
Histone Methylases and Demethylases Regulating Antagonistic Methyl Marks: Changes Occurring in Cancer

Abstract
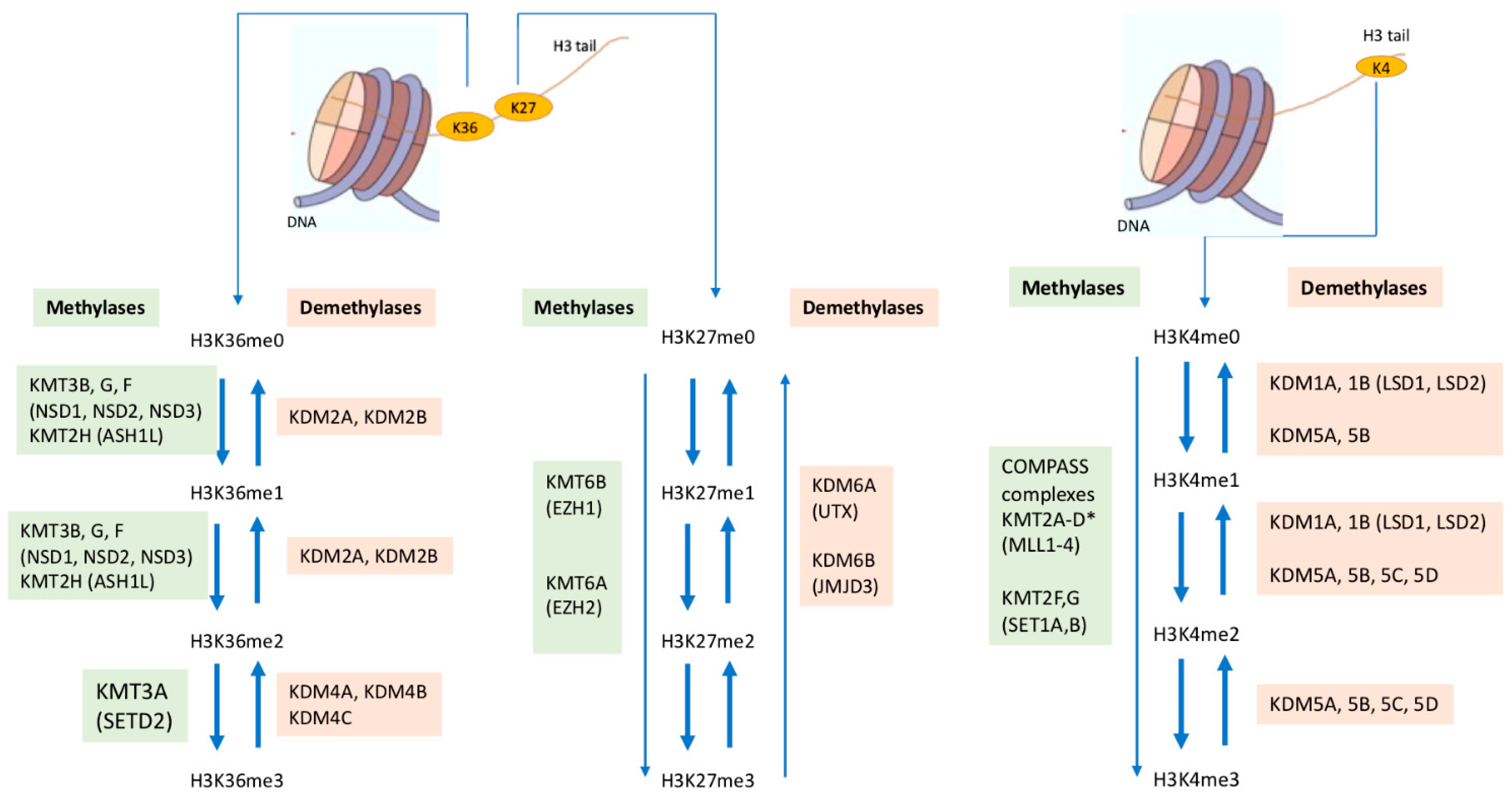
:1. Introduction
2. Regulation of Methylation of Lysine 27 on Histone 3
2.1. The PRC1 and PRC2 Complexes Methylating H3K27
2.2. Demethylation of H3K27me3
3. K27 Methylases and Demethylases in Cancer
3.1. The EZH2 Methylase: Expression, Mutations, and a Therapeutic Target

{kind=link}
{kind=link}
{kind=link}
| Type of Mutation or Translocation | Gene | Cancer Type | References |
|---|---|---|---|
| Inactivating Commonly found and high in bladder cancer | UTX/KDM6A Affecting H3K27 methylation and activation of enhancers | Common in T-ALL | [41] |
| KMT2C and KMT2D Affecting H3K4 mono-methylation at enhancers. | Bladder cancer ccRenal Carciomas non-Hodgkin’s lymphoma neuro-ectodermal tumours | [54,55,56] | |
| SETD2/KMT3A Inhibiting synthesis of H3K36me3 and therefore DNA damage response | ccRenal Carcinoma 32% Enteropathy-associated T cell lymphoma (EATL) | [47,54,57,58,59] | |
| EZH2/KMT6A Affects H3K27 methylation | Acute T cell leukaemia and myeloid disorders | [40,45,46,47] | |
| Activating mutations | EZH2/KMT6A Enhancing EZH2 function | Lymphomas | [48] |
| NSD2/KMT3B p.E1099K Increased enzyme activity | 10% of all ALL and especially paediatric B-ALL | [60,61] | |
| Duplication | MLL1/KMT2A partial tandem duplication (PTD) of exons 5 to 11 | 4–7% of AML | [62] |
| Translocations making fusion proteins Found in specific cancers | KMT2A-with 3′ domain from multiple genes including SEC components | 70% of infant leukaemia (ALL and AML) | [63,64] |
| NUP98-NSD1/KMT3B | 5% of AML | [65] | |
| NUP98-NSD3/KMT3F | AML (rare) | [65] | |
| NSD2 placed under the control of the strong IGH intronic Em enhancer t [4,5,6,7,8,9,10,11,12,13,14] | 15–20% Multiple myeloma 5–10% Paediatric ALL | [58,60,66] | |
| SX18/SSX | 90% Synovial sarcomas | [67] | |
| Mutations in non-canonical histones Found in specific cancers | H3F3A K27K to M G34 to R/V/D G34 to W/V | Childhood glioblastomas >90% Giant Cell tumours | [68,69,70,71] |
| H3F3B K36 to Methionine | >90% Chondroblastoma | [68] |
3.2. Mutations in Cancer Converting H3K27 to Methionine
3.3. KDM6A and KDM6B Demethylases in Cancer
- Clinical trials targeting EZH2 methylase (KMT6A) activity for cancer therapy are ongoing. Pre-clinical studies indicate that inhibiting interactions of compo-nents of PRC2 to destabilise the complex could provide an alternative targeting strategy.
- The mutation of lysine 27 to methionine in a non-canonical histone is seen in 70% of paediatric gliomas and inhibitors of EZH2 or BET proteins are therapeu-tic options.
- The main body of data indicates that the KDM6A/UTX demethylase protein acts as a tumour suppressor, and that tumours lacking or carrying mutations in KDM6A may, in some cancers, be targeted with either EZH2 or BET inhibitors.
| Functions in Normal Mouse Embryo Development and Viability Independent of Enzyme Activity | References | Enzyme Independent Effects in Cancer | References |
|---|---|---|---|
| KMT2A/MLL1 Progeny from mating of KMT2A heterozygous methylase null mice are produced at the expected Mendelian ratios. | [80] | EZH2/KMT6A Can activate transcription of genes targeted by the androgen receptor (AR) independently of the Polycomb complex which is required for methylase activity (Figure 3). | [50] |
| KDM6A(UTX) and UTY Male mice mutant for UTX can survive with UTY even though it has no demethylase activity. | [37] | LSD1/KDM1A Interaction of LSD1 with transcription factors activates expression of a network of genes favouring growth of CRPC prostate cancers (Figure 3). | [81,82] |
| KDM2B The ncPRC1.1 complex is recruited to chromatin in mouse ESCs by KDM2B through its CxxC-zinc finger (CxxC-ZF) domain, independently of its demethylase activity. | [25,26] | KDM2B Synovial Cancer: KDM2B is required for proliferation independently of demethylase activity (>90% of these cancers). Pancreatic Cancer: KDM2B can activate genes involved in ribosomal and mitochondrial function. | [67] [83,84] |
| KDM5B The ΔARID mice expressing KDM5B with no demethylase activity are viable and fertile. | [85,86] | KDM6A/UTX Mutations in domains other than the SET domain are seen in cancer suggesting a role of these domains in the tumour suppressor function. | [41] |
4. Regulation of Methylation of Lysine 36 on Histone 3
4.1. H3K36 Methylation: Methylases Associated with SET2 Domain
4.2. Demethylation of Methylated H3K36
5. Changes in Cancer Related to H3K36 Methylases
5.1. SETD2 (KMT3A): A Tumour Suppressor
5.2. Translocations and Mutations of NSD (KMT3B, G, F) Methylases
5.3. ASH1L in Leukaemia
5.4. Mutations in Non-Canonical Histones Affecting K36 Methylation
6. Changes in Cancer Related to H3K36 Demethylases
6.1. KDM2B Over-Expression in Cancer
6.2. KDM4 A–C Proteins Demethylating H3K36me3
7. Regulation of Methylation of Lysine 4 on Histone 3
7.1. Methylases in COMPASS Complexes (Complex Proteins Associated with SET1)
7.2. The KDM1 and KDM5 Demethylases Active on Methylated H3K4
8. Changes Associated with H3K4 Methylation in Cancer
8.1. KMT2A/MLL1 and Development of BET Inhibitors
8.2. Mutations in KMT2C and KMT2D
8.3. LSD1 (KDM1A) in Cancer
8.4. KDM5 Demethylases and Cancer
9. Perspectives
- There is considerable data arising from pre-clinical studies showing that disturbance of the methylation state of the histone lysines K4, K27, and K36 on histone 3 is a common event in cancer. These changes can occur as a result of mutations or translocations of the enzymes modifying the methylation state (Table 1), or to changes in their level of expression. While mutations in tumour suppressor genes, and changes of levels of expression, are found in a range of cancers, including the common carcinomas, other mutations and all translocations are associated with specific cancers, which are less common (see Table 1). Thus, inhibitors of the relevant KMTs or KDMs have been developed for potential clinical application in cancer therapy. However, only one methylase, EZH2 (KMT6A), and one demethylase, LSD1 (KDM1), are currently under extensive evaluation in the clinic for cancer therapy using inhibitors which target the enzyme activity.
- It is becoming clear that functions of the KMT and KDM proteins, which are independent of the enzyme activity, also play a role in development and in the changes occurring in some cancers (see Table 2 and Figure 3). However, pre-clinical research developing inhibitors for these histone methylases and demethylases has targeted the enzymatic activity. This approach makes it difficult to selectively inhibit specific homologues in families using the same chemical mechanism for enzymic activity. In evolution, the expansion of the number of genes with a common function from lower organisms to mammals (e.g., six homologues of the KMT2 methylase family, four dimethylating H3K36 and four of the KDM5 demethylases) is associated with increasing tissue complexity. This in turn emphasises the importance of cell phenotype in the expression and function of epigenetic factors. Obtaining more information regarding the differences in expression and function of the individual homologues in a family of KMTs or KDMs which may be seen in different tissues and in cancers developing from them is now a pressing area of research. Studies documenting the detailed structure of the homologues in a particular family could also give clues for identifying differential targeting. Taking this approach, inhibitors of the ASH1L methylase have recently been developed [117] which are selective in inhibiting ASH1L enzyme activity, but not the other methylases, which dimethylate H3K36. Such studies could lead to the development of not only enzymatic inhibitors specific for a particular homologue, but also for a new class of inhibitors targeting a function which is independent of the enzyme activity, and may be operative in specific cancers.
Author Contributions
Funding
Conflicts of Interest
References
- Lewis, P.H. New mutants report. Drosoph. Inf. Serv. 1947, 21, 69. [Google Scholar]
- Lewis, E.B. A gene complex controlling segmentation in Drosophila. Nature 1978, 276, 565–570. [Google Scholar] [CrossRef] [PubMed]
- Ingham, P.W. Differential expression of bithorax complex genes in the absence of the extra sex combs and trithorax genes. Nature 1983, 306, 591–593. [Google Scholar] [CrossRef] [PubMed]
- Rea, S.; Eisenhaber, F.; O’Carroll, D.; Strahl, B.D.; Sun, Z.W.; Schmid, M.; Opravil, S.; Mechtler, K.; Ponting, C.P.; Allis, C.D.; et al. Regulation of chromatin structure by site-specific histone H3 methyltrans-ferases. Nature 2000, 406, 593–599. [Google Scholar] [CrossRef]
- Cao, R.; Wang, L.; Wang, H.; Xia, L.; Erdjument-Bromage, H.; Tempst, P.; Jones, R.S.; Zhang, Y. Role of Histone H3 Lysine 27 Methylation in Polycomb-Group Silencing. Science 2002, 298, 1039–1043. [Google Scholar] [CrossRef] [Green Version]
- Miller, T.; Krogan, N.J.; Dover, J.; Erdjument-Bromage, H.; Tempst, P.; Johnston, M.; Greenblatt, J.F.; Shilatifard, A. COMPASS: A complex of proteins associated with a trithorax-related SET domain protein. Proc. Natl. Acad. Sci. USA 2001, 98, 12902–12907. [Google Scholar] [CrossRef] [Green Version]
- Yuan, W.; Xu, M.; Huang, C.; Liu, N.; Chen, S.; Zhu, B. H3K36 methylation antagonizes PRC2-mediated H3K27 methylation. J. Biol. Chem. 2011, 286, 7983–7989. [Google Scholar] [CrossRef] [Green Version]
- Schmähling, S.; Meiler, A.; Lee, Y.; Mohammed, A.; Finkl, K.; Tauscher, K.; Israel, L.; Wirth, M.; Philippou-Massier, J.; Blum, H.; et al. Regulation and function of H3K36 di-methylation by the trithorax group protein complex AMC. Development 2018, 145, dev163808. [Google Scholar] [CrossRef] [Green Version]
- Piunti, A.; Shilatifard, A. Epigenetic balance of gene expression by Polycomb and COMPASS families. Science 2016, 352, aad9780. [Google Scholar] [CrossRef] [Green Version]
- Schuettengruber, B.; Bourbon, H.M.; Di Croce, L.; Cavalli, G. Genome regulation by polycomb and Thrithorax: 70 Years and Counting. Cell 2017, 171, 34–37. [Google Scholar] [CrossRef] [Green Version]
- Geislee, S.J.; Paro, R. Trithorax and Polycomb group-dependent regulation: A tale of opposing activities. Development 2015, 142, 2876–2887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cenik, B.K.; Shilatifard, A. COMPASS and SWI/SNF complexes in development and disease. Nat. Rev. Genet. 2021, 22, 38–58. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Lan, F.; Matson, C.; Mulligan, P.; Whetstine, J.R.; Cole, P.; Casero, R.A.; Shi, Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 2004, 119, 941–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsukada, Y.-i.; Fang, J.; Erdjument-Bromage, H.; Warren, M.E.; Borchers, C.; Tempst, P.; Zhang, Y. Histone demethylation by a family of JmjC domain-containing proteins. Nature 2006, 439, 811–816. [Google Scholar] [CrossRef]
- Dimitrova, E.; Turberfield, A.; Klose, R.J. Histone demethylases in chromatin biology and beyond. EMBO Rep. 2015, 16, 1620–1639. [Google Scholar] [CrossRef]
- Hojfeldt, J.; Agger, K.; Helin, K. Histone lysine demethylases as targets for anticancer therapy. Nat. Rev. Drug Discov. 2013, 12, 917–930. [Google Scholar] [CrossRef]
- Pedersen, M.T.; Helin, K. Histone demethylases in development and disease. Trends Cell Biol. 2010, 20, 662–671. [Google Scholar] [CrossRef]
- Wang, S.-P.; Tang, Z.; Chen, C.-W.; Shimada, M.; Koche, R.; Wang, L.-H.; Nakadai, T.; Chramiec, A.; Krivtsov, A.V.; Armstrong, S.A.; et al. A UTX-MLL4-p300 Transcriptional Regulatory Network Coordinately Shapes Active Enhancer Landscapes for Eliciting Transcription. Mol. Cell 2017, 67, 308–321.e6. [Google Scholar] [CrossRef] [Green Version]
- Aranda, S.; Mas, G.; Di Croce, L. Regulation of gene transcription by Polycomb proteins. Sci. Adv. 2015, 1, e1500737. [Google Scholar] [CrossRef] [Green Version]
- Varlet, E.; Ovejero, S.; Martinez, A.-M.; Cavalli, G.; Moreaux, J. Role of Polycomb Complexes in Normal and Malignant Plasma Cells. Int. J. Mol. Sci. 2020, 21, 8047. [Google Scholar] [CrossRef]
- Healy, E.; Mucha, M.; Glancy, E.; Fitzpatrick, D.J.; Conway, E.; Neikes, H.K.; Monger, C.; Van Mierlo, G.; Baltissen, M.P.; Koseki, Y.; et al. PRC2.1 and PRC2.2 Synergize to Coordinate H3K27 Trimethylation. Mol. Cell 2019, 76, 437–452.e6. [Google Scholar] [CrossRef] [PubMed]
- Levine, S.S.; Weiss, A.; Erdjument-Bromage, H.; Shao, Z.; Tempst, P.; Kingston, R.E. The core of the polycomb repressive complex is compositionally and functionally conserved in flies and humans. Mol. Cell. Biol. 2002, 22, 6070–6078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; Wang, M.; Chang, L.; Yu, J.; Song, A.; Liu, C.; Huang, W.; Zhang, T.; Wu, X.; Shen, X.; et al. RYBP/YAF2-PRC1 complexes and histone H1-dependent chromatin compaction mediate propagation of H2AK119ub1 during cell division. Nat. Cell Biol. 2020, 22, 439–452. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Kallin, E.M.; Tsukada, Y.I.; Zhang, Y. The H3K36 demethylase Jhdm1b/Kdm2b regulates cell proliferation and senescence through p15Ink4b. Nat. Struct. Mol. Biol. 2008, 15, 1169–1175. [Google Scholar] [CrossRef]
- Farcas, A.M.; Blackledge, N.P.; Sudbery, I.; Long, H.K.; McGouran, J.F.; Rose, N.R.; Lee, S.; Sims, D.; Cerase, A.; Sheahan, T.W.; et al. KDM2B links the Polycomb Repressive Complex 1 (PRC1) to recognition of CpG islands. Elife 2012, 1, e00205. [Google Scholar] [CrossRef]
- He, J.; Shen, L.; Wan, M.; Taranova, O.; Wu, H.; Zhang, Y. Kdm2b maintains murine embryonic stem cell status by recruiting PRC1 complex to CpG islands of developmental genes. Nat. Cell Biol. 2013, 15, 373–384. [Google Scholar] [CrossRef] [Green Version]
- Wong, S.J.; Gearhart, M.A.; Taylor, A.B.; Nanyes, D.R.; Ha, D.J.; Robinson, A.K.; Artigas, J.A.; Lee, O.J.; Demeler, D.; Hart, P.J.; et al. KDM2B recruitment of the Polycomb Group Complex, PRC1.1, requires co-operation between PCGF1 and BCORL1. Structure 2016, 24, 1795–1801. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Vilstrup Johansen, J.; Helin, K. Fbxl10/Kdm2b recruits polycomb repressive complex 1 to CpG islands and regulates H2A ubiquitylation. Mol. Cell 2013, 49, 1134–1146. [Google Scholar] [CrossRef] [Green Version]
- Blackledge, N.P.; Farcas, A.M.; Kondo, T.; King, H.W.; McGouran, J.F.; Hanssen, L.L.P.; Ito, S.; Cooper, S.; Kondo, K.; Koseki, Y.; et al. Variant PRC1 complex-dependent H2A ubiquitylation drives PRC2 recruitment and polycomb domain formation. Cell 2014, 157, 1445–1459. [Google Scholar] [CrossRef] [Green Version]
- Kalb, R.; Latwiel, S.; Baymaz, H.I.; Jansen, P.W.; Muller, C.W.; Vermeulen, M.; Muller, J. Histone H2A monoubiquitination promotes histone H3 methylation in Polycomb repression. Nat. Struct. Mol. Biol. 2014, 21, 569–571. [Google Scholar] [CrossRef]
- Cooper, S.; Grijzenhout, A.; Underwood, E.; Ancelin, K.; Zhang, T.; Nesterova, T.B.; Anil-Kirmizitas, B.; Bassett, A.; Kooistra, S.M.; Agger, K.; et al. Jarid2 binds mono-ubiquitylated H2A lysine 119 to mediate crosstalk between Polycomb complexes PRC1 and PRC2. Nat. Commun. 2016, 7, 13661. [Google Scholar] [CrossRef] [Green Version]
- Sugishita, H.; Kondo, T.; Ito, S.; Nakayama, M.; Yakushiji-Kaminatsui, N.; Kawakami, E.; Koseki, Y.; Ohi-nata, Y.; Sharif, J.; Harachi, M.; et al. Variant PCGF1-PRC1 links PRC2 recruitment with differentia-tion-associated transcriptional inactivation at target genes. Nat. Commun. 2021, 12, 5341. [Google Scholar] [CrossRef]
- Cao, R.; Zhang, Y. SUZ12 is required for both the histone methyltransferase activity and the silencing function of the EED-EZH2 complex. Mol. Cell 2004, 15, 57–67. [Google Scholar] [CrossRef]
- Hojfeldt, J.; Hedehus, L.; Laugesen, A.; Tatar, T.; Wiehle, L.; Helin, K. Non-core subunits of the PRC2 complex are collectively required for its target-site specificity. Mol. Cell 2019, 76, 423–436.e3. [Google Scholar] [CrossRef]
- Conway, E.; Jerman, E.; Healy, E.; Ito, S.; Holoch, D.; Oliviero, G.; Deevy, O.; Glancy, E.; Fitzpatrick, D.J.; Mucha, M.; et al. A family of vertebrate- specific Polycombs encoded by the LCOR/LCORL genes balance PRC2 sub- type activities. Mol. Cell 2018, 70, 408–421. [Google Scholar] [CrossRef] [Green Version]
- Agger, K.; Cloos, P.A.C.; Christensen, J.; Pasini, D.; Rose, S.; Rappsilber, J.; Issaeva, I.; Canaani, E.; Salcini, A.E.; Helin, K. UTX and JMJD3 are histone H3K27 demethylases involved in HOX gene regulation and development. Nature 2007, 449, 731–734. [Google Scholar] [CrossRef]
- Shpargel, K.B.; Sengoku, T.; Yokoyama, S.; Magnuson, T. UTX and UTY demonstrate histone demethylase-independent function in mouse embryonic development. PLoS Genet. 2012, 8, e1002964. [Google Scholar] [CrossRef] [Green Version]
- Kleer, C.G.; Cao, Q.; Varambally, S.; Shen, R.; Ota, I.; Tomlins, S.A.; Ghosh, D.; Sewalt, R.G.A.B.; Otte, A.P.; Hayes, D.F.; et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc. Natl. Acad. Sci. USA 2003, 100, 11606–11611. [Google Scholar] [CrossRef] [Green Version]
- Varambally, S.; Dhanasekaran, S.M.; Zhou, M.; Barrette, T.R.; Kumar-Sinha, C.; Sanda, M.G.; Ghosh, D.; Pienta, K.J.; Sewalt, R.G.A.B.; Otte, A.P.; et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 2002, 4519, 624–629. [Google Scholar] [CrossRef]
- Hernando, H.; Gelato, K.A.; Lesche, R.; Beckmann, G.; Koehr, S.; Otto, S.; Steigemann, P.; Stresemann, C. EZH2 Inhibition Blocks Multiple Myeloma Cell Growth through Upregulation of Epithelial Tumor Suppressor Genes. Mol. Cancer Ther. 2016, 15, 287–298. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Shilatifard, A. UTX Mutations in Human Cancer. Cancer Cell 2019, 35, 168–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bitler, B.G.; Aird, K.M.; Garipov, A.; Li, H.; Amatangelo, M.; Kossenkov, A.V.; Schultz, D.C.; Liu, Q.; Shih, I.M.; Conejo-Garcia, J.R.; et al. Synthetic lethality by targeting EZH2 methyl transferase activity in ARID1A mutated cancers. Nat. Med. 2015, 21, 231–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.H.; Kim, W.; Howard, T.P.; Vazquez, F.; Tsherniak, A.; Wu, J.N.; Wang, W.; Haswell, J.R.; Walensky, L.D.; Hahn, W.C.; et al. SWI/SNF mutant cancers depend upon catalytic and non–catalytic activity of EZH2. Nat. Med. 2015, 21, 1491–1496. [Google Scholar] [CrossRef] [Green Version]
- Ezponda, T.; Dupéré-Richer, D.; Will, C.M.; Small, E.C.; Varghese, N.; Patel, T.; Nabet, B.; Popovic, R.; Oyer, J.; Bulic, M.; et al. UTX/KDM6A loss enhances the malignant phenotype of multiple myeloma and sensitizes cells to EZH2 inhibition. Cell Rep. 2017, 21, 628–640. [Google Scholar] [CrossRef] [Green Version]
- Ernst, T.; Chase, A.J.; Score, J.; Hidalgo-Curtis, C.E.; Bryant, C.; Jones, A.V.; Waghorn, K.; Zoi, K.; Ross, F.M.; Reiter, A.; et al. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid dis-orders. Nat. Genet. 2010, 42, 722–726. [Google Scholar] [CrossRef]
- Nikoloski, G.; Langemeijer, S.M.C.; Kuiper, R.P.; Knops, R.; Massop, M.; Tönnissen, E.R.L.T.M.; Van Der Heijden, A.; Scheele, T.N.; Vandenberghe, P.; De Witte, T.; et al. Somatic mutations of the histone methyltransferase gene EZH2 in myelodysplastic syndromes. Nat. Genet. 2010, 42, 665–667. [Google Scholar] [CrossRef]
- Ntziachristos, P.; Tsirigos, A.; Van Vlierberghe, P.; Nedjic, J.; Trimarchi, T.; Flaherty, M.S.; Ferres-Marco, D.; Da Ros, V.G.; Tang, Z.; Siegle, J.; et al. Genetic inactivation of the polycomb repressive complex 2 in T cell acute lymphoblastic leukemia. Nat. Med. 2012, 18, 298–302. [Google Scholar] [CrossRef]
- Skoda, R.C.; Schwaller, J. Dual roles of EZH2 in acute myeloid leukemia. J. Exp. Med. 2019, 216, 725–727. [Google Scholar] [CrossRef] [Green Version]
- Basheer, F.; Giotopoulos, G.; Meduri, E.; Yun, H.; Mazan, M.; Sasca, D.; Gallipoli, P.; Marando, L.; Asby, R.; Sheppard, O.; et al. Contrasting requirements during disease evolution identify EZH2 as a therapeutic target in AML. J. Exp. Med. 2019, 216, 966–998. [Google Scholar] [CrossRef] [Green Version]
- Xu, K.; Wu, Z.J.; Groner, A.C.; He, H.H.; Cai, C.; Lis, R.T.; Wu, X.; Stack, E.C.; Loda, M.; Liu, T.; et al. EZH2 oncogenic activity in castration-resistant prostate cancer cells is polycomb-independent. Science 2012, 338, 1465–1469. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Lee, Y.; Lu, X.; Song, B.; Fong, K.W.; Cao, Q.; Licht, J.D.; Zhao, J.C.; Yu, J. Polycomb-and Methylation-Independent Roles of EZH2 as a Transcription Activator. Cell Rep. 2018, 25, 2808–2820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Y.; He, C.; Wang, M.; Ma, X.; Mo, F.; Yang, S.; Han, J.; Wei, X. Targeting epigenetic regulators for cancer therapy: Mechanisms and advances in clinical trials. Signal Transduct. Target. Ther. 2019, 4, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, R.; Du, W.; Guo, W. EZH2: A novel target for cancer treatment. J. Hematol. Oncol. 2020, 13, 104. [Google Scholar] [CrossRef] [PubMed]
- Dalgliesh, G.L.; Furge, K.; Greenman, C.; Chen, L.; Bignell, G.; Butler, A.; Davies, H.; Edkins, S.; Hardy, C.; Latimer, C.; et al. Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature 2010, 463, 360–363. [Google Scholar] [CrossRef] [Green Version]
- Rao, R.C.; Dou, Y. Hijacked in cancer: The KMT2 (MLL) family of methyltransferases. Nat. Rev. Cancer 2015, 15, 334–346. [Google Scholar] [CrossRef] [Green Version]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [Green Version]
- Moffitt, A.B.; Ondrejka, S.L.; McKinney, M.; Rempel, R.E.; Goodlad, J.R.; The, C.H.; Leppa, S.; Mannisto, S.; Kovanen, P.E.; Tse, E.; et al. Enteropathy-associated T cell lymphoma subtypes are characterised by loss of function of SETD2. J. Exp. Med. 2017, 215, 1371–1386. [Google Scholar] [CrossRef]
- Kuo, A.J.; Cheung, P.; Chen, K.; Zee, B.M.; Kioi, M.; Lauring, J.; Xi, Y.; Park, B.H.; Shi, X.; Garcia, B.A.; et al. NSD2 links demethylation of histone H3 at lysine 36 to oncogenic programming. Mol Cell 2011, 44, 609–620. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Han, S.; Peng, R.; Jiao, C.; Wang, X.; Yang, X.; Yang, R.; Li, X. Li Depletion of histone de-methylase KDM5B inhibits cell proliferation of hepatocellular carcinoma by regulation of cell cycle check-point proteins p15 and p27. J. Exp. Clin. Cancer Res. 2016, 35, 37. [Google Scholar] [CrossRef] [Green Version]
- Jaffe, J.D.; Wang, Y.; Chan, H.M.; Zhang, J.; Huether, R.; Kryukov, G.V.; Bhang, H.-E.C.; Taylor, J.E.; Hu, M.; Englund, N.P.; et al. Global chromatin profiling reveals NSD2 mutations in pediatric acute lymphoblastic leukemia. Nat. Genet. 2013, 45, 1386–1391. [Google Scholar] [CrossRef]
- Pierro, J.; Saliba, J.; Narang, S.; Sethia, G.; Saint Fleur-Lominy, S.; Chowdhury, A.; Qualls, A.; Fay, H.; Kilberg, H.L.; Moriyama, T.; et al. The NSD2p.E1099K Mutation is Enriched at Relapse and Confers Druig Resistance in a Cell Context-Dependent Manner in Pediatric Acute Lymphoblastic Leukemia. Mol. Cancer Res. 2002, 18, 1153–1165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorrance, A.M.; Liu, S.; Yuan, W.; Becknell, B.; Arnoczky, K.J.; Guimond, M.; Strout, M.P.; Feng, L.; Nakamura, T.; Yu, L.; et al. MII partial tandem duplication induces aberrant Hox expression in vivo via specific epigenetic alterations. J. Clin. Investig. 2006, 116, 2707–2716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krivtsov, A.V.; Armstrong, S.A. MLL translocations, histone modifications and leukaemia stem-cell development. Nat. Rev. Cancer 2007, 7, 823–833. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.A.; Prinjha, R.K.; Dittmann, A.; Giotopoulos, G.; Bantscheff, M.; Chan, W.I.; Robson, S.C.; Chung, C.; Hopf, C.; Savitski, M.M.; et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature 2011, 478, 529–533. [Google Scholar] [CrossRef] [Green Version]
- Morishita, M.; di Luccio, E. Cancers and the NSD family of histone methyltransferases. Biochim. Biophys. Acta 2011, 1816, 158–163. [Google Scholar] [CrossRef]
- Lhoumaud, P.; Badri, S.; Rodriguez-Hernaez, J.; Sakellaropoulos, T.; Sethia, G.; Kloetgen, A.; Cornwell, M.; Bhattacharyya, S.; Ay, F.; Bonneau, R.; et al. NSD2 overexpression drives clustered chromatin and transcriptional changes in a subset of insulated domains. Nat. Commun. 2019, 10, 48431. [Google Scholar] [CrossRef] [Green Version]
- Banito, A.; Li, X.; Laporte, A.N.; Roe, J.-S.; Sanchez-Vega, F.; Huang, C.-H.; Dancsok, A.R.; Hatzi, K.; Chen, C.-C.; Tschaharganeh, D.F.; et al. The SS18-SSX oncoprotein hijacks KDM2B-PRC1.1 to drive synovial sarcoma. Cancer Cell 2018, 33, 527–541.e8. [Google Scholar] [CrossRef] [Green Version]
- Nacev, B.A.; Feng, L.; Bagert, J.D.; Lemiesz, A.E.; Gao, J.; Soshnev, A.A.; Kundra, R.; Schultz, N.; Muir, T.W.; Allis, C.D. The expanding landscape of “oncohistone” mutations in human cancers. Nature 2019, 567, 473–478. [Google Scholar] [CrossRef]
- Wu, G.; Broniscer, A.; McEachron, T.A.; Lu, C.; Paugh, B.S.; Becksfort, J.; Qu, C.; Ding, L.; Huether, R.; Parker, M.; et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat. Genet. 2012, 44, 251–253. [Google Scholar]
- Schwartzentruber, J.; Korshunov, A.; Liu, X.Y.; Jones, D.T.; Pfaff, E.; Jacob, K.; Sturm, D.; Fontebasso, A.M.; Quang, D.A.; Tönjes, M.; et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012, 482, 226–231. [Google Scholar] [CrossRef]
- Lewis, P.W.; Müller, M.M.; Koletsky, M.S.; Cordero, F.; Lin, S.; Banaszynski, L.A.; Garcia, B.A.; Muir, T.W.; Becher, O.J.; Allis, C.D. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 2013, 340, 857–861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vire, E.; Brenner, C.; Deplus, R.; Blanchon, L.; Fraga, M.; Didelot, C.M.; Morey, L.; Van Eynde, A.; Bernard, D.; Vanderwinden, J.-M.; et al. The Polycomb group protein EZH2 directly controls DNA methylation. Nature 2006, 439, 871–874. [Google Scholar] [CrossRef] [PubMed]
- Van der Vlag, J.; Otte, A.P. Transcriptional repression mediated by the human polycomb-group protein EED involves histone deacetylation. Nat. Genet. 1999, 23, 474–478. [Google Scholar] [CrossRef]
- Ren, Y.; Wang, Y.; Zhang, J.; Wang, Q.-X.; Han, L.; Mei, M.; Kang, C.-S. Targeted design and identification of AC1NOD4Q to block activity of HOTAIR by abrogating the scaffold interaction with EZH2. Clin. Epigenetics 2019, 11, 29. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, F.; Weissmann, S.; Leblanc, B.; Pandey, D.P.; Højfeldt, J.W.; Comet, I.; Zheng, C.; Johansen, J.V.; Rapin, N.; Porse, B.T.; et al. EZH2 is a potential therapeutic target for H3K27M-mutant pediatric gliomas. Nat. Med. 2017, 23, 483–492. [Google Scholar] [CrossRef] [PubMed]
- Gozdecka, M.; Meduri, E.; Mazan, M.; Tzelepis, K.; Dudek, M.; Knights, A.J.; Pardo, M.; Yu, L.; Choudhary, J.S.; Metzakopian, E.; et al. UTX-mediated enhancer and chromatin remodeling suppresses myeloid leukemogenesis through noncatalytic inverse regulation of ETS and GATA programs. Nat. Genet. 2018, 50, 883–894. [Google Scholar] [CrossRef] [PubMed]
- Ntziachristos, P.; Tsirigos, A.; Welstead, G.G.; Trimarchi, T.; Bakogianni, S.; Xu, L.; Loizou, E.; Holmfeldt, L.; Strikoudis, A.; King, B.; et al. Contrasting roles of histone 3 lysine 27 demethylases in acute lymphoblastic leukaemia. Nature 2014, 514, 513–517. [Google Scholar] [CrossRef]
- Andricovich, J.; Perkail, S.; Kai, Y.; Casasanta, N.; Peng, W.; Tzatsos, A. Loss of KDM6A activates super-enhancers to induce gender-specific squamous-like pancreatic cancer and confers sensitivity to BET inhibitors. Cancer Cell 2018, 33, 512–526. [Google Scholar] [CrossRef] [Green Version]
- Choi, H.-J.; Park, J.-H.; Park, M.; Won, H.-Y.; Joo, H.-S.; Lee, C.H.; Lee, J.-Y.; Kong, G. UTX inhibits EMT-induced breast CSC properties by epigenetic repression of EMT genes in cooperation with LSD1 and HDAC1. EMBO Rep. 2015, 16, 1288–1298. [Google Scholar] [CrossRef] [Green Version]
- Terranova, R.; Agherbi, H.; Boned, A.; Meresse, S.; Djabali, M. Histone and DNA methylation defects at Hox genes in mice expressing a SET domain-truncated form of Mll. Proc. Natl. Acad. Sci. USA 2006, 103, 6629–6634. [Google Scholar] [CrossRef] [Green Version]
- Fang, Y.; Liao, G.; Yu, B. LSD1/KDM1A inhibitors in clinical trials: Advances and prospect. J. Hematol. Oncol. 2019, 12, 129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sehrawat, A.; Gao, L.; Wang, Y.; Bankhead, A., 3rd; McWeeney, S.K.; King, C.J.; Schwartzman, J.; Urrutia, J.; Bisson, W.H.; Coleman, D.J.; et al. LSD1 activates a lethal prostate cancer gene network independently of its demethylase activity. Proc. Natl. Acad. Sci. USA 2018, 115, E4179–E4188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuang, Y.; Lu, F.; Guo, J.; Xu, H.; Wang, Q.; Xu, C.; Zeng, L.; Yi, S. Histone demethylase KDM2B up-regulates histone methyltransferase EZH2 expression and contributes to the progression of ovarian cancer in vitro and in vivo. Onco. Targets Ther. 2017, 10, 3131–3144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tzatsos, A.; Paskaleva, P.; Ferrari, F.; Deshpande, V.; Stoykova, S.; Contino, G.; Wong, K.K.; Lan, F.; Trojer, P.; Park, P.J.; et al. KDM2B promotes pancreatic cancer via Polycomb-dependent and -independent transcriptional programs. J. Clin. Investig. 2013, 123, 727–739. [Google Scholar] [CrossRef] [Green Version]
- Catchpole, S.; Spencer-Dene, B.; Hall, D.; Santangelo, S.; Rosewell, I.; Guenatri, M.; Beatson, R.; Scibetta, A.G.; Burchell, J.M.; Taylor-Papadimitriou, J. PLU-1/JARID1B/KDM5B is required for embryonic survival and contributes to cell proliferation in the mammary gland and in ER+ breast cancer cells. Int. J. Oncol. 2011, 38, 1267–1277. [Google Scholar]
- Jamshidi, S.; Catchpole, S.; Chen, J.; So, C.W.E.; Burchell, J.M.; Rahman, K.M.; Taylor-Papadimitriou, J. The KDM5B protein expressed in the viable and fertile ΔARID mouse exhibits no demethylase activity. Int. J. Oncol. 2021, 59, 96. [Google Scholar] [CrossRef]
- Schmitges, F.W.; Prusty, A.B.; Faty, M.; Stützer, A.; Lingaraju, G.M.; Aiwazian, J.; Sack, R.; Hess, D.; Li, L.; Zhou, S.; et al. Histone Methylation by PRC2 is inhibited by active chromatin marks. Mol. Cell 2011, 42, 330–341. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Yang, F.; Zhang, Z.; Zhang, J.; Cai, G.; Li, L.; Zheng, Y.; Chen, S.; Xi, R.; Zhu, B. Mrg15 stimulates Ash1 H3K36 methyltransferase activity and facilitates Ash1 Trithorax group protein function in Drosophila. Nat. Commun. 2017, 8, 1649. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.G.; Cai, L.; Pasillas, M.P.; Kamps, M.P. NUP98–NSD1 links H3K36 methylation to Hox-A gene activation and leukaemogenesis. Nat. Cell Biol. 2007, 9, 804–812. [Google Scholar] [CrossRef]
- Strahl, B.D.; Grant, P.A.; Briggs, S.D.; Sun, Z.W.; Bone, J.R.; Caldwell, J.A.; Mollah, S.; Cook, R.G.; Sha-banowitz, J.; Hunt, D.F.; et al. Set2 is a nucleosomal histone H3-selective methyltransferase that medi-ates transcriptional repression. Mol. Cell. Biol. 2002, 22, 1298–1306. [Google Scholar] [CrossRef] [Green Version]
- Wagner, E.J.; Carpenter, P.B. Understanding the language of Lys36 methylation at histone H3. Nat. Rev. Mol. Cell Biol. 2012, 13, 115–126. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Zhu, B. Roles of H3K36-specific histone methyltransferases in transcription: Antagonizing silencing and safeguarding transcription fidelity. Biophys. Rep. 2018, 4, 170–177. [Google Scholar] [CrossRef]
- Venkatesh, S.; Workman, J.L. Set2 mediated H3 lysine 36 methylation: Regulation of transcription elonga-tion and implications in organismal development. Wiley Interdiscip. Rev. Dev. Biol. 2013, 2, 685–700. [Google Scholar] [CrossRef] [Green Version]
- Edmunds, J.W.; Mahadevan, L.C.; Clayton, A.L. Dynamic histone H3 methylation during gene induction: HYPB/Setd2 mediates all H3K36 trimethylation. EMBO J. 2008, 27, 406–420. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Mao, G.; Tong, D.; Huang, J.; Gu, L.; Yang, W.; Li, G.-M. The histone mark h3k36me3 regulates human dna mismatch repair through its interaction with MutSα. Cell 2013, 153, 590–600. [Google Scholar] [CrossRef] [Green Version]
- Sun, Z.; Zhang, Y.; Jia, J.; Fang, Y.; Tang, Y.; Weu, H.; Fang, D. H3K36me3, message from chromatin to DNA damage repair. Cell Biosci. 2020, 10, 9. [Google Scholar] [CrossRef]
- Douglas, J.; Hanks, S.; Temple, I.K.; Davies, S.; Murray, A.; Upadhyaya, M.; Tomkins, S.; Hughes, H.E.; Cole, R.T.; Rahman, N. NSD1 mutations are the major cause of sotos syndrome and occur in some cases of weaver syndrome but are rare in other overgrowth phenotypes. Am. J. Hum. Genet. 2003, 72, 132–143. [Google Scholar] [CrossRef] [Green Version]
- Rayasam, G.V.; Wendling, O.; Angrand, P.-O.; Mark, M.; Niederreither, K.; Song, L.; Lerouge, T.; Hager, G.L.; Chambon, P.; Losson, R. NSD1 is essential for early post-implantation development and has a catalytically active SET domain. EMBO J. 2003, 22, 3153–3163. [Google Scholar] [CrossRef] [Green Version]
- Klymenko, T.; Muller, J. The histone methyltransferases Trithorax and Ash1 prevent transcriptional si-lencing by Polycomb group proteins. EMBO Rep. 2004, 5, 373–377. [Google Scholar] [CrossRef] [Green Version]
- Dorighi, K.M.; Tamkun, J.W. The trithorax group proteins Kismet and ASH1 promote H3K36 dimethylation to counteract Polycomb group repression in Drosophila. Development 2013, 140, 4182–4192. [Google Scholar] [CrossRef] [Green Version]
- Neri, F.; Rapelli, S.; Krepelova, A.; Incarnato, D.; Parlato, C.; Basile, G.; Maldotti, M.; Anselmi, F.; Oliviero, S. Intragenic DNA methylation prevents spurious transcription initiation. Nature 2017, 543, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Boulard, M.; Edwards, J.R.; Bestor, T.H. FBXL10 protects Polycomb-bound genes from hypermethylation. Nat. Genet. 2015, 47, 479–485. [Google Scholar] [CrossRef] [PubMed]
- Andricovich, J.; Kai, Y.; Peng, W.; Foudi, A.; Tzatsos, A. Histone demethylase KDM2B regulates lineage commitment in normal and malignant hematopoiesis. J. Clin. Investig. 2016, 126, 905–920. [Google Scholar] [CrossRef]
- Tzatsos, A.; Paskaleva, P.; Lymperi, S.; Contino, G.; Stoykova, S.; Chen, Z.; Wong, K.K.; Bardeesy, N. Lysine-specific demethylase 2B (KDM2B)-let-7-enhancer of zester homolog 2 (EZH2) pathway regulates cell cycle progression and senescence in primary cells. J. Biol. Chem. 2011, 286, 33061–33069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfau, R.; Tzatsos, A.; Kampranis, S.C.; Serebrennikova, O.B.; Bear, S.E.; Tsichlis, P.N. Members of a family of JmjC domain-containing oncoproteins immortalize embryonic fibroblasts via a JmjC domain-dependent process. Proc. Natl. Acad. Sci. USA 2008, 105, 1907–1912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, D.H.; Kim, G.W.; Jeon, Y.H.; Yoo, J.; Lee, S.W.; Kwon, S.H. Advances in histone demethylase KDM4 as cancer therapeutic targets. FASEB J. 2020, 34, 3461–3484. [Google Scholar] [CrossRef] [Green Version]
- Hillringhaus, L.; Yue, W.W.; Rose, N.R.; Ng, S.S.; Gileadi, C.; Loenarz, C.; Bello, S.H.; Bray, J.E.; Schofield, C.J.; Oppermann, U. Structural and Evolutionary Basis for the Dual Substrate Selectivity of Human KDM4 Histone Demethylase Family. J. Biol. Chem. 2011, 286, 41616–41625. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.K.; Bonaldi, T.; Cuomo, A.; Del Rosario, J.R.; Hosfield, D.J.; Kanouni, T.; Kao, S.C.; Lai, C.; Lobo, N.A.; Matuszkiewicz, J.; et al. Design of KDM4 inhibitors with antiproliferative effects in cancer models. ACS Med. Chem. Lett. 2017, 8, 869–874. [Google Scholar] [CrossRef]
- Pedersen, M.T.; Kooistra, S.M.; Radzisheuskaya, A.; Laugesen, A.; Johansen, J.V.; Hayward, D.G.; Nilsson, J.; Agger, K.; Helin, K. Continual removal of H3K9 promoter methylation by Jmjd2 demethylases is vital for ESC self-renewal and early development. EMBO J. 2016, 35, 1550–1564. [Google Scholar] [CrossRef] [Green Version]
- Duns, G.; Berg, E.V.D.; Van Duivenbode, I.; Osinga, J.; Hollema, H.; Hofstra, R.; Kok, K. Histone Methyltransferase Gene SETD2 Is a Novel Tumor Suppressor Gene in Clear Cell Renal Cell Carcinoma. Cancer Res. 2010, 70, 4287–4291. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Duns, G.; Westers, H.; Sijmons, R.; Berg, A.V.D.; Kok, K. SETD2: An epigenetic modifier with tumor suppressor functionality. Oncotarget 2016, 7, 50719–50734. [Google Scholar] [CrossRef] [Green Version]
- Al Sarakbi, W.; Sasi, W.; Jiang, W.G.; Roberts, T.; Newbold, R.F.; Mokbel, K. The mRNA expression of SETD2 in human breast cancer: Correlation with clinico-pathological parameters. BMC Cancer 2009, 9, 290. [Google Scholar] [CrossRef] [Green Version]
- Xie, P.; Tian, C.; An, L.; Nie, J.; Lu, K.; Xing, G.; Zhang, L.; He, F. Histone methyltransferase protein SETD2 interacts with p53 and selectively regulates its downstream genes. Cell. Signal. 2008, 20, 1671–1678. [Google Scholar] [CrossRef]
- Rosati, R.; La Starza, R.; Veronese, A.; Aventin, A.; Schwienbacher, C.; Vallespi, T.; Negrini, M.; Martelli, M.F.; Mecucci, C. NUP98 is fused to the NSD3 gene in acute myeloid leukemia associated with t(8;11)(p11.2;p15). Blood 2002, 99, 3857–3860. [Google Scholar] [CrossRef] [Green Version]
- La Starza, R.; Gorello, P.; Rosati, R.; Riezzo, A.; Veronese, A.; Ferrazzi, E.; Martelli, M.F.; Negrini, M.; Mecucci, C. Cryptic insertion producing two NUP98/NSD1 chimeric transcripts in adult refractor anemia with an excess of blasts. Genes Chromosomes Cancer 2004, 41, 395–399. [Google Scholar] [CrossRef]
- Zhu, L.; Li, Q.; Wong, S.H.; Huang, M.; Klein, B.J.; Shen, J.; Ikenouye, L.; Onishi, M.; Schneidawind, D.; Buechele, C.; et al. ASH1L links histone H3 lysine 36 dimethylation to MLL leukemia. Cancer Discov. 2016, 6, 770–783. [Google Scholar] [CrossRef] [Green Version]
- Rogawski, D.S.; Deng, J.; Li, H.; Miao, H.; Borkin, D.; Purohit, T.; Song, J.; Chase, J.; Li, S.; Ndoj, J.; et al. Discovery of first-in-class inhibitors of ASH1L histone methyltransferase with anti-leukemic activity. Nat. Commun. 2021, 12, 2792. [Google Scholar] [CrossRef]
- Hou, P.; Huang, C.; Liu, C.-P.; Yang, N.; Yu, T.; Yin, Y.; Zhu, B.; Xu, R.-M. Structural Insights into Stimulation of Ash1L’s H3K36 Methyltransferase Activity through Mrg15 Binding. Structure 2019, 27, 837–845. [Google Scholar] [CrossRef]
- Lee, Y.; Yoon, E.; Cho, S.; Schmähling, S.; Müller, J.; Song, J.-J. Structural Basis of MRG15-Mediated Activation of the ASH1L Histone Methyltransferase by Releasing an Autoinhibitory Loop. Structure 2019, 27, 846–852.e3. [Google Scholar] [CrossRef]
- Fang, J.; Huang, Y.; Mao, G.; Yang, S.; Rennert, G.; Gu, L.; Li, H.; Li, G.-M. Cancer-driving H3G34V/R/D mutations block H3K36 methylation and H3K36me3–MutSα interaction. Proc. Natl. Acad. Sci. USA 2018, 115, 9598–9603. [Google Scholar] [CrossRef] [Green Version]
- Jain, S.U.; Khazaei, S.; Marchione, D.M.; Lundgren, S.M.; Wang, X.; Weinberg, D.N.; Deshmukh, S.; Juretic, N.; Lu, C.; Allis, C.D.; et al. Histone H3.3 G34 mutations promote aberrant PRC2 activity and drive tumor progression. Proc. Natl. Acad. Sci. USA 2020, 117, 27354–27364. [Google Scholar] [CrossRef]
- Voon, H.; Udugama, M.; Lin, W.; Hii, L.; Law, R.; Steer, D.L.; Das, P.P.; Mann, J.R.; Wong, L.H. Inhibition of a K9/K36 demethylase by an H3.3 point mutation found in paediatric glioblastoma. Nat. Commun. 2018, 9, 3142. [Google Scholar] [CrossRef] [Green Version]
- Behjati, S.; Tarpey, P.S.; Presneau, N.; Scheipl, S.; Pillay, N.; Van Loo, P.; Wedge, D.; Cooke, S.L.; Gundem, G.; Davies, H.; et al. Distinct H3F3A and H3F3B driver mutations define chondroblastoma and giant cell tumor of bone. Nat. Genet. 2013, 45, 1479–1482. [Google Scholar] [CrossRef]
- Lu, C.; Jain, S.U.; Hoelper, D.; Bechet, D.; Molden, R.C.; Ran, L.; Murphy, D.; Venneti, S.; Hameed, M.; Pawel, B.R.; et al. Histone H3K36 mutations promote sarcomagenesis through altered histone methylation landscape. Science 2016, 352, 844–849. [Google Scholar] [CrossRef] [Green Version]
- Yan, M.; Yang, X.; Wang, H.; Shao, Q. The critical role of histone lysine demethylase KDM2B in cancer. Am. J. Transl. Res. 2018, 10, 2222–2233. [Google Scholar]
- He, J.; Nguyen, A.T.; Zhang, Y. KDM2b/JHDM1b, an H3K36me2-specific demethylase, is required for initiation and maintenance of acute myeloid leukemia. Blood 2011, 117, 3869–3880. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Zang, J.; Zhang, D.; Sun, Z.; Qiu, B.; Wang, X. KDM2B overexpression correlates with poor prognosis and regulates glioma cell growth. OncoTargets Ther. 2018, 11, 201–209. [Google Scholar] [CrossRef] [Green Version]
- Heinemann, B.; Nielsen, J.M.; Hudlesbusch, H.R.; Lees, M.J.; Larsen, D.V.; Boesen, T.; Labelle, M.; Ger-lach, L.O.; Birk, P.; Helin, K. Inhibtion of demethylase by GSK-J1/J4. Nature 2014, 514, E1–E2. [Google Scholar] [CrossRef]
- Mannironi, C.; Proietto, M.; Bufalieri, F.; Cundari, E.; Alagia, A.; Danovska, S.; Rinaldi, T.; Famiglini, V.; Coluccia, A.; La Regina, G.; et al. An high-throughput in vivo screening system to select H3K4-specific histone demethylase inhibitors. PLoS ONE 2014, 9, e86002. [Google Scholar] [CrossRef]
- Agger, K.; Miyagi, S.; Pedersen, M.T.; Kooistra, S.M.; Johansen, J.V.; Helin, K. Jmjd2/Kdm4 demethylases are required for expression of Il3ra and survival of acute myeloid leukemia cells. Genes Dev. 2016, 30, 1278–1288. [Google Scholar] [CrossRef] [Green Version]
- Agger, K.; Nishimura, K.; Miyagi, S.; Messling, J.E.; Rasmussen, K.D.; Helin, K. The KDM4/JMJD2 histone demethylases are required for hematopoietic stem cell maintenance. Blood 2019, 134, 1154–1158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morera, L.; Lübbert, M.; Jung, M. Targeting histone methyltransferases and demethylases in clinical trials for cancer therapy. Clin. Epigenetics 2016, 8, 57. [Google Scholar] [CrossRef] [Green Version]
- Shilatifard, A. The COMPASS family of histone H3K4 methylases: Mechanisms of regulation in development and disease pathogenesis. Annu. Rev. Biochem. 2012, 81, 65–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milne, T.A.; Briggs, S.D.; Brock, H.W.; Martin, M.E.; Gibbs, D.; Allis, C.D.; Hess, J.L. MLL targets SET domain methyltransferase activity to hox gene promoters. Mol. Cell 2002, 10, 1107–1117. [Google Scholar] [CrossRef]
- Sze, C.C.; Shilatifard, A. MLL3/MLL4/COMPASS Family on Epigenetic Regulation of Enhancer Function and Cancer. Cold Spring Harb. Perspect. Med. 2016, 6, a026427. [Google Scholar] [CrossRef] [Green Version]
- Denissov, S.; Hofemeister, H.; Marks, H.; Kranz, A.; Ciotta, G.; Singh, S.; Anas-Tassiadis, K.; Stunnenberg, H.G.; Stewart, A.F. Mll2 is required for H3K4 trimethylation on bivalent promoters in embryonic stem cells, whereas Mll1 is redundant. Development 2014, 141, 526–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamane, K.; Tateishi, K.; Klose, R.J.; Fang, J.; Fabrizio, L.A.; Erdjument-Bromage, H.; Taylor-Papadimitriou, J.; Tempst, P.; Zhang, Y. PLU-1 is an H3K4 demethylase involved in transcriptional repression and breast cancer cell proliferation. Mol. Cell 2007, 25, 801–812. [Google Scholar] [CrossRef]
- Xiang, Y.; Zhu, Z.; Han, G.; Ye, X.; Xu, B.; Peng, Z.; Ma, Y.; Yu, Y.; Lin, H.; Chen, A.P.; et al. JARID1B is a histone H3 lysine 4 demethylase up-regulated in prostate cancer. Proc. Natl. Acad. Sci. USA 2007, 104, 19226–19231. [Google Scholar] [CrossRef] [Green Version]
- Christensen, J.; Agger; Cloos, P.A.; Pasini, D.; Rose, S.; Sennels, L.; Rappsilber, J.; Hansen, K.H.; Salcini, A.E.; Helin, K. RBP2 belongs to a family of demethylases, specific for tri-and dimethylated lysine 4 on Histone 3. Cell 2007, 128, 1063–1076. [Google Scholar] [CrossRef] [Green Version]
- Metzger, E.; Wissmann, M.; Yin, N.; Müller, J.M.; Schneider, R.; Peters, A.H.F.M.; Günther, T.; Buettner, R.; Schüle, R. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature 2005, 437, 436–439. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, H.; Chen, Y.; Sun, Y.; Yang, F.; Yu, W.; Liang, J.; Sun, L.; Yang, X.; Shi, L.; et al. LSD1 Is a Subunit of the Nu RD Complex and Targets the Metastasis Programs in Breast Cancer. Cell 2009, 138, 660–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, R.; Chen, F.; Dong, Z.; Hu, D.; Barbera, A.J.; Clark, E.A.; Fang, J.; Yang, Y.; Mei, P.; Rutenberg, M.; et al. LSD2/KDM1B and its cofactor NPAC/GLYR1 endow a structural and molecular model for regulation of H3K4 demethylation. Mol. Cell 2013, 49, 558–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adamo, A.; Sesé, B.; Boue, S.; Castaño, J.; Paramonov, I.; Barrero, M.; Belmonte, J.C.I. LSD1 regulates the balance between self-renewal and differentiation in human embryonic stem cells. Nat. Cell Biol. 2011, 13, 652–659. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Hevi, S.; Kurash, J.K.; Lei, H.; Gay, F.; Bajko, J.; Su, H.; Sun, W.; Chang, H.; Xu, G.; et al. The lysine de-methylase LSD1 (KDM1) is required for maintenance of global DNA methylation. Nat. Genet. 2009, 41, 125–129. [Google Scholar] [CrossRef] [PubMed]
- Whyte, W.A.; Bilodeau, S.; Orlando, D.A.; Hoke, H.A.; Frampton, G.M.; Foster, C.T.; Cowley, S.M.; Young, R.A. Enhancer decommissioning by LSD1 during embryonic stem cell differentiation. Nature 2012, 482, 221–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maiques-Diaz, A.; Somervaille, T.C. LSD1: Biologic roles and therapeutic targeting. Epigenomics 2016, 8, 1103–1116. [Google Scholar] [CrossRef] [Green Version]
- Defeo-Jones, D.; Huang, P.S.; Jones, R.E.; Haskell, K.M.; Vuocolo, G.A.; Hanobik, M.G.; Huber, H.E.; Oliff, A. Cloning of cDNAs for cellular proteins that bind to the retinoblastoma gene product. Nature 1991, 352, 251–254. [Google Scholar] [CrossRef]
- Klose, R.J.; Yan, Q.; Tothova, Z.; Yamane, K.; Erdjument-Bromage, H.; Tempst, P.; Gilliland, D.G.; Zhang, Y.; Kaelin, W.G. The Retinoblastoma Binding Protein RBP2 Is an H3K4 Demethylase. Cell 2007, 128, 889–900. [Google Scholar] [CrossRef] [Green Version]
- Lu, P.J.; Sundquist, K.; Baeckstrom, D.; Poulsom, R.; Hanby, A.; Meier-Ewert, S.; Jones, T.; Mitchell, M.; Pitha-Rowe, P.; Freemont, P.; et al. A Novel Gene (PLU-1) Containing Highly Conserved Putative DNA/Chromatin Binding Motifs Is Specifically Up-regulated in Breast Cancer. J. Biol. Chem. 1999, 274, 15633–15645. [Google Scholar] [CrossRef] [Green Version]
- Barrett, A.; Madsen, B.; Copier, J.; Lu, P.J.; Cooper, L.; Scibetta, A.G.; Burchell, J.; Taylor-Papadimitriou, J. PLU-1 nuclear protein, which is upregulated in breast cancer, shows restricted expression in normal human adult tissues: A new cancer/testis antigen? Int. J. Cancer 2002, 101, 581–588. [Google Scholar] [CrossRef]
- Iwase, S.; Lan, F.; Bayliss, P.; de la Torre-Ubieta, L.; Huarte, M.; Qi, H.H.; Whetstine, J.R.; Bonni, A.; Roberts, T.M.; Shi, Y. The X-linked mental retardation gene SMCX/JARID1C defines a family of histone H3 lysine 4 demethylases. Cell 2007, 128, 1077–1088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwase, S.; Brookes, E.; Agarwal, S.; Badeaux, A.I.; Ito, H.; Vallianatos, C.N.; Tomassy, G.S.; Kasza, T.; Lin, G.; Thompson, A.; et al. A mouse model of X-linked intellectual disability associated with impaired removal of histone methylation. Cell Rep. 2016, 14, 1000–1009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonçalves, T.F.; Gonçalves, A.P.; Rodrigues, N.F.; dos Santos, J.M.; Pimentel, M.M.G.; Santos-Rebouças, C.B. KDM5C mutational screening among males with intellectual disability suggestive of X-Linked inheritance and review of the literature. Eur. J. Med Genet. 2014, 57, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Darvishi, E.; Slemmons, K.; Wan, Z.; Mitra, S.; Hou, X.; Hugues Parmentier, J.; Eddie Loh, Y.H.; Helman, L.J. Molecular mechanisms of Guadecitabine induced FGFR4 down regulation in alveolar rhabdomyosarcomas. Neoplasia 2020, 22, 274–282. [Google Scholar] [CrossRef] [PubMed]
- Scibetta, A.G.; Santangelo, S.; Coleman, J.; Hall, D.; Chaplin, T.; Copier, J.; Catchpole, S.; Burchell, J.; Taylor-Papadimitriou, J. Functional analysis of the transcription repressor PLU-1/JARID1B. Mol. Cell Biol. 2007, 27, 7220–7235. [Google Scholar] [CrossRef] [Green Version]
- Tu, S.; Teng, Y.-C.; Yuan, C.; Wu, Y.-T.; Chan, M.-Y.; Cheng, A.-N.; Lin, P.-H.; Juan, L.-J.; Tsai, M.-D. The ARID domain of the H3K4 demethylase RBP2 binds to a DNA CCGCCC motif. Nat. Struct. Mol. Biol. 2008, 15, 419–421. [Google Scholar] [CrossRef]
- Torres, I.O.; Kuchenbecker, K.M.; Nnadi, C.I.; Fletterick, R.J.; Kelly, M.J.S.; Fujimori, D.G. Histone demethylase KDM5A is regulated by its reader domain through a positive-feedback mechanism. Nat. Commun. 2015, 6, 6204. [Google Scholar] [CrossRef]
- Zhang, Y.; Yang, H.; Guo, X.; Rong, N.; Song, Y.; Xu, Y.; Lan, W.; Zhang, X.; Liu, M.; Xu, Y.; et al. The PHD1 finger of KDM5B recognizes unmodified H3K4 during the demethylation of histone H3K4me2/3 by KDM5B. Protein Cell 2014, 5, 837–850. [Google Scholar] [CrossRef] [Green Version]
- Klein, B.J.; Piao, L.; Xia, Z.; Rincon-Arano, H.; Rothbart, S.B.; Peng, D.; Wen, H.; Larsson, C.; Zhang, X.; Zheng, X.; et al. The histone-H3K4-specific demethylase KDM5B binds to its substrate and product through distinct PHD fingers. Cell Rep. 2014, 6, 325–335. [Google Scholar] [CrossRef] [Green Version]
- Horton, J.R.; Engstrom, A.; Zoeller, E.L.; Liu, X.; Shanks, J.R.; Zhang, X.; Johns, M.A.; Vertino, P.M.; Fu, H.; Cheng, X. Characterization of a Linked Jumonji Domain of the KDM5/JARID1 Family of Histone H3 Lysine 4 Demethylases. J. Biol. Chem. 2016, 291, 2631–2646. [Google Scholar] [CrossRef] [Green Version]
- Longbotham, J.E.; Chio, C.; Dharmarajan, V.; Trnka, M.; Torres, I.O.; Goswami, D.; Ruiz, K.; Burlingame, A.L.; Griffin, P.R.; Fujimori, D.G. Histone H3 binding to the PHD1 domain of histone demethylase KDM5A enables active site remodeling. Nat. Commun. 2019, 10, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Hayek, L.; Tuncay, I.O.; Nijem, N.; Russell, J.; Ludwig, S.; Kaur, K.; Li, X.; Anderton, P.; Tang, M.; Gerard, A.; et al. KDM5A mutations identified in autism spectrum disorder using forward genetics. eLife 2020, 9, e56883. [Google Scholar] [CrossRef] [PubMed]
- Mizukami, H.; Kim, J.-D.; Tabara, S.; Lu, W.; Kwon, C.; Nakashima, M.; Fukamizu, A. KDM5D-mediated H3K4 demethylation is required for sexually dimorphic gene expression in mouse embryonic fibroblasts. J. Biochem. 2018, 165, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Kosugi, M.; Otani, M.; Kikkawa, Y.; Itakura, Y.; Sakai, K.; Ito, T.; Toyoda, M.; Sekita, Y.; Kimura, T. Mutations of histone demethylase genes encoded by X and Y chromosomes, Kdm5c and Kdm5d, lead to noncompaction cardiomyopathy in mice. Biochem. Biophys. Res. Commun. 2020, 525, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Kim, C.; Bakken, T.E.; Gillentine, M.A.; Henning, B.; Mao, Y.; Gilissen, C.; The SPARK Consortium; Nowakowski, T.J.; Eichler, E.E. Integrated analyses of de novo mutations from 46,612 trios with autism and developmental disorders. bioRxiv 2021. [Google Scholar] [CrossRef]
- Albert, M.; Schmitz, S.U.; Kooistra, S.M.; Malatesta, M.; Morales Torres, C.; Rekling, J.C.; Johansen, J.V.; Abarrategui, I.; Helin, K. The histone demethylase Jarid1b ensures faithful mouse development by protecting developmental genes from aberrant H3K4me3. PLoS Genet. 2013, 9, e1003461. [Google Scholar] [CrossRef] [Green Version]
- Barrett, A.; Santangelo, S.; Tan, K.; Catchpole, S.; Roberts, K.; Spencer-Dene, B.; Hall, D.; Scibetta, A.; Burchell, J.; Verdin, E.; et al. Breast cancer associated transcriptional repressor PLU-1/JARID1B interacts directly with histone deacetylases. Int. J. Cancer 2007, 121, 265–275. [Google Scholar] [CrossRef]
- Li, Q.; Shi, L.; Gui, B.; Yu, W.; Wang, J.; Zhang, D.; Han, X.; Yao, Z.; Shang, Y. Binding of the JmjC demethylase JARID1B to LSD1/NuRD suppresses angiogenesis and metastasis in breast cancer cells by repressing chemokine CCL14. Cancer Res. 2011, 71, 6899–6908. [Google Scholar] [CrossRef] [Green Version]
- Tahiliani, M.; Mei, P.; Fang, R.; Leonor, T.; Rutenberg, M.; Shimizu, F.; Li, J.; Rao, A.; Shi, Y. The histone H3K4 demethylase SMCX links REST target genes to X-linked mental retardation. Nature 2007, 447, 601–605. [Google Scholar] [CrossRef]
- Nishibuchi, G.; Shibata, Y.; Hayakawa, T.; Hayakawa, N.; Ohtani, Y.; Sinmyozu, K.; Tagami, H.; Nakayama, J.-I. Physical and functional interactions between the histone H3K4 demethylase KDM5A and the nucleosome remodeling and deacetylase (NuRD) complex. J. Biol. Chem. 2014, 289, 28956–28970. [Google Scholar] [CrossRef] [Green Version]
- Penterling, C.; Drexler, G.A.; Böhland, C.; Stamp, R.; Wilke, C.; Braselmann, H.; Caldwell, R.B.; Reindl, J.; Girst, S.; Greubel, C.; et al. Depletion of histone demethylase Jarid1A resulting in histone hyperacetylation and radiation sensitivity does not affect DNA double-strand break repair. PLoS ONE 2016, 11, e0156599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasini, D.; Hansen, K.H.; Christensen, J.; Agger, K.; Cloos, P.A.; Helin, K. Coordinated regulation of transcriptional repression by the RBP2 H3K4 demethylase and Polycomb-Repressive Complex 2. Genes Dev. 2008, 22, 1345–1355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Liang, J.; Li, Q. Coordinated regulation of retinoic acid signaling pathway by KDM5B and polycomb repressive complex 2. J. Cell. Biochem. 2014, 115, 1528–1538. [Google Scholar] [CrossRef] [PubMed]
- Vedadi, M.; Blazer, L.; Eram, M.S.; Barsyte-Lovejoy, D.; Arrowsmith, C.H.; Hajian, T. Targeting human SET1/MLL family of proteins. Protein. Sci. 2017, 26, 662–676. [Google Scholar] [CrossRef] [Green Version]
- Ford, D.J.; Dingwall, A.K. The cancer COMPASS: Navigating the functions of MLL complexes in cancer. Cancer Genet. 2015, 208, 1780–1791. [Google Scholar] [CrossRef]
- Muntean, A.G.; Tan, J.; Sitwala, K.; Huang, Y.; Bronstein, J.; Connelly, J.A.; Basrur, V.; Elenitoba-Johnson, K.S.; Hess, J.L. The PAF complex synergizes with MLL fusion proteins at HOX loci to promote leukemo-genesis. Cancer Cell 2010, 17, 609–621. [Google Scholar] [CrossRef] [Green Version]
- Fong, C.Y.; Gilan, O.; Lam, E.Y.N.; Rubin, A.F.; Ftouni, S.; Tyler, D.; Stanley, K.; Sinha, D.; Yeh, P.; Morison, J.; et al. BET inhibitor resistance emerges from leukaemia stem cells. Nature 2015, 525, 538–542. [Google Scholar] [CrossRef]
- Cao, F.; Townsend, E.C.; Karatas, H.; Xu, J.; Li, L.; Lee, S.; Liu, L.; Chen, Y.; Ouillette, P.; Zhu, J.; et al. Targeting MLL1 H3K4 methyltransferase activity in mixed-lineage leukemia. Mol. Cell 2014, 53, 247–261. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Kim, D.H.; Lee, S.; Yang, Q.H.; Lee, D.K.; Lee, S.K.; Roeder, R.G.; Lee, J.W. A tumor suppressive coactivator complex of p53 containing ASC-2 and histone H3-lysine-4 methyltransferase MLL3 or its paralogue MLL4. Proc. Natl. Acad. USA 2009, 106, 8513–8518. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Zhao, Z.; Ozark, P.A.; Fantini, D.; Marshall, S.A.; Rendleman, E.J.; Cozzolino, K.A.; Louis, N.; He, X.; Morgan, M.A.; et al. Resetting the epigenetic balance of polycomab and COMPASS function at enhancers for cancer therapy. Nat. Med. 2018, 24, 758–769. [Google Scholar] [CrossRef]
- Mohammad, H.P.; Smitheman, K.N.; Kamat, C.D.; Soong, D.; Federowicz, K.E.; Van Aller, G.S.; Schneck, J.L.; Carson, J.D.; Liu, Y.; Butticello, M.; et al. A DNA Hypomethylation Signature Predicts Antitumor Activity of LSD1 Inhibitors in SCLC. Cancer Cell 2015, 28, 57–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellis, L.; Loda, M. LSD1: A single target to combat lineage plasticity in lethal prostate cancer. Proc. Natl. Acad. Sci. USA 2018, 115, 4530–4531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banck, M.S.; Li, S.; Nishio, H.; Wang, C.; Beutler, A.S.; Walsh, M.J. The ZNF217 oncogene is a candidate organizer of repressive histone modifiers. Epigenetics 2009, 4, 100–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maes, T.; Tirapu, I.; Mascaró, C.; Ortega, A.; Estiarte, A.; Valls, N.; Castro-Palomino, J.; Arjol, C.B.; Kurz, G. Preclinical characterization of a potent and selective inhibitor of the histone demethylase KDM1A for MLL leukemia. J. Clin. Oncol. 2013, 31, e13543. [Google Scholar] [CrossRef]
- Salamero, O.; Montesinos, P.; Willekens, C.; Pérez-Simón, J.A.; Pigneux, A.; Récher, C.; Popat, R.; Carpio, C.; Molinero, C.; Mascaró, C.; et al. First-in-Human Phase I Study of Iadademstat (ORY-1001): A First-in-Class Lysine-Specific Histone Demethylase 1A Inhibitor, in Relapsed or Refractory Acute Myeloid Leukemia. J. Clin. Oncol. 2020, 38, 4260–4273. [Google Scholar] [CrossRef]
- Harris, W.J.; Huang, X.; Lynch, J.T.; Spencer, G.J.; Hitchin, J.R.; Li, Y.; Ciceri, F.; Blaser, J.G.; Greystoke, B.F.; Jordan, A.M.; et al. The histone demethylase KDM1A sustains the oncogenis potential of MLL-AF9 leukemia stem cells. Cancer Cell 2012, 21, 473–487. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, Y.; Gamo, K.; Yabuki, M.; Takagi, S.; Toyoshima, K.; Nakayama, K.; Nakayama, A.; Morimoto, M.; Miyashita, H.; Dairiki, R.; et al. A Novel LSD1 inhibitor T-3775440 disrupts GFI1B-containing complex leading to transdifferentiation and impaired growth of AML cells. Mol. Cancer Ther. 2017, 16, 273–284. [Google Scholar] [CrossRef] [Green Version]
- Maiques-Diaz, A.; Spencer, G.J.; Lynch, J.T.; Ciceri, F.; Williams, E.L.; Amaral, F.M.; Wiseman, D.; Harris, W.J.; Li, Y.; Sahoo, S.; et al. Enhancer Activation by Pharmacologic Displacement of LSD1 from GFI1 Induces Differentiation in Acute Myeloid Leukemia. Cell Rep. 2018, 22, 3641–3659. [Google Scholar] [CrossRef] [Green Version]
- Vinyard, M.E.; Su, C.; Siegenfeld, A.P.; Waterbury, A.L.; Freedy, A.M.; Gosavi, P.M.; Park, Y.; Kwan, E.E.; Senzer, B.; Doench, J.G.; et al. CRISPR-suppressor scanning reveals a nonenzymatic role of LSD1 in AML. Nat. Chem. Biol. 2019, 15, 529–539. [Google Scholar] [CrossRef]
- Tsai, M.-C.; Manor, O.; Wan, Y.; Mosammaparast, N.; Wang, J.K.; Lan, F.; Shi, Y.; Segal, E.; Chang, H.Y. Long noncoding RNA as modular scaffold of histone modification complexes. Science 2010, 329, 689–693. [Google Scholar] [CrossRef] [Green Version]
- Gupta, R.A.; Shah, N.; Wang, K.C.; Kim, J.; Horlings, H.M.; Wong, D.J.; Tsai, M.-C.; Hung, T.; Argani, P.; Rinn, J.L.; et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 2010, 464, 1071–1076. [Google Scholar] [CrossRef] [PubMed]
- Pastori, C.; Kapranov, P.; Penas, C.; Peschansky, V.; Volmar, C.-H.; Sarkaria, J.N.; Bregy, A.; Komotar, R.; St Laurent, G.; Ayad, N.G.; et al. The Bromodomain protein BRD4 controls HOTAIR, a long noncoding RNA essential for glioblastoma proliferation. Proc. Natl. Acad. Sci. USA 2015, 112, 8326–8331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harmeyer, K.M.; Facompre, N.D.; Herlyn, M.; Basu, D. JARID1 Histone Demethylases: Emerging Targets in Cancer. Trends Cancer 2017, 3, 713–725. [Google Scholar] [CrossRef] [PubMed]
- Han, M.; Xu, W.; Cheng, P.; Jin, H.; Wang, X. al Histone demethylase lysine demethylase 5B in development and cancer. Oncotarget 2017, 8, 8980–8991. [Google Scholar] [CrossRef] [Green Version]
- Xhabija, B.; Kidder, B.L. KDM5B is a master regulator of the H3K4-methylome in stem cells, development and cancer. Semin. Cancer Biol. 2018, 57, 79–85. [Google Scholar] [CrossRef]
- Yamamoto, S.; Wu, Z.; Russnes, H.G.; Takagi, S.; Peluffo, G.; Vaske, C.; Zhao, X.; Vollan, H.K.M.; Maruyama, R.; Ekram, M.B.; et al. JARID1B is a luminal lineage-driving oncogene in breast cancer. Cancer Cell 2014, 25, 762–777. [Google Scholar] [CrossRef] [Green Version]
- Montano, M.M.; Yeh, I.-J.; Chen, Y.; Hernandez, C.; Kiselar, J.G.; De La Fuente, M.; Lawes, A.M.; Nieman, M.T.; Kiser, P.D.; Jacobberger, J.; et al. Inhibition of the histone demethylase, KDM5B, directly induces re-expression of tumor suppressor protein HEXIM1 in cancer cells. Breast Cancer Res. 2019, 21, 138. [Google Scholar] [CrossRef]
- Dai, B.; Hu, Z.; Huang, H.; Zhu, G.; Xiao, Z.; Wan, W.; Zhang, P.; Jia, W.; Zhang, L. Overexpressed KDM5B is associated with the progression of glioma and promotes glioma cell growth via downregulating p21. Biochem. Biophys. Res. Commun. 2014, 454, 221–227. [Google Scholar] [CrossRef]
- Ricketts, C.J.; Lineham, W.M. Gender Specific Mutation Incidence and Survival Associations in Clear Cell Renal Cell Carcinoma (CCRCC). PLoS ONE 2015, 10, e0140257. [Google Scholar] [CrossRef] [Green Version]
- Smith, J.A.; Haberstroh, F.S.; White, E.A.; Livingston, D.M.; DeCaprio, J.A.; Howley, P.M. SMCX and components of the TIP60 complex contribute to E2 regulation of the HPV E6/E7 promoter. Virology 2014, 468–470, 311–321. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Dhar, S.S.; Chen, T.-Y.; Kan, P.-Y.; Wei, Y.; Kim, J.-H.; Chan, C.-H.; Lin, H.-K.; Hung, M.-C.; Lee, M.G. JARID1D Is a Suppressor and Prognostic Marker of Prostate Cancer Invasion and Metastasis. Cancer Res. 2016, 76, 831–843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komura, K.; Jeong, S.H.; Hinohara, K.; Qu, F.; Wang, X.; Hiraki, M.; Azuma, H.; Lee, G.S.; Kantoff, P.W.; Sweeney, C.J. Resistance to docetaxel in prostate cancer is associated with androgen receptor ac-tivation and loss of KDM5D expression. Proc. Natl. Acad. Sci. USA 2016, 113, 6259–6264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, S.H.; Goode, D.L.; Iwasaki, M.; Wei, M.C.; Kuo, H.-P.; Zhu, L.; Schneidawind, D.; Duque-Afonso, J.; Weng, Z.; Cleary, M.L. The H3K4-Methyl Epigenome Regulates Leukemia Stem Cell Oncogenic Potential. Cancer Cell 2015, 28, 198–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, S.V.; Lee, D.Y.; Li, B.; Quinlan, M.P.; Takahashi, F.; Maheswaran, S.; McDermott, U.; Azizian, N.; Zou, L.; Fischbach, M.A.; et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 2010, 141, 69–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roesch, A.; Fukunaga-Kalabis, M.; Schmidt, E.C.; Zabierowski, S.E.; Brafford, P.A.; Vultur, A.; Basu, D.; Gimotty, P.; Vogt, T.; Herlyn, M. A Temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth. Cell 2010, 141, 583–594. [Google Scholar] [CrossRef] [Green Version]
- Vinogradova, M.; Gehling, V.S.; Gustafson, A.; Arora, S.; Tindell, C.A.; Wilson, C.; Williamson, K.E.; Guler, G.D.; Gangurde, P.; Manieri, W.; et al. An inhibitor of KDM5 demethylases reduces survival of drug-tolerant cancer cells. Nat. Chem. Biol. 2016, 12, 531–538. [Google Scholar] [CrossRef]
- Johansson, C.; Velupillai, S.; Tumber, A.; Szykowska, A.; Hookway, E.S.; Nowak, R.; Strain-Damerell, C.; Gileadi, C.; Philpott, M.; Burgess-Brown, N.; et al. Structural analysis of human KDM5B guides histone demethylase inhibitor development. Nat. Chem. Biol. 2016, 12, 539–545. [Google Scholar] [CrossRef]
- Sayegh, J.; Cao, J.; Zou, M.R.; Morales, A.; Blair, L.P.; Norcia, M.; Hoyer, D.; Tackett, A.J.; Merkel, J.S.; Yan, Q. Identification of small molecule inhibitors of jumonji AT-rich interactive domain 1B (JARID1B) histone demethylase by a sensitive high throughput screen. J. Biol. Chem. 2013, 288, 9408–9417. [Google Scholar] [CrossRef] [Green Version]
- Bavetsias, V.; Lanigan, R.M.; Ruda, G.F.; Atrash, B.; McLaughlin, M.G.; Tumber, A.; Mok, N.Y.; Le Bihan, Y.-V.; Dempster, S.; Boxall, K.J.; et al. 8-Substituted Pyrido[3,4-d]pyrimidin-4(3H)-one Derivatives As Potent, Cell Permeable, KDM4 (JMJD2) and KDM5 (JARID1) Histone Lysine Demethylase Inhibitors. J. Med. Chem. 2016, 59, 1388–1409. [Google Scholar] [CrossRef]
- Leadem, B.R.; Kagiampakis, I.; Wilson, C.; Cheung, T.K.; Arnott, D.; Trojer, P.; Classon, M.; Easwaran, H.; Baylin, S.B. A KDM5 Inhibitor Increases Global H3K4 Trimethylation Occupancy and Enhances the Bi-ological Efficacy of 5-Aza-2’-Deoxycytidine. Cancer Res. 2018, 78, 1127–1139. [Google Scholar] [CrossRef] [Green Version]
- Madsen, B.; Spencer-Dene, B.; Poulsom, R.; Hall, D.; Lu, P.J.; Scott, K.; Shaw, A.T.; Burchell, J.M.; Freemont, P.; Taylor-Papadimitriou, J. Characterisation and developmental expression of mouse Plu-1, a homologue of a human nuclear protein (PLU-1) which is specifically up-regulated in breast cancer. Mech. Dev. 2002, 119, s239–s246. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Taylor-Papadimitriou, J.; Burchell, J.M. Histone Methylases and Demethylases Regulating Antagonistic Methyl Marks: Changes Occurring in Cancer. Cells 2022, 11, 1113. https://doi.org/10.3390/cells11071113
Taylor-Papadimitriou J, Burchell JM. Histone Methylases and Demethylases Regulating Antagonistic Methyl Marks: Changes Occurring in Cancer. Cells. 2022; 11(7):1113. https://doi.org/10.3390/cells11071113
Chicago/Turabian StyleTaylor-Papadimitriou, Joyce, and Joy M. Burchell. 2022. "Histone Methylases and Demethylases Regulating Antagonistic Methyl Marks: Changes Occurring in Cancer" Cells 11, no. 7: 1113. https://doi.org/10.3390/cells11071113