
Sevoflurane Dampens Acute Pulmonary Inflammation via the Adenosine Receptor A2B and Heme Oxygenase-1

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. LPS-Induced Pulmonary Inflammation and Pharmacological Application
2.3. In Vivo Migration Assay
2.4. Chemokine Release
2.5. Gene Expression
2.6. Microvascular Leakage
2.7. Generation of Chimeric Mice
2.8. Immunohistochemistry
2.9. Immunofluorescence
2.10. In Vitro Experiments
2.11. Statistical Analysis and Software
3. Results
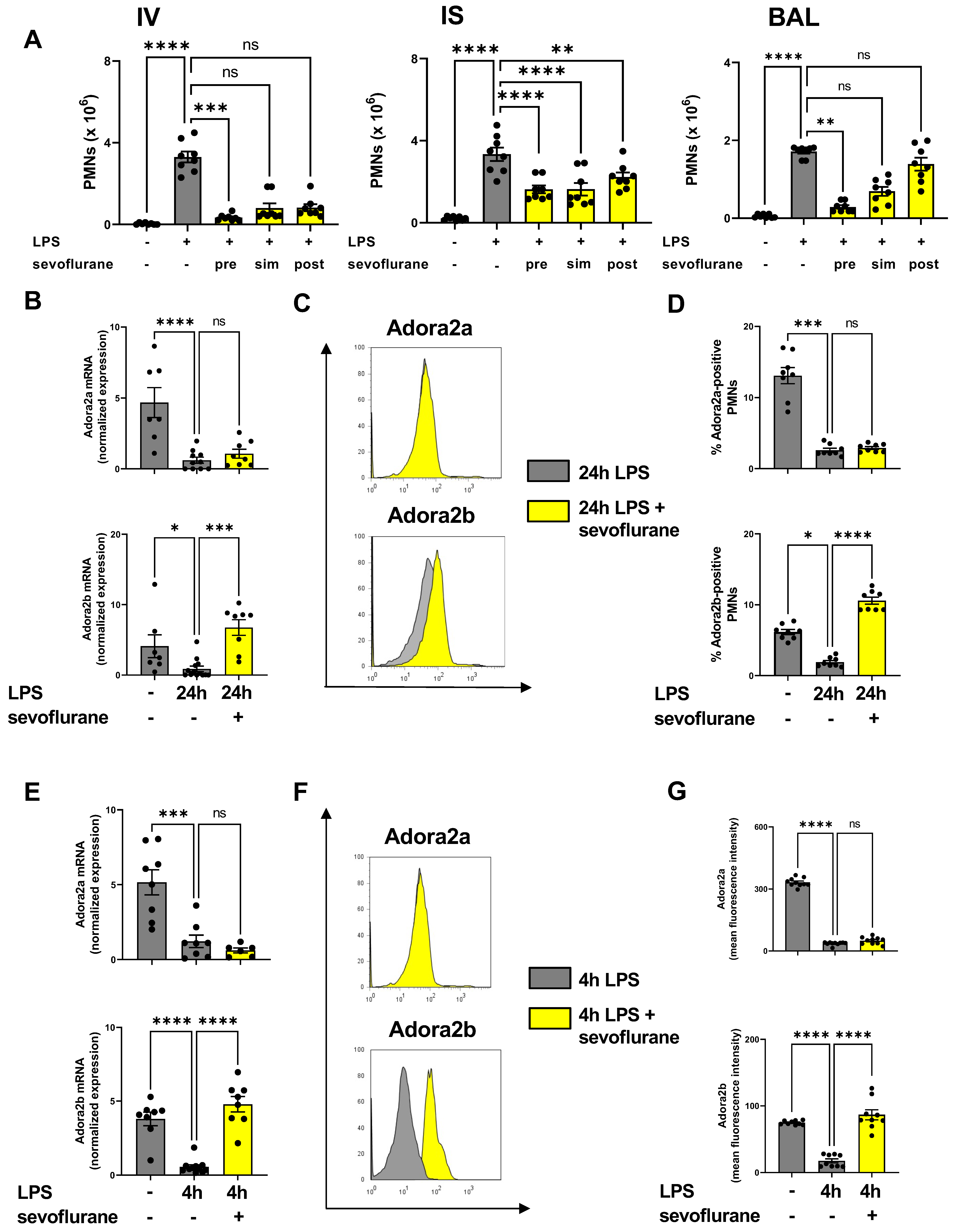
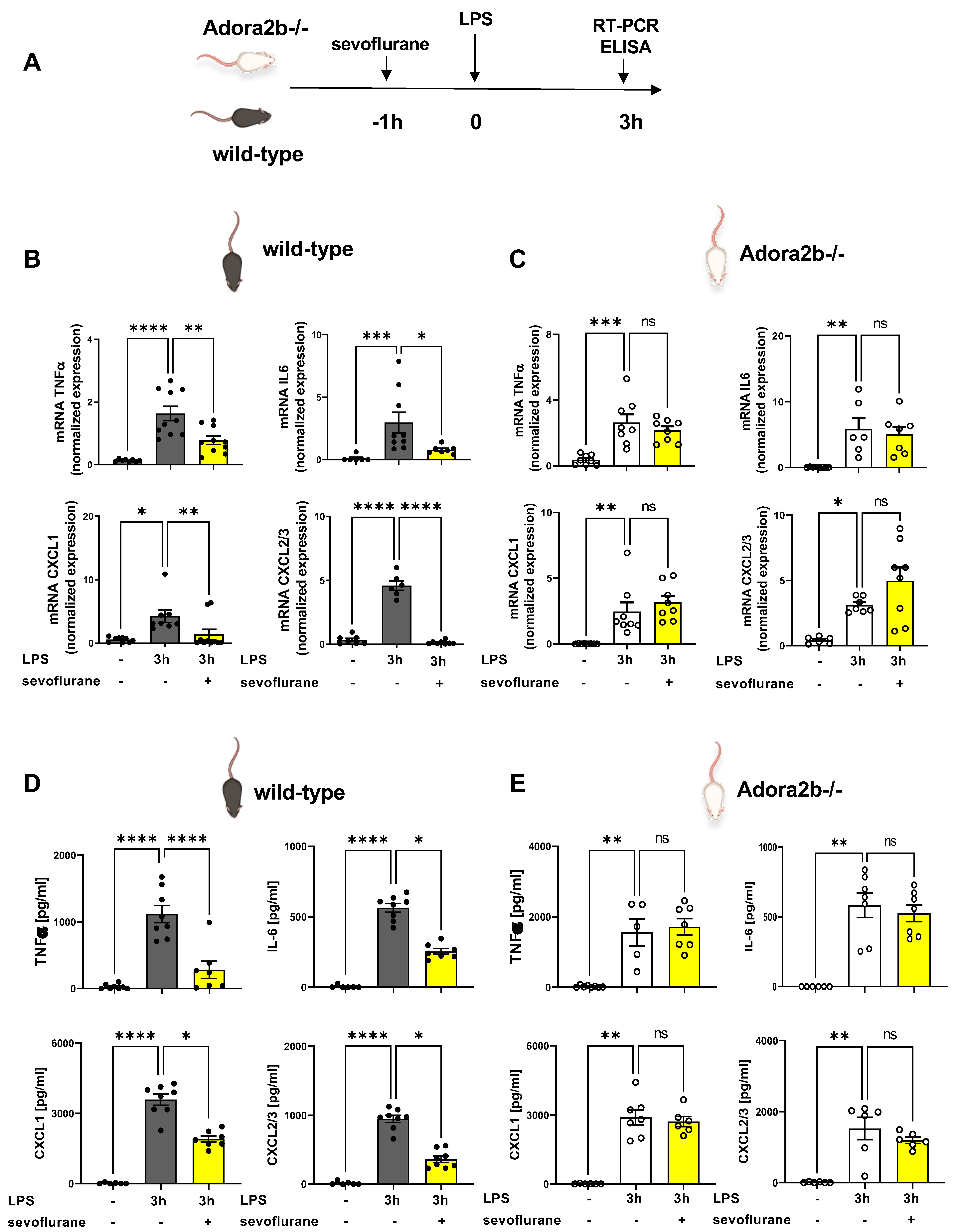
3.1. Time-Dependent Anti-Inflammatory Effects of Sevoflurane
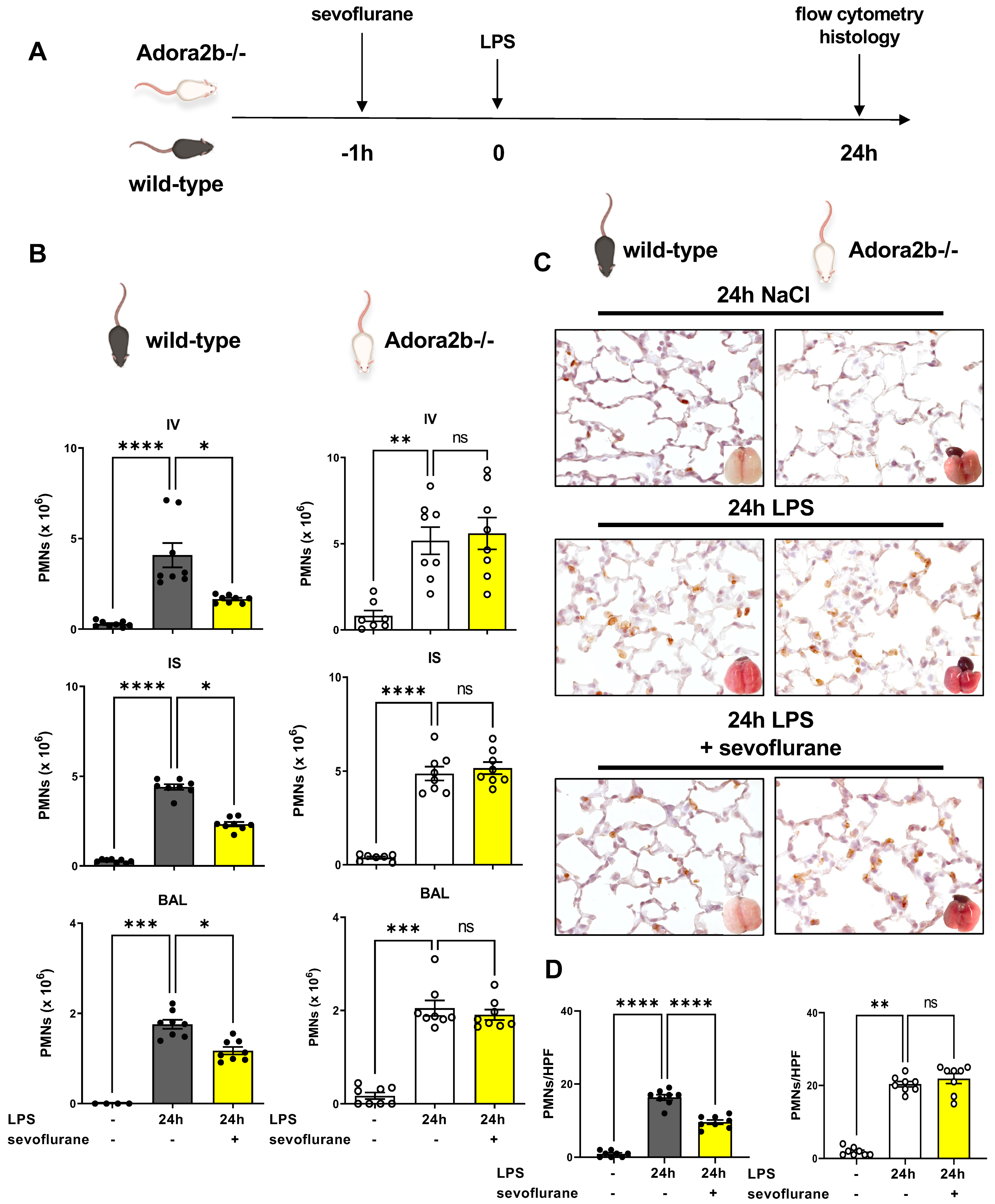
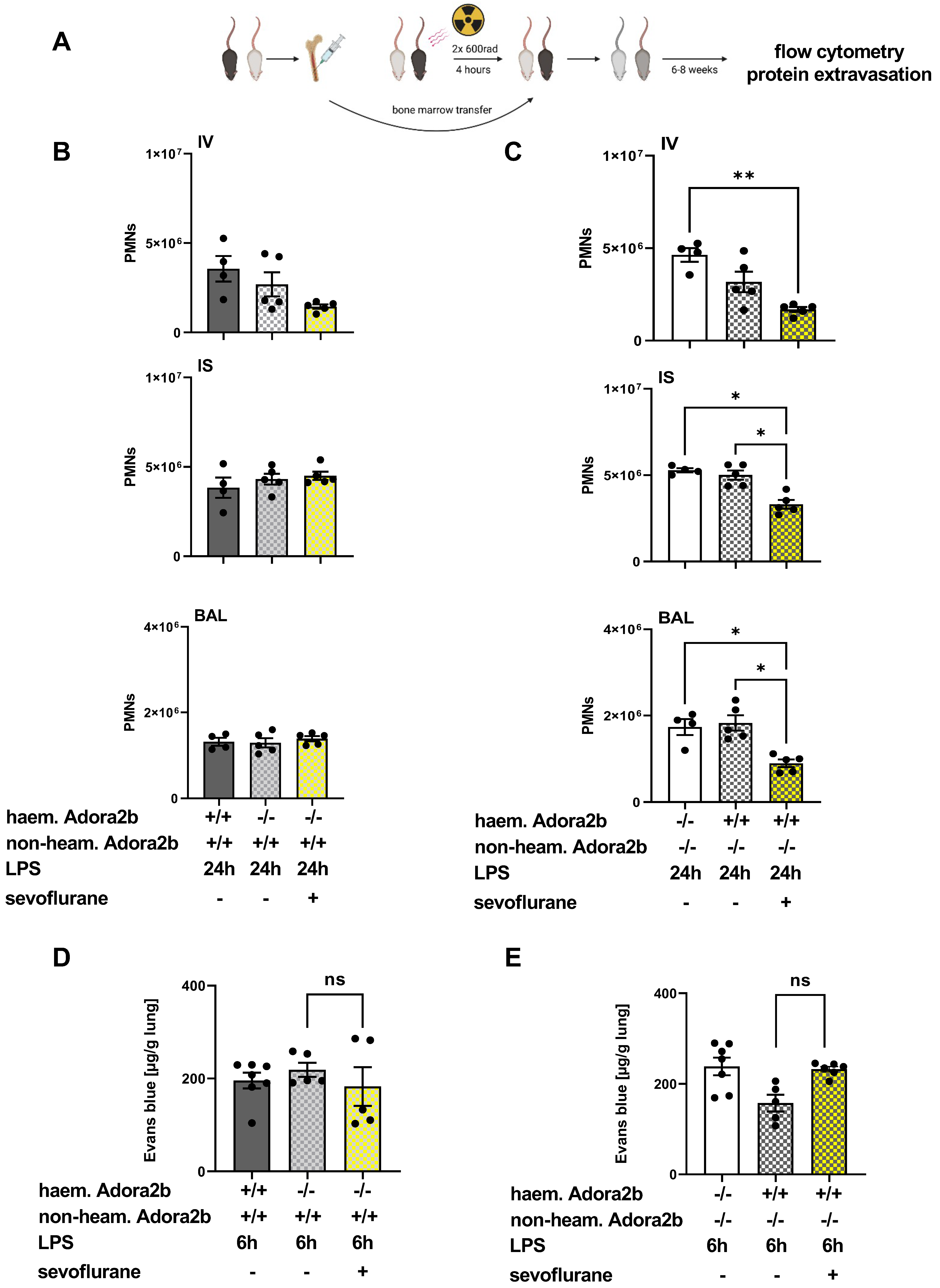
3.2. Protective Effects of Sevoflurane on the Influx of PMNs Depend on a Functional Adora2b
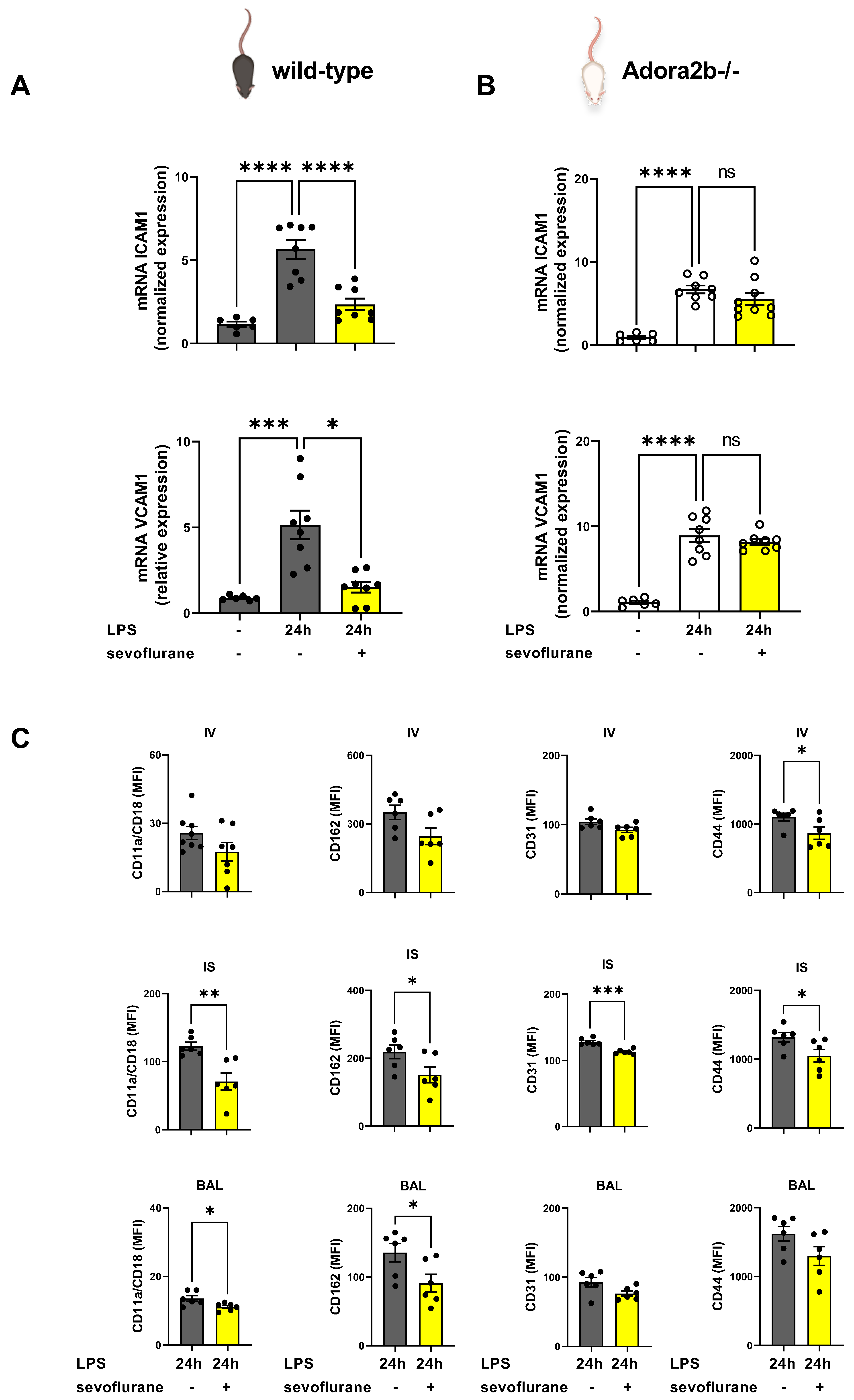
3.3. The Impact of Sevoflurane on Adhesion Molecules during Acute Pulmonary Inflammation
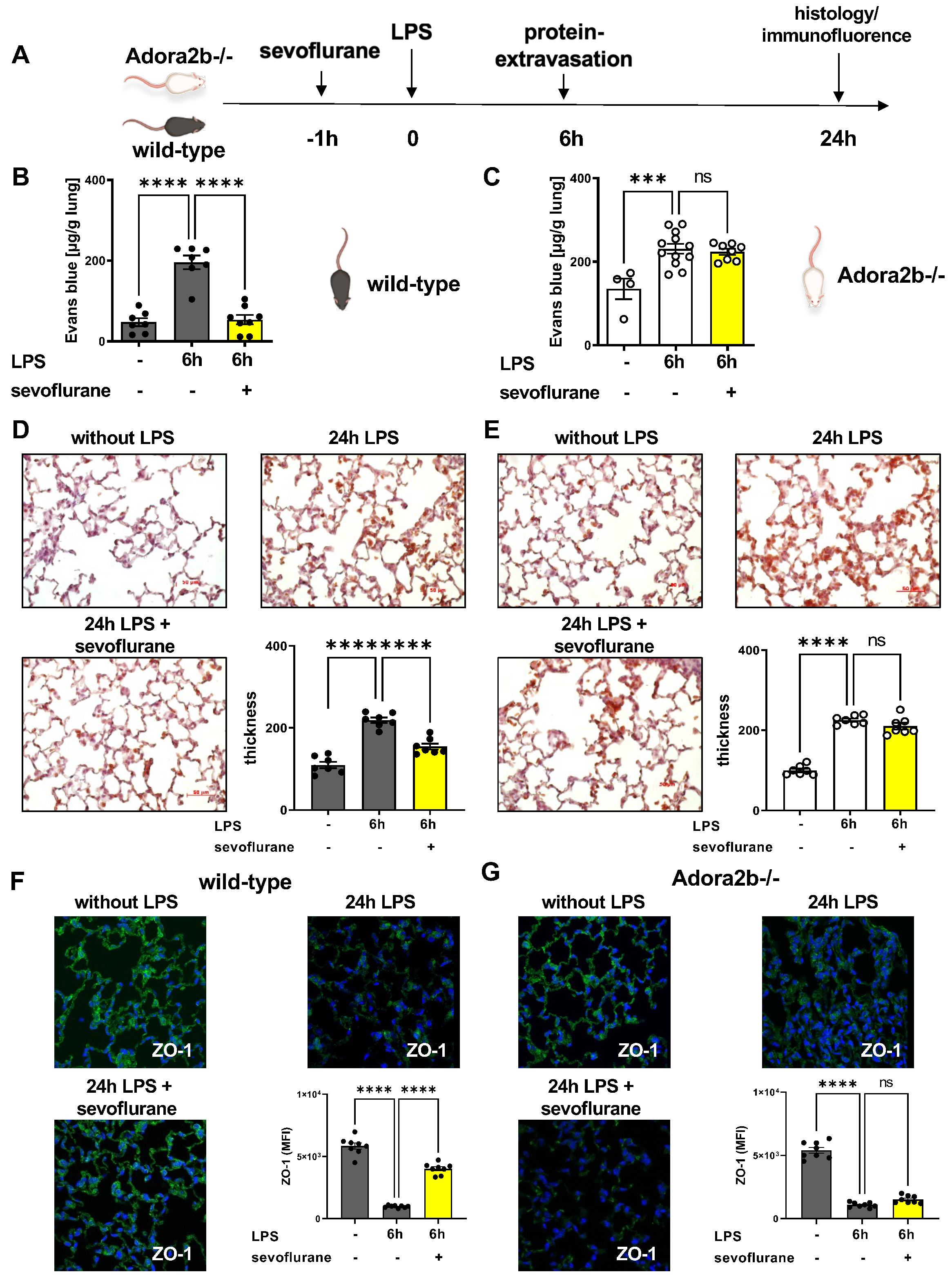
3.4. Protective Effects of Sevoflurane on Microvascular Permeability
3.5. Sevoflurane Dampens the Expression of Inflammatory Mediators
3.6. The Impact of Sevoflurane on Hematopoietic and Non-Hematopoietic Adora2b In Vivo
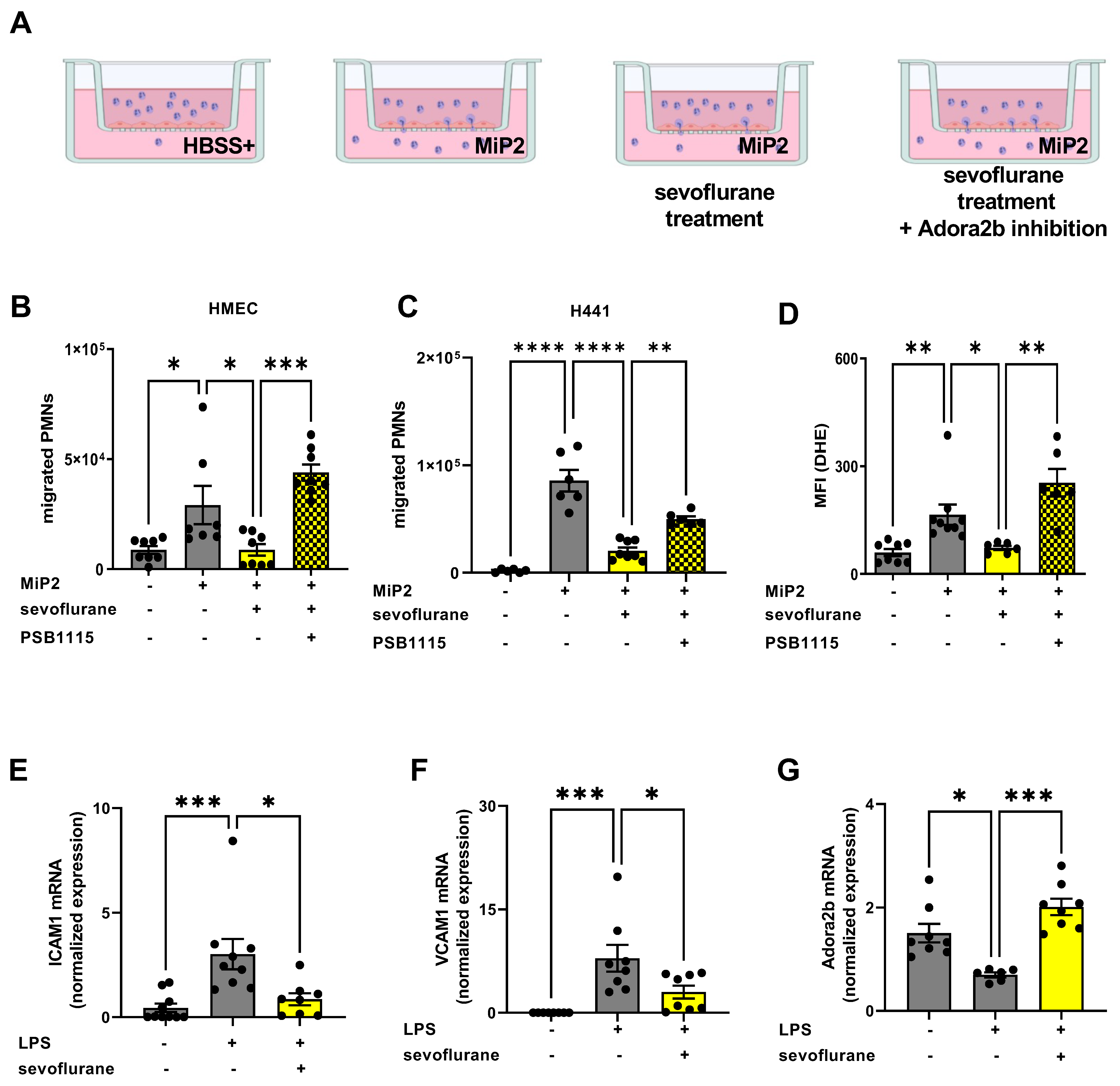
3.7. Protective Effects of Sevoflurane In Vitro Depend on a Functional Adora2b
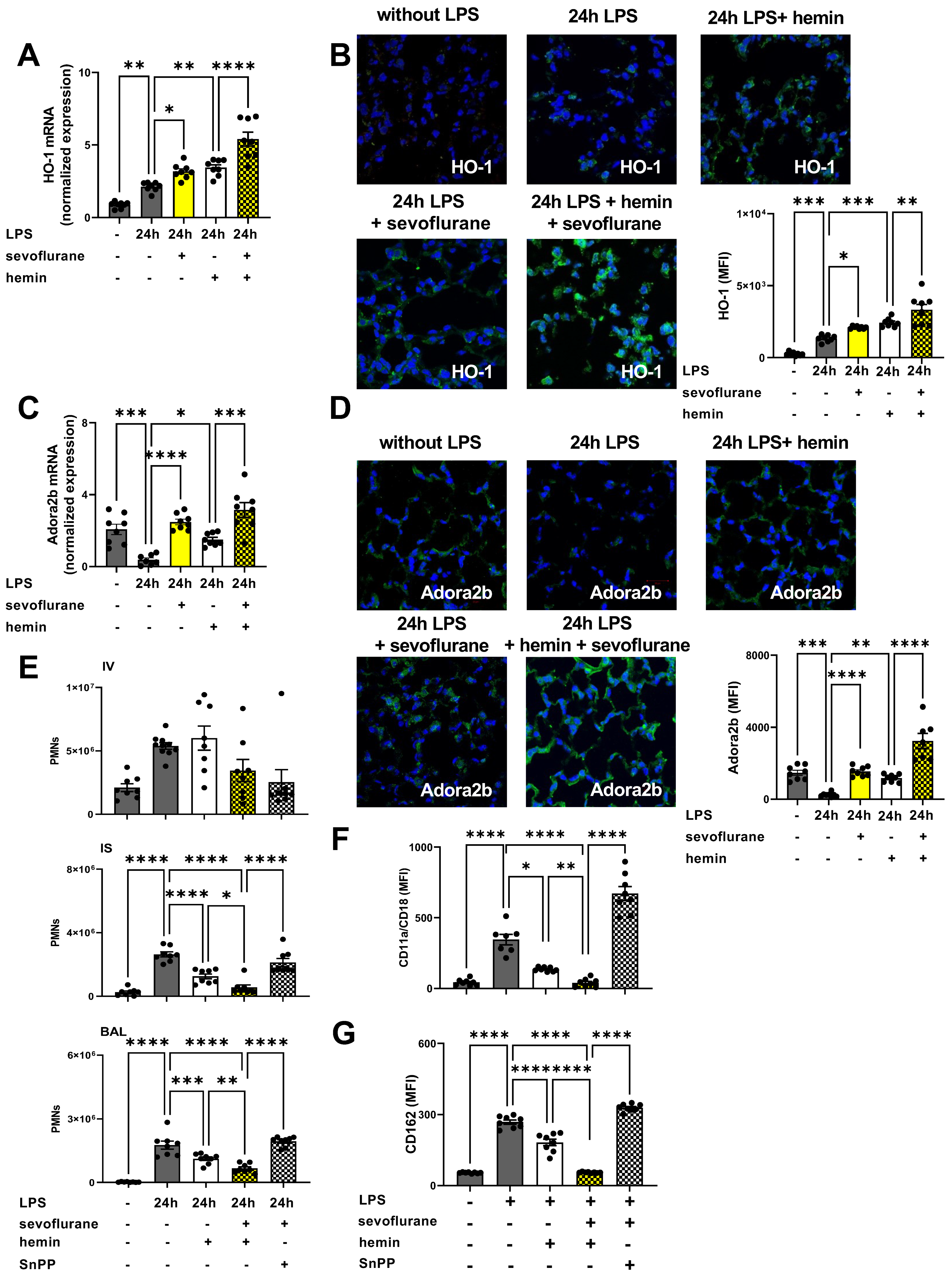
3.8. The Impact of the Anesthetic Agent Sevoflurane on HO-1 Modulation during Acute Pulmonary Inflammation
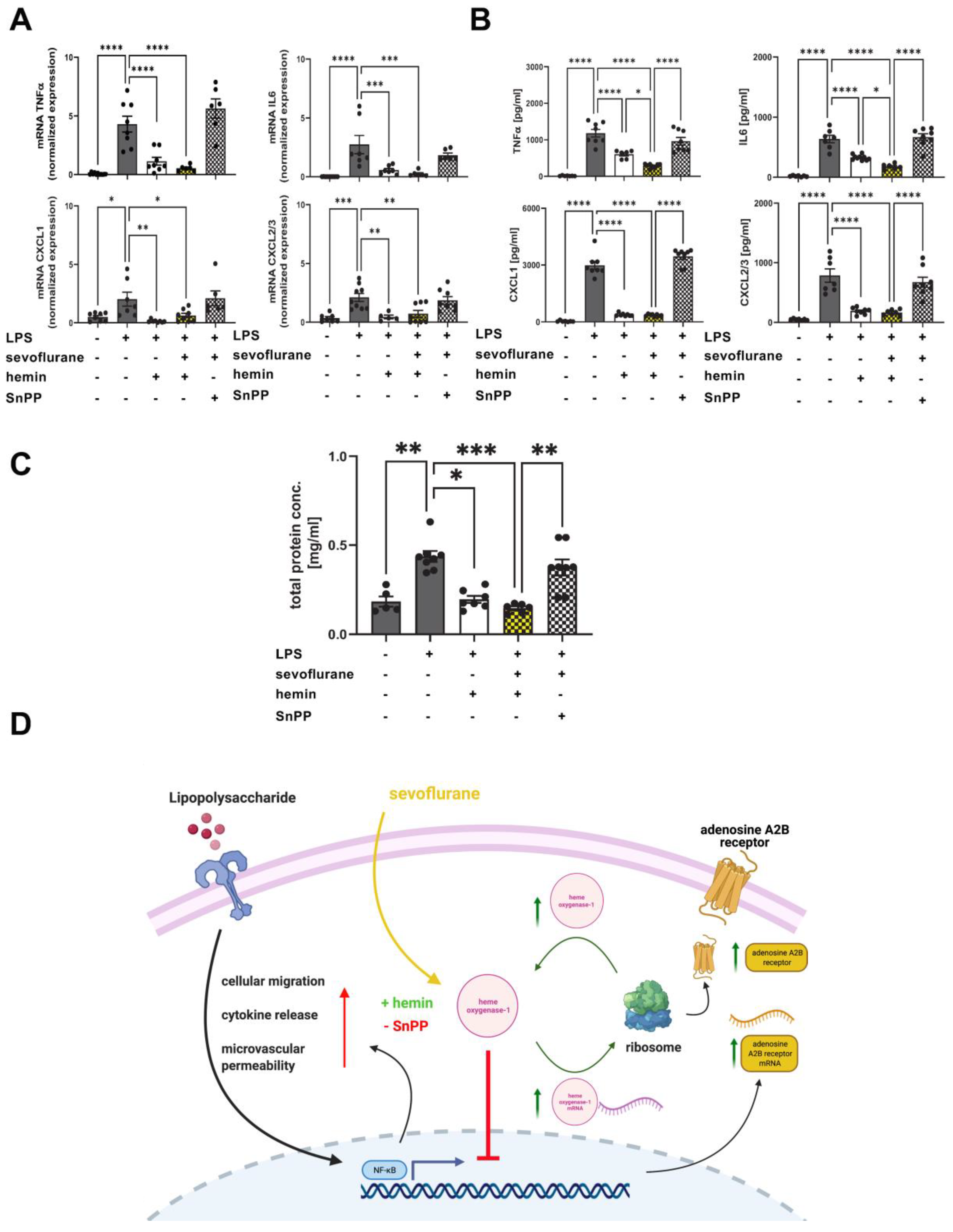
3.9. Protective Effects of Combined Treatment with Sevoflurane and Hemin on the Release of Inflammatory Cytokines and Microvascular Permeability
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Matthay, M.A.; Zemans, R.L.; Zimmerman, G.A.; Arabi, Y.M.; Beitler, J.R.; Mercat, A.; Herridge, M.; Randolph, A.G.; Calfee, C.S. Acute respiratory distress syndrome. Nat. Rev. Dis. Primers 2019, 5, 18. [Google Scholar] [CrossRef]
- Huppert, L.A.; Matthay, M.A.; Ware, L.B. Pathogenesis of Acute Respiratory Distress Syndrome. Semin. Respir. Crit. Care Med. 2019, 40, 31–39. [Google Scholar] [CrossRef] [Green Version]
- Juss, J.; Herre, J.; Begg, M.; Bradley, G.; Lennon, M.; Amour, A.; House, D.; Hessel, E.M.; Summers, C.; Condliffe, A.M.; et al. Genome-wide transcription profiling in neutrophils in acute respiratory distress syndrome. Lancet 2015, 385 (Suppl. S1), S55. [Google Scholar] [CrossRef]
- Artigas, A.; Camprubi-Rimblas, M.; Tantinya, N.; Bringue, J.; Guillamat-Prats, R.; Matthay, M.A. Inhalation therapies in acute respiratory distress syndrome. Ann. Transl. Med. 2017, 5, 293. [Google Scholar] [CrossRef] [Green Version]
- Festic, E.; Carr, G.E.; Cartin-Ceba, R.; Hinds, R.F.; Banner-Goodspeed, V.; Bansal, V.; Asuni, A.T.; Talmor, D.; Rajagopalan, G.; Frank, R.D.; et al. Randomized Clinical Trial of a Combination of an Inhaled Corticosteroid and Beta Agonist in Patients at Risk of Developing the Acute Respiratory Distress Syndrome. Crit. Care Med. 2017, 45, 798–805. [Google Scholar] [CrossRef]
- Bellani, G.; Laffey, J.G.; Pham, T.; Fan, E.; Brochard, L.; Esteban, A.; Gattinoni, L.; van Haren, F.; Larsson, A.; McAuley, D.F.; et al. Epidemiology, Patterns of Care, and Mortality for Patients With Acute Respiratory Distress Syndrome in Intensive Care Units in 50 Countries. JAMA 2016, 315, 788–800. [Google Scholar] [CrossRef]
- Ware, L.B.; Koyama, T.; Zhao, Z.; Janz, D.R.; Wickersham, N.; Bernard, G.R.; May, A.K.; Calfee, C.S.; Matthay, M.A. Biomarkers of lung epithelial injury and inflammation distinguish severe sepsis patients with acute respiratory distress syndrome. Crit. Care 2013, 17, R253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlapfer, M.; Piegeler, T.; Dull, R.O.; Schwartz, D.E.; Mao, M.; Bonini, M.G.; Z’Graggen, B.R.; Beck-Schimmer, B.; Minshall, R.D. Propofol increases morbidity and mortality in a rat model of sepsis. Crit. Care 2015, 19, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krannich, A.; Leithner, C.; Engels, M.; Nee, J.; Petzinka, V.; Schroder, T.; Jorres, A.; Kruse, J.; Storm, C. Isoflurane Sedation on the ICU in Cardiac Arrest Patients Treated With Targeted Temperature Management: An Observational Propensity-Matched Study. Crit. Care Med. 2017, 45, e384–e390. [Google Scholar] [CrossRef]
- Jerath, A.; Panckhurst, J.; Parotto, M.; Lightfoot, N.; Wasowicz, M.; Ferguson, N.D.; Steel, A.; Beattie, W.S. Safety and Efficacy of Volatile Anesthetic Agents Compared With Standard Intravenous Midazolam/Propofol Sedation in Ventilated Critical Care Patients: A Meta-analysis and Systematic Review of Prospective Trials. Anesth. Analg. 2017, 124, 1190–1199. [Google Scholar] [CrossRef] [PubMed]
- Granja, T.F.; Kohler, D.; Schad, J.; de Oliveira, C.B.; Konrad, F.; Hoch-Gutbrod, M.; Streienberger, A.; Rosenberger, P.; Straub, A. Adenosine Receptor Adora2b Plays a Mechanistic Role in the Protective Effect of the Volatile Anesthetic Sevoflurane during Liver Ischemia/Reperfusion. Anesthesiology 2016, 125, 547–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Q.Y.; Wu, L.; Wei, C.S.; Zhou, Y.N.; Wu, H.M. Sevoflurane Prevents Airway Remodeling via Downregulation of VEGF and TGF-beta1 in Mice with OVA-Induced Chronic Airway Inflammation. Inflammation 2019, 42, 1015–1022. [Google Scholar] [CrossRef] [PubMed]
- Burburan, S.M.; Silva, J.D.; Abreu, S.C.; Samary, C.S.; Guimaraes, I.H.; Xisto, D.G.; Morales, M.M.; Rocco, P.R. Effects of inhalational anaesthetics in experimental allergic asthma. Anaesthesia 2014, 69, 573–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugasawa, Y.; Yamaguchi, K.; Kumakura, S.; Murakami, T.; Suzuki, K.; Nagaoka, I.; Inada, E. Effects of sevoflurane and propofol on pulmonary inflammatory responses during lung resection. J. Anesth. 2012, 26, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Bowser, J.L.; Lee, J.W.; Yuan, X.; Eltzschig, H.K. The Hypoxia-Adenosine Link during Inflammation. J. Appl. Physiol. (1985) 2017, 123, 1303–1320. [Google Scholar] [CrossRef] [Green Version]
- Yuan, X.; Lee, J.W.; Bowser, J.L.; Neudecker, V.; Sridhar, S.; Eltzschig, H.K. Targeting Hypoxia Signaling for Perioperative Organ Injury. Anesth. Analg. 2017, 126, 308–321. [Google Scholar] [CrossRef]
- Idzko, M.; Ferrari, D.; Riegel, A.K.; Eltzschig, H.K. Extracellular nucleotide and nucleoside signaling in vascular and blood disease. Blood 2014, 124, 1029–1037. [Google Scholar] [CrossRef] [Green Version]
- Ferrari, D.; McNamee, E.N.; Idzko, M.; Gambari, R.; Eltzschig, H.K. Purinergic Signaling During Immune Cell Trafficking. Trends Immunol. 2016, 37, 399–411. [Google Scholar] [CrossRef]
- Borg, N.; Alter, C.; Gorldt, N.; Jacoby, C.; Ding, Z.; Steckel, B.; Quast, C.; Bonner, F.; Friebe, D.; Temme, S.; et al. CD73 on T Cells Orchestrates Cardiac Wound Healing After Myocardial Infarction by Purinergic Metabolic Reprogramming. Circulation 2017, 136, 297–313. [Google Scholar] [CrossRef]
- Eckle, T.; Hughes, K.; Ehrentraut, H.; Brodsky, K.S.; Rosenberger, P.; Choi, D.S.; Ravid, K.; Weng, T.; Xia, Y.; Blackburn, M.R.; et al. Crosstalk between the equilibrative nucleoside transporter ENT2 and alveolar Adora2b adenosine receptors dampens acute lung injury. FASEB J. 2013, 27, 3078–3089. [Google Scholar] [CrossRef] [Green Version]
- Zimmerman, M.A.; Tak, E.; Ehrentraut, S.F.; Kaplan, M.; Giebler, A.; Weng, T.; Choi, D.S.; Blackburn, M.R.; Kam, I.; Eltzschig, H.K.; et al. Equilibrative nucleoside transporter (ENT)-1-dependent elevation of extracellular adenosine protects the liver during ischemia and reperfusion. Hepatology 2013, 58, 1766–1778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoegl, S.; Brodsky, K.S.; Blackburn, M.R.; Karmouty-Quintana, H.; Zwissler, B.; Eltzschig, H.K. Alveolar Epithelial A2B Adenosine Receptors in Pulmonary Protection during Acute Lung Injury. J. Immunol. 2015, 195, 1815–1824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aherne, C.M.; Saeedi, B.; Collins, C.B.; Masterson, J.C.; McNamee, E.N.; Perrenoud, L.; Rapp, C.R.; Curtis, V.F.; Bayless, A.; Fletcher, A.; et al. Epithelial-specific A2B adenosine receptor signaling protects the colonic epithelial barrier during acute colitis. Mucosal Immunol. 2015, 8, 1324–1338. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, M.A.; Grenz, A.; Tak, E.; Kaplan, M.; Ridyard, D.; Brodsky, K.S.; Mandell, M.S.; Kam, I.; Eltzschig, H.K. Signaling through hepatocellular A2B adenosine receptors dampens ischemia and reperfusion injury of the liver. Proc. Natl. Acad. Sci. USA 2013, 110, 12012–12017. [Google Scholar] [CrossRef] [Green Version]
- Eckle, T.; Hartmann, K.; Bonney, S.; Reithel, S.; Mittelbronn, M.; Walker, L.A.; Lowes, B.D.; Han, J.; Borchers, C.H.; Buttrick, P.M.; et al. Adora2b-elicited Per2 stabilization promotes a HIF-dependent metabolic switch crucial for myocardial adaptation to ischemia. Nat. Med. 2012, 18, 774–782. [Google Scholar] [CrossRef] [Green Version]
- Konrad, F.M.; Neudeck, G.; Vollmer, I.; Ngamsri, K.C.; Thiel, M.; Reutershan, J. Protective effects of pentoxifylline in pulmonary inflammation are adenosine receptor A2A dependent. FASEB J. 2013, 27, 3524–3535. [Google Scholar] [CrossRef]
- Hasko, G.; Xu, D.Z.; Lu, Q.; Nemeth, Z.H.; Jabush, J.; Berezina, T.L.; Zaets, S.B.; Csoka, B.; Deitch, E.A. Adenosine A2A receptor activation reduces lung injury in trauma/hemorrhagic shock. Crit. Care Med. 2006, 34, 1119–1125. [Google Scholar] [CrossRef]
- Ngamsri, K.C.; Fabian, F.; Fuhr, A.; Gamper-Tsigaras, J.; Straub, A.; Fecher, D.; Steinke, M.; Walles, H.; Reutershan, J.; Konrad, F.M. Sevoflurane Exerts Protective Effects in Murine Peritonitis-induced Sepsis via Hypoxia-inducible Factor 1alpha/Adenosine A2B Receptor Signaling. Anesthesiology 2021, 135, 136–150. [Google Scholar] [CrossRef]
- Gross, C.M.; Kovacs-Kasa, A.; Meadows, M.L.; Cherian-Shaw, M.; Fulton, D.J.; Verin, A.D. Adenosine and ATPgammaS protect against bacterial pneumonia-induced acute lung injury. Sci. Rep. 2020, 10, 18078. [Google Scholar] [CrossRef]
- Kreth, S.; Kaufmann, I.; Ledderose, C.; Luchting, B.; Thiel, M. Reduced ligand affinity leads to an impaired function of the adenosine A2A receptor of human granulocytes in sepsis. J. Cell. Mol. Med. 2009, 13, 985–994. [Google Scholar] [CrossRef] [Green Version]
- Hoetzel, A.; Schmidt, R. Regulatory role of anesthetics on heme oxygenase-1. Curr. Drug Targets 2010, 11, 1495–1503. [Google Scholar] [CrossRef] [PubMed]
- Campbell, N.K.; Fitzgerald, H.K.; Dunne, A. Regulation of inflammation by the antioxidant haem oxygenase 1. Nat. Rev. Immunol. 2021, 21, 411–425. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Yu, M.; Fang, Q.; Zhang, L.; You, C.; Wang, X.; Liu, Y.; Han, C. Heme oxygenase-1 induction mitigates burn-associated early acute kidney injury via the TLR4 signaling pathway. Burns 2021, 48, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.F.; Ward, P.A. Role of oxidants in lung injury during sepsis. Antioxid. Redox Signal. 2007, 9, 1991–2002. [Google Scholar] [CrossRef]
- Konrad, F.M.; Zwergel, C.; Ngamsri, K.C.; Reutershan, J. Anti-inflammatory Effects of Heme Oxygenase-1 Depend on Adenosine A2A- and A2B-Receptor Signaling in Acute Pulmonary Inflammation. Front. Immunol. 2017, 8, 1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konrad, F.M.; Knausberg, U.; Hone, R.; Ngamsri, K.C.; Reutershan, J. Tissue heme oxygenase-1 exerts anti-inflammatory effects on LPS-induced pulmonary inflammation. Mucosal Immunol. 2016, 9, 98–111. [Google Scholar] [CrossRef]
- Ngamsri, K.C.; Muller, A.; Bosmuller, H.; Gamper-Tsigaras, J.; Reutershan, J.; Konrad, F.M. The Pivotal Role of CXCR7 in Stabilization of the Pulmonary Epithelial Barrier in Acute Pulmonary Inflammation. J. Immunol. 2017, 198, 2403–2413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ngamsri, K.C.; Wagner, R.; Vollmer, I.; Stark, S.; Reutershan, J. Adenosine receptor A1 regulates polymorphonuclear cell trafficking and microvascular permeability in lipopolysaccharide-induced lung injury. J. Immunol. 2010, 185, 4374–4384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konrad, F.M.; Meichssner, N.; Bury, A.; Ngamsri, K.C.; Reutershan, J. Inhibition of SDF-1 receptors CXCR4 and CXCR7 attenuates acute pulmonary inflammation via the adenosine A2B-receptor on blood cells. Cell Death Dis. 2017, 8, e2832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagata, K.; Browne, K.D.; Suto, Y.; Kumasaka, K.; Cognetti, J.; Johnson, V.E.; Marks, J.; Smith, D.H.; Pascual, J.L. Early heparin administration after traumatic brain injury: Prolonged cognitive recovery associated with reduced cerebral edema and neutrophil sequestration. J. Trauma Acute Care Surg. 2017, 83, 406–412. [Google Scholar] [CrossRef]
- Ali, R.A.; Gandhi, A.A.; Meng, H.; Yalavarthi, S.; Vreede, A.P.; Estes, S.K.; Palmer, O.R.; Bockenstedt, P.L.; Pinsky, D.J.; Greve, J.M.; et al. Adenosine receptor agonism protects against NETosis and thrombosis in antiphospholipid syndrome. Nat. Commun. 2019, 10, 1916. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ying, L.; Jin, K.K.; Nan, Y.; Hu, S.; Wu, X.; Qi, R.; Luo, X.; Wang, L. Adenosine A(2A) receptor activation reverses hypoxiainduced rat pulmonary artery smooth muscle cell proliferation via cyclic AMPmediated inhibition of the SDF1CXC4 signaling pathway. Int. J. Mol. Med. 2018, 42, 607–614. [Google Scholar] [CrossRef]
- Filippi, M.D. Neutrophil transendothelial migration: Updates and new perspectives. Blood 2019, 133, 2149–2158. [Google Scholar] [CrossRef]
- Konrad, F.M.; Wohlert, J.; Gamper-Tsigaras, J.; Ngamsri, K.C.; Reutershan, J. How Adhesion Molecule Patterns Change While Neutrophils Traffic through the Lung during Inflammation. Mediat. Inflamm. 2019, 2019, 1208086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amalakuhan, B.; Habib, S.A.; Mangat, M.; Reyes, L.F.; Rodriguez, A.H.; Hinojosa, C.A.; Soni, N.J.; Gilley, R.P.; Bustamante, C.A.; Anzueto, A.; et al. Endothelial adhesion molecules and multiple organ failure in patients with severe sepsis. Cytokine 2016, 88, 267–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyun, Y.M.; Choe, Y.H.; Park, S.A.; Kim, M. LFA-1 (CD11a/CD18) and Mac-1 (CD11b/CD18) distinctly regulate neutrophil extravasation through hotspots I and II. Exp. Mol. Med. 2019, 51, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrero, R.; Prados, L.; Ferruelo, A.; Puig, F.; Pandolfi, R.; Guillamat-Prats, R.; Moreno, L.; Matute-Bello, G.; Artigas, A.; Esteban, A.; et al. Fas activation alters tight junction proteins in acute lung injury. Thorax 2018, 74, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Konrad, F.M.; Witte, E.; Vollmer, I.; Stark, S.; Reutershan, J. Adenosine receptor A2b on hematopoietic cells mediates LPS-induced migration of PMNs into the lung interstitium. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 303, L425–L438. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.R.; Shaefi, S.; Otterbein, L.E. HO-1 and CD39: It Takes Two to Protect the Realm. Front. Immunol. 2019, 10, 1765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Wang, L.; Xing, Q.; Li, K.; Si, J.; Ma, X.; Mao, L. Sevoflurane inhibits ferroptosis: A new mechanism to explain its protective role against lipopolysaccharide-induced acute lung injury. Life Sci. 2021, 275, 119391. [Google Scholar] [CrossRef] [PubMed]
- Sima, L.J.; Ma, X.W. Effect of sevoflurane on hepatic ischemia-reperfusion injury in rats via JAK2-STAT3 pathway. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 1350–1356. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Yang, Y.; Li, Y.; Li, J.; Ma, Q.; Zhao, Y.; Wang, D. Effects of sevoflurane on pulmonary cytosolic phospholipase A(2) and clara cell secretory protein expressions in rabbits with one-lung ventilation-induced lung injury. Nan Fang Yi Ke Da Xue Xue Bao 2013, 33, 469–473. [Google Scholar] [PubMed]
- Herrmann, I.K.; Castellon, M.; Schwartz, D.E.; Hasler, M.; Urner, M.; Hu, G.; Minshall, R.D.; Beck-Schimmer, B. Intravenous application of a primary sevoflurane metabolite improves outcome in murine septic peritonitis: First results. PLoS ONE 2013, 8, e72057. [Google Scholar] [CrossRef] [Green Version]
- Kortekaas, K.A.; van der Baan, A.; Aarts, L.P.; Palmen, M.; Cobbaert, C.M.; Verhagen, J.C.; Engbers, F.H.; Klautz, R.J.; Lindeman, J.H. Cardiospecific sevoflurane treatment quenches inflammation but does not attenuate myocardial cell damage markers: A proof-of-concept study in patients undergoing mitral valve repair. Br. J. Anaesth. 2014, 112, 1005–1014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, H.; Zhou, D.; Lu, J.; Wu, C.; Zhu, Z. Effects of Pre-Cardiopulmonary Bypass Administration of Dexmedetomidine on Cardiac Injuries and the Inflammatory Response in Valve Replacement Surgery With a Sevoflurane Postconditioning Protocol: A Pilot Study. J. Cardiovasc. Pharmacol. 2019, 74, 91–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de la Gala, F.; Pineiro, P.; Reyes, A.; Vara, E.; Olmedilla, L.; Cruz, P.; Garutti, I. Postoperative pulmonary complications, pulmonary and systemic inflammatory responses after lung resection surgery with prolonged one-lung ventilation. Randomized controlled trial comparing intravenous and inhalational anaesthesia. Br. J. Anaesth. 2017, 119, 655–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.L.; Wang, D.; Zhang, G.Y.; Guo, X.L. Comparison of the myocardial protective effect of sevoflurane versus propofol in patients undergoing heart valve replacement surgery with cardiopulmonary bypass. BMC Anesthesiol. 2017, 17, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jabaudon, M.; Boucher, P.; Imhoff, E.; Chabanne, R.; Faure, J.S.; Roszyk, L.; Thibault, S.; Blondonnet, R.; Clairefond, G.; Guerin, R.; et al. Sevoflurane for Sedation in Acute Respiratory Distress Syndrome. A Randomized Controlled Pilot Study. Am. J. Respir. Crit. Care Med. 2017, 195, 792–800. [Google Scholar] [CrossRef] [PubMed]
- Zheng, B.; Zhan, Q.; Chen, J.; Xu, H.; He, Z. Sevoflurane pretreatment enhance HIF-2alpha expression in mice after renal ischemia/reperfusion injury. Int. J. Clin. Exp. Pathol. 2015, 8, 13114–13119. [Google Scholar] [PubMed]
- Kong, H.Y.; Zhu, S.M.; Wang, L.Q.; He, Y.; Xie, H.Y.; Zheng, S.S. Sevoflurane protects against acute kidney injury in a small-size liver transplantation model. Am. J. Nephrol. 2010, 32, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Lucchinetti, E.; Ambrosio, S.; Aguirre, J.; Herrmann, P.; Harter, L.; Keel, M.; Meier, T.; Zaugg, M. Sevoflurane inhalation at sedative concentrations provides endothelial protection against ischemia-reperfusion injury in humans. Anesthesiology 2007, 106, 262–268. [Google Scholar] [CrossRef] [PubMed]
- Jabaudon, M.; Audard, J.; Pereira, B.; Jaber, S.; Lefrant, J.Y.; Blondonnet, R.; Godet, T.; Futier, E.; Lambert, C.; Bazin, J.E.; et al. Early Changes over Time in the Radiographic Assessment of Lung Edema Score Are Associated with Survival in ARDS. Chest 2020, 158, 2394–2403. [Google Scholar] [CrossRef] [PubMed]
- Kellner, P.; Muller, M.; Piegeler, T.; Eugster, P.; Booy, C.; Schlapfer, M.; Beck-Schimmer, B. Sevoflurane Abolishes Oxygenation Impairment in a Long-Term Rat Model of Acute Lung Injury. Anesth. Analg. 2017, 124, 194–203. [Google Scholar] [CrossRef] [Green Version]
- Beck-Schimmer, B.; Restin, T.; Muroi, C.; Roth Z’Graggen, B.; Keller, E.; Schlapfer, M. Sevoflurane sedation attenuates early cerebral oedema formation through stabilisation of the adherens junction protein beta catenin in a model of subarachnoid haemorrhage: A randomised animal study. Eur. J. Anaesthesiol. 2020, 37, 402–412. [Google Scholar] [CrossRef] [PubMed]
- Eltzschig, H.K. Extracellular adenosine signaling in molecular medicine. J. Mol. Med. 2013, 91, 141–146. [Google Scholar] [CrossRef]
- Eckle, T.; Fullbier, L.; Wehrmann, M.; Khoury, J.; Mittelbronn, M.; Ibla, J.; Rosenberger, P.; Eltzschig, H.K. Identification of ectonucleotidases CD39 and CD73 in innate protection during acute lung injury. J. Immunol. 2007, 178, 8127–8137. [Google Scholar] [CrossRef]
- Kiers, D.; Wielockx, B.; Peters, E.; van Eijk, L.T.; Gerretsen, J.; John, A.; Janssen, E.; Groeneveld, R.; Peters, M.; Damen, L.; et al. Short-Term Hypoxia Dampens Inflammation in vivo via Enhanced Adenosine Release and Adenosine 2B Receptor Stimulation. EBioMedicine 2018, 33, 144–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanei, Y.; Hanon, S.; Van-Tosh, A.; Schweitzer, P. Adenosine-induced atrial fibrillation during pharmacologic stress testing: Report of eight cases and review of the literature. Int. J. Cardiol. 2008, 129, e15–e17. [Google Scholar] [CrossRef] [PubMed]
- Ohta, A.; Sitkovsky, M. Role of G-protein-coupled adenosine receptors in downregulation of inflammation and protection from tissue damage. Nature 2001, 414, 916–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobson, K.A.; Tosh, D.K.; Jain, S.; Gao, Z.G. Historical and Current Adenosine Receptor Agonists in Preclinical and Clinical Development. Front. Cell. Neurosci. 2019, 13, 124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okuno, T.; Koutsogiannaki, S.; Hou, L.; Bu, W.; Ohto, U.; Eckenhoff, R.G.; Yokomizo, T.; Yuki, K. Volatile anesthetics isoflurane and sevoflurane directly target and attenuate Toll-like receptor 4 system. FASEB J. 2019, 33, 14528–14541. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Zhang, X.; Tian, J.; Liu, G.; Li, X.; Shen, D. Sevoflurane alleviates LPSinduced acute lung injury via the microRNA27a3p/TLR4/MyD88/NFkappaB signaling pathway. Int. J. Mol. Med. 2019, 44, 479–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryter, S.W. Significance of Heme and Heme Degradation in the Pathogenesis of Acute Lung and Inflammatory Disorders. Int. J. Mol. Sci. 2021, 22, 5509. [Google Scholar] [CrossRef] [PubMed]
- Konrad, F.M.; Braun, S.; Ngamsri, K.C.; Vollmer, I.; Reutershan, J. Heme oxygenase-1 attenuates acute pulmonary inflammation by decreasing the release of segmented neutrophils from the bone marrow. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 307, L707–L717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Vijayan, V.; Jang, M.S.; Thorenz, A.; Greite, R.; Rong, S.; Chen, R.; Shushakova, N.; Tudorache, I.; Derlin, K.; et al. Labile Heme Aggravates Renal Inflammation and Complement Activation After Ischemia Reperfusion Injury. Front. Immunol. 2019, 10, 2975. [Google Scholar] [CrossRef] [PubMed]
- Han, S.J.; Kim, M.; D’Agati, V.D.; Lee, H.T. 6-Shogaol protects against ischemic acute kidney injury by modulating NF-kappaB and heme oxygenase-1 pathways. Am. J. Physiol. Renal. Physiol. 2019, 317, F743–F756. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.W.; Liu, X.; Macias, A.A.; Baron, R.M.; Perrella, M.A. Heme oxygenase-1-derived carbon monoxide enhances the host defense response to microbial sepsis in mice. J. Clin. Investig. 2008, 118, 239–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwasashi, H.; Suzuki, M.; Unno, M.; Utiyama, T.; Oikawa, M.; Kondo, N.; Matsuno, S. Inhibition of heme oxygenase ameliorates sepsis-induced liver dysfunction in rats. Surg. Today 2003, 33, 30–38. [Google Scholar] [CrossRef]
- Bauer, I.; Raupach, A. The Role of Heme Oxygenase-1 in Remote Ischemic and Anesthetic Organ Conditioning. Antioxidants 2019, 8, 403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faller, S.; Strosing, K.M.; Ryter, S.W.; Buerkle, H.; Loop, T.; Schmidt, R.; Hoetzel, A. The volatile anesthetic isoflurane prevents ventilator-induced lung injury via phosphoinositide 3-kinase/Akt signaling in mice. Anesth. Analg. 2012, 114, 747–756. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.Q.; Lin, L.N.; Wang, L.R.; Jin, L.D. Sevoflurane attenuates pulmonary inflammation and ventilator-induced lung injury by upregulation of HO-1 mRNA expression in mice. Int. J. Nanomed. 2013, 6, 1075–1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ngamsri, K.-C.; Fuhr, A.; Schindler, K.; Simelitidis, M.; Hagen, M.; Zhang, Y.; Gamper-Tsigaras, J.; Konrad, F.M. Sevoflurane Dampens Acute Pulmonary Inflammation via the Adenosine Receptor A2B and Heme Oxygenase-1. Cells 2022, 11, 1094. https://doi.org/10.3390/cells11071094
Ngamsri K-C, Fuhr A, Schindler K, Simelitidis M, Hagen M, Zhang Y, Gamper-Tsigaras J, Konrad FM. Sevoflurane Dampens Acute Pulmonary Inflammation via the Adenosine Receptor A2B and Heme Oxygenase-1. Cells. 2022; 11(7):1094. https://doi.org/10.3390/cells11071094
Chicago/Turabian StyleNgamsri, Kristian-Christos, Anika Fuhr, Katharina Schindler, Mariana Simelitidis, Michelle Hagen, Yi Zhang, Jutta Gamper-Tsigaras, and Franziska M. Konrad. 2022. "Sevoflurane Dampens Acute Pulmonary Inflammation via the Adenosine Receptor A2B and Heme Oxygenase-1" Cells 11, no. 7: 1094. https://doi.org/10.3390/cells11071094