. [Google Scholar] [CrossRef]
- D’Arcy, M.S. Cell death: A review of the major forms of apoptosis, necrosis and autophagy. Cell Biol. Int. 2019, 43, 582–592. [Google Scholar] [CrossRef]
- Holler, N.; Zaru, R.; Micheau, O.; Thome, M.; Attinger, A.; Valitutti, S.; Bodmer, J.L.; Schneider, P.; Seed, B.; Tschopp, J. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat. Immunol. 2000, 1, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Kepp, O.; Chan, F.K.; Kroemer, G. Necroptosis: Mechanisms and Relevance to Disease. Annu. Rev. Pathol. 2017, 12, 103–130. [Google Scholar] [CrossRef] [PubMed]
- Dondelinger, Y.; Darding, M.; Bertrand, M.J.; Walczak, H. Poly-ubiquitination in TNFR1-mediated necroptosis. Cell Mol. Life Sci. 2016, 73, 2165–2176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tortola, L.; Nitsch, R.; Bertrand, M.J.M.; Kogler, M.; Redouane, Y.; Kozieradzki, I.; Uribesalgo, I.; Fennell, L.M.; Daugaard, M.; Klug, H.; et al. The Tumor Suppressor Hace1 Is a Critical Regulator of TNFR1-Mediated Cell Fate. Cell Rep. 2016, 16, 3414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, K.X.; Firus, J.; Prieur, B.; Jia, W.; Rennie, P.S. To die or to survive, a fatal question for the destiny of prostate cancer cells after androgen deprivation therapy. Cancers 2011, 3, 1498–1512. [Google Scholar] [CrossRef] [Green Version]
- Chan, F.K.; Luz, N.F.; Moriwaki, K. Programmed necrosis in the cross talk of cell death and inflammation. Annu. Rev. Immunol. 2015, 33, 79–106. [Google Scholar] [CrossRef]
- Ting, A.T.; Bertrand, M.J.M. More to Life than NF-κB in TNFR1 Signaling. Trends Immunol. 2016, 37, 535–545. [Google Scholar] [CrossRef] [Green Version]
- Dillon, C.P.; Oberst, A.; Weinlich, R.; Janke, L.J.; Kang, T.B.; Ben-Moshe, T.; Mak, T.W.; Wallach, D.; Green, D.R. Survival function of the FADD-CASPASE-8-cFLIP(L) complex. Cell Rep. 2012, 1, 401–407. [Google Scholar] [CrossRef] [Green Version]
- Dondelinger, Y.; Declercq, W.; Montessuit, S.; Roelandt, R.; Goncalves, A.; Bruggeman, I.; Hulpiau, P.; Weber, K.; Sehon, C.A.; Marquis, R.W.; et al. MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell Rep. 2014, 22, 971–981. [Google Scholar] [CrossRef] [Green Version]
- Quarato, G.; Guy, C.S.; Grace, C.R.; Llambi, F.; Nourse, A.; Rodriguez, D.A.; Wakefield, R.; Frase, S.; Moldoveanu, T.; Green, D.R. Sequential Engagement of Distinct MLKL Phosphatidylinositol-Binding Sites Executes Necroptosis. Mol. Cell 2016, 61, 589–601. [Google Scholar] [CrossRef] [Green Version]
- Cho, Y.S.; Challa, S.; Moquin, D.; Genga, R.; Ray, T.D.; Guildford, M.; Chan, F.K. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 2009, 137, 1112–1123. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Wang, H.; Wang, Z.; He, S.; Chen, S.; Liao, D.; Wang, L.; Yan, J.; Liu, W.; Lei, X.; et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 2012, 148, 213–227. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Jiang, H.; Chen, S.; Du, F.; Wang, X. The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways. Cell 2012, 148, 228–243. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Wang, Y.; Zhang, Y.; He, X.; Zhong, C.Q.; Ni, H.; Chen, X.; Liang, Y.; Wu, J.; Zhao, S.; et al. RIP3 targets pyruvate dehydrogenase complex to increase aerobic respiration in TNF-induced necroptosis. Nat. Cell Biol. 2018, 20, 186–197. [Google Scholar] [CrossRef]
- Tait, S.W.; Oberst, A.; Quarato, G.; Milasta, S.; Haller, M.; Wang, R.; Karvela, M.; Ichim, G.; Yatim, N.; Albert, M.L.; et al. Widespread mitochondrial depletion via mitophagy does not compromise necroptosis. Cell Rep. 2013, 5, 878–885. [Google Scholar] [CrossRef]
- Zhou, W.; Yuan, J. Necroptosis in health and diseases. Semin. Cell Dev. Biol. 2014, 35, 14–23. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.J.; Wang, K.Y.; Zhang, H.Z.; Meng, X.Y.; Chen, J.F.; Wang, P.; Jiang, J.H.; Ma, Q. Up-Regulation of RIP3 Alleviates Prostate Cancer Progression by Activation of RIP3/MLKL Signaling Pathway and Induction of Necroptosis. Front. Oncol. 2020, 10, 1720. [Google Scholar] [CrossRef]
- Lu, Z.; Wu, C.; Zhu, M.; Song, W.; Wang, H.; Wang, J.; Guo, J.; Li, N.; Liu, J.; Li, Y.; et al. Ophiopogonin D' induces RIPK1-dependent necroptosis in androgen-dependent LNCaP prostate cancer cells. Int J. Oncol. 2020, 56, 439–447. [Google Scholar] [CrossRef]
- Fu, W.; Li, H.; Fu, H.; Zhao, S.; Shi, W.; Sun, M.; Li, Y. The SIRT3 and SIRT6 Promote Prostate Cancer Progression by Inhibiting Necroptosis-Mediated Innate Immune Response. J. Immunol. Res. 2020, 2020, 8820355. [Google Scholar] [CrossRef]
- Lin, H.Y.; Lin, Y.S.; Shih, S.P.; Lee, S.B.; El-Shazly, M.; Chang, K.M.; Yang, Y.S.H.; Lee, Y.L.; Lu, M.C. The Anti-Proliferative Activity of Secondary Metabolite from the Marine Streptomyces sp. against Prostate Cancer Cells. Life 2021, 11, 1414. [Google Scholar] [CrossRef]
- Lee, Y.J.; Park, K.S.; Lee, S.H. Curcumin Targets Both Apoptosis and Necroptosis in Acidity-Tolerant Prostate Carcinoma Cells. Biomed. Res. Int. 2021, 2021, 8859181. [Google Scholar] [CrossRef]
- Lee, Y.J.; Nam, H.S.; Cho, M.K.; Lee, S.H. Arctigenin induces necroptosis through mitochondrial dysfunction with CCN1 upregulation in prostate cancer cells under lactic acidosis. Mol. Cell Biochem. 2020, 467, 45–56. [Google Scholar] [CrossRef]
- Markowitsch, S.D.; Juetter, K.M.; Schupp, P.; Hauschulte, K.; Vakhrusheva, O.; Slade, K.S.; Thomas, A.; Tsaur, I.; Cinatl, J., Jr.; Michaelis, M.; et al. Shikonin Reduces Growth of Docetaxel-Resistant Prostate Cancer Cells Mainly through Necroptosis. Cancers 2021, 13, 882. [Google Scholar] [CrossRef]
- Rizzi, F.; Naponelli, V.; Silva, A.; Modernelli, A.; Ramazzina, I.; Bonacini, M.; Tardito, S.; Gatti, R.; Uggeri, J.; Bettuzzi, S. Polyphenon E(R), a standardized green tea extract, induces endoplasmic reticulum stress, leading to death of immortalized PNT1a cells by anoikis and tumorigenic PC3 by necroptosis. Carcinogenesis 2014, 35, 828–839. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Huang, Q.A.J.; Li, L.; Li, X.; Zhang, Z.; Dong, J.T. The Cardiac Glycoside Deslanoside Exerts Anticancer Activity in Prostate Cancer Cells by Modulating Multiple Signaling Pathways. Cancers 2021, 13, 5809. [Google Scholar] [CrossRef]
- Zaffaroni, N.; Beretta, G.L. Resveratrol and Prostate Cancer: The Power of Phytochemicals. Curr. Med. Chem. 2021, 28, 4845–4862. [Google Scholar] [CrossRef]
- Lee, S.H.; Lee, Y.J. Synergistic anticancer activity of resveratrol in combination with docetaxel in prostate carcinoma cells. Nutr. Res. Pract. 2021, 15, 12–25. [Google Scholar] [CrossRef]
- Endo, S.; Hoshi, M.; Matsunaga, T.; Inoue, T.; Ichihara, K.; Ikari, A. Autophagy inhibition enhances anticancer efficacy of artepillin C, a cinnamic acid derivative in Brazilian green propolis. Biochem. Biophys. Res. Commun. 2018, 497, 437–443. [Google Scholar] [CrossRef]
- Gomes, A.R.; Pires, A.S.; Abrantes, A.M.; Gonçalves, A.C.; Costa, S.C.; Varela, C.L.; Silva, E.T.; Botelho, M.F.; Roleira, F.M.F. Design, synthesis, and antitumor activity evaluation of steroidal oximes. Bioorg. Med. Chem. 2021, 46, 116360. [Google Scholar] [CrossRef]
- Sun, Y.; Gao, P.; Zhu, L.; Li, Z.; Zhao, R.; Li, C.; Shan, L. Synthesis and biological evaluation of 17-cyanopyridine derivatives of pregnenolone as potential anti-prostate cancer agents. Steroids 2021, 171, 108841. [Google Scholar] [CrossRef]
- Nishizaki, T.; Kanno, T.; Tsuchiya, A.; Kaku, Y.; Shimizu, T.; Tanaka, A. 1-[2-(2-Methoxyphenylamino)ethylamino]-3-(naphthalene-1- yloxy)propan-2-ol may be a promising anticancer drug. Molecules 2014, 19, 21462–21472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deeraksa, A.; Pan, J.; Sha, Y.; Liu, X.D.; Eissa, N.T.; Lin, S.H.; Yu-Lee, L.Y. Plk1 is upregulated in androgen-insensitive prostate cancer cells and its inhibition leads to necroptosis. Oncogene 2013, 32, 2973–2983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kharaziha, P.; Chioureas, D.; Baltatzis, G.; Fonseca, P.; Rodriguez, P.; Gogvadze, V.; Lennartsson, L.; Björklund, A.C.; Zhivotovsky, B.; Grandér, D.; et al. Oncotarget. 2015 Sorafenib-induced defective autophagy promotes cell death by necroptosis. Oncotarget 2015, 6, 37066–37082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, J.; Yan, M.; Liu, X.; Yin, S.; Lu, S.; Fan, L.; Hu, H. Inorganic Selenium Induces Nonapoptotic Programmed Cell Death in PC-3 Prostate Cancer Cells Associated with Inhibition of Glycolysis. J. Agric. Food Chem. 2019, 67, 10637–10645. [Google Scholar] [CrossRef]
- Martínez-Esquivias, F.; Gutiérrez-Angulo, M.; Pérez-Larios, A.; Sánchez-Burgos, J.; Becerra-Ruiz, J.; Guzmán-Flores, J.M. Anticancer Activity of Selenium Nanoparticles In Vitro Studies. Anticancer Agents Med. Chem. 2021. [Google Scholar] [CrossRef]
- Sonkusre, P. Specificity of Biogenic Selenium Nanoparticles for Prostate Cancer Therapy With Reduced Risk of Toxicity: An in vitro and in vivo Study. Front. Oncol 2020, 9, 1541. [Google Scholar] [CrossRef] [Green Version]
- Sonkusre, P.; Cameotra, S.S.J. Biogenic selenium nanoparticles induce ROS-mediated necroptosis in PC-3 cancer cells through TNF activation. Nanobiotechnology 2017, 15, 43. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Characteristic | Necroptosis | Necrosis | Apoptosis | |
|---|---|---|---|---|
| Involved proteins | RIP3 | + | / | / |
| MLKL | + | / | / | |
| Caspase 3 | / | / | + | |
| Cell properties | Membrane perforation | + | + | / |
| Membrane blebbing | / | / | + | |
| DNA fragmentation | + | + | + | |
| Cell lysis and swelling | + | + | / | |
| Inflammation | + | + | / |
| Class of Compounds | Compound | Cell Lines | In Vivo Studies | Necroptosis Inhibitors | Necroptosis Markers |
|---|---|---|---|---|---|
| Compounds of natural origin | Lu01-M | DU145, PC3, LNCaP | No | Yes | TNFR2, p-RIP1, p-MLKL |
| Curcumin | PC3, PC3AcT | No | Yes | p-RIP3, p-MLKL | |
| Arctigenin | PC3, PC3AcT | No | Yes | p-RIP3, p-MLKL | |
| Shikonin | Pc3, DU145, LNCaP, 22Rv1 | No | Yes | p-RIP1, p-RIP3 | |
| Ophiopogonin D’ | PC3, LNCaP | No | Yes | p-RIP1, p-RIP3, p-MLKL | |
| Polyphenon E | PC3, PNT1a | No | No | Cell morphological changes | |
| Deslanoside | PC3, DU145, 22Rv1 | PC3, 22Rv1 | No | Genome wide expression profile | |
| Resveratrol | LNCaP | No | No | p-RIP3, p-MLKL | |
| Artepillin C | CW22Rv1 | No | No | p-RIP3 | |
| Synthetic and semisynthetic small molecules | Steroid-oxime derivatives | PC3 | No | No | Decreased mitochondrial membrane potential |
| 17-cyanopyridine pregnenolone derivative | PC3 | No | No | p-RIP1, p-RIP3, p-MLKL | |
| Naftopidil derivative (HUHS1015) | PC3, DU145, LNCaP | No a | yes | In vitro drug combination studies with necrostatin 1 | |
| BI2536 | LNCaP, LNCaP-AI | No | Yes | In vitro drug combination studies with necrostatin 1 | |
| Sorafenib | DU145 | No | Yes | p-RIP1, p-RIP3, p-MLKL | |
| Selenium and selenium-based nanoparticles | Selenite | PC3, DU145, LNCaP | No | Yes | In vitro drug combination studies with dabrafenib and N-acetyl cysteine |
| Selenium-based nanoparticles | PC3, LNCaP | No | Yes | In vitro drug combination studies with necrostatin 1 |
| Compound | NCT Number | Markers | Phase |
|---|---|---|---|
| Curcumin | NCT03769766 | PSA | III |
| NCT03211104 | PSA | na | |
| NCT02064673 | PSA | III | |
| Curcumin and piperine | NCT04731844 | nd | II |
| Curcumin and ursolic acid | NCT04403568 | p65, NF-kB | I |
| Curcumin and Vitamin D, omega 3, turmeric | NCT03290417 | PSA | na |
| Curcumin and taxotere | NCT02095717 | PSA | III |
| Curcumin and radiotherapy | NCT01917890 | TNF-α, NF-kB | na |
| NCT02724618 | PSA | II | |
| NCT03493997 | nd | II | |
| Polyphenon E | NCT00596011 | PSA | II |
| NCT00676780 | PSA, VEGF, HGF | II | |
| NCT01340599 | PSA, Ki67, Bcl2, Cyclin D, p27, VEGF, CD31, MMP2 and 9, IGF1 | II | |
| NCT00459407 | MMP2, MMP9, IGF1 | I | |
| NCT00253643 | FASN, Ki67 | na | |
| NCT04597359 | PSA, Ki67 | II | |
| BI2536 | NCT00706498 | PSA | II |
| Sorafenib | NCT00090545 | PSA | II |
| NCT00694291 | PSA | II | |
| NCT00466752 | PSA, p-ERK, p-AKT, p-S6-kinase, caspase 3, Ki67 | I | |
| NCT00093457 | PSA | II | |
| Sorafenib and leuprolide or bicalutamide | NCT00924807 | PSA | I-II |
| Sorafenib and docetaxel | NCT00589420 | PSA | II |
| NCT00619996 | PSA, p-ERK, VEGF-R2 | II | |
| Sorafenib and taxotere | NCT00405210 | PSA | I |
| Sorafenib and mitoxantrone | NCT00452387 | PSA | II |
| Sorafenib and gleevec | NCT00424385 | PSA | I |
| Selenite and docetaxel | NCT01155791 | PSA | I |
| Selenite and radiotherapy | NCT02184533 | PSA | I |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beretta, G.L.; Zaffaroni, N. Necroptosis and Prostate Cancer: Molecular Mechanisms and Therapeutic Potential. Cells 2022, 11, 1221. https://doi.org/10.3390/cells11071221
Beretta GL, Zaffaroni N. Necroptosis and Prostate Cancer: Molecular Mechanisms and Therapeutic Potential. Cells. 2022; 11(7):1221. https://doi.org/10.3390/cells11071221
Chicago/Turabian StyleBeretta, Giovanni Luca, and Nadia Zaffaroni. 2022. "Necroptosis and Prostate Cancer: Molecular Mechanisms and Therapeutic Potential" Cells 11, no. 7: 1221. https://doi.org/10.3390/cells11071221