
Extracellular Lipids in the Lung and Their Role in Pulmonary Fibrosis

 , and
, and
Abstract
:1. The Physiopathology of Pulmonary Fibrosis
2. Overview of Pulmonary Lipids and Their Metabolism
2.1. Phospholipids
2.2. Fatty Acids
2.3. Non-Glyceride Lipids and Lipoproteins
3. Dysregulated Lipids and Their Metabolism during Lung Fibrosis
4. Extracellular Lipids as Important Regulators of Fibrosis Progression
5. Lipids and Pulmonary Fibrosis, The Hope for Potential Biomarkers
6. Lipid-Focused Therapies
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lederer, D.J.; Martinez, F.J. Idiopathic pulmonary fibrosis. N. Engl. J. Med. 2018, 378, 1811–1823. [Google Scholar] [CrossRef]
- Richeldi, L.; Cottin, V.; du Bois, R.M.; Selman, M.; Kimura, T.; Bailes, Z.; Schlenker-Herceg, R.; Stowasser, S.; Brown, K.K. Nintedanib in patients with idiopathic pulmonary fibrosis: Combined evidence from the TOMORROW and INPULSIS(®) trials. Respir. Med. 2016, 113, 74–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, T.E.; Bradford, W.Z.; Castro-Bernardini, S.; Fagan, E.A.; Glaspole, I.; Glassberg, M.K.; Gorina, E.; Hopkins, P.M.; Kardatzke, D.; Lancaster, L.; et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014, 370, 2083–2092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez, I.E.; Eickelberg, O. New cellular and molecular mechanisms of lung injury and fibrosis in idiopathic pulmonary fibrosis. Lancet 2012, 380, 680–688. [Google Scholar] [CrossRef]
- Burgy, O.; Bellaye, P.-S.; Beltramo, G.; Goirand, F.; Bonniaud, P. Pathogenesis of fibrosis in interstitial lung disease. Curr. Opin. Pulm. Med. 2020, 26, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Phan, T.H.G.; Paliogiannis, P.; Nasrallah, G.K.; Giordo, R.; Eid, A.H.; Fois, A.G.; Zinellu, A.; Mangoni, A.A.; Pintus, G. Emerging cellular and molecular determinants of idiopathic pulmonary fibrosis. Cell. Mol. Life Sci. 2021, 78, 2031–2057. [Google Scholar] [CrossRef] [PubMed]
- Strunz, M.; Simon, L.M.; Ansari, M.; Kathiriya, J.J.; Angelidis, I.; Mayr, C.H.; Tsidiridis, G.; Lange, M.; Mattner, L.F.; Yee, M.; et al. Alveolar regeneration through a Krt8+ transitional stem cell state that persists in human lung fibrosis. Nat. Commun. 2020, 11, 3559. [Google Scholar] [CrossRef]
- Jiang, P.; Gil de Rubio, R.; Hrycaj, S.M.; Gurczynski, S.J.; Riemondy, K.A.; Moore, B.B.; Omary, M.B.; Ridge, K.M.; Zemans, R.L. Ineffectual type 2-to-type 1 alveolar epithelial cell differentiation in idiopathic pulmonary fibrosis: Persistence of the KRT8hi transitional state. Am. J. Respir. Crit. Care Med. 2020, 201, 1443–1447. [Google Scholar] [CrossRef]
- Kathiriya, J.J.; Wang, C.; Zhou, M.; Brumwell, A.; Cassandras, M.; Le Saux, C.J.; Cohen, M.; Alysandratos, K.-D.; Wang, B.; Wolters, P.; et al. Human alveolar type 2 epithelium transdifferentiates into metaplastic KRT5+ basal cells. Nat. Cell. Biol. 2022, 24, 10–23. [Google Scholar] [CrossRef] [PubMed]
- Tsukui, T.; Sun, K.-H.; Wetter, J.B.; Wilson-Kanamori, J.R.; Hazelwood, L.A.; Henderson, N.C.; Adams, T.S.; Schupp, J.C.; Poli, S.D.; Rosas, I.O.; et al. Collagen-producing lung cell atlas identifies multiple subsets with distinct localization and relevance to fibrosis. Nat. Commun. 2020, 11, 1920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kheirollahi, V.; Wasnick, R.M.; Biasin, V.; Vazquez-Armendariz, A.I.; Chu, X.; Moiseenko, A.; Weiss, A.; Wilhelm, J.; Zhang, J.-S.; Kwapiszewska, G.; et al. Metformin induces lipogenic differentiation in myofibroblasts to reverse lung fibrosis. Nat. Commun. 2019, 10, 2987. [Google Scholar] [CrossRef] [PubMed]
- Xie, T.; Wang, Y.; Deng, N.; Huang, G.; Taghavifar, F.; Geng, Y.; Liu, N.; Kulur, V.; Yao, C.; Chen, P.; et al. Single-cell deconvolution of fibroblast heterogeneity in mouse pulmonary fibrosis. Cell Rep. 2018, 22, 3625–3640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.K.; Kugler, M.C.; Wolters, P.J.; Robillard, L.; Galvez, M.G.; Brumwell, A.N.; Sheppard, D.; Chapman, H.A. Alveolar epithelial cell mesenchymal transition develops in vivo during pulmonary fibrosis and is regulated by the extracellular matrix—PubMed. Proc. Natl. Acad. Sci. USA 2006, 103, 13180–13185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rock, J.R.; Barkauskas, C.E.; Cronce, M.J.; Xue, Y.; Harris, J.R.; Liang, J.; Noble, P.W.; Hogan, B.L. Multiple stromal populations contribute to pulmonary fibrosis without evidence for epithelial to mesenchymal transition—PubMed. Proc. Natl. Acad. Sci. USA 2011, 108, E1475–E1483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Decologne, N.; Kolb, M.; Margetts, P.J.; Menetrier, F.; Artur, Y.; Garrido, C.; Gauldie, J.; Camus, P.; Bonniaud, P. TGF-Beta1 induces progressive pleural scarring and subpleural Fibrosis—PubMed. J. Immunol. 2007, 179, 6043–6451. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, N.; Phan, S.H.; Imaizumi, K.; Matsuo, M.; Nakashima, H.; Kawabe, T.; Shimokata, K.; Hasegawa, Y. Endothelial-mesenchymal transition in bleomycin-induced pulmonary fibrosis—PubMed. Am. J. Respir. Cell Mol. Biol. 2010, 43, 161–172. [Google Scholar] [CrossRef] [Green Version]
- Jia, W.; Wang, Z.; Gao, C.; Wu, J.; Wu, Q. Trajectory modeling of endothelial-to-mesenchymal transition reveals galectin-3 as a mediator in pulmonary fibrosis. Cell Death Dis. 2021, 12, 327. [Google Scholar] [CrossRef]
- Lim, W.W.; Corden, B.; Ng, B.; Vanezis, K.; D’Agostino, G.; Widjaja, A.A.; Song, W.H.; Xie, C.; Su, L.; Kwek, X.Y.; et al. Interleukin-11 is important for vascular smooth muscle phenotypic switching and aortic inflammation, fibrosis and remodeling in mouse models—PubMed. Sci. Rep. 2020, 10, 17853. [Google Scholar] [CrossRef]
- Fois, A.G.; Posadino, A.M.; Giordo, R.; Cossu, A.; Agouni, A.; Rizk, N.M.; Pirina, P.; Carru, C.; Zinellu, A.; Pintus, G. Antioxidant Activity Mediates Pirfenidone Antifibrotic Effects in Human Pulmonary Vascular Smooth Muscle Cells Exposed to Sera of Idiopathic Pulmonary Fibrosis Patients. Oxidative Med. Cell. Longev. 2018, 2018, 2639081. [Google Scholar] [CrossRef]
- Reyfman, P.A.; Walter, J.M.; Joshi, N.; Anekalla, K.R.; McQuattie-Pimentel, A.C.; Chiu, S.; Fernandez, R.; Akbarpour, M.; Chen, C.-I.; Ren, Z.; et al. Single-cell transcriptomic analysis of human lung provides insights into the pathobiology of pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2019, 199, 1517–1536. [Google Scholar] [CrossRef]
- Morse, C.; Tabib, T.; Sembrat, J.; Buschur, K.L.; Bittar, H.T.; Valenzi, E.; Jiang, Y.; Kass, D.J.; Gibson, K.; Chen, W.; et al. Proliferating SPP1/MERTK-expressing macrophages in idiopathic pulmonary fibrosis. Eur. Respir. J. 2019, 54, 1802441. [Google Scholar] [CrossRef] [PubMed]
- Adams, T.S.; Schupp, J.C.; Poli, S.; Ayaub, E.A.; Neumark, N.; Ahangari, F.; Chu, S.G.; Raby, B.A.; DeIuliis, G.; Januszyk, M.; et al. Single-Cell RNA-seq reveals ectopic and aberrant lung-resident cell populations in idiopathic pulmonary fibrosis. Sci. Adv. 2020, 6, eaba1983. [Google Scholar] [CrossRef] [PubMed]
- Misharin, A.V.; Morales-Nebreda, L.; Reyfman, P.A.; Cuda, C.M.; Walter, J.M.; McQuattie-Pimentel, A.C.; Chen, C.-I.; Anekalla, K.R.; Joshi, N.; Williams, K.J.N.; et al. Monocyte-derived alveolar macrophages drive lung fibrosis and persist in the lung over the life span. J. Exp. Med. 2017, 214, 2387–2404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mould, K.J.; Moore, C.M.; McManus, S.A.; McCubbrey, A.L.; McClendon, J.D.; Griesmer, C.L.; Henson, P.M.; Janssen, W.J. Airspace macrophages and monocytes exist in transcriptionally distinct subsets in healthy adults. Am. J. Respir. Crit. Care Med. 2021, 203, 946–956. [Google Scholar] [CrossRef] [PubMed]
- Schiller, H.B.; Mayr, C.H.; Leuschner, G.; Strunz, M.; Staab-Weijnitz, C.; Preisendörfer, S.; Eckes, B.; Moinzadeh, P.; Krieg, T.; Schwartz, D.A.; et al. Deep proteome profiling reveals common prevalence of MZB1-positive plasma B cells in human lung and skin fibrosis. Am. J. Respir. Crit. Care Med. 2017, 196, 1298–1310. [Google Scholar] [CrossRef] [PubMed]
- Mould, K.J.; Jackson, N.D.; Henson, P.M.; Seibold, M.; Janssen, W.J. Single cell RNA sequencing identifies unique inflammatory airspace macrophage subsets. JCI Insight 2019, 4, 126556. [Google Scholar] [CrossRef]
- McQuattie-Pimentel, A.C.; Ren, Z.; Joshi, N.; Watanabe, S.; Stoeger, T.; Chi, M.; Lu, Z.; Sichizya, L.; Aillon, R.P.; Chen, C.-I.; et al. The lung microenvironment shapes a dysfunctional response of alveolar macrophages in aging. J. Clin. Investig. 2021, 131, 140299. [Google Scholar] [CrossRef] [PubMed]
- Hackett, T.L.; Osei, E.T. Modeling extracellular matrix-cell interactions in lung repair and chronic disease. Cells 2021, 10, 2145. [Google Scholar] [CrossRef] [PubMed]
- Burgy, O.; Königshoff, M. The WNT signaling pathways in wound healing and fibrosis. Matrix Biol. 2018, 68–69, 67–80. [Google Scholar] [CrossRef]
- Sime, P.J.; Xing, Z.; Graham, F.L.; Csaky, K.G.; Gauldie, J. Adenovector-mediated gene transfer of active transforming growth factor-beta1 induces prolonged severe fibrosis in rat lung—PubMed. J. Clin. Investig. 1997, 100, 768–776. [Google Scholar] [CrossRef] [Green Version]
- Bonniaud, P.; Margetts, P.J.; Kolb, M.; Schroeder, J.A.; Kapoun, A.M.; Damm, D.; Murphy, A.; Chakravarty, S.; Dugar, S.; Higgins, L.; et al. Progressive transforming growth factor beta1-induced lung fibrosis is blocked by an orally active ALK5 kinase inhibitor. Am. J. Respir. Crit. Care Med. 2005, 171, 889–898. [Google Scholar] [CrossRef] [PubMed]
- Nabhan, A.N.; Brownfield, D.G.; Harbury, P.B.; Krasnow, M.A.; Desai, T.J. Single-cell Wnt signaling niches maintain stemness of alveolar type 2 cells. Science 2018, 359, 1118–1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, F.; Lagares, D.; Choi, K.M.; Stopfer, L.; Marinković, A.; Vrbanac, V.; Probst, C.K.; Hiemer, S.E.; Sisson, T.H.; Horowitz, J.C.; et al. Mechanosignaling through YAP and TAZ drives fibroblast activation and fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L344–L357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, T.; Huang, Z.; Zhang, H.; Posner, C.; Jia, G.; Ramalingam, T.R.; Xu, M.; Brightbill, H.; Egen, J.G.; Dey, A.; et al. TAZ is required for lung alveolar epithelial cell differentiation after injury. JCI Insight 2019, 5, e128674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meiners, S.; Eickelberg, O.; Königshoff, M. Hallmarks of the ageing lung. Eur. Respir. J. 2015, 45, 807–827. [Google Scholar] [CrossRef] [PubMed]
- Schafer, M.J.; White, T.A.; Iijima, K.; Haak, A.J.; Ligresti, G.; Atkinson, E.J.; Oberg, A.L.; Birch, J.; Salmonowicz, H.; Zhu, Y.; et al. Cellular senescence mediates fibrotic pulmonary disease. Nat. Commun. 2017, 8, 14532. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, M.; Korfei, M.; Mutze, K.; Klee, S.; Skronska-Wasek, W.; Alsafadi, H.N.; Ota, C.; Costa, R.; Schiller, H.B.; Lindner, M.; et al. Senolytic drugs target alveolar epithelial cell function and attenuate experimental lung fibrosis ex vivo. Eur. Respir. J. 2017, 50, 1602367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kellogg, D.L.; Musi, N.; Nambiar, A.M. Cellular Senescence in Idiopathic Pulmonary Fibrosis. Curr. Mol. Biol. Rep. 2021, 7, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Justice, J.N.; Nambiar, A.M.; Tchkonia, T.; LeBrasseur, N.K.; Pascual, R.; Hashmi, S.K.; Prata, L.; Masternak, M.M.; Kritchevsky, S.B.; Musi, N.; et al. Senolytics in idiopathic pulmonary fibrosis: Results from a first-in-human, open-label, pilot study. EBioMedicine 2019, 40, 554–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellaye, P.-S.; Beltramo, G.; Burgy, O.; Collin, B.; Cochet, A.; Bonniaud, P. Measurement of hypoxia in the lung in idiopathic pulmonary fibrosis: A matter of control. Eur. Respir. J. 2022, 59, 2102711. [Google Scholar] [CrossRef] [PubMed]
- Porter, J.C.; Win, T.; Erlandsson, K.; Fraioli, F.; Rashidnasab, A.; Holman, B.; Ganeshan, B.; Screaton, N.J.; Maher, T.M.; Endozo, R.; et al. Measurement of hypoxia in the lung in idiopathic pulmonary fibrosis: An F-MISO PET/CT study. Eur. Respir. J. 2021, 58, 2004584. [Google Scholar] [CrossRef] [PubMed]
- Tanguy, J.; Goirand, F.; Bouchard, A.; Frenay, J.; Moreau, M.; Mothes, C.; Oudot, A.; Helbling, A.; Guillemin, M.; Bonniaud, P.; et al. [18F]FMISO PET/CT imaging of hypoxia as a non-invasive biomarker of disease progression and therapy efficacy in a preclinical model of pulmonary fibrosis: Comparison with the [18F]FDG PET/CT approach. Eur. J. Pediatr. 2021, 48, 3058–3074. [Google Scholar] [CrossRef] [PubMed]
- Gille, T.; Didier, M.; Boubaya, M.; Moya, L.; Sutton, A.; Carton, Z.; Baran-Marszak, F.; Sadoun-Danino, D.; Israël-Biet, D.; Cottin, V.; et al. Obstructive sleep apnoea and related comorbidities in incident idiopathic pulmonary fibrosis. Eur. Respir. J. 2017, 49, 1601934. [Google Scholar] [CrossRef]
- Gille, T.; Didier, M.; Rotenberg, C.; Delbrel, E.; Marchant, D.; Sutton, A.; Dard, N.; Haine, L.; Voituron, N.; Bernaudin, J.-F.; et al. Intermittent Hypoxia Increases the Severity of Bleomycin-Induced Lung Injury in Mice. Oxidative Med. Cell. Longev. 2018, 2018, 1240192. [Google Scholar] [CrossRef] [PubMed]
- Agudelo, C.W.; Samaha, G.; Garcia-Arcos, I. Alveolar Lipids in Pulmonary Disease. A Review. Lipids Health Dis. 2020, 19, 122. [Google Scholar] [CrossRef] [PubMed]
- Castillo-Sánchez, J.C.; Cruz, A.; Pérez-Gil, J. Structural hallmarks of lung surfactant: Lipid-protein interactions, membrane structure and future challenges. Arch. Biochem. Biophys. 2021, 703, 108850. [Google Scholar] [CrossRef] [PubMed]
- Nuñez-Garcia, M.; Gomez-Santos, B.; Buqué, X.; García-Rodriguez, J.L.; Romero, M.R.; Marin, J.J.G.; Arteta, B.; García-Monzón, C.; Castaño, L.; Syn, W.-K.; et al. Osteopontin regulates the cross-talk between phosphatidylcholine and cholesterol metabolism in mouse liver. J. Lipid Res. 2017, 58, 1903–1915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, X.; Wang, J.; Zhu, Y.; Feng, W.; Zhai, C.; Liu, L.; Shi, W.; Wang, Q.; Zhang, Q.; Chai, L.; et al. S1P induces pulmonary artery smooth muscle cell proliferation by activating calcineurin/NFAT/OPN signaling pathway. Biochem. Biophys. Res. Commun. 2019, 516, 921–927. [Google Scholar] [CrossRef]
- Romani, P.; Brian, I.; Santinon, G.; Pocaterra, A.; Audano, M.; Pedretti, S.; Mathieu, S.; Forcato, M.; Bicciato, S.; Manneville, J.-B.; et al. Extracellular matrix mechanical cues regulate lipid metabolism through Lipin-1 and SREBP. Nat. Cell Biol. 2019, 21, 338–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, L.T.Y.; Skiba, N.; Ullmer, C.; Rao, P.V. Lysophosphatidic Acid Induces ECM Production via Activation of the Mechanosensitive YAP/TAZ Transcriptional Pathway in Trabecular Meshwork Cells. Investig. Opthalmol. Vis. Sci. 2018, 59, 1969–1984. [Google Scholar] [CrossRef] [Green Version]
- Mamazhakypov, A.; Schermuly, R.T.; Schaefer, L.; Wygrecka, M. Lipids—Two sides of the same coin in lung fibrosis. Cell Signal. 2019, 60, 65–80. [Google Scholar] [CrossRef] [PubMed]
- Suryadevara, V.; Ramchandran, R.; Kamp, D.W.; Natarajan, V. Lipid mediators regulate pulmonary fibrosis: Potential mechanisms and signaling pathways. Int. J. Mol. Sci. 2020, 21, 4257. [Google Scholar] [CrossRef] [PubMed]
- Hollenbeck, C.B. An Introduction to the Nutrition and Metabolism of Choline. Cent. Nerv. Syst. Agents Med. Chem. 2012, 12, 100–113. [Google Scholar] [CrossRef]
- Ridgway, N.D. The role of phosphatidylcholine and choline metabolites to cell proliferation and survival. Crit. Rev. Biochem. Mol. Biol. 2013, 48, 20–38. [Google Scholar] [CrossRef] [PubMed]
- Zeisel, S.H. Choline. A nutrient that is involved in the regulation of cell proliferation, cell death, and cell transformation. Adv. Exp. Med. Biol. 1996, 399, 131–141. [Google Scholar] [PubMed]
- Hirai, K.; Watanabe, S.; Nishijima, N.; Shibata, K.; Hase, A.; Yamanaka, T.; Inazu, M. Molecular and functional analysis of choline transporters and antitumor effects of choline transporter-like protein 1 inhibitors in human pancreatic cancer cells. Int. J. Mol. Sci. 2020, 21, 5190. [Google Scholar] [CrossRef] [PubMed]
- Inazu, M.; Yamada, T.; Kubota, N.; Yamanaka, T. Functional expression of choline transporter-like protein 1 (CTL1) in small cell lung carcinoma cells: A target molecule for lung cancer therapy. Pharmacol. Res. 2013, 76, 119–131. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Tontonoz, P. Phospholipid remodeling in physiology and disease. Annu. Rev. Physiol. 2019, 81, 165–188. [Google Scholar] [CrossRef] [PubMed]
- Gibellini, F.; Smith, T.K. The Kennedy pathway - De novo synthesis of phosphatidylethanolamine and phosphatidylcholine. IUBMB Life 2010, 62, 414–428. [Google Scholar] [CrossRef] [PubMed]
- Ishiguro, N.; Oyabu, M.; Sato, T.; Maeda, T.; Minami, H.; Tamai, I. Decreased biosynthesis of lung surfactant constituent phosphatidylcholine due to inhibition of choline transporter by gefitinib in lung alveolar cells. Pharm. Res. 2008, 25, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Dodia, C.; Fisher, A.B.; Chander, A.; Kleinzeller, A. Inhibitors of choline transport in alveolar type ii epithelial cells. Am. J. Respir. Cell Mol. Biol. 1992, 6, 426–429. [Google Scholar] [CrossRef] [PubMed]
- Lockman, P.R.; Allen, D.D. The transport of choline. Drug Dev. Ind. Pharm. 2002, 28, 749–771. [Google Scholar] [CrossRef]
- Oelberg, D.G.; Xu, F. Conductive choline transport by alveolar epithelial plasma membrane vesicles. Mol. Genet. Metab. 1998, 65, 220–228. [Google Scholar] [CrossRef]
- Agostoni, C.; Bruzzese, M.G. Fatty acids: Their biochemical and functional classification. Pediatr. Med. Chir. 1992, 14, 473–479. [Google Scholar]
- Tvrzicka, E.; Kremmyda, L.-S.; Stankova, B.; Zak, A. Fatty acids as biocompounds: Their role in human metabolism, health and disease—A review. Part 1: Classification, dietary sources and biological functions. Biomed. Pap. Med. Fac. Univ. Palacky Olomouc Czech Repub. 2011, 155, 117–130. [Google Scholar] [CrossRef] [PubMed]
- Beld, J.; Lee, D.J.; Burkart, M.D. Fatty acid biosynthesis revisited: Structure elucidation and metabolic engineering. Mol. Biosyst. 2015, 11, 38–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohno, Y.; Suto, S.; Yamanaka, M.; Mizutani, Y.; Mitsutake, S.; Igarashi, Y.; Sassa, T.; Kihara, A. ELOVL1 production of C24 Acyl-CoAs is linked to C24 sphingolipid synthesis. Proc. Natl. Acad. Sci. USA 2010, 107, 18439–18444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guillou, H.; Zadravec, D.; Martin, P.G.P.; Jacobsson, A. The key roles of elongases and desaturases in mammalian fatty acid metabolism: Insights from transgenic mice. Prog. Lipid Res. 2010, 49, 186–199. [Google Scholar] [CrossRef] [PubMed]
- Jakobsson, A.; Westerberg, R.; Jacobsson, A. Fatty acid elongases in mammals: Their regulation and roles in metabolism. Prog. Lipid Res. 2006, 45, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Bach, L.; Faure, J.-D. Role of very-long-chain fatty acids in plant development, when chain length does matter. Comptes Rendus. Biol. 2010, 333, 361–370. [Google Scholar] [CrossRef] [PubMed]
- McMaster, C.R. From yeast to humans—Roles of the Kennedy pathway for phosphatidylcholine synthesis. FEBS Lett. 2018, 592, 1256–1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perrakis, A.; Moolenaar, W.H. Autotaxin: Structure-function and signaling. J. Lipid Res. 2014, 55, 1010–1018. [Google Scholar] [CrossRef] [Green Version]
- Song, J.; Clair, T.; Noh, J.H.; Eun, J.W.; Ryu, S.Y.; Lee, S.N.; Ahn, Y.M.; Kim, S.Y.; Lee, S.H.; Park, W.S.; et al. Autotaxin (LysoPLD/NPP2) protects fibroblasts from apoptosis through its enzymatic product, lysophosphatidic acid, utilizing albumin-bound substrate. Biochem. Biophys. Res. Commun. 2005, 337, 967–975. [Google Scholar] [CrossRef] [PubMed]
- Onono, F.O.; Morris, A.J. Phospholipase D and choline metabolism. Handb. Exp. Pharmacol. 2020, 259, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Wu, L.; Chen, J.; Dong, L.; Chen, C.; Wen, Z.; Hu, J.; Fleming, I.; Wang, D.W. Metabolism pathways of arachidonic acids: Mechanisms and potential therapeutic targets. Signal. Transduct. Target. Ther. 2021, 6, 94. [Google Scholar] [CrossRef] [PubMed]
- Peters-Golden, M.; Henderson, W.R. Leukotrienes. N. Engl. J. Med. 2007, 357, 1841–1854. [Google Scholar] [CrossRef] [PubMed]
- Schildknecht, S.; Daiber, A.; Ghisla, S.; Cohen, R.A.; Bachschmid, M.M. Acetaminophen inhibits prostanoid synthesis by scavenging the PGHS-activator peroxynitrite. FASEB J. 2007, 22, 215–224. [Google Scholar] [CrossRef] [Green Version]
- Moore, B.B.; Peters-Golden, M. Opposing roles of leukotrienes and prostaglandins in fibrotic lung disease. Expert Rev. Clin. Immunol. 2006, 2, 87–100. [Google Scholar] [CrossRef]
- Chandrasekharan, J.A.; Sharma-Walia, N. Lipoxins: Nature’s way to resolve inflammation. J. Inflamm. Res. 2015, 8, 181–192. [Google Scholar] [CrossRef] [Green Version]
- Beers, M.F.; Mulugeta, S. The biology of the ABCA3 lipid transporter in lung health and disease. Cell Tissue Res. 2017, 367, 481–493. [Google Scholar] [CrossRef]
- Zhao, L.; Li, Y.; Ding, Q.; Li, Y.; Chen, Y.; Ruan, X.Z. CD36 senses dietary lipids and regulates lipids homeostasis in the intestine. Front. Physiol. 2021, 12, 669279. [Google Scholar] [CrossRef] [PubMed]
- Silverstein, R.L.; Febbraio, M. CD36, a scavenger receptor involved in immunity, metabolism, angiogenesis, and behavior. Sci. Signal. 2009, 2, re3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, S.; Chaudhary, O.; Rodríguez-Morales, P.; Sun, X.; Chen, D.; Zappasodi, R.; Xu, Z.; Pinto, A.F.; Williams, A.; Schulze, I.; et al. Uptake of oxidized lipids by the scavenger receptor CD36 promotes lipid peroxidation and dysfunction in CD8+ T cells in tumors. Immunity 2021, 54, 1561–1577.e7. [Google Scholar] [CrossRef] [PubMed]
- Banesh, S.; Trivedi, V. Therapeutic potentials of scavenger receptor CD36 Mediated innate immune responses against infectious and non-infectious diseases. Curr. Drug. Discov. Technol. 2020, 17, 299–317. [Google Scholar] [CrossRef] [PubMed]
- Dobri, A.-M.; Dudău, M.; Enciu, A.-M.; Hinescu, M.E. CD36 in Alzheimer’s disease: An overview of molecular mechanisms and therapeutic targeting. Neuroscience 2021, 453, 301–311. [Google Scholar] [CrossRef]
- Ogretmen, B. Sphingolipid metabolism in cancer signalling and therapy. Nat. Rev. Cancer 2018, 18, 33–50. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Larrauri, A.; Presa, N.; Dominguez-Herrera, A.; Ouro, A.; Trueba, M.; Gomez-Muñoz, A. Role of bioactive sphingolipids in physiology and pathology. Essays Biochem. 2020, 64, 579–589. [Google Scholar] [CrossRef]
- Levy, M.; Futerman, A.H. Mammalian ceramide synthases. IUBMB Life 2010, 62, 347–356. [Google Scholar] [CrossRef]
- Mullen, T.D.; Hannun, Y.A.; Obeid, L.M. Ceramide synthases at the centre of sphingolipid metabolism and biology. Biochem. J. 2012, 441, 789–802. [Google Scholar] [CrossRef] [Green Version]
- Wang, E.; He, X.; Zeng, M. The Role of S1P and the related signaling pathway in the development of tissue fibrosis. Front. Pharmacol. 2018, 9, 1504. [Google Scholar] [CrossRef]
- Cuvillier, O. Sphingosine 1-phosphate receptors: From biology to physiopathology. Med. Sci. 2012, 28, 951–957. [Google Scholar] [CrossRef] [Green Version]
- Cuvillier, O. Sphingosine in apoptosis signaling. Biochim. Biophys. Acta 2002, 1585, 153–162. [Google Scholar] [CrossRef]
- Brizuela, L.; Ader, I.; Mazerolles, C.; Bocquet, M.; Malavaud, B.; Cuvillier, O. First evidence of sphingosine 1-phosphate lyase protein expression and activity downregulation in human neoplasm: Implication for resistance to therapeutics in prostate cancer. Mol. Cancer Ther. 2012, 11, 1841–1851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceccom, J.; Loukh, N.; Lauwers-Cances, V.; Touriol, C.; Nicaise, Y.; Gentil, C.; Uro-Coste, E.; Pitson, S.; Maurage, C.A.; Duyckaerts, C.; et al. Reduced sphingosine kinase-1 and enhanced sphingosine 1-phosphate lyase expression demonstrate deregulated sphingosine 1-phosphate signaling in Alzheimer’s disease. Acta Neuropathol. Commun. 2014, 2, 12. [Google Scholar] [CrossRef]
- Holthuis, J.; Menon, A. Lipid landscapes and pipelines in membrane homeostasis. Nature 2014, 510, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Endo, A. A historical perspective on the discovery of statins. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2010, 86, 484–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elshourbagy, N.A.; Meyers, H.V.; Abdel-Meguid, S.S. Cholesterol: The good, the bad, and the ugly—Therapeutic targets for the treatment of dyslipidemia. Med. Princ. Pract. 2014, 23, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Summer, R.; Mora, A.L. Lipid metabolism: A new player in the conundrum of lung fibrosis. Am. J. Respir. Cell Mol. Biol. 2019, 61, 669–670. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Mizuno, T.; Sridharan, A.; Du, Y.; Guo, M.; Tang, J.; Wikenheiser-Brokamp, K.A.; Perl, A.-K.T.; Funari, V.A.; Gokey, J.J.; et al. Single-Cell RNA sequencing identifies diverse roles of epithelial cells in idiopathic pulmonary fibrosis. JCI Insight 2016, 1, e90558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manali, E.D.; Legendre, M.; Nathan, N.; Kannengiesser, C.; Coulomb-L’Hermine, A.; Tsiligiannis, T.; Tomos, P.; Griese, M.; Borie, R.; Clement, A.; et al. Bi-allelic missense ABCA3 mutations in a patient with childhood ILD who reached adulthood. ERJ Open Res. 2019, 5, 00066–02019. [Google Scholar] [CrossRef] [Green Version]
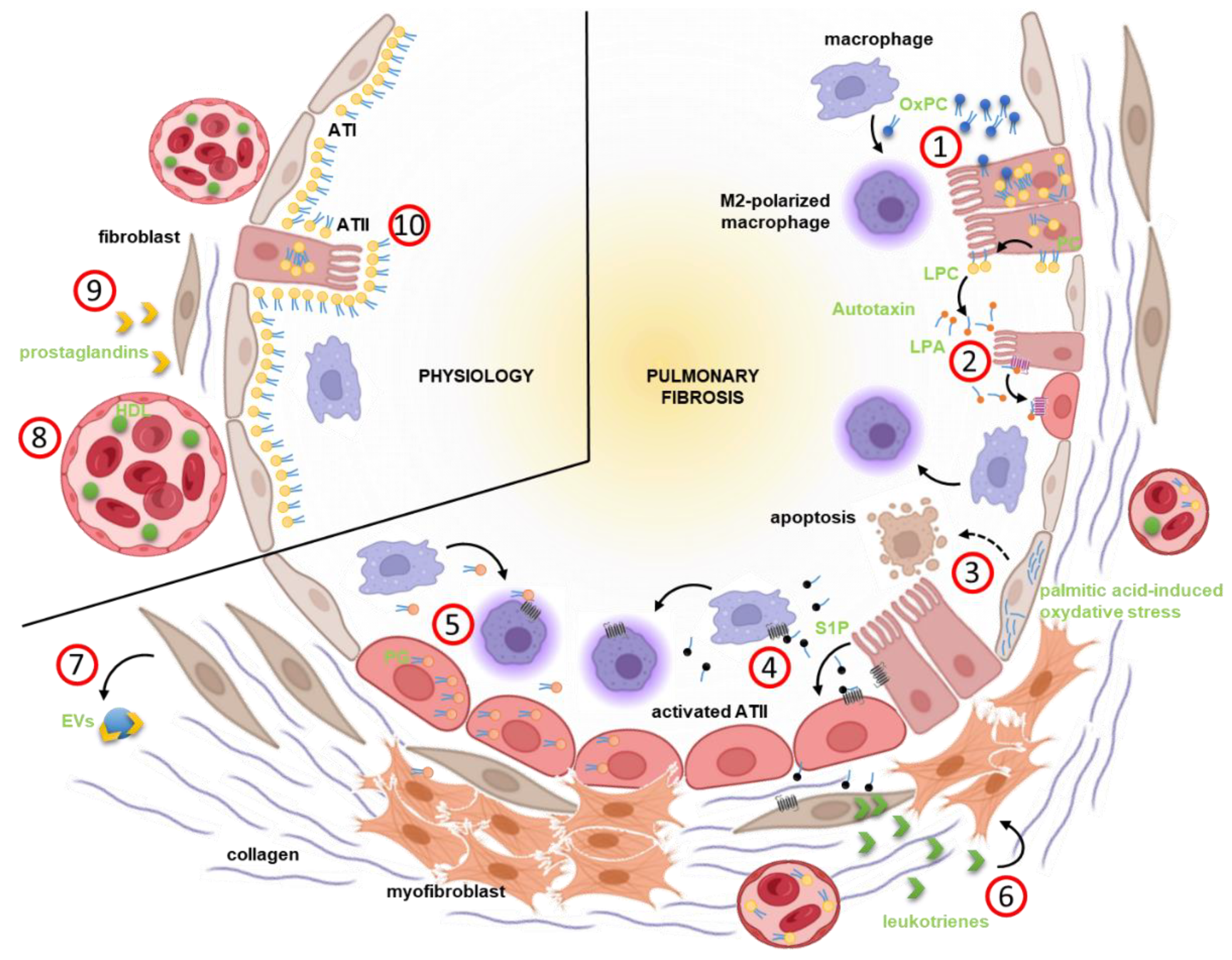
- Romero, F.; Shah, D.; Duong, M.; Penn, R.B.; Fessler, M.B.; Madenspacher, J.; Stafstrom, W.; Kavuru, M.; Lu, B.; Kallen, C.B.; et al. A pneumocyte-macrophage paracrine lipid axis drives the lung toward fibrosis. Am. J. Respir. Cell Mol. Biol. 2015, 53, 74–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angelidis, I.; Simon, L.M.; Fernandez, I.E.; Strunz, M.; Mayr, C.H.; Greiffo, F.R.; Tsitsiridis, G.; Ansari, M.; Graf, E.; Strom, T.-M.; et al. An atlas of the aging lung mapped by single cell transcriptomics and deep tissue proteomics. Nat. Commun. 2019, 10, 963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Agha, E.; Moiseenko, A.; Kheirollahi, V.; De Langhe, S.; Crnkovic, S.; Kwapiszewska, G.; Szibor, M.; Kosanovic, D.; Schwind, F.; Schermuly, R.T.; et al. Two-way conversion between lipogenic and myogenic fibroblastic phenotypes marks the progression and resolution of lung fibrosis. Cell Stem Cell 2017, 20, 571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
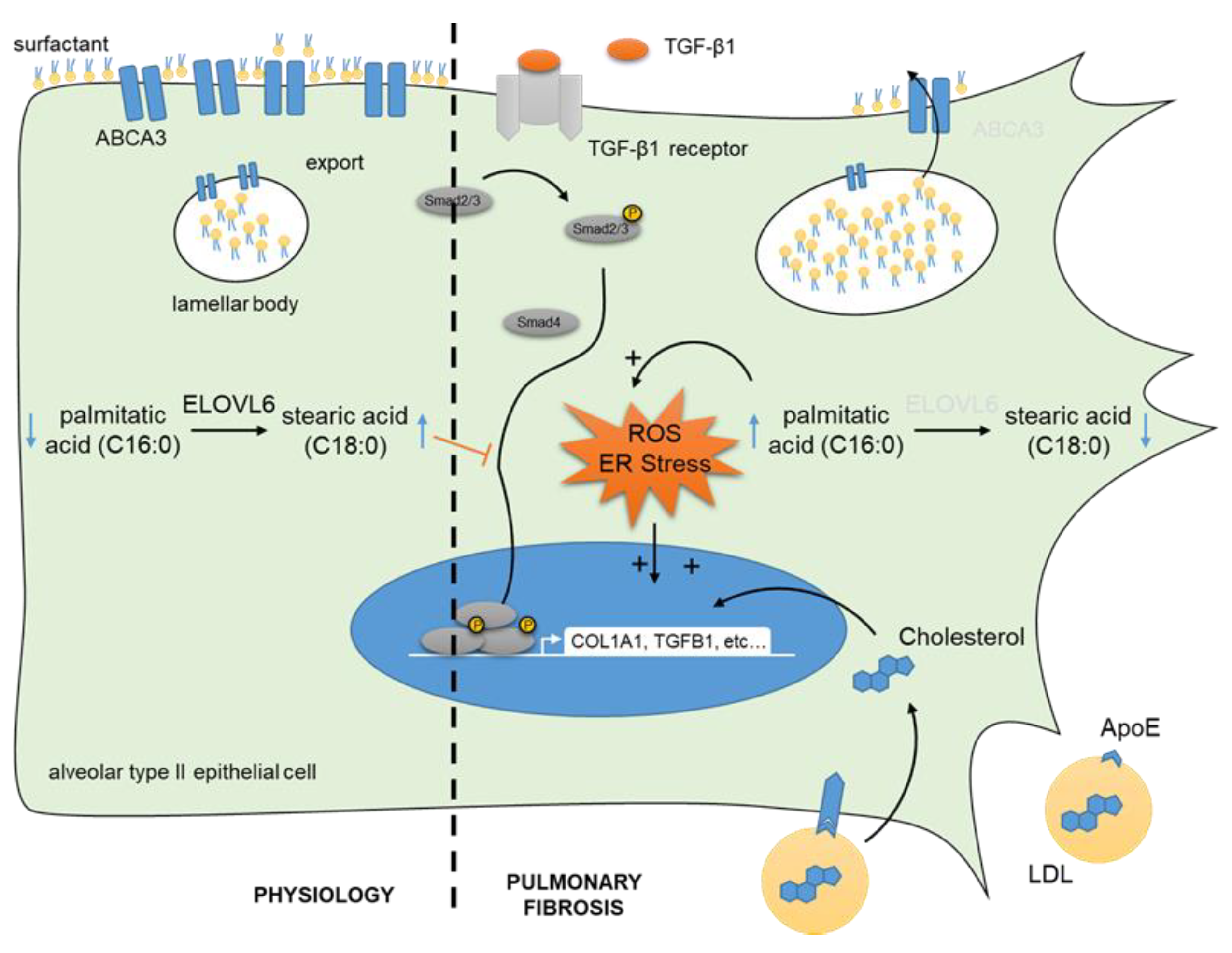
- Sunaga, H.; Matsui, H.; Ueno, M.; Maeno, T.; Iso, T.; Syamsunarno, M.R.A.A.; Anjo, S.; Matsuzaka, T.; Shimano, H.; Yokoyama, T.; et al. Deranged fatty acid composition causes pulmonary fibrosis in Elovl6-deficient mice. Nat. Commun. 2013, 4, 2563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oikonomou, N.; Mouratis, M.-A.; Tzouvelekis, A.; Kaffe, E.; Valavanis, C.; Vilaras, G.; Karameris, A.; Prestwich, G.D.; Bouros, D.; Aidinis, V. Pulmonary autotaxin expression contributes to the pathogenesis of pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2012, 47, 566–574. [Google Scholar] [CrossRef] [PubMed]
- Wiley, C.D.; Brumwell, A.N.; Davis, S.S.; Jackson, J.R.; Valdovinos, A.; Calhoun, C.; Alimirah, F.; Castellanos, C.A.; Ruan, R.; Wei, Y.; et al. Secretion of leukotrienes by senescent lung fibroblasts promotes pulmonary fibrosis. JCI Insight 2019, 4, 130056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S.K.; Wettlaufer, S.H.; Hogaboam, C.M.; Flaherty, K.R.; Martinez, F.J.; Myers, J.L.; Colby, T.V.; Travis, W.D.; Toews, G.B.; Peters-Golden, M. Variable prostaglandin E2 resistance in fibroblasts from patients with usual interstitial pneumonia. Am. J. Respir. Crit. Care Med. 2008, 177, 66–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berhan, A.; Harris, T.; Jaffar, J.; Jativa, F.; Langenbach, S.; Lönnstedt, I.; Alhamdoosh, M.; Ng, M.; Lee, P.; Westall, G.; et al. Cellular microenvironment stiffness regulates eicosanoid production and signaling pathways. Am. J. Respir. Cell Mol. Biol. 2020, 63, 819–830. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.S.; Berdyshev, E.V.; Tran, J.T.; Xie, L.; Chen, J.; Ebenezer, D.L.; Mathew, B.; Gorshkova, I.; Zhang, W.; Reddy, S.P.; et al. Sphingosine-1-phosphate lyase is an endogenous suppressor of pulmonary fibrosis: Role of S1P signalling and autophagy. Thorax 2015, 70, 1138–1148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zemski Berry, K.A.; Murphy, R.C.; Kosmider, B.; Mason, R.J. Lipidomic characterization and localization of phospholipids in the human lung. J. Lipid Res. 2017, 58, 926–933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, K.; Tanaka, N.; Ikari, J.; Suzuki, M.; Anazawa, R.; Abe, M.; Saito, Y.; Tatsumi, K. Comprehensive lipid profiling of bleomycin-induced lung injury. J. Appl. Toxicol. 2019, 39, 658–671. [Google Scholar] [CrossRef]
- Kulkarni, Y.M.; Dutta, S.; Iyer, A.K.V.; Wright, C.A.; Ramesh, V.; Kaushik, V.; Semmes, O.J.; Azad, N. A lipidomics approach to identifying key lipid species involved in VEGF-inhibitor mediated attenuation of bleomycin-induced pulmonary fibrosis. Proteom. Clin. Appl. 2018, 12, e1700086. [Google Scholar] [CrossRef] [PubMed]
- Chu, S.G.; Villalba, J.A.; Liang, X.; Xiong, K.; Tsoyi, K.; Ith, B.; Ayaub, E.A.; Tatituri, R.V.; Byers, D.E.; Hsu, F.-F.; et al. Palmitic acid-rich high-fat diet exacerbates experimental pulmonary fibrosis by modulating endoplasmic reticulum stress. Am. J. Respir. Cell Mol. Biol. 2019, 61, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-S.; Yoo, H.J.; Lee, K.M.; Song, H.E.; Kim, S.J.; Lee, J.O.; Hwang, J.J.; Song, J.W. Stearic acid attenuates profibrotic signalling in idiopathic pulmonary fibrosis. Respirology 2021, 26, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.-P.; Hsu, C.-L.; Fan, L.-C.; Huang, Z.; Bhatia, D.; Chen, Y.-J.; Hisata, S.; Cho, S.J.; Nakahira, K.; Imamura, M.; et al. Mitofusins regulate lipid metabolism to mediate the development of lung fibrosis. Nat. Commun. 2019, 10, 3390. [Google Scholar] [CrossRef]
- Romero, F.; Hong, X.; Shah, D.; Kallen, C.B.; Rosas, I.; Guo, Z.; Schriner, D.; Barta, J.; Shaghaghi, H.; Hoek, J.B.; et al. Lipid synthesis is required to resolve endoplasmic reticulum stress and limit fibrotic responses in the lung. Am. J. Respir. Cell Mol. Biol. 2018, 59, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, T.; Sugiura, H.; Koarai, A.; Ichikawa, T.; Minakata, Y.; Matsunaga, K.; Nakanishi, M.; Hirano, T.; Akamatsu, K.; Yanagisawa, S.; et al. Increase of 27-hydroxycholesterol in the airways of patients with COPD: Possible role of 27-hydroxycholesterol in tissue fibrosis. Chest 2012, 142, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, T.; Sugiura, H.; Koarai, A.; Kikuchi, T.; Hiramatsu, M.; Kawabata, H.; Akamatsu, K.; Hirano, T.; Nakanishi, M.; Matsunaga, K.; et al. 25-hydroxycholesterol promotes fibroblast-mediated tissue remodeling through NF-ΚB dependent pathway. Exp. Cell Res. 2013, 319, 1176–1186. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Gordon, E.M.; Figueroa, D.M.; Barochia, A.V.; Levine, S.J. Emerging roles of apolipoprotein E and apolipoprotein A-I in the pathogenesis and treatment of lung disease. Am. J. Respir. Cell Mol. Biol. 2016, 55, 159–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Z.; Jin, J.; Wang, Y.; Sun, J. High density lipoprotein promoting proliferation and migration of type II alveolar epithelial cells during inflammation state. Lipids Health Dis. 2017, 16, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pison, U.; Max, M.; Neuendank, A.; Weissbach, S.; Pietschmann, S. Host defence capacities of pulmonary surfactant: Evidence for “non-Surfactant” functions of the surfactant system. Eur. J. Clin. Investig. 1994, 24, 586–599. [Google Scholar] [CrossRef] [PubMed]
- Gille, C.; Spring, B.; Bernhard, W.; Gebhard, C.; Basile, D.; Lauber, K.; Poets, C.F.; Orlikowsky, T.W. Differential effect of surfactant and its saturated phosphatidylcholines on human blood macrophages. J. Lipid Res. 2007, 48, 307–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griese, M.; Bonella, F.; Costabel, U.; de Blic, J.; Tran, N.-B.; Liebisch, G. Quantitative lipidomics in pulmonary alveolar proteinosis. Am. J. Respir. Crit. Care Med. 2019, 200, 881–887. [Google Scholar] [CrossRef] [PubMed]
- Esteves, P.; Blanc, L.; Celle, A.; Dupin, I.; Maurat, E.; Amoedo, N.; Cardouat, G.; Ousova, O.; Gales, L.; Bellvert, F.; et al. Crucial role of fatty acid oxidation in asthmatic bronchial smooth muscle remodelling. Eur. Respir. J. 2021, 58, 2004252. [Google Scholar] [CrossRef] [PubMed]
- Balijepalli, P.; Sitton, C.C.; Meier, K.E. Lysophosphatidic acid signaling in cancer cells: What makes LPA so special? Cells 2021, 10, 2059. [Google Scholar] [CrossRef] [PubMed]
- Tager, A.M.; LaCamera, P.; Shea, B.S.; Campanella, G.S.; Selman, M.; Zhao, Z.; Polosukhin, V.; Wain, J.; Karimi-Shah, B.A.; Kim, N.D.; et al. The lysophosphatidic acid receptor LPA1 links pulmonary fibrosis to lung injury by mediating fibroblast recruitment and vascular leak. Nat. Med. 2008, 14, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Tang, N.; Zhao, Y.; Feng, R.; Liu, Y.; Wang, S.; Wei, W.; Ding, Q.; An, M.S.; Wen, J.; Li, L. Lysophosphatidic acid accelerates lung fibrosis by inducing differentiation of mesenchymal stem cells into myofibroblasts. J. Cell Mol. Med. 2014, 18, 156–169. [Google Scholar] [CrossRef] [PubMed]
- Magkrioti, C.; Galaris, A.; Kanellopoulou, P.; Stylianaki, E.-A.; Kaffe, E.; Aidinis, V. Autotaxin and chronic inflammatory diseases. J. Autoimmun. 2019, 104, 102327. [Google Scholar] [CrossRef] [PubMed]
- Funke, M.; Knudsen, L.; Lagares, D.; Ebener, S.; Probst, C.K.; Fontaine, B.A.; Franklin, A.; Kellner, M.; Kühnel, M.; Matthieu, S.; et al. Lysophosphatidic acid signaling through the lysophosphatidic acid-1 receptor is required for alveolarization. Am. J. Respir. Cell Mol. Biol. 2016, 55, 105–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taleb, S.J.; Wei, J.; Mialki, R.K.; Dong, S.; Li, Y.; Zhao, J.; Zhao, Y. A blocking peptide stabilizes lysophosphatidic acid receptor 1 and promotes lysophosphatidic acid-induced cellular responses. J. Cell. Biochem. 2021, 122, 827–834. [Google Scholar] [CrossRef] [PubMed]
- Charbeneau, R.P.; Peters-Golden, M. Eicosanoids: Mediators and therapeutic targets in fibrotic lung disease. Clin. Sci. 2005, 108, 479–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zannikou, M.; Barbayianni, I.; Fanidis, D.; Grigorakaki, T.; Vlachopoulou, E.; Konstantopoulos, D.; Fousteri, M.; Nikitopoulou, I.; Kotanidou, A.; Kaffe, E.; et al. MAP3K8 regulates Cox-2-mediated prostaglandin E2 production in the lung and suppresses pulmonary inflammation and fibrosis. J. Immunol. 2021, 206, 607–620. [Google Scholar] [CrossRef] [PubMed]
- Evans, I.C.; Barnes, J.L.; Garner, I.M.; Pearce, D.R.; Maher, T.M.; Shiwen, X.; Renzoni, E.A.; Wells, A.U.; Denton, C.P.; Laurent, G.J.; et al. Epigenetic regulation of cyclooxygenase-2 by methylation of C8orf4 in pulmonary fibrosis. Clin. Sci. 2016, 130, 575–586. [Google Scholar] [CrossRef] [Green Version]
- Hirata, H.; Arima, M.; Fukushima, Y.; Sugiyama, K.; Tokuhisa, T.; Fukuda, T. Leukotriene C4 aggravates bleomycin-induced pulmonary fibrosis in mice. Respirology 2013, 18, 674–681. [Google Scholar] [CrossRef] [PubMed]
- Peters-Golden, M.; Bailie, M.; Marshall, T.; Wilke, C.; Phan, S.H.; Toews, G.B.; Moore, B.B. Protection from pulmonary fibrosis in leukotriene-deficient mice. Am. J. Respir. Crit. Care Med. 2002, 165, 229–235. [Google Scholar] [CrossRef]
- Shimbori, C.; Shiota, N.; Okunishi, H. Effects of montelukast, a cysteinyl-leukotriene type 1 receptor antagonist, on the pathogenesis of bleomycin-induced pulmonary fibrosis in mice. Eur. J. Pharmacol. 2011, 650, 424–430. [Google Scholar] [CrossRef]
- Izumo, T.; Kondo, M.; Nagai, A. Effects of a leukotriene B4 receptor antagonist on bleomycin-induced pulmonary fibrosis. Eur. Respir. J. 2009, 34, 1444–1451. [Google Scholar] [CrossRef]
- Hur, J.; Kang, J.Y.; Rhee, C.K.; Kim, Y.K.; Lee, S.Y. The leukotriene receptor antagonist pranlukast attenuates airway remodeling by suppressing TGF-β signaling. Pulm. Pharmacol. Ther. 2018, 48, 5–14. [Google Scholar] [CrossRef]
- Kida, T.; Ayabe, S.; Omori, K.; Nakamura, T.; Maehara, T.; Aritake, K.; Urade, Y.; Murata, T. Prostaglandin D2 attenuates bleomycin-induced lung inflammation and pulmonary fibrosis. PLoS ONE 2016, 11, e0167729. [Google Scholar] [CrossRef] [Green Version]
- Dackor, R.T.; Cheng, J.; Voltz, J.W.; Card, J.W.; Ferguson, C.D.; Garrett, R.C.; Bradbury, J.A.; DeGraff, L.M.; Lih, F.B.; Tomer, K.B.; et al. Prostaglandin E₂ protects murine lungs from bleomycin-induced pulmonary fibrosis and lung dysfunction. Am. J. Physiol. Lung Cell Mol. Physiol. 2011, 301, L645–L655. [Google Scholar] [CrossRef] [Green Version]
- Lovgren, A.K.; Jania, L.A.; Hartney, J.M.; Parsons, K.K.; Audoly, L.P.; Fitzgerald, G.A.; Tilley, S.L.; Koller, B.H. COX-2-derived prostacyclin protects against bleomycin-induced pulmonary fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2006, 291, L144–L156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kountz, T.S.; Jairaman, A.; Kountz, C.D.; Stauderman, K.A.; Schleimer, R.P.; Prakriya, M. Differential regulation of ATP- and UTP-evoked prostaglandin E2 and IL-6 production from human airway epithelial cells. J. Immunol. 2021, 207, 1275–1287. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, E.; Zeldich, E.; Weinreb, M.M.; Moses, O.; Nemcovsky, C.; Weinreb, M. Prostaglandin E2 inhibits the proliferation of human gingival fibroblasts via the EP2 Receptor and Epac. J. Cell Biochem. 2009, 108, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Wettlaufer, S.H.; Hogaboam, C.; Aronoff, D.M.; Peters-Golden, M. Prostaglandin E(2) inhibits collagen expression and proliferation in patient-derived normal lung fibroblasts via E Prostanoid 2 receptor and CAMP signaling. Am. J. Physiol. Lung Cell Mol. Physiol. 2007, 292, L405–L413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolodsick, J.E.; Peters-Golden, M.; Larios, J.; Toews, G.B.; Thannickal, V.J.; Moore, B.B. Prostaglandin E2 inhibits fibroblast to myofibroblast transition via E. prostanoid receptor 2 signaling and cyclic adenosine monophosphate elevation. Am. J. Respir. Cell Mol. Biol. 2003, 29, 537–544. [Google Scholar] [CrossRef]
- Mukherjee, S.; Sheng, W.; Michkov, A.; Sriarm, K.; Sun, R.; Dvorkin-Gheva, A.; Insel, P.A.; Janssen, L.J. Prostaglandin E2 inhibits profibrotic function of human pulmonary fibroblasts by disrupting Ca2+ signaling. Am. J. Physiol. Lung Cell Mol. Physiol. 2019, 316, L810–L821. [Google Scholar] [CrossRef] [PubMed]
- Epa, A.P.; Thatcher, T.H.; Pollock, S.J.; Wahl, L.A.; Lyda, E.; Kottmann, R.M.; Phipps, R.P.; Sime, P.J. Normal human lung epithelial cells inhibit transforming growth factor-β induced myofibroblast differentiation via prostaglandin E2. PLoS ONE 2015, 10, e0135266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lacy, S.H.; Epa, A.P.; Pollock, S.J.; Woeller, C.F.; Thatcher, T.H.; Phipps, R.P.; Sime, P.J. Activated human T lymphocytes inhibit TGFβ-induced fibroblast to myofibroblast differentiation via prostaglandins D2 and E2. Am. J. Physiol. Lung Cell Mol. Physiol. 2018, 314, L569–L582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S.K.; Fisher, A.S.; Scruggs, A.M.; White, E.S.; Hogaboam, C.M.; Richardson, B.C.; Peters-Golden, M. Hypermethylation of PTGER2 confers prostaglandin E2 resistance in fibrotic fibroblasts from humans and mice. Am. J. Pathol. 2010, 177, 2245–2255. [Google Scholar] [CrossRef]
- Bauman, K.A.; Wettlaufer, S.H.; Okunishi, K.; Vannella, K.M.; Stoolman, J.S.; Huang, S.K.; Courey, A.J.; White, E.S.; Hogaboam, C.M.; Simon, R.H.; et al. The antifibrotic effects of plasminogen activation occur via prostaglandin E2 synthesis in humans and mice. J. Clin. Investig. 2010, 120, 1950–1960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okunishi, K.; Sisson, T.H.; Huang, S.K.; Hogaboam, C.M.; Simon, R.H.; Peters-Golden, M. Plasmin overcomes resistance to prostaglandin E2 in fibrotic lung fibroblasts by reorganizing protein kinase a signaling. J. Biol. Chem. 2011, 286, 32231–32243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cartier, A.; Hla, T. Sphingosine 1-phosphate: Lipid signaling in pathology and therapy. Science 2019, 366, eaar5551. [Google Scholar] [CrossRef]
- Petrache, I.; Berdyshev, E.V. Ceramide signaling and metabolism in pathophysiological states of the lung. Annu. Rev. Physiol. 2016, 78, 463–480. [Google Scholar] [CrossRef] [PubMed]
- Schwalm, S.; Pfeilschifter, J.; Huwiler, A. Sphingosine-1-phosphate: A janus-faced mediator of fibrotic diseases. Biochim. Biophys. Acta 2013, 1831, 239–250. [Google Scholar] [CrossRef] [PubMed]
- Milara, J.; Navarro, R.; Juan, G.; Peiró, T.; Serrano, A.; Ramón, M.; Morcillo, E.; Cortijo, J. Sphingosine-1-phosphate is increased in patients with idiopathic pulmonary fibrosis and mediates epithelial to mesenchymal transition. Thorax 2012, 67, 147–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhami, R.; He, X.; Schuchman, E.H. Acid sphingomyelinase deficiency attenuates bleomycin-induced lung inflammation and fibrosis in mice. Cell Physiol. Biochem. 2010, 26, 749–760. [Google Scholar] [CrossRef] [PubMed]
- Tatler, A.L.; Jenkins, G. Sphingosine-1-phosphate metabolism: Can its enigmatic lyase promote the autophagy of fibrosis? Thorax 2015, 70, 1106–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, L.; Wu, X.; Li, X.; Ni, X.; Gu, W.; Wang, X.; Ji, H.; Hu, L.; Zhu, L. S1PR3 deficiency alleviates radiation-induced pulmonary fibrosis through the regulation of epithelial-mesenchymal transition by targeting MiR-495-3p. J. Cell Physiol. 2020, 235, 2310–2324. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.S.; Sudhadevi, T.; Fu, P.; Punathil-Kannan, P.-K.; Ebenezer, D.L.; Ramchandran, R.; Putherickal, V.; Cheresh, P.; Zhou, G.; Ha, A.W.; et al. Sphingosine kinase 1/S1P signaling contributes to pulmonary fibrosis by activating Hippo/YAP pathway and mitochondrial reactive oxygen species in lung fibroblasts. Int. J. Mol. Sci. 2020, 21, 2064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knipe, R.S.; Spinney, J.J.; Abe, E.A.; Probst, C.K.; Franklin, A.; Logue, A.; Giacona, F.; Drummond, M.; Griffith, J.; Brazee, P.L.; et al. Endothelial-specific loss of sphingosine-1-phosphate receptor 1 increases vascular permeability and exacerbates bleomycin-induced pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2021, 66, 38–52. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-J.; Im, D.-S. Deficiency of sphingosine-1-phosphate receptor 2 (S1P2) attenuates bleomycin-induced pulmonary fibrosis. Biomol. Ther. 2019, 27, 318–326. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Okamoto, Y.; Asano, Y.; Ishimaru, K.; Aki, S.; Yoshioka, K.; Takuwa, N.; Wada, T.; Inagaki, Y.; Takahashi, C.; et al. Sphingosine-1-phosphate receptor-2 facilitates pulmonary fibrosis through potentiating IL-13 pathway in macrophages. PLoS ONE 2018, 13, e0197604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Dyken, S.J.; Locksley, R.M. Interleukin-4- and Interleukin-13-mediated alternatively activated macrophages: Roles in homeostasis and disease. Annu. Rev. Immunol. 2013, 31, 317–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin-Medina, A.; Lehmann, M.; Burgy, O.; Hermann, S.; Baarsma, H.A.; Wagner, D.E.; De Santis, M.M.; Ciolek, F.; Hofer, T.P.; Frankenberger, M.; et al. Increased extracellular vesicles mediate WNT5A signaling in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2018, 198, 1527–1538. [Google Scholar] [CrossRef]
- Shaba, E.; Landi, C.; Carleo, A.; Vantaggiato, L.; Paccagnini, E.; Gentile, M.; Bianchi, L.; Lupetti, P.; Bargagli, E.; Prasse, A.; et al. Proteome characterization of BALF extracellular vesicles in idiopathic pulmonary fibrosis: Unveiling undercover molecular pathways. Int J. Mol. Sci. 2021, 22, 5696. [Google Scholar] [CrossRef]
- Njock, M.-S.; Guiot, J.; Henket, M.A.; Nivelles, O.; Thiry, M.; Dequiedt, F.; Corhay, J.-L.; Louis, R.E.; Struman, I. Sputum Exosomes: Promising biomarkers for idiopathic pulmonary fibrosis. Thorax 2019, 74, 309–312. [Google Scholar] [CrossRef] [Green Version]
- Parimon, T.; Yao, C.; Habiel, D.M.; Ge, L.; Bora, S.A.; Brauer, R.; Evans, C.M.; Xie, T.; Alonso-Valenteen, F.; Medina-Kauwe, L.K.; et al. Syndecan-1 promotes lung fibrosis by regulating epithelial reprogramming through extracellular vesicles. JCI Insight 2019, 5, 129359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, G.; Maremanda, K.P.; Campos, M.; Chand, H.S.; Li, F.; Hirani, N.; Haseeb, M.A.; Li, D.; Rahman, I. Distinct Exosomal MiRNA Profiles from BALF and lung tissue of COPD and IPF patients. Int. J. Mol. Sci. 2021, 22, 11830. [Google Scholar] [CrossRef] [PubMed]
- d’Alessandro, M.; Soccio, P.; Bergantini, L.; Cameli, P.; Scioscia, G.; Foschino Barbaro, M.P.; Lacedonia, D.; Bargagli, E. Extracellular vesicle surface signatures in IPF patients: A multiplex bead-based flow cytometry approach. Cells 2021, 10, 1045. [Google Scholar] [CrossRef] [PubMed]
- Guiot, J.; Cambier, M.; Boeckx, A.; Henket, M.; Nivelles, O.; Gester, F.; Louis, E.; Malaise, M.; Dequiedt, F.; Louis, R.; et al. macrophage-derived exosomes attenuate fibrosis in airway epithelial cells through delivery of antifibrotic MiR-142-3p. Thorax 2020, 75, 870–881. [Google Scholar] [CrossRef] [PubMed]
- Kadota, T.; Fujita, Y.; Araya, J.; Watanabe, N.; Fujimoto, S.; Kawamoto, H.; Minagawa, S.; Hara, H.; Ohtsuka, T.; Yamamoto, Y.; et al. Human bronchial epithelial cell-derived extracellular vesicle therapy for pulmonary fibrosis via inhibition of TGF-β-WNT crosstalk. J. Extracell. Vesicles 2021, 10, e12124. [Google Scholar] [CrossRef] [PubMed]
- Hough, K.P.; Wilson, L.S.; Trevor, J.L.; Strenkowski, J.G.; Maina, N.; Kim, Y.-I.; Spell, M.L.; Wang, Y.; Chanda, D.; Dager, J.R.; et al. Unique lipid signatures of extracellular vesicles from the airways of asthmatics. Sci. Rep. 2018, 8, 10340. [Google Scholar] [CrossRef] [PubMed]
- Torregrosa Paredes, P.; Esser, J.; Admyre, C.; Nord, M.; Rahman, Q.K.; Lukic, A.; Rådmark, O.; Grönneberg, R.; Grunewald, J.; Eklund, A.; et al. Bronchoalveolar lavage fluid exosomes contribute to cytokine and leukotriene production in allergic asthma. Allergy 2012, 67, 911–919. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Chong, S.G.; Upagupta, C.; Yanagihara, T.; Saito, T.; Shimbori, C.; Bellaye, P.-S.; Nishioka, Y.; Kolb, M.R. Fibrotic extracellular matrix induces release of extracellular vesicles with pro-fibrotic MiRNA from fibrocytes. Thorax 2021, 76, 895–906. [Google Scholar] [CrossRef]
- Lacy, S.H.; Woeller, C.F.; Thatcher, T.H.; Pollock, S.J.; Small, E.M.; Sime, P.J.; Phipps, R.P. Activated Human Lung Fibroblasts Produce Extracellular Vesicles with Antifibrotic Prostaglandins. Am. J. Respir. Cell Mol. Biol. 2019, 60, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Mayr, C.H.; Simon, L.M.; Leuschner, G.; Ansari, M.; Schniering, J.; Geyer, P.E.; Angelidis, I.; Strunz, M.; Singh, P.; Kneidinger, N.; et al. Integrative analysis of cell state changes in lung fibrosis with peripheral protein biomarkers. EMBO Mol. Med. 2021, 13, e12871. [Google Scholar] [CrossRef]
- Barochia, A.V.; Kaler, M.; Weir, N.; Gordon, E.M.; Figueroa, D.M.; Yao, X.; Lemma WoldeHanna, M.; Sampson, M.; Remaley, A.T.; Grant, G.; et al. Serum levels of small HDL particles are negatively correlated with death or lung transplantation in an observational study of idiopathic pulmonary fibrosis. Eur. Respir. J. 2021, 58, 2004053. [Google Scholar] [CrossRef] [PubMed]
- Podolanczuk, A.J.; Raghu, G.; Tsai, M.Y.; Kawut, S.M.; Peterson, E.; Sonti, R.; Rabinowitz, D.; Johnson, C.; Barr, R.G.; Hinckley Stukovsky, K.; et al. Cholesterol, lipoproteins and subclinical interstitial lung disease: The MESA study. Thorax 2017, 72, 472–474. [Google Scholar] [CrossRef] [Green Version]
- Mallampalli, R.K.; Ryan, A.J.; Carroll, J.L.; Osborne, T.F.; Thomas, C.P. Lipid deprivation increases surfactant phosphatidylcholine synthesis via a sterol-sensitive regulatory element within the CTP: Phosphocholine cytidylyltransferase promoter. Biochem. J. 2002, 362, 81–88. [Google Scholar] [CrossRef]
- Ryan, A.J.; Medh, J.D.; McCoy, D.M.; Salome, R.G.; Mallampalli, R.K. Maternal loading with very low-density lipoproteins stimulates fetal surfactant synthesis. Am. J. Physiol. Lung Cell Mol. Physiol. 2002, 283, L310–L318. [Google Scholar] [CrossRef] [Green Version]
- Yan, F.; Wen, Z.; Wang, R.; Luo, W.; Du, Y.; Wang, W.; Chen, X. Identification of the lipid biomarkers from plasma in idiopathic pulmonary fibrosis by lipidomics. BMC Pulm. Med. 2017, 17, 174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nambiar, S.; Clynick, B.; How, B.S.; King, A.; Walters, E.H.; Goh, N.S.; Corte, T.J.; Trengove, R.; Tan, D.; Moodley, Y. There is detectable variation in the lipidomic profile between stable and progressive patients with idiopathic pulmonary fibrosis (IPF). Respir. Res. 2021, 22, 105. [Google Scholar] [CrossRef] [PubMed]
- Rangarajan, S.; Bone, N.B.; Zmijewska, A.A.; Jiang, S.; Park, D.W.; Bernard, K.; Locy, M.L.; Ravi, S.; Deshane, J.; Mannon, R.B.; et al. Metformin reverses established lung fibrosis in a bleomycin model. Nat. Med. 2018, 24, 1121–1127. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Cai, Y.; Zhang, W.; Chen, X. Quercetin ameliorates pulmonary fibrosis by inhibiting SphK1/S1P Signaling. Biochem. Cell Biol. 2018, 96, 742–751. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Ma, A.-Q.; Yang, L.; Dang, X.-M. Atorvastatin attenuates bleomycin-induced pulmonary fibrosis via suppressing INOS expression and the CTGF (CCN2)/ERK signaling pathway. Int. J. Mol. Sci. 2013, 14, 24476–24491. [Google Scholar] [CrossRef]
- Mammoliti, O.; Palisse, A.; Joannesse, C.; El Bkassiny, S.; Allart, B.; Jaunet, A.; Menet, C.; Coornaert, B.; Sonck, K.; Duys, I.; et al. Discovery of the S1P2 antagonist GLPG2938 (1-[2-Ethoxy-6-(Trifluoromethyl)-4-Pyridyl]-3-[[5-Methyl-6-[1-Methyl-3-(Trifluoromethyl)Pyrazol-4-Yl]Pyridazin-3-Yl]Methyl]Urea), a preclinical candidate for the treatment of idiopathic pulmonary fibrosis. J. Med. Chem. 2021, 64, 6037–6058. [Google Scholar] [CrossRef] [PubMed]
- Kreuter, M.; Bonella, F.; Maher, T.M.; Costabel, U.; Spagnolo, P.; Weycker, D.; Kirchgaessler, K.-U.; Kolb, M. Effect of statins on disease-related outcomes in patients with idiopathic pulmonary fibrosis. Thorax 2017, 72, 148–153. [Google Scholar] [CrossRef] [Green Version]
- M Lambert, E.; A Wuyts, W.; Yserbyt, J.; De Sadeleer, L.J. Statins: Cause of fibrosis or the opposite? Effect of cardiovascular drugs in idiopathic pulmonary fibrosis. Respir. Med. 2021, 176, 106259. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.; Xu, G.; Xiong, C.; Yang, X.; Yan, S.; Tao, Y.; Li, H.; Li, Y.; Yao, S.; Zhao, Y. Alpha-lipoic acid attenuates silica-induced pulmonary fibrosis by improving mitochondrial function via AMPK/PGC1α pathway activation in C57BL/6J Mice. Toxicol. Lett. 2021, 350, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Hao, Y.; Zhang, H.; Ying, W.; Li, D.; Ge, Y.; Ying, B.; Cheng, B.; Lian, Q.; Jin, S. Posttreatment with Protectin DX ameliorates bleomycin-induced pulmonary fibrosis and lung dysfunction in mice. Sci. Rep. 2017, 7, 46754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Q.; Wu, Y.; Zhao, F.; Wang, J. Maresin 1 inhibits transforming growth factor-Β1-induced proliferation, migration and differentiation in human lung fibroblasts. Mol. Med. Rep. 2017, 16, 1523–1529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, H.; Chan-Li, Y.; Collins, S.L.; Zhang, Y.; Hallowell, R.W.; Mitzner, W.; Horton, M.R. Pulmonary delivery of docosahexaenoic acid mitigates bleomycin-induced pulmonary fibrosis. BMC Pulm. Med. 2014, 14, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Xie, M.; Lu, C.; Mao, J.; Cao, Y.; Yang, Y.; Wei, Y.; Liu, X.; Cao, S.; Song, Y.; et al. Design and synthesis of leukotriene A4 hydrolase inhibitors to alleviate idiopathic pulmonary fibrosis and acute lung injury. Eur. J. Med. Chem. 2020, 203, 112614. [Google Scholar] [CrossRef]
- Ji, Y.-D.; Luo, Z.-L.; Chen, C.-X.; Li, B.; Gong, J.; Wang, Y.-X.; Chen, L.; Yao, S.-L.; Shang, Y. BML-111 suppresses TGF-Β1-Induced lung fibroblast activation in vitro and decreases experimental pulmonary fibrosis in vivo. Int. J. Mol. Med. 2018, 42, 3083–3092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ninou, I.; Kaffe, E.; Müller, S.; Budd, D.C.; Stevenson, C.S.; Ullmer, C.; Aidinis, V. Pharmacologic targeting of the ATX/LPA axis attenuates bleomycin-induced pulmonary fibrosis. Pulm. Pharmacol. Ther. 2018, 52, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Matralis, A.N.; Afantitis, A.; Aidinis, V. Development and therapeutic potential of autotaxin small molecule inhibitors: From bench to advanced clinical trials. Med. Res. Rev. 2019, 39, 976–1013. [Google Scholar] [CrossRef] [PubMed]
- Pascual, G.; Avgustinova, A.; Mejetta, S.; Martín, M.; Castellanos, A.; Attolini, C.S.-O.; Berenguer, A.; Prats, N.; Toll, A.; Hueto, J.A.; et al. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature 2017, 541, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Nath, A.; Chan, C. Genetic alterations in fatty acid transport and metabolism genes are associated with metastatic progression and poor prognosis of human cancers. Sci. Rep. 2016, 6, 18669. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Li, Y. CD36 tango in cancer: Signaling pathways and functions. Theranostics 2019, 9, 4893–4908. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chen, Y.; Lv, L.; Chen, J. Silencing CD36 gene expression results in the inhibition of latent-TGF-Beta1 activation and suppression of silica-induced lung fibrosis in the rat. Respir. Res. 2009, 10, 36. [Google Scholar] [CrossRef] [Green Version]
- Trappe, A.; Donnelly, S.C.; McNally, P.; Coppinger, J.A. Role of extracellular vesicles in chronic lung disease. Thorax 2021, 76, 1047–1056. [Google Scholar] [CrossRef] [PubMed]
- Yamada, M. Extracellular vesicles: Their emerging roles in the pathogenesis of respiratory diseases. Respir. Investig. 2021, 59, 302–311. [Google Scholar] [CrossRef]
- Abreu, S.C.; Lopes-Pacheco, M.; Weiss, D.J.; Rocco, P.R.M. Mesenchymal stromal cell-derived extracellular vesicles in lung diseases: Current status and perspectives. Front. Cell Dev. Biol. 2021, 9, 600711. [Google Scholar] [CrossRef] [PubMed]
- Massa, M.; Croce, S.; Campanelli, R.; Abbà, C.; Lenta, E.; Valsecchi, C.; Avanzini, M.A. Clinical Applications of Mesenchymal Stem/Stromal Cell Derived Extracellular Vesicles: Therapeutic Potential of an Acellular Product. Diagnostics 2020, 10, 999. [Google Scholar] [CrossRef] [PubMed]
- Dinh, P.C.; Paudel, D.; Brochu, H.; Popowski, K.D.; Gracieux, M.C.; Cores, J.; Huang, K.; Hensley, M.T.; Harrell, E.; Vandergriff, A.C.; et al. Inhalation of lung spheroid cell secretome and exosomes promotes lung repair in pulmonary fibrosis. Nat Commun. 2020, 11, 1064. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.L.; Lau, S.N.; Leaw, B.; Nguyen, H.P.T.; Salamonsen, L.A.; Saad, M.I.; Chan, S.T.; Zhu, D.; Krause, M.; Kim, C.; et al. Amnion epithelial cell-derived exosomes restrict lung injury and enhance endogenous lung repair. Stem Cells Transl. Med. 2018, 7, 180–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmer, S.M.; Snyder, L.; Todd, J.L.; Soule, B.; Christian, R.; Anstrom, K.; Luo, Y.; Gagnon, R.; Rosen, G. Randomized, double-blind, placebo-controlled, phase 2 trial of BMS-986020, a lysophosphatidic acid receptor antagonist for the treatment of idiopathic pulmonary fibrosis. Chest 2018, 154, 1061–1069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalil, N.; Manganas, H.; Ryerson, C.J.; Shapera, S.; Cantin, A.M.; Hernandez, P.; Turcotte, E.E.; Parker, J.M.; Moran, J.E.; Albert, G.R.; et al. Phase 2 clinical trial of PBI-4050 in patients with idiopathic pulmonary fibrosis. Eur. Respir. J. 2019, 53, 1800663. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, Q.T.; Nsaibia, M.J.; Sirois, M.G.; Calderone, A.; Tardif, J.-C.; Fen Shi, Y.; Ruiz, M.; Daneault, C.; Gagnon, L.; Grouix, B.; et al. PBI-4050 reduces pulmonary hypertension, lung fibrosis, and right ventricular dysfunction in heart failure. Cardiovasc. Res. 2020, 116, 171–182. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Proteins Associated with Lipid Metabolism | Regulation in Lung Fibrosis | Evidence | Disease/Model | Cell Type(s) | Reference |
|---|---|---|---|---|---|
| ATP Binding Cassette Subfamily A Member 3 | down | scRNAseq | patient, bleomycin model | ATII | [99] |
| Sterol Regulatory Element Binding Transcription Factor 2 | up | scRNAseq | bleomycin model | ATII, lipofibroblasts | [102] |
| Peroxisome Proliferator Activated Receptor Gamma | down | qPCR | IPF | lung tissue | [103] |
| Elongation of Long Chain Fatty Acids 6 | down | qPCR, IHC | patient, bleomycin model | ATII | [104] |
| Autotaxin | up | IHC, qPCR, ELISA | patient, bleomycin model | hyperplastic bronchiolar and alveolar epithelium, fibroblasts, macrophages | [105] |
| Arachidonate 5-Lipoxygenase | up | qPCR | bleomycin model | senescent cells | [106] |
| Leukotriene C4 Synthase | up | qPCR | bleomycin model | senescent cells | [106] |
| Prostaglandin D2 Synthase | up | qPCR | bleomycin model | senescent cells | [106] |
| Prostaglandin-Endoperoxide Synthase 2 | up | qPCR | bleomycin model | senescent cells | [106] |
| Prostaglandin E Synthase | up | qPCR | bleomycin model | senescent cells | [106] |
| Prostaglandin E Receptor 2 | down | western blot | IPF | fibroblasts | [107] |
| Prostaglandin E Synthase | down | IHC | IPF | epithelial cells, fibroblasts | [108] |
| Sphingosine-1-Phosphate Lyase 1 | up | IHC, western blot, qPCR | patient, bleomycin model | fibrotic tissue, fibroblasts, PBMCs | [109] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burgy, O.; Loriod, S.; Beltramo, G.; Bonniaud, P. Extracellular Lipids in the Lung and Their Role in Pulmonary Fibrosis. Cells 2022, 11, 1209. https://doi.org/10.3390/cells11071209
Burgy O, Loriod S, Beltramo G, Bonniaud P. Extracellular Lipids in the Lung and Their Role in Pulmonary Fibrosis. Cells. 2022; 11(7):1209. https://doi.org/10.3390/cells11071209
Chicago/Turabian StyleBurgy, Olivier, Sabrina Loriod, Guillaume Beltramo, and Philippe Bonniaud. 2022. "Extracellular Lipids in the Lung and Their Role in Pulmonary Fibrosis" Cells 11, no. 7: 1209. https://doi.org/10.3390/cells11071209